

Abstract
The N-terminal (1–83) fragment of the major constituent of plasma high-density lipoprotein, apolipoprotein A-I (apoA-I), strongly tends to form amyloid fibrils, leading to systemic amyloidosis. Here, using a series of deletion variants, we examined the roles of two major amyloidogenic segments (residues 14–22 and 50–58) in the aggregation and fibril formation of an amyloidogenic G26R variant of the apoA-I 1–83 fragment (apoA-I 1–83/G26R). Thioflavin T fluorescence assays and atomic force microscopy revealed that elimination of residues 14–22 completely inhibits fibril formation of apoA-I 1–83/G26R, whereas Δ32–40 and Δ50–58 variants formed fibrils with markedly reduced nucleation and fibril growth rates. CD measurements revealed structural transitions from random coil to β-sheet structures in all deletion variants except for the Δ14–22 variant, indicating that residues 14–22 are critical for the β-transition and fibril formation. Thermodynamic analysis of the kinetics of fibril formation by apoA-I 1–83/G26R indicated that both nucleation and fibril growth are enthalpically unfavorable, whereas entropically, nucleation is favorable, but fibril growth is unfavorable. Interestingly, the nucleation of the Δ50–58 variant was entropically unfavorable, indicating that residues 50–58 entropically promote the nucleation step in fibril formation of apoA-I 1–83/G26R. Moreover, a residue-level structural investigation of apoA-I 1–83/G26R fibrils with site-specific pyrene labeling indicated that the two amyloidogenic segments are in close proximity to form an amyloid core structure, whereas the N- and C-terminal tail regions are excluded from the amyloid core. These results provide critical insights into the aggregation mechanism and fibril structure of the amyloidogenic N-terminal fragment of apoA-I.
Keywords: amyloid, fibril, apolipoprotein, protein structure, protein aggregation, activation energy, apolipoprotein A-I, G26R variant, hereditary systemic amyloidosis, pyrene labeling
Introduction
Apolipoprotein (apoA-I)3 is the major structural and functional constituent of plasma high-density lipoprotein (HDL) that plays a critical role in the formation and metabolism of HDL particles (1, 2). Many naturally occurring mutations in human apoA-I are associated with reduced plasma HDL levels and hereditary systemic amyloidosis (3). To date, ∼20 naturally occurring mutations in human apoA-I associated with familial amyloid polyneuropathy have been reported (4, 5) in which the majority of the amyloidogenic mutations are clustered in two regions of the N-terminal residues 26–90 and 154–178 (4, 6, 7). Hereditary apoA-I amyloidosis is characterized by deposition of the N-terminal 80–100-residue fragments of the variant protein as amyloid fibrils in peripheral organs such as heart, liver, kidneys, or gastrointestinal tract, causing organ damage (6, 8, 9). It has been hypothesized that the specific amyloidogenic mutations perturb the native protein structure, increasing susceptibility to proteolysis and thereby releasing the N-terminal amyloidogenic fragment (4, 10). The molecular basis for the onset and development of apoA-I systemic amyloidosis is largely unknown.
Sequence-based analyses of N-terminal residues 1–100 in apoA-I predict that there are two major aggregation-prone segments (residues 14–22 and 50–58) together with a minor segment (residues 69–72) in which the rank order of aggregation propensity is residues 14–22 > residues 50–58 > residues 69–72 (10, 11). In agreement with this prediction, the N-terminal 1–83 or 1–93 fragments of apoA-I were shown to have a strong propensity to form amyloid fibrils (11, 12). Studies of synthetic apoA-I fragment peptides demonstrated that the peptides containing either the first (residues 14–22) or the second (residues 50–58) amyloidogenic segment have the ability to form amyloid-like fibrils with β-transition, whereas the peptide only containing residues 69–72 does not form fibrils at neutral pH (13, 14). Interestingly, because the two major amyloidogenic segments (residues 14–22 and 50–58) overlap with the hydrophobic α-helix–forming regions upon lipid binding (15, 16), lipid membrane environments significantly affect the fibril-forming properties of the N-terminal fragments or peptides of apoA-I (17–19).
We previously reported that the Iowa (G26R) point mutation, the first and most common amyloidogenic mutation found in apoA-I (20, 21), greatly facilitates fibril formation by the N-terminal 1–83 fragment of apoA-I in solution (12) as well as on lipid membranes (18, 19). Secondary structure examinations of the G26R variant of full-length apoA-I by EPR spectroscopy (21) and hydrogen–deuterium exchange MS (22) demonstrated that the G26R mutation induces widespread α-helix destabilization in the N-terminal helix bundle domain and promotes a transition of residues 27–56 to β-strand–rich structure. However, structural information on amyloid fibrils formed by the N-terminal fragment of apoA-I G26R variant is lacking.
In the present study, we asked how each amyloidogenic segment in apoA-I modulates aggregation and fibril formation of the N-terminal 1–83 fragment of the G26R variant using a series of deletion variants (Δ14–22, Δ32–40, Δ50–58, and Δ68–76) that lack different amyloidogenic regions along the molecule. We also examined the intermolecular contacts of apoA-I 1–83/G26R fibrils at a residue level by employing site-directed cysteine mutagenesis and fluorescence labeling to gain structural insights into amyloid fibrils formed by the N-terminal fragment of apoA-I G26R.
Results
Design of deletion or cysteine-substituted variants of apoA-I 1–83/G26R
We designed two types of mutations in the N-terminal 1–83 fragment of apoA-I G26R variant: deletion mutations that remove nine amino acid residues in the amyloidogenic regions of the protein and single amino acid mutations that introduce cysteine substitutions for labeling by pyrene maleimide at different positions along the molecule (Fig. 1A). The deletion variants, Δ14–22, Δ50–58, and Δ68–76, lack the highly amyloidogenic segments of residues 14–22, 50–58, and 69–72, respectively (10), whereas the Δ32–40 variant lacks the putative loop region between two β-strands from residues 14–31 to 41–58 (23) (Fig. 1B). In the cysteine variants, cysteine residues were introduced into the highly amyloidogenic segments together with the N- and C-terminal tail regions that have low amyloid propensity (Fig. 1B). The Cys-substituted positions were selectively labeled with pyrene maleimide to probe the polarity and intermolecular proximity of different segments of apoA-I 1–83/G26R fragment in the fibril form. We note that cysteine mutations and pyrene labeling did not significantly inhibit the fibril-forming ability of the protein, although several mutants appeared to have somewhat enhanced fibril-forming propensity (Fig. S1).
Figure 1.
