

Abstract
Ryanodine receptor type I (RYR1)-related myopathies (RYR1 RM) are a clinically and histopathologically heterogeneous group of conditions that represent the most common subtype of childhood onset non-dystrophic muscle disorders. There are no treatments for this severe group of diseases. A major barrier to therapy development is the lack of an animal model that mirrors the clinical severity of pediatric cases of the disease. To address this, we used CRISPR/Cas9 gene editing to generate a novel recessive mouse model of RYR1 RM. This mouse (Ryr1TM/Indel) possesses a patient-relevant point mutation (T4706M) engineered into 1 allele and a 16 base pair frameshift deletion engineered into the second allele. Ryr1TM/Indel mice exhibit an overt phenotype beginning at 14 days of age that consists of reduced body/muscle mass and myofibre hypotrophy. Ryr1TM/Indel mice become progressively inactive from that point onward and die at a median age of 42 days. Histopathological assessment shows myofibre hypotrophy, increased central nuclei and decreased triad number but no clear evidence of metabolic cores. Biochemical analysis reveals a marked decrease in RYR1 protein levels (20% of normal) as compared to only a 50% decrease in transcript. Functional studies at end stage show significantly reduced electrically evoked Ca2+ release and force production. In summary, Ryr1TM/Indel mice exhibit a post-natal lethal recessive form of RYR1 RM that pheno-copies the severe congenital clinical presentation seen in a subgroup of RYR1 RM children. Thus, Ryr1TM/Indel mice represent a powerful model for both establishing the pathomechanisms of recessive RYR1 RM and pre-clinical testing of therapies for efficacy.
Introduction
Ryanodine receptor type 1 (RYR1)-related myopathies (RYR1 RM) are a clinically and histopathologically heterogeneous group of neuromuscular disorders united by the presence of mutations in the RYR1 gene (1,2). RYR1 encodes a large, homotetrameric Ca2+ release channel, located at the terminal cisternae of the sarcoplasmic reticulum, which is required for excitation–contraction coupling (3,4). RYR1 RMs typically present at birth, are slowly progressive and are defined by morphological abnormalities in skeletal muscle (5,6). RYR1 RMs are the most common subgroup of a larger set of conditions called congenital myopathies (7), which are historically defined by muscle biopsy features and are typically caused by mutations in structural, contractile or regulatory muscle proteins (8). At present, care for congenital myopathies is strictly supportive as there are no approved therapies for this clinically severe group of debilitating disorders (9).
Both heterozygous/dominant and bi-allelic/recessive mutations have been identified in RYR1 (10–12). Dominant mutations are nearly always missense variants that impact channel function (3,13); they are typically associated with increased susceptibility to malignant hyperthermia (MH) and/or milder clinical features with a central core disease histological phenotype (12). Recessive RYR1 mutations are usually observed in patients with a severe childhood onset disease and are associated with multiminicore disease, centronuclear myopathy or congenital fibre-type disproportion (11,14). The most frequent mutational pattern observed with recessive RYR1 RM is a missense mutation on one allele coupled with a ‘hypomorphic’ (nonsense, frameshift, etc.) mutation on the other alelle (14).
Currently, the pathogenesis underlying RYR1 RM is only partially understood. In particular, a large knowledge gap exists concerning recessive forms of the disease. Existing mouse models of RYR1 RM have contributed to our understanding of disease pathogenesis and have identified potential therapies, including N-acetylcysteine (15,16), AICAR (17) and 4-phenylbutyrate (18). These models include the Ryr1 knockout (or dyspedic) mouse (19) and several knock-in mouse lines for select dominantly inherited human RYR1 mutations (R163C, Y522S, G2434R, T4826I and I4898T; using human RYR1 numbering) (15,20–23). The dyspedic mouse dies at birth, an attribute that makes it unsuitable for in vivo drug development. The dominantly inherited knock-in models have provided valuable insight into effects of specific mutations on Ca2+ dynamics and how these alterations lead to increased susceptibility to MH with or without a mild, late-onset myopathy. However, as a group, all dominantly inherited RYR1 knock-in mice described to date exhibit, if any, only late onset and extremely mild clinical phenotypes and genetically only model heterozygous/dominant cases of RYR1 RM. There is thus a pressing need to develop mouse models that pheno-copy the early onset and clinical severity of congenital, recessive RYR1 RM.
To address this gap, we developed a genetically relevant mouse model of recessive RYR1-RM. This model is a compound heterozygote with one allele containing a T4709M missense mutation in exon 96 of RYR1 (referred to as the ‘TM’ allele and equivalent to T4706M in human RYR1) and the second allele possessing a 16 bp frameshift deletion (also located in exon 96 and referred to as the ‘indel’ allele). The TM substitution is a recurrent mutation reported in patients with recessive RYR1 RM (14,24). For example, Zhou et al. (24) reported that a child with reputed monoallelic expression of T4709M exhibited markedly reduced RYR1 protein expression in skeletal muscle and presented with pronounced facial/proximal weakness, scoliosis, opthalmoplegia and respiratory impairment. While parental Ryr1TM/+ and Ryr1indel/+ mice do not exhibit an overt phenotype, Ryr1TM/indel mice present a highly reproducible phenotype characterized by reduced body/muscle mass, progressive muscle weakness, small myofibre size, markedly reduced RYR1 expression and early death. Thus, heterozygous Ryr1TM/Indel mice exhibit a severe, early-onset recessive RYR1-related myopathy. As the first mouse model of post-natal lethal recessive RYR1 RM, Ryr1TM/Indel mice represent an ideal model for future pre-clinical testing of potential therapies.
Results
Generation of the Ryr1TM/Indel mice
Using a CRISPR/Cas9 gene-editing strategy, we generated a compound heterozygous mouse model of RYR1 RM in which one allele contains the TM missense mutation in the 96th exon of RYR1 with the second allele containing a 16 bp frame-shift deletion in the same exon (Fig. 1A). The threonine-to-methionine missense mutation was previously reported in multiple cases of RYR1 RM, including a compound heterozygous case where the patient exhibited childhood onset and progressive muscle weakness (25) and a second heterozygotic case where the patient showed a severe phenotype (24). In the latter case, the TM mutation was reported as ‘monoallelic’ since expression of the other allele in muscle was undetectable. Both patients showed markedly reduced RyR1 expression, a common feature of recessive RYR1 RM (14). The second mutation of this mouse model, a 16 bp deletion in exon 96, causes a premature stop codon resulting in a null allele. Both mutations were confirmed by Sanger sequencing (Fig. 1B). We established germ line transmission from multiple founders and subsequently outcrossed the resulting F1 mice to wild-type (WT) C57BL6J animals. All analyses were performed on F3 generation mice and beyond. Of note, parental Ryr1TM/+ and Ryr1Indel/+ mice are visibly indistinguishable from WT mice, exhibiting normal body weight, fertility and survival.
Figure 1.