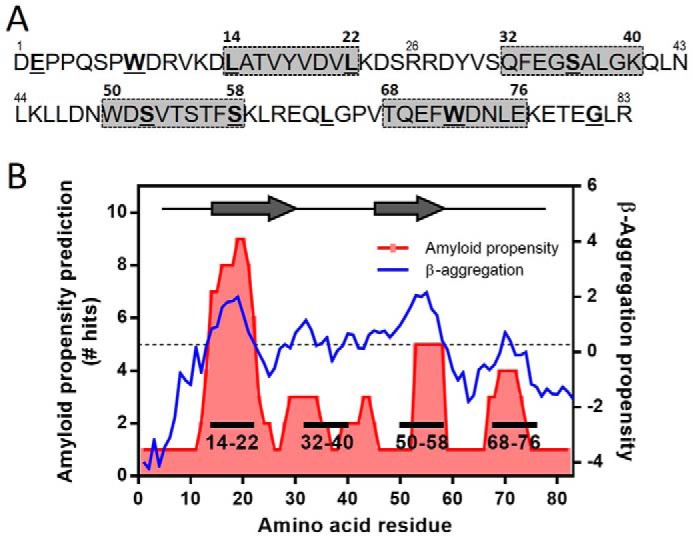
Design of deletion and cysteine variants of apoA-I 1–83/G26R. A, primary sequence of the N-terminal residues 1–83 of human apoA-I G26R variant. Deleted regions in the deletion variants are highlighted with shaded boxes. The amino acids substituted with cysteine for pyrene labeling are underlined and shown in bold. B, amyloid propensity prediction and β-aggregation propensity of the N-terminal residues 1–83 of apoA-I G26R variant. Amyloid propensity prediction was generated using the consensus algorithm AmylPred2 (54). β-Aggregation propensity was calculated with the Zyggregator method (55). Residues 14–31 (21) and 46–59 (13) predicted to have high amyloid-forming propensity are shown as arrows.
Effect of deletion on fibril-forming property of apoA-I 1–83/G26R
We first examined the fibril-forming propensities of the deletion variants of apoA-I 1–83/G26R using the amyloid-sensitive fluorescent dye thioflavin T (ThT). As reported previously (12), apoA-I 1–83/G26R exhibited a large increase in ThT fluorescence at neutral pH (Fig. 2A). In contrast, there was no increase in ThT fluorescence for the Δ14–22 variant, indicating that residues 14–22 are crucial for fibril formation of apoA-I 1–83/G26R. Significantly delayed increases in ThT fluorescence for the Δ32–40 and Δ50–58 variants suggest that the segments spanning residues 32–40 as well as 50–58 are important for nucleation in fibril formation of the 1–83/G26R variant. Interestingly, the Δ68–76 variant exhibited greatly enhanced ThT fluorescence intensity (Fig. 2A, inset), suggesting the possibility that residues 68–76 inhibit the fibril-forming ability of apoA-I 1–83/G26R. However, precipitation analyses of fibrils formed by the 1–83/G26R deletion variants indicated that the fibril-forming ability of the Δ68–76 variant is somewhat less than that of the other apoA-I 1–83/G26R variants except for Δ14–22 (Fig. S2). Such an inhibited or delayed increase in ThT fluorescence for the Δ14–22, Δ32–40, or Δ50–58 variant and the enhanced ThT fluorescence for the Δ68–76 variant were also observed at different protein concentrations (Fig. S3).
Figure 2.
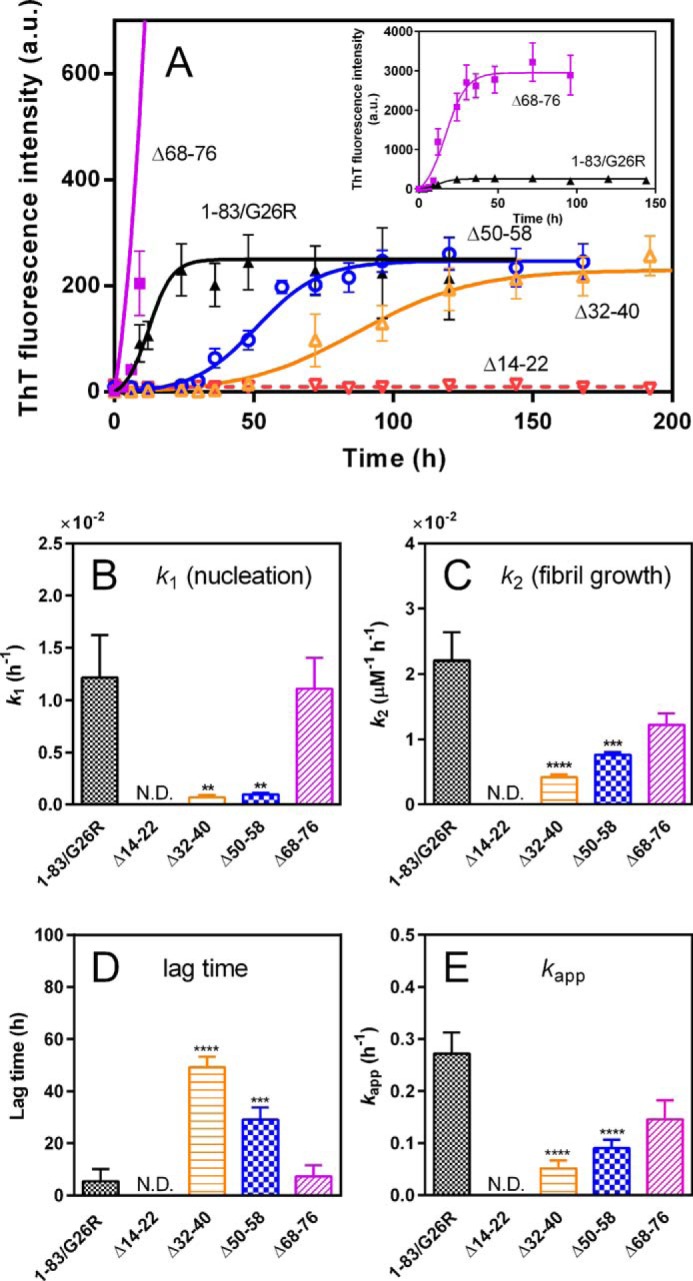
Formation of amyloid-like structure was monitored by ThT fluorescence for apoA-I 1–83/G26R and deletion variants at 37 °C. A, 1–83/G26R (▴), 1–83/G26R Δ14–22 (▿), 1–83/G26R Δ32–40 (▵), 1–83/G26R Δ50–58 (○), and 1–83/G26R Δ68–76 (■). The inset shows comparison of 1–83/G26R and 1–83/G26R Δ68–76. ApoA-I 1–83 variants were incubated at 37 °C with agitation on an orbital rotator in the presence of 10 μm ThT. The data were from at least three independent experiments. Error bars represent S.E. The solid lines are the fitted curves by the Finke–Watzky two-step model. Protein concentration was 0.1 mg/ml. a.u., arbitrary units. B and C, comparison of rate constants of nucleation (k1) and fibril growth (k2) for fibril formation of 1–83/G26R variants according to Finke–Watzky Equation 2. D and E, comparison of lag time (D) and apparent rate constant (E) for the growth of fibrils of apoA-I 1–83/G26R variants according to sigmoidal Equation 1. Error bars represent S.E. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 versus 1–83/G26R. N.D., not determined.
The kinetics of the ThT fluorescence increase were analyzed by the Finke–Watzky two-step model of a homogeneous nucleation followed by autocatalytic heterogeneous fibril growth, which has been applied to a broad range of protein aggregation kinetics (24–27). Comparison of rate constants of the nucleation (k1) and fibril growth (k2) for fibril formation by the deletion variants (Fig. 2, B and C) clearly demonstrates that the Δ32–40 and Δ50–58 variants have significantly decreased rate constants of both nucleation and fibril growth, whereas the Δ68–76 exhibits similar kinetic parameters compared with 1–83/G26R. Kinetic parameters obtained by the empirical sigmoidal equation (Fig. 2, D and E) provide similar conclusions: the Δ32–40 and Δ50–58 variants significantly increase the lag time and decrease the apparent rate constant for fibril growth. We note that the Finke–Watzky model is based on two pseudoelementary reaction steps of nucleation and fibril growth without further considerations on the nuclei size and fibril propagation mechanisms such as secondary pathways (27), although it has been proposed that the secondary nucleation process occurs during fibril formation of β-amyloid peptides and α-synuclein (28–30).
We next performed circular dichroism (CD) measurements of the 1–83/G26R deletion variants to determine the secondary structural changes during incubation. As shown in Fig. 3, all spectra of the deletion variants except for Δ14–22 displayed a single minimum at around 216 nm after incubation for 120 h, implying conversion from random coil to β-sheet–rich structure. Consistent with the delayed increase in ThT fluorescence (Fig. 2A), such apparent transition to β-structure was not observed for the Δ32–40 and Δ50–58 variants at shorter incubation for 60 h (Fig. S4A). No secondary structural change occurred for the Δ14–22 variant during incubation.
Figure 3.

Far-UV CD spectra of apoA-I 1–83/G26R and deletion variants before (0 h; dashed line) and after incubation for 120 h (solid line). A, 1–83/G26R; B, 1–83/G26R Δ14–22; C, 1–83/G26R Δ32–40; D, 1–83/G26R Δ50–58; E, 1–83/G26R Δ68–76. Protein concentration was 50 μg/ml. deg, degrees.
Fig. 4 shows atomic force microscopy (AFM) and transmission EM (TEM) images of the 1–83/G26R deletion variants after 120-h incubation. AFM images (Fig. 4A) show that the 1–83/G26R as well as Δ32–40, Δ50–58, and Δ68–76 variants formed straight fibrils, whereas Δ14–22 did not form apparent fibrils but rather formed small spherical aggregates. TEM observations also confirm the formation of straight fibrils by the 1–83/G26R as well as the deletion variants except for the Δ14–22 variant, in which it rarely formed fibrils (Fig. 4B). In contrast, apparent straight fibrils were not observed for the Δ32–40 and Δ50–58 variants at incubation for 60 h (Fig. S4B). These results indicate that the highly amyloidogenic segment spanning residues 14–22 is necessary for the formation of amyloid fibril structure by apoA-I 1–83/G26R, whereas the segments of residues 32–40 and 50–58 are not necessary for fibril formation but modulate the kinetics of nucleation and fibril formation. Consistently, the fibrils formed by the deletion variants after 120-h incubation exhibited stability against urea denaturation similar to that of 1–83/G26R fibrils (Fig. S5). In addition, the fibrils formed by the deletion variants induced strong cytotoxicity with HEK293 cells similarly to 1–83/G26R fibrils, whereas the Δ14–22 exhibited reduced cytotoxicity (Fig. S6). This indicates that the formation of the fibril structure is critical to the cytotoxicity of apoA-I 1–83/G26R variants (12).
Figure 4.

AFM (A) and TEM (B) images of apoA-I 1–83/G26R and deletion variants after 120-h incubation. Scale bars represent 1 μm (AFM) and 0.5 μm or 100 nm (TEM), respectively.
Thermodynamic analysis of fibril formation by apoA-I 1–83/G26R
To understand the thermodynamic aspects of the aggregation of apoA-I 1–83/G26R into amyloid fibrils, we explored the effect of temperature on the kinetics of formation of amyloid-like fibrils. We note that CD measurements confirmed that there are no secondary structural changes across the temperature range of 25–42 °C (data not shown). Fig. 5A shows the time course of ThT fluorescence intensity with the fitted curves by the Finke–Watzky two-step model at different temperatures. Comparison of rate constants of the nucleation (k1) and fibril growth (k2) at each temperature indicates that both rate constants greatly increase with temperature (Fig. 5B). From the linear relationship of ln(k1/T) or ln(k2/T) with 1/T based on the Eyring equation (Fig. 5C), the activation enthalpy ΔH* and entropy ΔS* for the nucleation and fibril growth steps in fibril formation by apoA-I 1–83/G26R were obtained. Table 1 summarizes the ΔH* and ΔS* values and the activation Gibbs free energy ΔG*. These parameters demonstrated that although the activation free energy barriers for nucleation and fibril growth are similar, the contributions of activation enthalpy and entropy to the free energy barrier are quite different: the nucleation step is enthalpically unfavorable but entropically favorable, whereas fibril growth is both enthalpically and entropically unfavorable. That is, the free energy barrier for nucleation is entirely enthalpic, whereas that for fibril growth consists of both enthalpic and entropic barriers.
Figure 5.

Thermodynamic analysis of amyloid fibril formation of apoA-I 1-83/G26R variants. A, kinetics of formation of amyloid-like structure monitored by ThT fluorescence for apoA-I 1–83/G26R at different temperatures. The data were from at least four independent experiments. Error bars represent S.E. The dashed lines are the fitted curves by the Finke–Watzky two-step model. Protein concentration was 50 μg/ml. a.u., arbitrary units. B, comparison of rate constants of nucleation (k1) and fibril growth (k2) for fibril formation of 1–83/G26R. Error bars represent S.E. C, Eyring plots of rate constants of k1 and k2 for fibril formation of 1–83/G26R. D, Eyring plots of rate constants k1 and k2 for fibril formation of 1–83/G26R Δ50–58.
Table 1.
Thermodynamic parameters for nucleation and fibril growth in fibril formation of apoA-I 1–83/G26R and deletion variants
The data were from at least three independent experiments.
| ΔH*a | ΔS*a | ΔG*b | |
|---|---|---|---|
| kJ/mol | J/mol K | kJ/mol | |
| ApoA-I 1–83/G26R | |||
| Nucleation (k1) | 126 ± 20 | 55 ± 14 | 109 |
| Fibril growth (k2) | 65 ± 6 | −122 ± 33 | 103 |
| Δ32–40 | |||
| Nucleation (k1) | 77 ± 12 | −122 ± 64 | 115 |
| Fibril growth (k2) | 87 ± 22 | −47 ± 14 | 102 |
| Δ50–58 | |||
| Nucleation (k1) | 22 ± 14 | −281 ± 160 | 110 |
| Fibril growth (k2) | 98 ± 6 | −20 ± 2 | 104 |
| Δ68–76 | |||
| Nucleation (k1) | 137 ± 16 | 70 ± 14 | 115 |
| Fibril growth (k2) | 47 ± 10 | −170 ± 45 | 100 |
a ΔH* and ΔS* were obtained from the slope and y-intercept of the linear plot, respectively, according to Equation 3.
b ΔG* was calculated from ΔH* and ΔS* according to ΔG* = ΔH* − TΔS* at 37 °C.