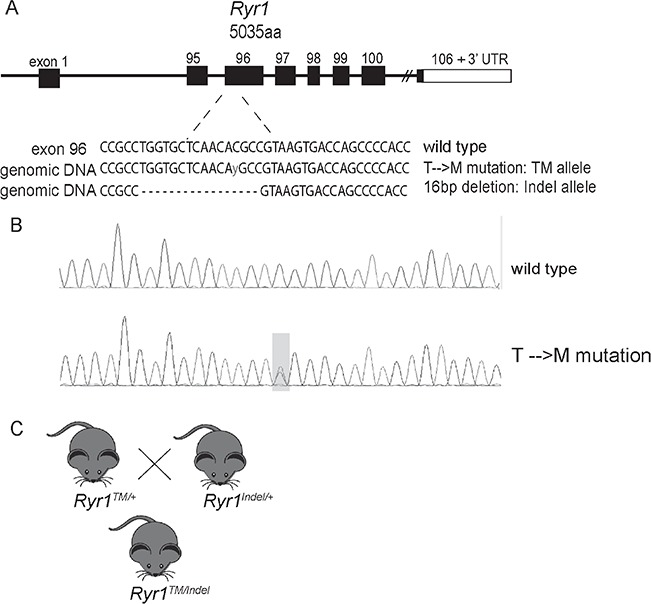
Generation of the Ryr1TM/Indel mouse line. (A) Using CRISPR/Cas9, a threonine-to-methionine missense mutation (T4709M) and a 16 bp deletion (Indel) were generated in exon 96 of the mouse Ryr1 locus. ‘y’ indicates a heterozygous base change from C to T. (B) Representative Sanger sequencing results showing the T4709M substitution and 16 bp deletion mutations. (C) Breeding strategy of crossing heterozygous Ryr1WT/TM mice with Ryr1WT/Indel mice to obtain compound Ryr1TM/Indel mice.
Ryr1TM/Indel mice exhibit reduced body weight and decreased postnatal survival
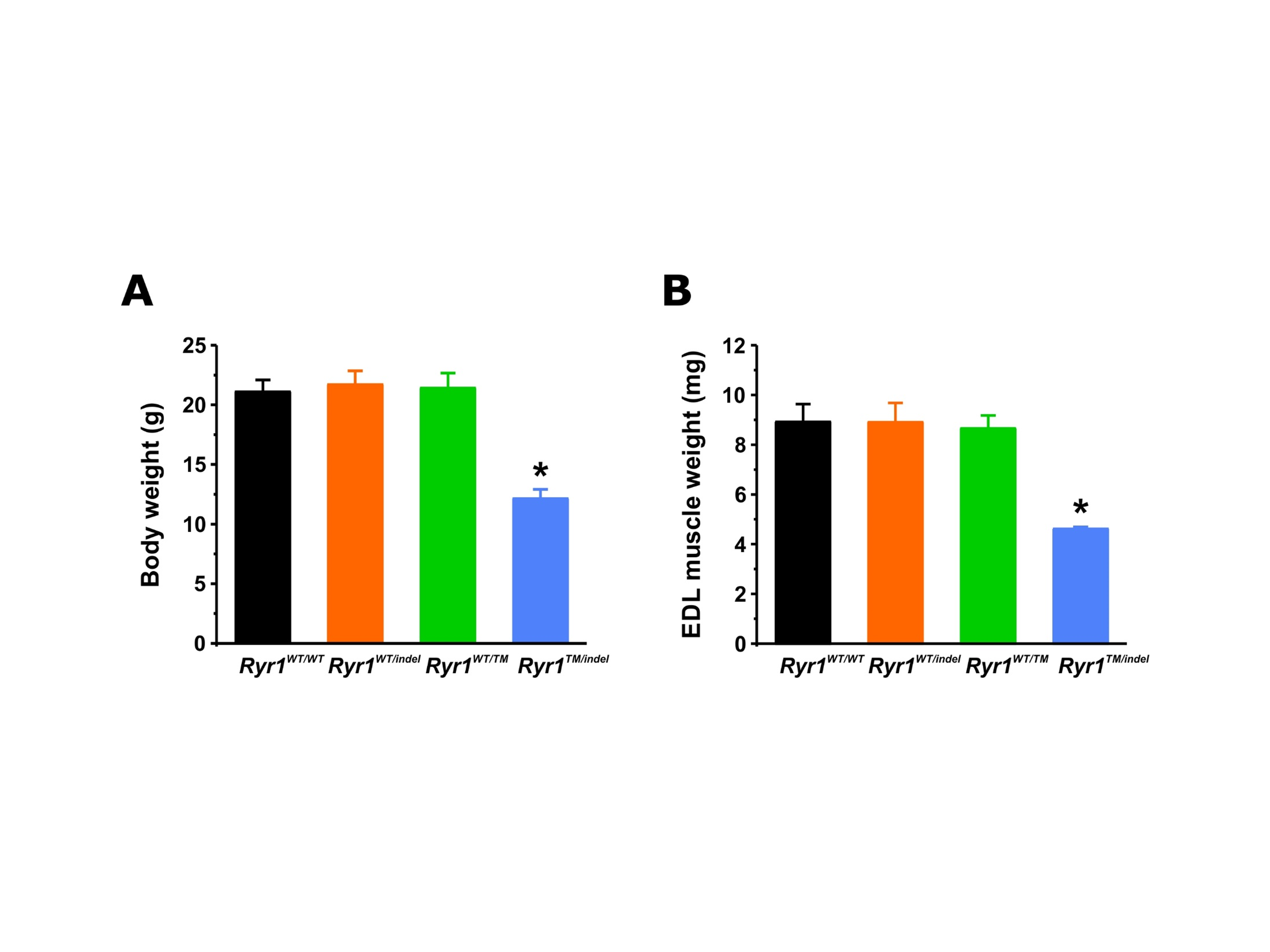
By intercrossing the Ryr1TM/+ and Ryr1Indel/+ lines, we obtained compound heterozygous mice containing both mutations (Ryr1TM/Indel) (Fig. 1C). Ryr1TM/Indel mice are born at the expected Mendelian frequency (25% Ryr1TM/Indel mice on average). While a small number of Ryr1TM/Indel mice died during the first 3 days of life (n = 3), for the majority of Ryr1TM/Indel mice, the first overt phenotype observed was reduced body weight (Fig. 2A). This size difference was noticeable as early as at 14 days postnatal and progressed with age in both genders thereafter (Fig. 2B). The weight of female Ryr1TM/Indel mice was ~ 63% that of WT littermates, while the weight of male Ryr1TM/Indel mice was ~ 52% that of WT littermates. Consistent with the whole body measurements, wet weight of extensor digitorum longus (EDL) muscles excised from 50-day-old mice was significantly decreased (~50%) compared to that of all other genotypes (Supplementary Material, Fig. S1). Thus, the reduced body weight of Ryr1TM/Indel mice reflects a substantial reduction of the myofibre compartment.
Figure 2.