We also performed thermodynamic analysis of fibril formation by the deletion variants to gain insight into the role of each segment in fibril formation by apoA-I 1–83/G26R (Figs. 5D and S7). The Δ14–22 variant did not form fibrils at any temperatures examined (Fig. S7A). In contrast, the Δ50–58 variant exhibited temperature-dependent increases in ThT fluorescence (Fig. S7C), and Eyring plots of rate constants of k1 and k2 (Fig. 5D) gave the thermodynamic parameters for fibril formation of the Δ50–58 variant (Table 1). Despite similar activation free energy barriers between apoA-I 1–83/G26R and the Δ50–58 variant, a large reduction in the unfavorable activation enthalpy and the concomitant unfavorable activation entropy for nucleation in fibril formation of Δ50–58 are observed, in sharp contrast to the favorable entropy of nucleation for 1–83/G26R. For fibril growth of the Δ50–58 variant, the activation enthalpy and entropy are both unfavorable. These results indicate that residues 50–58 entropically drive the nucleation in fibril formation by apoA-I 1–83/G26R. Similar to Δ50–58, the Δ32–40 variant exhibits the unfavorable activation enthalpy and entropy for nucleation in which the reduction in the unfavorable activation enthalpy from 1–83/G26R is much smaller than that for Δ50–58 (Table 1). Regarding the Δ68–76 variant, the thermodynamic parameters for nucleation are similar, but the unfavorable activation enthalpy of fibril growth is significantly reduced compared with 1–83/G26R (Table 1). This indicates that residues 68–76 inhibit fibril growth of apoA-I 1–83/G26R by increasing the unfavorable activation enthalpy.
Determination of amyloid core regions in apoA-I 1–83/G26R fibrils
We next performed fluorescence measurements of pyrene-labeled apoA-I 1–83/G26R variants to map intermolecular proximities of different segments in the fibril form. Pyrene fluorescence emission spectra display unique features that give information on the spatial proximity and polarity of the environment. When two pyrene rings are within 10 Å of each other, a broad and red-shifted emission peak appears generally at around 470 nm, attributed to formation of an excited-state dimer, excimer (31). Thus, the excimer ratio (ratio of fluorescence intensity of excimer at 470 nm to that of monomer at 385 nm) indicates the extent of excimer formation and, therefore, the intermolecular proximity of pyrene-labeled positions of the protein in oligomers and fibrils (32–36). In addition, we also determined the pyrene scale as the intensity ratio between bands at 375 and 385 nm to evaluate the local hydrophobicity around the probe at the labeled positions (33, 37).
Fig. 6, A and B, show typical fluorescence emission spectra of pyrene-labeled apoA-I 1–83/G26R variants before and after incubation. Before incubation, no excimer peaks at around 470 nm were observed for all pyrene-labeled variants (all fluorescence emission spectra of pyrene-labeled variants are shown in Fig. S8). In fibrils, a large excimer peak appeared when the label was at position 22, whereas only a small excimer peak was seen with the label at position 81. Fig. 6C shows the profile of excimer ratios as well as the pyrene scale in fibrils as a function of residue number. Positions 22 and 52 exhibit high excimer ratios, consistent with two regions around the highly amyloidogenic segments (residues 14–22 and 50–58) having strong intermolecular contacts. In contrast, low excimer ratios at positions 2, 8, 72, and 81 suggest sequestration of the N- and C-terminal tail regions from intermolecular contacts. The relatively low excimer ratio at position 36 agrees with the prediction that residues 32–40 form a loop region between two β-strands (23). We note that pyrene scale values for all labeled positions are similarly low (Fig. 6C), indicating that pyrene molecules at all labeled positions in fibrils are located in hydrophobic environments.
Figure 6.

Mapping of amyloid core regions in apoA-I 1–83/G26R fibrils. A and B, fluorescence emission spectra of pyrene (Py)-labeled apoA-I 1–83/G26R variants before (0 h; dashed line) and after incubation for 120 h (solid line). A, 1–83/L22C-Py/G26R; B, 1–83/G26R/G81C-Py. Protein concentration was 10 μg/ml. C, excimer ratio profile for pyrene-labeled apoA-I 1–83/G26R variants in amyloid fibrils. The ratio of excimer fluorescence to monomer fluorescence is plotted. The pyrene scale as the ratio of pyrene fluorescence intensity at 375 nm to that at 385 nm is also plotted. Residues 14–31 and 46–59 predicted to have high amyloid-forming propensity are shown as arrows. Error bars represent S.E. a.u., arbitrary units.
Discussion
Sequence-based predictions of the β-aggregation (11) and amyloid propensity (10) indicate that there are two major aggregation-prone segments of residues 14–22 and 50–58 in the N-terminal 1–83 residues of apoA-I. Based on the secondary structure examination of the aggregates of apoA-I G26R variant (21), together with a finding that a peptide comprising residues 46–59 of apoA-I forms amyloid-like fibrils (13), it was considered that two segments spanning residues 14–31 and 46–59 in apoA-I 1–83/G26R are highly amyloidogenic regions (Fig. 1B). Indeed, we previously demonstrated that synthetic apoA-I fragment peptides containing these two amyloidogenic segments have the ability to form amyloid-like fibrils (14, 18, 38). However, details on the roles of each amyloidogenic segment in amyloid fibril formation by apoA-I 1–83/G26R are unknown.
The present study demonstrated that the deletion of residues 14–22 completely inhibits β-transition and fibril formation of apoA-I 1–83/G26R, whereas the deletion of residues 50–58 markedly decreases the rate constants of nucleation and fibril elongation (Fig. 2, B and C). This information clearly indicates that the first amyloidogenic segment containing residues 14–22 is crucial for fibril formation of apoA-I 1–83/G26R, whereas the second amyloidogenic segment containing residues 50–58 is largely involved in the nucleation step of fibril formation. Indeed, a Y18P mutation that impairs formation of β-sheet structure in residues 14–22 strongly inhibited fibril formation by apoA-I 1–83/G26R (18). Interestingly, the effects of deletions on the fibril-forming ability of apoA-I 1–83/G26R bound to lipid membranes are quite different from those in solution (Fig. S9). Because α-helix formation of the amyloidogenic segments upon lipid binding strongly inhibits β-transition and fibril formation by the N-terminal fragment of apoA-I (18, 19), it is likely that the second amyloidogenic segment containing residues 50–58 is not available for fibril formation of apoA-I 1–83/G26R on the membrane surface (19). The inhibitory effect of the deletion of residues 32–40 on the kinetics of nucleation and fibril growth (Fig. 2) is possibly because the lack of residues 32–40 may alter the appropriate intramolecular interaction between two putative β-strands consisting of highly amyloidogenic residues of 14–22 and 50–58 in fibril formation. Deletion of residues 68–76 that are rich in negatively charged amino acids has small effects on the kinetics of fibril formation but causes great increases in ThT fluorescence intensity (Fig. 2). This indicates that the C-terminal tail region that is rich in negatively charged amino acids has inhibitory effects on aggregation and fibril formation of apoA-I 1–83/G26R as observed for α-synuclein (39).
The thermodynamic analyses of ThT fluorescence kinetics (Figs. 5 and S7) give further insights into the molecular mechanism of aggregation and fibril formation of apoA-I 1–83/G26R and deletion variants. In fibril formation of apoA-I 1–83/G26R, the nucleation process is enthalpically unfavorable but entropically favorable, whereas the fibril elongation process is enthalpically and entropically unfavorable (Table 1). The unfavorable activation enthalpies of nucleation and fibril growth are generally observed for many amyloidogenic proteins (40–43), likely due to the net unfavorable formation and breakage of many weak interactions necessary to reach the transition state (42). In contrast, the favorable activation entropies of nucleation observed for 1–83/G26R (Table 1) as well as a fungal prion-forming domain (40) and β-amyloid 42 (43) peptides are thought to come from the desolvation of hydrophobic regions of the protein/peptide molecule in the transition state (42, 43). The unfavorable activation entropy of fibril growth for 1–83/G26R may arise because resolvation of the fibril surface dominates over desolvation of the existing nucleus (43).
The activation energy parameters for nucleation in fibril formation of the Δ50–58 variant are quite different from those of 1–83/G26R (Table 1). Based on the X-ray crystal structure of C-terminally truncated human apoA-I Δ185–243 showing that residues 44–55 are in an extended nonhelical conformation (44), it was proposed that residues 44–55 provide a template for nucleation of intermolecular β-aggregation (7). In this regard, it is possible that the favorable activation entropy of nucleation of apoA-I 1–83/G26R comes from desolvation of the exposed segment in residues 44–55 in the nucleation process. Consistent with this idea, deletion of residues 50–58 causes the activation entropy of nucleation to be highly negative (Table 1), indicating that the segment in residues 50–58 entropically promotes nucleation in fibril formation of the 1–83/G26R molecule. In addition, a large reduction in the unfavorable activation enthalpy of nucleation observed for the Δ50–58 variant (Table 1) suggests that desolvation of the segment in residues 50–58 seems to contribute to the unfavorable activation enthalpy in forming a nucleus (42).
The Δ32–40 variant also exhibits the unfavorable activation enthalpy and entropy for nucleation similar to the case of Δ50–58 variant (Table 1). However, the reduction in the unfavorable activation enthalpy from 1–83/G26R is much smaller than that for Δ50–58. This may suggest that the deletion of residues 32–40 inhibits the nucleation step, possibly through altering the appropriate intermolecular interaction of the second amyloidogenic segment containing residues 50–58. In contrast, the thermodynamic parameters of nucleation for the Δ68–78 variant are similar to those of 1–83/G26R, but the unfavorable activation enthalpy of fibril growth is significantly reduced (Table 1). This indicates that residues 68–76 enthalpically inhibit fibril growth of apoA-I 1–83/G26R and may explain why the Δ68–78 variant exhibits the great increases in ThT fluorescence intensity (Fig. 2). That is, the lower unfavorable activation enthalpy of fibril growth is likely to decrease the critical concentration at which fibrils appear from nuclei (45, 46), leading to the formation of a large number of fibrils. Indeed, the great enhancement of ThT fluorescence was also observed for 1–83/G26R and the Δ50–58 variant with increasing concentration of protein (Fig. S3).
The use of pyrene to probe the spatial proximity (<10 Å) between pairs of labeled residues in the proteins has been successfully employed to gain structural details of oligomers and fibrils formed by a yeast prion (32), α-synuclein (35, 36), and nondisease-related HypF-N protein (34). Based on the FRET study, we previously proposed a hypothetical model of fibril structure of the N-terminal fragment of apoA-I G26R variant in which β-strands from residues 14–31 and 41–58 form a self-complementary steric zipper stabilized by van der Waals and hydrophobic interactions (23). In addition, solid-state NMR analysis of aggregates formed by methionine-oxidized full-length apoA-I revealed that several amino acids within residues 13–22 and 50–59 are detected as β-sheet structure (47). The results of pyrene excimer fluorescence of apoA-I 1–83/G26R fibrils (Fig. 6) demonstrate for the first time that the two segments with high amyloid-forming propensity (residues 14–22 and 50–58) are indeed in close proximity to form amyloid core structure, whereas the N-terminal (residues 1–10) and C-terminal (residues 70–83) tail regions as well as the central (around residues 32–40) region are likely to be excluded from the amyloid core. Because C-terminal residues 70–83 (EFWDNLEKETEGLR) are rich in negatively charged amino acids at neutral pH, electrostatic repulsions would inhibit the intermolecular aggregation of this region. In addition, the presence of proline residues at the extreme N-terminal region is likely to prevent the β-structure–rich aggregation (48, 49). It should be noted that, in amyloid fibrils, apoA-I molecules are proposed to be packed in a parallel, in-register β-sheet structure (10, 23).
In summary, we have demonstrated that the two highly amyloidogenic segments of residues 14–22 and 50–58 play crucial roles in aggregation and fibril formation of the G26R variant of the N-terminal 1–83 fragment of apoA-I in which residues 14–22 are necessary for fibril formation, whereas residues 50–58 entropically drive the nucleation process. In addition, structural investigation of apoA-I 1–83/G26R fibrils indicates that these two amyloidogenic segments are in close proximity to form amyloid core structure, whereas the N- and C-terminal tail regions are excluded from the amyloid core. The present findings provide novel molecular insights into the aggregation mechanism and fibril structure of the amyloidogenic N-terminal fragment of apoA-I as summarized in Fig. 7.
Figure 7.

Schematic representation of the aggregation mechanism of apoA-I 1–83/G26R. In aggregation and fibril formation of apoA-I 1–83/G26R, the two highly amyloidogenic segments of residues 14–22 (shown in red) and 50–58 (shown in blue) play crucial roles: residues 14–22 are necessary for fibril formation, whereas residues 50–58 entropically drive the nucleation process. The nucleation process is enthalpically unfavorable (activation enthalpy ΔH* > 0) but entropically favorable (activation entropy ΔS* > 0), whereas the fibril elongation process is enthalpically and entropically unfavorable (ΔH* > 0, ΔS* < 0). The favorable activation entropy of nucleation is thought to come from desolvation of residues 50–58. In a fibrillar state, the two amyloidogenic segments are in close proximity to form amyloid core structure, whereas the N- and C-terminal tail regions are excluded from the amyloid core. It has been proposed that apoA-I molecules are packed in a parallel, in-register β-sheet structure in amyloid fibrils (10, 23). See the text for more detail.
Experimental procedures
Preparation of recombinant apoA-I proteins
The N-terminal fragment 1–83 of apoA-I with G26R substitution (apoA-I 1–83/G26R) and its engineered variants with deletions of Δ14–22, Δ32–40, Δ50–58, and Δ68–76 or cysteine substitutions of E2C, W8C, L14C, L22C, S36C, S52C, S58C, L64C, W72C, and G81C were expressed in Escherichia coli as thioredoxin fusion proteins and isolated and purified as described (12, 18). Cleavage of the thioredoxin fusion protein with thrombin leaves the target apoA-I with two extra amino acids, Gly-Ser, at the N terminus. The apoA-I preparations were at least 95% pure as assessed by SDS-PAGE. These apoA-I variants were dialyzed from 6 m guanidine HCl with or without 1% β-mercaptoethanol solution into the appropriate buffer before use. Labeling of cysteine-containing apoA-I 1–83/G26R variants with N-(1-pyrene)maleimide (Thermo Fisher Scientific, Rockford, IL) was performed as described (50, 51).
Preparation of small unilamellar vesicles
Small unilamellar vesicles were prepared as described previously (18, 52). Briefly, a dried film of egg phosphatidylcholine (Kewpie, Tokyo, Japan) was hydrated in 10 mm Tris-HCl buffer (150 mm NaCl, 0.02% NaN3, pH 7.4) and sonicated on ice under nitrogen. After removing titanium debris, the samples were centrifuged in a Beckman MLA-55 rotor for 1.5 h at 15 °C at 40,000 rpm to separate any remaining large vesicles.