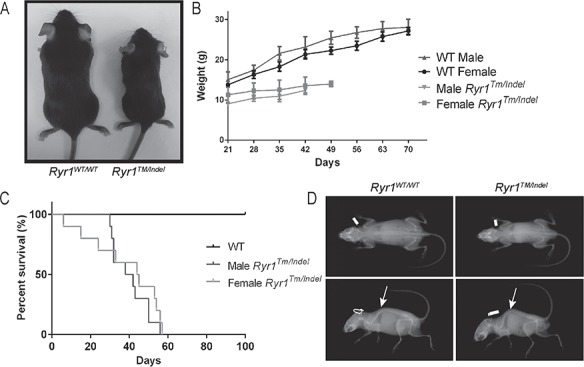
Ryr1TM/Indel mice exhibit reduced body weight and premature death. (A) Ryr1TM/Indel mice (40 days old) exhibit reduced size. Ryr1TM/Indel mice also show hunched posture and splayed hindlimbs compared to Ryr1WT/WT mice. Depicted is a photograph of representative Ryr1WT/WT and Ryr1TM/Indel littermate mice at 40 days. (B) Reduced body weight of Ryr1TM/Indel mice, noticeable at 14 days postnatal, progresses with age (n = 8–9). (C) Ryr1TM/Indel mice exhibit decreased survival. Kaplan–Meier survival analysis reveals that Ryr1TM/Indel mice do not survive longer than 57 days with a median survival of 42 days (n = 25 for each group). (D) X-ray analysis reveals both scoliosis (lateral curve, top panel) and spinal kyphosis (arrow, bottom panels) in Ryr1TM/Indel mice compared to Ryr1WT/WT mice.
The progression of disease in Ryr1TM/Indel mice was rapid. Most of these mice did not survive longer than 57 days of age, with an overall median survival of 33 days (Fig. 2C). When the small number of Ryr1TM/Indel mice that died during the first few days of life was removed, median survival was 42 days. At endpoint, Ryr1TM/Indel mice exhibited reduced movement, hindlimb paralysis and a hunched posture (Supplementary Material, Video S1). X-ray analysis of Ryr1TM/Indel mice at 40 days showed severe kyphoscoliosis (Fig. 2D). These spine changes, combined with muscle weakness, likely resulted in respiratory failure as the cause of death.
Ryr1TM/Indel mice exhibit reduced muscle force generation
While the Ryr1TM/Indel mice were smaller in size and appeared weak by end point, results from functional motor analyses were inconsistent. Grip strength (normalized for body weight), performed at 55 days of age, was not significantly different between Ryr1WT/WT and Ryr1TM/Indel mice (Fig. 3A). However, wire hang testing performed at the same age was significantly decreased in Ryr1TM/Indel mice (Fig. 3B). To more directly assess specific muscle force generation capacity, we performed ex vivo muscle contractility studies in freshly isolated EDL muscles. Both peak twitch (Fig. 4A and C) and tetanic-specific (Fig. 4B and D) force (normalized for physiological cross-sectional area) were significantly reduced in EDL muscles from Ryr1TM/Indel mice in the absence of major changes in the force–frequency relationship (Fig. 4E and F).
Figure 3.
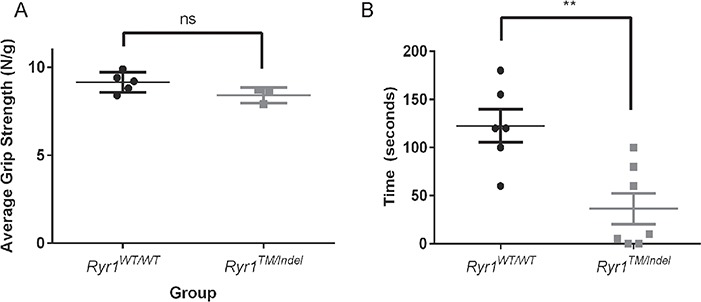
Ryr1TM/Indel mice exhibit in vivo muscle weakness assessed by wire hang test. (A) Average combined forelimb and hindlimb grip strength (normalized to body weight) in 55-day-old Ryr1WT/WT (circles) and Ryr1TM/Indel mice (squares). No statistically significant difference was observed; Ryr1TM/Indel exhibit an overall grip strength of 8.40 ± 0.25 (N/g) (n = 5, 3 male and 2 female) and Ryr1WT/WT an average of 9.14 ± 0.25 (N/g) (n = 4, 3 male and 2 female). (B) Wire hang test of 55-day-old Ryr1WT/WT (circles) and Ryr1TM/Indel mice (squares). Ryr1TM/Indel exhibit significantly decreased average [±standard error of the mean (SEM)] wire hang time in spite of a lower body weight (average = 36.4 ± 16.1 s, n = 6) compared to Ryr1WT/WT mice (average = 122.5 ± 17.1 s, n = 7) (**P = 0.0039).
Figure 4.
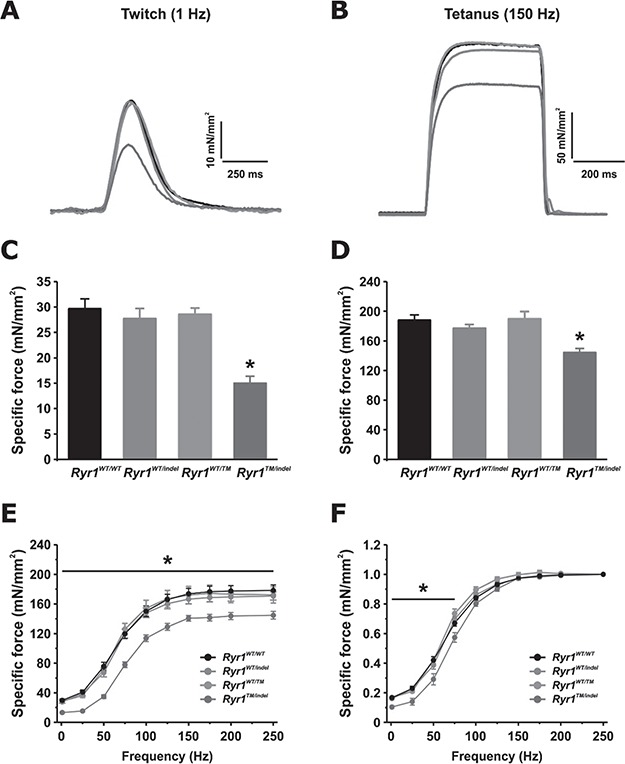
Reduced twitch and tetanic-specific force production in EDL muscles from Ryr1TM/Indel mice. (A, B) Representative superimposed specific force traces elicited during twitch (A) and tetanic (B) stimulation. (C, D) Average (±SEM) peak twitch (C) and tetanic-specific (D) force for each genotype. (E, F) Analysis of specific (E) and relative (F) force–frequency curves from the same muscles shown in (A)–(D). Number of muscles analysed: Ryr1WT/WT = 12 (6 mice); Ryr1WT/indel = 16 (8 mice); Ryr1WT/TM = 6 (3 mice); Ryr1TM/indel = 8 (4 mice).