CD spectroscopy
Far-UV CD spectra were recorded from 190 to 260 nm at 25 °C using a Jasco J-1500 spectropolarimeter (Jasco, Tokyo, Japan). The apoA-I variants of 50 μg/ml in 10 mm Tris-HCl buffer (pH 7.4) were subjected to CD measurements in a 1-mm quartz cuvette, and the results were corrected by subtracting the buffer baseline.
Fluorescence measurements
Fluorescence measurements were carried out with an F-2700 or F-7000 fluorescence spectrophotometer (Hitachi High-Technologies, Tokyo, Japan) at 25 °C. Kinetics of formation of amyloid-like structure were monitored using ThT. ApoA-I 1–83/G26R variants (100 μg/ml) in 10 mm Tris-HCl buffer (150 mm NaCl, 0.02% NaN3, pH 7.4) were incubated at 37 °C with agitation on an orbital rotator in the presence of 10 μm ThT. ThT fluorescence was recorded at 485 nm with an excitation wavelength of 445 nm. The time-dependent increase in ThT fluorescence intensity was fitted to a sigmoidal equation (12, 53),
| (Eq. 1) |
where F is the fluorescence intensity, F0 is the initial baseline during the lag phase, and Fmax is the final baseline after the growth phase has ended. kapp is the apparent rate constant for the growth of fibrils, and tm is the time to 50% of maximal fluorescence. The lag time is calculated as tm − 2/k.
ThT fluorescence data were also analyzed by the Finke–Watzky two-step model equation for nucleation followed by autocatalytic growth (24, 41),
| (Eq. 2) |
where [A]0 is initial concentration of monomer protein and k1 and k2 are the rate constants corresponding to the nucleation and growth of fibrils, respectively. Thermodynamic parameters for nucleation and fibril growth were determined from the Eyring equations,
| (Eq. 3) |
| (Eq. 4) |
where kB and h are the Boltzmann and Planck constants, respectively. The activation enthalpy (ΔH*) and entropy (ΔS*) were obtained from the slope and y-intercept of the linear plot according to Equation 3, respectively. The activation Gibbs free energy (ΔG*) was calculated from the rate constant k according to Equation 4 or from ΔH* and ΔS* according to ΔG* = ΔH* − TΔS*.
For proximity analysis, pyrene emission fluorescence of pyrene-labeled apoA-I 1–83/G26R variants were monitored before and after incubation. To obtain optimal pyrene excimer signals, mixtures of 25 mol % of pyrene-labeled and 75 mol % of unlabeled proteins were used (32, 35). Pyrene emission fluorescence was recorded from 360 to 600 nm using a 342-nm excitation wavelength. The pyrene scale was calculated as the ratio of fluorescence intensity at 375 nm to that at 385 nm (33, 37). Excimer to monomer ratio was calculated as the ratio of fluorescence intensity at 470 nm (excimer signal) to that at 385 nm (monomer signal).
For chemical denaturation experiments, fibrils of apoA-I 1–83/G26R deletion variants (50 μg/ml) in 10 mm Tris-HCl buffer (150 mm NaCl, 0.02% NaN3, pH 7.4) were incubated overnight at 4 °C with urea at various concentrations in the presence of 10 μm ThT. Denaturation of fibrils was monitored from the change in ThT fluorescence intensity.
AFM
Each sample solution in 10 mm Tris buffer was diluted to 10 μg/ml with distilled water, and 10 μl of the mixture was deposited on freshly cleaved mica (The Nilaco Co., Tokyo, Japan). After washing three times with distilled water (20 μl), samples were imaged under ambient conditions at room temperature using a NanoScope® IIIa tapping-mode AFM (Veeco, Plainview, NY) and Micro cantilever OMCL-AC160TS-R3 (Olympus, Tokyo, Japan).
TEM
A 5-μl droplet of the sample suspension was placed on a glow-discharged Cu grid (300 mesh) coated with carbon, and then a 150-μl droplet of 2% phosphotungstic acid solution (adjusted at pH 7.0 with NaOH) was added. After excess staining solution was blotted with a filter paper after 1-min incubation, the grid was dried under illumination of an incandescent lamp. TEM images were obtained at 120 kV on an FEI Tecnai F20 transmission electron microscope.
Cytotoxicity assay
Cytotoxicity was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described (12). Briefly, HEK293 cells were plated on poly-l-lysine–coated 24-well plates in Dulbecco's modified Eagle's medium containing 2% fetal bovine serum. After 24-h incubation, apoA-I 1–83/G26R deletion variants in 10 mm PBS were added, and cells were further incubated for 24 h. Cell viability was quantitatively determined by reduction of MTT.
Statistical analysis
Data were analyzed via one-way analysis of variance followed by Dunnett's test by means of Prism software (GraphPad Software, La Jolla, CA). Results were regarded as significant for p < 0.05.
Author contributions
C. M. and H. S. conceptualization; C. M., M. N., N. N., M. S., N. K., A. S., K. F., S. H., T. B., and T. O. data curation; C. M. methodology; C. M. and H. S. writing-original draft; M. N., N. N., M. S., N. K., A. S., and T. O. investigation; K. F. and S. H. resources; T. B. and T. O. visualization; K. N. formal analysis; K. N. and H. S. writing-review and editing; H. S. supervision; H. S. funding acquisition; H. S. project administration.
Supplementary Material
Acknowledgments
TEM observations were performed by K. Yan and Dr. K. Hirose (EM group, Tsukuba Innovation Arena, AIST, Tsukuba, Japan). We thank Drs. Michael C. Phillips and Sissel Lund-Katz (University of Pennsylvania) and Dr. W. Sean Davidson (University of Cincinnati) for valuable advice.
This work was partly supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant JP17H03979 (to H. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S9.