Ryr1TM/Indel muscle exhibits decreased myofibre size but preserved muscle structure
We examined muscle structure using light microscopic analyses. We focused investigations on tibialis anterior (TA), comparing the common muscle stains haematoxylin and eosin (H/E) and succinate dehydrogenase (SDH) at 55 days of age. By H/E staining (Fig. 5A), two consistent abnormalities were observed. The most notable was a significant reduction in myofibre size (Fig. 5A and B). Myofibre size, quantified as average fibre diameter, was significantly reduced in muscle from Ryr1TM/Indel mice compared to that of WT littermates (Fig. 5C, Supplementary Material, Fig. S2C). The second histological abnormality observed was a modest, but highly significant (P < 0.0001), increase in the percentage of fibres with central/internal nuclei (Fig. 5A) in TA fibres from Ryr1TM/Indel mice (0.8%) compared to that observed in WT mice (0.2%) (Fig. 5D). SDH staining is used to reveal cores and core-like lesions (8), abnormalities commonly seen in a subset of RYR1 RM patients. However, SDH staining of TA muscle from Ryr1TM/Indel mice did not reveal evidence of cores (Fig. 5B). In addition, neither H/E nor SDH staining revealed changes typical of dystrophic muscle disease (e.g. fibrosis, fatty infiltrate or evidence of degenerating or regenerating fibres).
Figure 5.
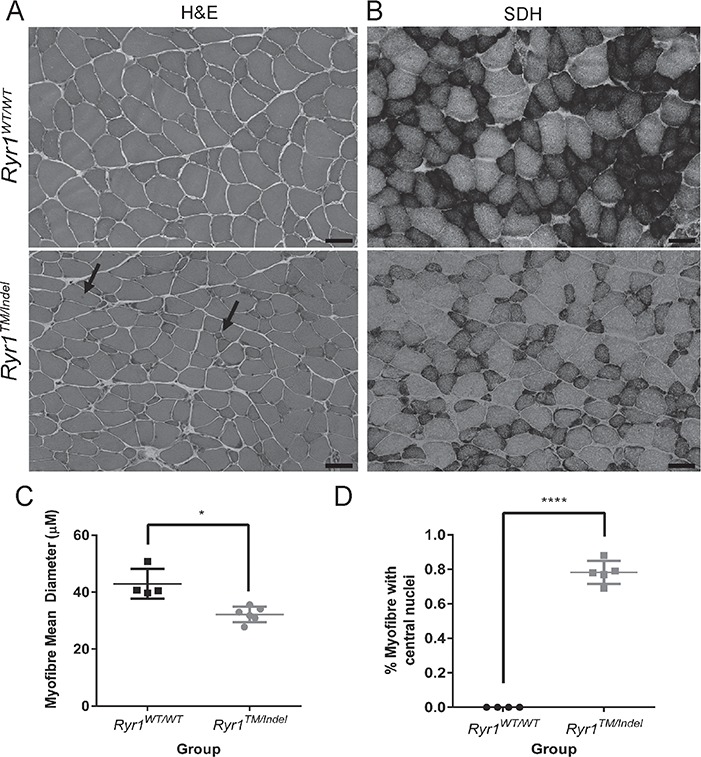
Histological analysis of Ryr1TM/Indel mice reveals reduced myofibre size and increased percent of central nuclei. Histopathologic analysis of cross sections for TA muscle of 55-day-old Ryr1WT/WT and Ryr1TM/Indel mice. (A) As seen with H/E stain, fibres from Ryr1TM/Indel mice compared to Ryr1WT/WT mice exhibit decreased fibre size and reveal the presence of central nuclei. Scale bar is 50 μm. (B) As seen with SDH stain, muscles from Ryr1TM/Indel mice do not exhibit cores or core-like lesions. Scale bar is 50 μm. (C) Mean (±S.E.M.) myofibre diameter of Ryr1TM/Indel (n = 4, male) and Ryr1WT/WT mice (n = 6, 4 male and 2 female). Average fibre diameter is 42.9 ± 2.6 μm versus 32.2 ± 1.1 μm in Ryr1WT/WT and Ryr1TM/Indel mice, respectively (*P = 0.0189). (D) Percent of myofibres containing central nuclei in Ryr1TM/Indel (n = 5) and Ryr1WT/WT mice (n = 5). Average (±SEM) percent central nuclei is 0.00% and 0.78% ± 0.03% in Ryr1WT/WT and Ryr1TM/Indel mice, respectively (****P < 0.0001).
Given the observation of myofibre hypotrophy, we assessed whether fibre type was altered in muscle from Ryr1TM/Indel mice and if the hypotrophy was restricted to a single fibre type. We assessed fibre type using fibre type-specific antibodies by both immunostaining and silver stain analyses. No differences in the distribution of Type I versus Type II fibres were observed in TA muscles of Ryr1TM/Indel mice (Supplementary Material, Fig. S2A and B). In addition, a similar ~ 25% reduction in fibre diameter was observed in both Type I and IIB fibres in muscles from Ryr1TM/Indel mice (Supplementary Material, Fig. S2C). Using silver stain analysis of Type II myosin isoforms, the relative fraction of Type IIA/IX and Type IIB isoforms were not significantly different in TA muscles for any genotype (Supplementary Material, Fig. S3).
RYR1 transcript and protein levels are reduced in muscle from Ryr1TM/Indel mice
The 16 bp frame-shift (indel) mutant allele is expected to produce a transcript that undergoes nonsense mediated decay and thus results in a 50% reduction in Ryr1 transcript levels. Using quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis, we indeed observed this expected reduction in Ryr1 transcript, but not in another muscle-specific transcript (dihydropyridine receptor, DHPR), in quadriceps muscle from both Ryr1WT/Indel and Ryr1TM/Indel mice (Fig. 6). However, western blot analysis revealed a ~ 80% reduction in RyR1 protein level in both TA (Fig. 7A and B) and quadriceps (data not shown) muscle from only Ryr1TM/Indel mice. Importantly, a truncated band was not identified using antibody 34C (which recognizes an epitope between RyR1 amino acids 2756–2803) in muscle samples from either Ryr1WT/Indel or Ryr1TM/Indel mice, consistent with the prediction that indel allele does not produce a stable truncated RyR1 fragment. In spite of a ~ 50% in Ryr1 transcript, RyR1 protein level was unaltered in muscle from Ryr1WT/Indel mice, consistent with the lack of an overt phenotype of these mice (and also heterozygous Ryr1-null mice) (26,27). A more modest, but still statistically significant, decrease in DHPR (which activates RyR1 during excitation–contraction coupling) protein level, but not the sarco-endoplasmic reticulum calcium ATPase (SERCA), was also observed in muscle from Ryr1TM/Indel mice (Fig. 7C and D). Interestingly, a smaller reduction in RyR1 and DHPR protein levels, in the absence of a change in Ryr1 transcript, was also observed in muscle from 7-month-old homozygous Ryr1TM/TM mice (Supplementary Material, Fig. S4).
Figure 6.