- apoA-I
- apolipoprotein A-I
- AFM
- atomic force microscopy
- TEM
- transmission EM
- HDL
- high-density lipoprotein
- ThT
- thioflavin T
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
References
- 1. Phillips M. C. (2013) New insights into the determination of HDL structure by apolipoproteins: thematic review series: high density lipoprotein structure, function, and metabolism. J. Lipid Res. 54, 2034–2048 10.1194/jlr.R034025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosenson R. S., Brewer H. B. Jr, Ansell B. J., Barter P., Chapman M. J., Heinecke J. W., Kontush A., Tall A. R., and Webb N. R. (2016) Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 13, 48–60 10.1038/nrcardio.2015.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sorci-Thomas M. G., and Thomas M. J. (2002) The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc. Med. 12, 121–128 10.1016/S1050-1738(01)00163-3 [DOI] [PubMed] [Google Scholar]
- 4. Arciello A., Piccoli R., and Monti D. M. (2016) Apolipoprotein A-I: the dual face of a protein. FEBS Lett. 590, 4171–4179 10.1002/1873-3468.12468 [DOI] [PubMed] [Google Scholar]
- 5. Tougaard B. G., Pedersen K. V., Krag S. R., Gilbertson J. A., Rowczenio D., Gillmore J. D., and Birn H. (2016) A case report of hereditary apolipoprotein A-I amyloidosis associated with a novel APOA1 mutation and variable phenotype. Eur. J. Med. Genet. 59, 474–477 10.1016/j.ejmg.2016.05.015 [DOI] [PubMed] [Google Scholar]
- 6. Obici L., Franceschini G., Calabresi L., Giorgetti S., Stoppini M., Merlini G., and Bellotti V. (2006) Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid 13, 191–205 10.1080/13506120600960288 [DOI] [PubMed] [Google Scholar]
- 7. Gursky O., Mei X., and Atkinson D. (2012) The crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on amyloid formation by the N-terminal fragment. Biochemistry 51, 10–18 10.1021/bi2017014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andreola A., Bellotti V., Giorgetti S., Mangione P., Obici L., Stoppini M., Torres J., Monzani E., Merlini G., and Sunde M. (2003) Conformational switching and fibrillogenesis in the amyloidogenic fragment of apolipoprotein A-I. J. Biol. Chem. 278, 2444–2451 10.1074/jbc.M204801200 [DOI] [PubMed] [Google Scholar]
- 9. Joy T., Wang J., Hahn A., and Hegele R. A. (2003) APOA1 related amyloidosis: a case report and literature review. Clin. Biochem. 36, 641–645 10.1016/S0009-9120(03)00110-3 [DOI] [PubMed] [Google Scholar]
- 10. Das M., Mei X., Jayaraman S., Atkinson D., and Gursky O. (2014) Amyloidogenic mutations in human apolipoprotein A-I are not necessarily destabilizing—a common mechanism of apolipoprotein A-I misfolding in familial amyloidosis and atherosclerosis. FEBS J. 281, 2525–2542 10.1111/febs.12809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raimondi S., Guglielmi F., Giorgetti S., Di Gaetano S., Arciello A., Monti D. M., Relini A., Nichino D., Doglia S. M., Natalello A., Pucci P., Mangione P., Obici L., Merlini G., Stoppini M., et al. (2011) Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of apolipoprotein A-I. J. Mol. Biol. 407, 465–476 10.1016/j.jmb.2011.01.044 [DOI] [PubMed] [Google Scholar]
- 12. Adachi E., Nakajima H., Mizuguchi C., Dhanasekaran P., Kawashima H., Nagao K., Akaji K., Lund-Katz S., Phillips M. C., and Saito H. (2013) Dual role of an N-terminal amyloidogenic mutation in apolipoprotein A-I: destabilization of helix bundle and enhancement of fibril formation. J. Biol. Chem. 288, 2848–2856 10.1074/jbc.M112.428052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong Y. Q., Binger K. J., Howlett G. J., and Griffin M. D. (2012) Identification of an amyloid fibril forming peptide comprising residues 46–59 of apolipoprotein A-I. FEBS Lett. 586, 1754–1758 10.1016/j.febslet.2012.05.007 [DOI] [PubMed] [Google Scholar]
- 14. Adachi E., Kosaka A., Tsuji K., Mizuguchi C., Kawashima H., Shigenaga A., Nagao K., Akaji K., Otaka A., and Saito H. (2014) The extreme N-terminal region of human apolipoprotein A-I has a strong propensity to form amyloid fibrils. FEBS Lett. 588, 389–394 10.1016/j.febslet.2013.11.031 [DOI] [PubMed] [Google Scholar]
- 15. Saito H., Lund-Katz S., and Phillips M. C. (2004) Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog. Lipid Res. 43, 350–380 10.1016/j.plipres.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 16. Chetty P. S., Mayne L., Lund-Katz S., Stranz D., Englander S. W., and Phillips M. C. (2009) Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 19005–19010 10.1073/pnas.0909708106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mendoza-Espinosa P., Montalvan-Sorrosa D., García-González V., Moreno A., Castillo R., and Mas-Oliva J. (2014) Microenvironmentally controlled secondary structure motifs of apolipoprotein A-I derived peptides. Mol. Cell Biochem. 393, 99–109 10.1007/s11010-014-2050-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mizuguchi C., Ogata F., Mikawa S., Tsuji K., Baba T., Shigenaga A., Shimanouchi T., Okuhira K., Otaka A., and Saito H. (2015) Amyloidogenic mutation promotes fibril formation of the N-terminal apolipoprotein A-I on lipid membranes. J. Biol. Chem. 290, 20947–20959 10.1074/jbc.M115.664227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizuguchi C., Nakamura M., Kurimitsu N., Ohgita T., Nishitsuji K., Baba T., Shigenaga A., Shimanouchi T., Okuhira K., Otaka A., and Saito H. (2018) Effect of phosphatidylserine and cholesterol on membrane-mediated fibril formation by the N-terminal amyloidogenic fragment of apolipoprotein A-I. Sci. Rep. 8, 5497 10.1038/s41598-018-23920-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nichols W. C., Dwulet F. E., Liepnieks J., and Benson M. D. (1988) Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem. Biophys. Res. Commun. 156, 762–768 10.1016/S0006-291X(88)80909-4 [DOI] [PubMed] [Google Scholar]
- 21. Lagerstedt J. O., Cavigiolio G., Roberts L. M., Hong H. S., Jin L. W., Fitzgerald P. G., Oda M. N., and Voss J. C. (2007) Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry 46, 9693–9699 10.1021/bi7005493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chetty P. S., Ohshiro M., Saito H., Dhanasekaran P., Lund-Katz S., Mayne L., Englander W., and Phillips M. C. (2012) Effects of the Iowa and Milano mutations on apolipoprotein A-I structure and dynamics determined by hydrogen exchange and mass spectrometry. Biochemistry 51, 8993–9001 10.1021/bi300926j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Girych M., Gorbenko G., Trusova V., Adachi E., Mizuguchi C., Nagao K., Kawashima H., Akaji K., Lund-Katz S., Phillips M. C., and Saito H. (2014) Interaction of thioflavin T with amyloid fibrils of apolipoprotein A-I N-terminal fragment: resonance energy transfer study. J. Struct. Biol. 185, 116–124 10.1016/j.jsb.2013.10.017 [DOI] [PubMed] [Google Scholar]
- 24. Morris A. M., Watzky M. A., Agar J. N., and Finke R. G. (2008) Fitting neurological protein aggregation kinetic data via a 2-step, minimal/“Ockham's razor” model: the Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry 47, 2413–2427 10.1021/bi701899y [DOI] [PubMed] [Google Scholar]
- 25. Watzky M. A., Morris A. M., Ross E. D., and Finke R. G. (2008) Fitting yeast and mammalian prion aggregation kinetic data with the Finke-Watzky two-step model of nucleation and autocatalytic growth. Biochemistry 47, 10790–10800 10.1021/bi800726m [DOI] [PubMed] [Google Scholar]