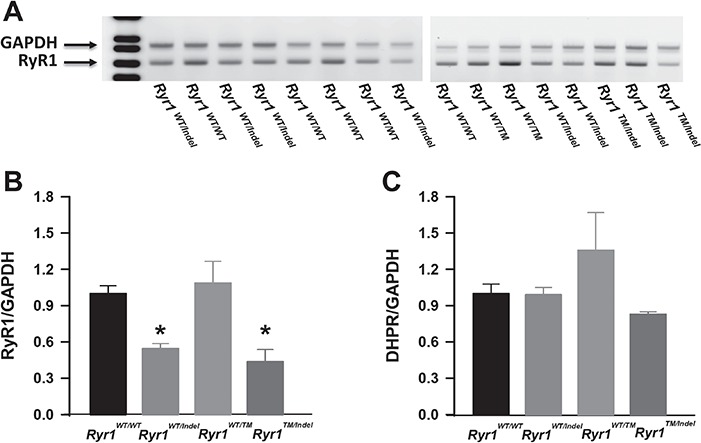
Ryr1 transcript levels are reduced in quadriceps muscle of Ryr1WT/Indel and Ryr1TM/Indel mice. (A) Representative agarose gel of Ryr1 (lower band) and Gapdh loading control (upper band) amplification products. (B, C) Quantitative analysis of Ryr1 (B) and DHPR (C) transcript levels in quadriceps muscles obtained by semiquantitative PCR using 6-FAM-labelled primers. Data are shown as mean ± SEM from five Ryr1WT/WT, six Ryr1WT/Indel, three RyR1WT/TM and three Ryr1TM/Indel mice (*P < 0.05).
Figure 7.
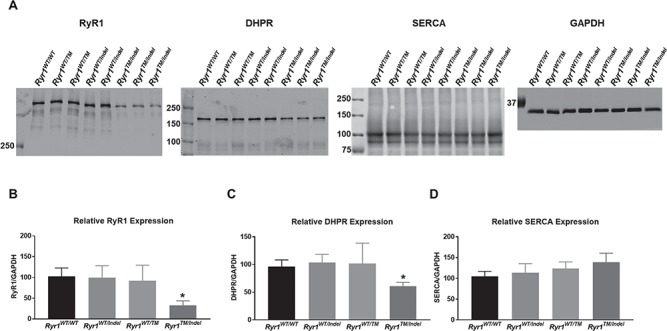
RyR1 protein level is markedly reduced in TA muscle from Ryr1TM/Indel mice. (A) Representative western blots gels for RyR1, DHPR, SERCA and GAPDH in TA muscle from each genotype. Loaded per lane was 5 μg of total TA lysate. (B) Quantitation of relative RyR1 level (normalized to GAPDH) for each genotype. (C) Quantitation of relative DHPR level (normalized to GAPDH) for each genotype. (D) Quantitation of relative SERCA level (normalized to GAPDH) for each genotype. Data are shown as mean ± SEM from seven Ryr1WT/WT, eight Ryr1WTInde, three Ryr1WT/TM and four Ryr1TM/Indel mice (*P < 0.001).
Ryr1TM/Indel mice display aberrant intracellular calcium dynamics
Given the reduction in both RyR1 and DHPR protein levels (Fig. 7), and specific force generation (Fig. 4) in muscle from Ryr1TM/Indel mice, we assessed the magnitude of electrically evoked Ca2+ release in single muscle fibres isolated from the flexor digitorum brevis (FDB) muscle fibres acutely dissociated from 50-day-old mice for each genotype (Fig. 8). For these experiments, FDB fibres were loaded with mag-fluo-4, a rapid, low-affinity Ca2+ dye that maximizes resolution of Ca2+ transient magnitude and kinetics (28). Representative traces of normalized mag-fluo-4 fluorescence (Fig. 5A and B) revealed a significant reduction in peak Ca2+ transient amplitude during both electrically evoked twitch (1 Hz; Fig. 5C) and tetanic (100 Hz; Fig. 5D) stimulation in FDB fibres from Ryr1TM/Indel mice compared to that observed for all other genotypes. In addition, a greater temperature-dependent increase in resting Ca2+ was observed in FDB fibres from Ryr1TM/Indel mice compared to that of all other genotypes (Supplementary Material, Fig. S5).
Figure 8.
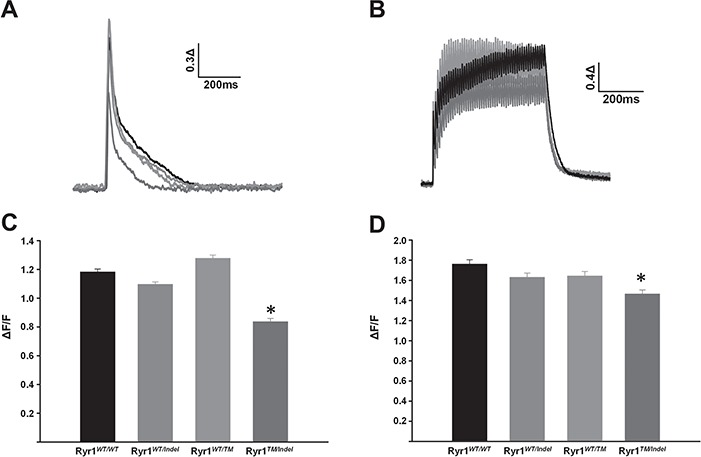
Reduced twitch and tetanic Ca2+ release in FDB fibres from Ryr1TM/Indel mice. (A, B) Representative superimposed changes in relative mag-fluo-4 fluorescence during electrically evoked twitch (A) and tetanic (B) stimulation. (C, D) Average (±SEM) peak twitch (C) and tetanic (D) change in relative mag-fluo-4 fluorescence for each genotype compared to Ryr1WT/WT. Data are shown as mean ± SEM in fibres from Ryr1WT/WT (n = 92), Ryr1WT/Indel (n = 107), RyR1WT/TM (n = 43) and Ryr1TM/Indel (n = 60) mice (*P < 0.001).
Homozygous Ryr1TM/TM mice exhibit increased sensitivity to isoflurane
A subset of patients with RYR1 mutations experience MH when exposed to volatile anaesthetics such as halothane and isoflurane (29). However, the MH status of most patients with recessive RYR1 myopathy is unknown. Given that knock-in mice with known MH-associated RYR1 mutations recapitulate this enhanced sensitivity to volatile anaesthetics (21,23,30,31), we tested whether adult 7-month-old Ryr1WT/TM or Ryr1TM/TM mice exhibit a hyperthermic MH response during 25 min exposure to isoflurane (similar studies in Ryr1TM/Indel mice was precluded by the early lethality of these mice). Ryr1WT/WT and MH-susceptible Ryr1WT/YS mice were used as negative and positive controls, respectively. Change in core temperature was used as an index of the animal’s relative response to isoflurane exposure (Fig. 9A). While the change in core temperature during isoflurane exposure was not different between Ryr1WT/WT and Ryr1WT/TM mice, Ryr1TM/TM mice exhibited a significant increase in core temperature during isoflurane exposure (Fig. 9B). In fact, the maximal increase in core temperature from baseline during 25 min isoflurane exposure was similar in magnitude to that observed in MH-susceptible Ryr1WT/YS mice. However, the rate of core temperature increase was faster in Ryr1WT/YS mice. Moreover, all three Ryr1WT/YS mice tested died during isoflurane exposure, while only one of three Ryr1TM/TM mice died at the end of the experiment. Thus, Ryr1TM/TM mice, but not Ryr1WT/TM mice, exhibit a potentially lethal hyperthermic response during isoflurane exposure.
Figure 9.
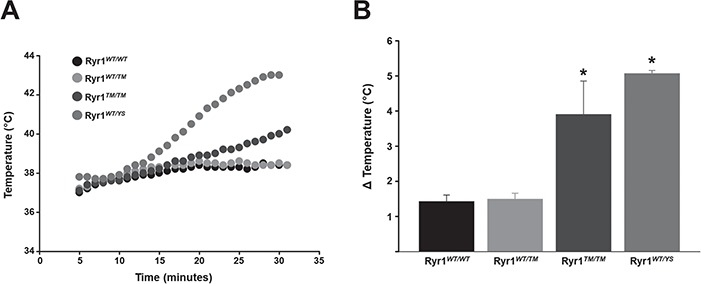
Ryr1TM/TM mice exhibit a hyperthermic response during isoflurane exposure. (A) Representative time course of core temperature changes in adult Ryr1WT/WT, Ryr1WT/TM, Ryr1TM/TM and Ryr1WT/YS mice during 25 min exposure to isoflurane. (B) Average (±SEM) maximal change in core temperature during isoflurane exposure for each of the genotypes tested. Data are shown as mean ± SEM from 4 Ryr1WT/WT, 11 Ryr1WT/TM, 3 Ryr1TM/TM and 3 Ryr1WT/YS mice (*P < 0.05 compared to Ryr1WT/WT).
Discussion
We generated and characterized the first mouse model of severe, recessive RYR1 RM (Ryr1TM/Indel mice). This model matches the most common genetic subtype of recessive RYR1 RM (i.e. a missense allele coupled with a hypomorphic allele) (11,14). The severe phenotype of Ryr1TM/Indel mice (early onset, muscle weakness/hypotrophy and premature death) resembles the clinical features seen in with such patients with severe, recessive RYR1 RM. Thus, Ryr1TM/Indel mice represent an ideal model for elucidating pathomechanisms related to recessive RYR1 RM and provide a powerful platform for testing of potential therapies for pre-clinical efficacy.
One of the most striking findings observed in the Ryr1TM/Indel mice is a marked reduction in RyR1 protein levels in skeletal muscle. With one missense and one frameshift allele, one would have predicted a 50% reduction in transcript and protein. While a ~ 50% reduction in RYR1 mRNA was observed in muscle from both Ryr1Indel/+ and Ryr1TM/Indel mice, ~ 80% reduction in RyR1 protein was observed only in Ryr1TM/Indel mice. Our finding of a ~ 50% reduction in RYR1 transcript with a minimal reduction in RYR1 protein in Ryr1Indel/+ mice is similar to that reported recently in mice with a single RYR1 null allele (~60% reduction in RYR1 transcript with only a ~ 15% reduction in RYR1 protein) (32). The precise reason for the marked (~80%) reduction in RYR1 protein level in Ryr1TM/Indel mice will require additional experimentation. However, we hypothesize that this likely involves a combination of a reduction in RYR1 transcript being coupled with increased misfolding/instability and degradation of RyR1 homotetramers formed from T4709M mutant subunits. Of relevance, changes in protein levels beyond what would be predicted by genomic mutation have been documented in patients with both compound heterozygous missense mutations and in patients with recessive RYR1 RM genotypes similar to our mouse (missense + nonsense) (13,14,24,33). Importantly, we previously established a significant correlation between RyR1 protein reduction in skeletal muscle and clinical severity in RYR1 RM patients (14). Thus, Ryr1TM/Indel mice provide a unique opportunity to establish why these protein changes occur and to develop strategies for preventing or reversing these.
A second important observation is that Ryr1TM/Indel mice exhibit a severe, rapidly progressive lethal phenotype. Ryr1TM/Indel mice are visually distinguishable from their Ryr1WT/WT, Ryr1WT/TM and Ryr1WT/Indel littermates starting at 14 days of age due to their smaller size. The Ryr1TM/Indel mice remain distinctly small, become progressively less active over the next 2–3 weeks, develop progressive spine curvature and die with a median age of 42 days. Physiologically, the mice exhibit reduced electrically evoked Ca2+ release and specific force generation, consistent with muscle weakness, likely due to a marked reduction in RyR1 (and DHPR) expression. The mice appear to die from respiratory compromise, likely through a combination of intrinsic respiratory muscle weakness and restrictive lung disease due to spine curvature. All of these features are consistent with clinical observations of patients with recessive RYR1 RM, who as a group typically exhibit a congenital/early childhood onset of symptoms including diffuse muscle weakness, progressive spinal deformities, respiratory failure and (in some cases) premature death (6,11,14,34,35).
The phenotypic features of Ryr1TM/Indel mice stand in stark contrast to those seen in other mouse models of RYR1 RM, which include Ryr1 knockout (dyspedic) mice and multiple dominant Ryr1 knock-in mouse lines (15,20–23). Dyspedic mice are embryonic lethal (in the homozygous state), while heterozygotes Ryr1-null mice develop normally, are fertile and do not exhibit an overt baseline phenotype (26,27). Of note, no patients have yet been described with two nonsense alleles, and humans with a single null allele also do not exhibit a myopathy. In terms of the Ryr1 knock-in mice generated, they possess dominant mutations observed in patients with MH susceptibility, environmental heat stress and/or central core disease (20–23). None of these models in the heterozygous condition exhibit early lethality or even an early-onset myopathy. The knock-in models of MH and environmental heat stress (R163C, G2434R and T4826I mice) either lack a myopathic phenotype or exhibit a relatively mild, late-onset myopathy (Y522S or YS mice). Mice with an I4898T Ryr1 mutation, which is relatively frequently observed in patients with central core disease, have been generated by two different groups (on different backgrounds) (18,20). Patients with the I4898T mutation exhibit an extremely broad range of clinical symptoms, from severe weakness and death in infancy, to a very mild late-onset weakness and normal life span (36,37). In concert with this, while most I4898T knock-in mice exhibit very mild weakness in the absence of an overt phenotype and normal survival, a small subset of these mice have been reported to exhibit a severe myopathy in adulthood (18,20).
The fact that the existing RYR1 knock-in mouse models lack obvious and severe phenotypic abnormalities has presented a challenge for drug development for patients with RYR1 RM. Nevertheless, these models have been used to identify aberrant cellular pathways associated with Ryr1 disease mutations and led to the identification of 4-PBA and N-acetylcysteine as potential treatments for RYR1 RM (15,18,38). In fact, based on mouse studies and supporting data from zebrafish and patient cell lines (15,16), N-acetylcysteine recently entered clinical trial for ambulant RYR1 RM patients. The trial has been completed, and the results are forthcoming. However, no study has examined long term efficacy, and the lack of an overt, early-onset phenotype in the existing animal models has hindered dose optimization and therapeutic window studies. Moving forward, as Ryr1TM/Indel mice exhibit an obvious, early-onset phenotype (reduced body size) and early lethality, these phenotypes can be used as robust and reproducible outcome measures ideal for pre-clinical efficacy studies and thus used to marshal dose optimization studies and examine efficacy at different disease stages.