- 26. Nakajima K., Ogi H., Adachi K., Noi K., Hirao M., Yagi H., and Goto Y. (2016) Nucleus factory on cavitation bubble for amyloid beta fibril. Sci. Rep. 6, 22015 10.1038/srep22015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iashchishyn I. A., Sulskis D., Nguyen Ngoc M., Smirnovas V., and Morozova-Roche L. A. (2017) Finke-Watzky two-step nucleation-autocatalysis model of S100A9 amyloid formation: protein misfolding as “nucleation” event. ACS Chem. Neurosci. 8, 2152–2158 10.1021/acschemneuro.7b00251 [DOI] [PubMed] [Google Scholar]
- 28. Cohen S. I., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., and Knowles T. P. (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 10.1073/pnas.1218402110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meisl G., Yang X., Hellstrand E., Frohm B., Kirkegaard J. B., Cohen S. I., Dobson C. M., Linse S., and Knowles T. P. (2014) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. U.S.A. 111, 9384–9389 10.1073/pnas.1401564111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tornquist M., Michaels T. C. T., Sanagavarapu K., Yang X., Meisl G., Cohen S. I. A., Knowles T. P. J., and Linse S. (2018) Secondary nucleation in amyloid formation. Chem. Commun. 54, 8667–8684 10.1039/C8CC02204F [DOI] [PubMed] [Google Scholar]
- 31. Bains G., Patel A. B., and Narayanaswami V. (2011) Pyrene: a probe to study protein conformation and conformational changes. Molecules 16, 7909–7935 10.3390/molecules16097909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krishnan R., and Lindquist S. L. (2005) Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 435, 765–772 10.1038/nature03679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thirunavukkuarasu S., Jares-Erijman E. A., and Jovin T. M. (2008) Multiparametric fluorescence detection of early stages in the amyloid protein aggregation of pyrene-labeled α-synuclein. J. Mol. Biol. 378, 1064–1073 10.1016/j.jmb.2008.03.034 [DOI] [PubMed] [Google Scholar]
- 34. Campioni S., Mannini B., Zampagni M., Pensalfini A., Parrini C., Evangelisti E., Relini A., Stefani M., Dobson C. M., Cecchi C., and Chiti F. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147 10.1038/nchembio.283 [DOI] [PubMed] [Google Scholar]
- 35. Gallea J. I., and Celej M. S. (2014) Structural insights into amyloid oligomers of the Parkinson disease-related protein α-synuclein. J. Biol. Chem. 289, 26733–26742 10.1074/jbc.M114.566695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haney C. M., and Petersson E. J. (2018) Fluorescence spectroscopy reveals N-terminal order in fibrillar forms of α-synuclein. Chem. Commun. 54, 833–836 10.1039/C7CC08601F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tamamizu-Kato S., Kosaraju M. G., Kato H., Raussens V., Ruysschaert J. M., and Narayanaswami V. (2006) Calcium-triggered membrane interaction of the α-synuclein acidic tail. Biochemistry 45, 10947–10956 10.1021/bi060939i [DOI] [PubMed] [Google Scholar]
- 38. Mikawa S., Mizuguchi C., Nishitsuji K., Baba T., Shigenaga A., Shimanouchi T., Sakashita N., Otaka A., Akaji K., and Saito H. (2016) Heparin promotes fibril formation by the N-terminal fragment of amyloidogenic apolipoprotein A-I. FEBS Lett. 590, 3492–3500 10.1002/1873-3468.12426 [DOI] [PubMed] [Google Scholar]
- 39. Levitan K., Chereau D., Cohen S. I., Knowles T. P., Dobson C. M., Fink A. L., Anderson J. P., Goldstein J. M., and Millhauser G. L. (2011) Conserved C-terminal charge exerts a profound influence on the aggregation rate of α-synuclein. J. Mol. Biol. 411, 329–333 10.1016/j.jmb.2011.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sabaté R., Castillo V., Espargaró A., Saupe S. J., and Ventura S. (2009) Energy barriers for HET-s prion forming domain amyloid formation. FEBS J. 276, 5053–5064 10.1111/j.1742-4658.2009.07202.x [DOI] [PubMed] [Google Scholar]
- 41. Morris A. M., and Finke R. G. (2009) α-Synuclein aggregation variable temperature and variable pH kinetic data: a re-analysis using the Finke-Watzky 2-step model of nucleation and autocatalytic growth. Biophys. Chem. 140, 9–15 10.1016/j.bpc.2008.11.003 [DOI] [PubMed] [Google Scholar]
- 42. Buell A. K., Dhulesia A., White D. A., Knowles T. P., Dobson C. M., and Welland M. E. (2012) Detailed analysis of the energy barriers for amyloid fibril growth. Angew. Chem. Int. Ed. Engl. 51, 5247–5251 10.1002/anie.201108040 [DOI] [PubMed] [Google Scholar]
- 43. Cohen S. I. A., Cukalevski R., Michaels T. C. T., Šarić A., Törnquist M., Vendruscolo M., Dobson C. M., Buell A. K., Knowles T. P. J., and Linse S. (2018) Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-β peptide. Nat. Chem. 10, 523–531 10.1038/s41557-018-0023-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mei X., and Atkinson D. (2011) Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J. Biol. Chem. 286, 38570–38582 10.1074/jbc.M111.260422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hortschansky P., Christopeit T., Schroeckh V., and Fändrich M. (2005) Thermodynamic analysis of the aggregation propensity of oxidized Alzheimer's β-amyloid variants. Protein Sci. 14, 2915–2918 10.1110/ps.051585905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arosio P., Knowles T. P., and Linse S. (2015) On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 17, 7606–7618 10.1039/C4CP05563B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Witkowski A., Chan G. K. L., Boatz J. C., Li N. J., Inoue A. P., Wong J. C., van der Wel P. C. A., and Cavigiolio G. (2018) Methionine oxidized apolipoprotein A-I at the crossroads of HDL biogenesis and amyloid formation. FASEB J. 32, 3149–3165 10.1096/fj.201701127R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams A. D., Portelius E., Kheterpal I., Guo J. T., Cook K. D., Xu Y., and Wetzel R. (2004) Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842 10.1016/j.jmb.2003.11.008 [DOI] [PubMed] [Google Scholar]
- 49. Abedini A., and Raleigh D. P. (2006) Destabilization of human IAPP amyloid fibrils by proline mutations outside of the putative amyloidogenic domain: is there a critical amyloidogenic domain in human IAPP? J. Mol. Biol. 355, 274–281 10.1016/j.jmb.2005.10.052 [DOI] [PubMed] [Google Scholar]
- 50. Mizuguchi C., Hata M., Dhanasekaran P., Nickel M., Phillips M. C., Lund-Katz S., and Saito H. (2012) Fluorescence analysis of the lipid binding-induced conformational change of apolipoprotein E4. Biochemistry 51, 5580–5588 10.1021/bi300672s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kono M., Okumura Y., Tanaka M., Nguyen D., Dhanasekaran P., Lund-Katz S., Phillips M. C., and Saito H. (2008) Conformational flexibility of the N-terminal domain of apolipoprotein A-I bound to spherical lipid particles. Biochemistry 47, 11340–11347 10.1021/bi801503r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saito H., Dhanasekaran P., Nguyen D., Deridder E., Holvoet P., Lund-Katz S., and Phillips M. C. (2004) α-Helix formation is required for high affinity binding of human apolipoprotein A-I to lipids. J. Biol. Chem. 279, 20974–20981 10.1074/jbc.M402043200 [DOI] [PubMed] [Google Scholar]
- 53. Nielsen L., Khurana R., Coats A., Frokjaer S., Brange J., Vyas S., Uversky V. N., and Fink A. L. (2001) Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 40, 6036–6046 10.1021/bi002555c [DOI] [PubMed] [Google Scholar]
- 54. Tsolis A. C., Papandreou N. C., Iconomidou V. A., and Hamodrakas S. J. (2013) A consensus method for the prediction of 'aggregation-prone' peptides in globular proteins. PLoS One 8, e54175 10.1371/journal.pone.0054175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tartaglia G. G., Pawar A. P., Campioni S., Dobson C. M., Chiti F., and Vendruscolo M. (2008) Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 380, 425–436 10.1016/j.jmb.2008.05.013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.