A final consideration related to the Ryr1TM/Indel mice is their histological phenotype. RYR1 RM patients have historically been defined based largely on biopsy muscle pathology findings. For heterozygous/de novo/dominant mutations, a strong correlation is found with central core disease pathology, and such patients tend to exhibit a consistent clinical picture, though this genotype–phenotype correlation is not predictive of clinical severity (11,39,40). On the other hand, patients with recessive RYR1 RM exhibit a broad range of histopathology with multi-minicore myopathy, centronuclear myopathy and congenital fibre-type disproportion being the most common (11,14). In the presence of at least one hypomorphic allele, the histology typically does not include cores. Importantly, in a large cohort study (14), we did not find an association between clinical severity and histological subtype with recessive RYR1 RM, with all variations of histological patterns being observed in patients with severe disease.
In a similar manner, the muscle pathology of Ryr1TM/Indel mice also does not conform to a specific histologic pattern. The most striking finding is reduced myofibre size in both Type I and Type II fibres. As there is no obvious predominance of Type I fibres, Ryr1TM/Indel mice do not meet the definition of congenital fibre-type disproportion (41,42). While a modest increase in central nuclei is observed, this was found in < 1% of fibres. Typically, 5–10% of fibres with central nuclei are observed in mouse models of other genetic subtypes of centronuclear myopathy. Lastly, we did not detect cores or core-like lesions using oxidative stains. This is consistent with our observation that patients with hypomorphic alleles tend not to exhibit cores on pathology (14). The significance of the fact that Ryr1TM/Indel mice do not exhibit a specific histopathology type is not clear. However, the finding that myofibre hypotrophy is consistently observed in muscle from both Ryr1TM/Indel mice and recessive RYR1 RM patients suggests that observations made in these mice should hold relevance for the majority of histological patterns in recessive RYR1 RM individuals. Consistent with this, missense/hypomorphic allele combinations are observed in patients with all different subtypes of pathology other than central core disease.
As mentioned above, myofibre hypotrophy is a consistent muscle pathology observed in both Ryr1TM/Indel mice and patients with RYR1 RM. While clearly a highly relevant finding, the mechanisms that underlie myofibre hypotrophy in Ryr1TM/Indel mice and RYR1 RM patients (or other similar myopathies) remain unknown. Aberrant intracellular calcium dynamics has been proposed as one possible initiating trigger, potentially through activation of downstream epigenetic/transcriptional changes (43,44), though how this works at the mechanistic level is not known. Alternations in sonic hedgehog signalling have been described in a loss of function ryr1 mutant in zebrafish (45), and such changes represent a plausible hypothesis for hypotrophy in mammalian muscle. Moving forward, Ryr1TM/Indel mice provide an excellent opportunity to explore these potential pathomechanisms in detail. In addition, therapeutic strategies aimed at increasing myofibre size have been considered as an intervention for other muscle diseases and could also be tested in Ryr1TM/Indel mice. The most frequently considered strategy involves myostatin inhibition. However, results of myostatin inhibition for treating other congenital myopathies have been mixed, with some positive findings in the myotubularin knockout mouse model of centronuclear myopathy and negative results in a model of nemaline myopathy (46–48). Thus, it is difficult a priori to predict the potential impact of such therapy in RYR1 RM.
In summary, we generated and characterized the first mouse model of severe, early-onset recessive RYR1 RM (Ryr1TM/Indel mice). These mice exhibit clearly observable, early-onset phenotypes, premature mortality and a consistent pattern of myofibre hypotrophy. Thus, Ryr1TM/Indel mice will be invaluable for studying pathomechanisms and pre-clinical drug development for this prevalent and severe form of muscle disease in children for which there currently is no treatment.
Materials and Methods
Animal care and treatment
All animal procedures were performed in compliance with the Animals for Research Act of Ontario and the Guidelines of the Canadian council on Animal Care. The Toronto Centre for Phenogenomics (TCP) Animal Care Committee reviewed and approved all procedures on the Dowling lab Animal Use Protocol for the mouse housed at the TCP. The University Committee on Animal Resources Committee at the University of Rochester reviewed and approved all animal procedures conducted at the University of Rochester. Mice were maintained under proper regulations: clean environment, regulated temperature and light cycles, unrestricted water access and proper food supply. Mouse tails were clipped, and mice were weaned and handled according to standard protocol, and all routine care, experiments and necessary euthanasia were performed following the international guidelines and animal protocols approved at the University of Toronto, TCP and the University of Rochester.
Generation and genotyping of Ryr1 strains
A CRISPR/Cas9 gene-editing strategy was used to generate separate Ryr1Indel/+ and Ryr1TM/+ lines of mice (on congenic C57BL6 background).
Ryr1 Indel/+ mice were generated according to protocol at TCP. The allele was generated by injecting Cas9 endonuclease and a guide RNA with the spacer sequence ACGGCGTGTTGAGCACCAGG and a single-strand oligonucleotide encoding the changes c.14117C>T, p.T4706M and the silent change c.14100C>T to inactivate the protospacer adjacent motif (PAM) sequence and prevent re-cutting of the repaired allele in ENSMUST00000179893. Subsequent non-homologous end joining-mediated repair introduced an indel consisted of a 16 bp deletion from Chr7:29013738 to 29013753 in C57BL6 background. Interbreeding Ryr1Indel/+ mice resulted in birth lethality of homozygous Ryr1Indel/Indel mice, similar to that observed for homozygous Ryr1 knockout (dyspedic) mice. The Ryr1Indel/+ mice are available from The Jackson Laboratory as Stock No. 033337.
RyrTM/+ mice were generated according to protocol at TCP. This allele was generated at TCP by injecting Cas9 endonuclease and a guide RNA with the spacer sequence ACGGCGTGTTGAGCACCAGG and a single-strand oligonucleotide encoding the changes c.14117C>T,p.T4706M and the silent change c.14100C>T to inactivate the PAM sequence and prevent re-cutting of the repaired allele in ENSMUST00000179893 in the C57BL background. Interbreeding Ryr1TMl/+ mice resulted in homozygous Ryr1TM/TM mice that exhibit no overt phenotype and live into adulthood. The Ryr1TM/+ mice are available from The Jackson Laboratory as Stock No. 033336.
Cryopreservation
Muscle tissue from TA was dissected and mounted into small balsawood pieces previously frozen with drops of optimal cutting temperature compound (Tissue-Tek; ThermoFisher, Waltham, MA). Mounted muscle tissues were then flash frozen in isopentane at −55°C. The 8 μm cross sections of flash frozen TA muscle were cut and mounted on Superfrost Plus slides (ThermoFisher, Waltham, MA) using a Leica cryostat (Leica Microsystems) at −20°C.
Tissue extraction
Total protein lysates were extracted from TA muscle of male and female mice. Until processing, TA was snap frozen in liquid nitrogen and stored at −80°C. The muscle was minced and homogenized for 3 min using the TissueLyserII (Qiagen, Hilden, Germany) in 1X cell lysis buffer supplemented with protease and phosphatase inhibitors. Lysates were chilled at 4°C for 10 min, then centrifuged at 12000× g for 10 min at 4°C. Supernatants were collected, and protein concentration determined using the Pierce™ BCA protein assay kit (Thermo Fisher Scientific).
Quantitative RT-PCR
Quadriceps muscles were excised and snap frozen in liquid nitrogen. Total RNA was isolated using Trizol reagent according to the manufacturer’s protocol (Thermo Fisher Scientific). RNA (1 μg) was treated with DNase (Thermo Fisher Scientific), and then cDNA libraries were created using Super Script III First-Strand Synthesis System primed with oligo (dT) (Thermo Fisher Scientific) as previously described (49). Ryr1 and Cacna1s transcript levels were determined by semiquantitative PCR using end fluorescein (6-FAM)-labeled forward primers (Integrated DNA Technologies, Coralville, IA) using 10 ng cDNA prepared as described above. Reactions were quantified at 32 cycles for Cacna1s and 35 cycles for Ryr1. Gapdh was used as a loading control within the linear range with primer concentrations diluted 10× compared to that for Cacna1s or Ryr1 primers. Primers used were
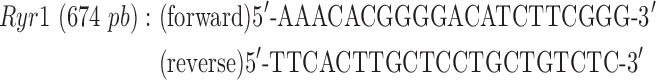 |
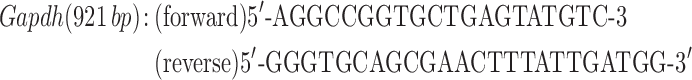 |
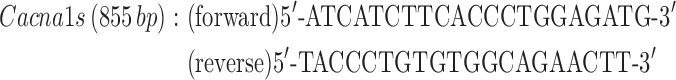 |
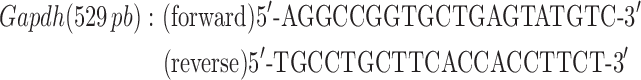 |
Western blot analysis
Protein was isolated in RIPA buffer from TA muscle of Ryr1TM/Indel mice and age-matched littermates. A total of 5 g of total protein was run on either 4.5% SDS-acrylyamide gel for RyR1, 7.5% SDS-acrylamide gel for DHPR and SERCA or 12% SDS-acrylamide gel for GAPDH. Blots were run for 2–2.5 h at 100 V and transferred overnight at 25 V. The membrane was blocked in 3% bovine serum albumin (BSA) in Tris Buffered Saline with Tween 20 (TBST) for 1 h at room temperature before incubating with primary antibodies overnight at 4°C for RyR1 (Mouse Anti-Ryr1 monoclonal supernatant of clone 34C from the Developmental Studies Hybridoma Bank) at 1:300 dilution DHPR (Mouse anti CaV1.1 from Thermo Fisher Scientific, Cat. #MA3-920) at 1:500 dilution, SERCA (Rabbit anti SERCA1/2/3 from Santa Cruz Biotechnology Cat No. sc30110) at 1:10000 dilution and GAPDH (Mouse anti GAPDH from Thermo Fisher Cat. No. AM 4300) at 1:50000 dilution all in 3% BSA in TBST. After three washes in TBST, blots were incubated with Anti-Mouse or Anti Rabbit IgG-800 at 1:10000 dilution in 5% nonfat milk, in TBST. Samples were visualized on the Odyssey Infra-red imager from LiCor. Indirect analysis of protein expression was performed using the Image Studio software from LiCor.
Histology analysis
Flash-frozen muscle tissue in isopentane from TA, 8 μm cross sections, was stained with Mayer’s H/E or SDH. Images were captured with an Infinity1 camera (Lumenera Corp., Ottawa, CA) using an Olympus BX43 light microscope. Quantification of the number of centrally nucleated fibres and myofibre size was performed manually from H/E photographs taken at 20× magnification.
Immunofluorescence
Mounted TA sections were washed and then blocked for 30 min in blocking buffer [1% Tris-buffered saline (TBS) solution, 0.1% BSA, 10% goat serum, 0.1% Triton X-100 and 0.1% Tween-20 (pH 7.9)]. Slides were then incubated overnight at 4°C with primary antibodies against myosin isotypes and with dystrophin at 1:150 and 1:1000, respectively. Secondary antibody (anti-rabbit Alexa Flour® 555 1:1000, Life Technologies, Carlsbad, CA) was applied for 1 h at room temperature the following day. Slides were washed several times with wash buffer (1% TBS solution, 0.1% Tween-20 and 0.1% Triton X-100) to remove excess secondary antibody and then mounted with VECTASHIELD and 4′,6-diamidino-2-phenylindole (Vectorlabs, Burlingame, CA). Micrographs were captured with an Infinity1 camera (Lumenera Corporation, Ottawa, CA) at 20× magnification with eponymous software visualized through an Olympus BX43 light microscope (Olympus, Center Valley, PA).
Immunostaining
RyR1 immunostaining was performed using routine immunohistochemistry on frozen sections using acetone fixation at −20°C for 20 min. Sections were blocked in 1% TBS solution and 10% goat serum and incubated in the primary RyR1 antibody (Abcam 2868 34C) at 1:50 for 1 h at 37°C. Biotinylated secondary antibody 1:100 (ABC kit; Vector Laboratories) was applied for 30 min at 37°C and incubated in the Avidin-Biotin detection system for 30 min at 37°C. Signal was detected using 3,3'-diaminobenzidine, counterstained in haematoxylin and mounted with Peramount (Thermo Fisher Scientific).
Ex vivo EDL muscle contractility measurements
Ex vivo assessment of muscle force production was made in intact, excised EDL muscles. Mice were anaesthetized by intra-peritoneal injection of anaesthetic (50). EDL muscles were isolated, tied using 4–0 surgical suture, excised and then attached to a servo motor and force transducer (Aurora Scientific, Ontario, CA) between two platinum electrode plates in a chamber continuously perfused with oxygenated Ringer’s solution. Optimal stimulation level and muscle length (L0) were determined using a series of 1 Hz twitch stimulation trains while stretching the muscle to a length that generated maximal force (F0). After establishing L0, muscles were first equilibrated using three tetani (500 ms, 150 Hz) given at 1 min intervals. After 3 min of additional equilibration, muscles were subjected to a standard force frequency protocol (from 1 to 250 Hz). All muscle contractility experiments were carried out at 30°C. Muscle force was recorded using Dynamic Muscle Control software (Aurora Scientific, Ontario, CA) and analysed using a combination of both Dynamic Muscle Analysis (Aurora Scientific, Ontario, CA) and Clampfit 10.0 (Molecular Devices, San Jose, CA) software. Specific force was calculated by normalizing the absolute force to the physiological cross sectional area as previously described (51,52).
Isolation of single FDB muscle fibres
Single FDB muscle fibres were dissociated from the footpad of mice by enzymatic digestion with collagenase A (1 mg/ml) in regular rodent Ringer solution (in mm: 146 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2 and 10 HEPES, pH 7.4) with gentle agitation for 1 h at 37°C. Single FDB muscle fibres were then plated on 35 mm glass-bottomed dishes (MatTek Corp., Ashland, MA) and allowed to settle for 20 min before experimentation.
Electrically evoked Ca2+ release in single FDB fibres
FDB fibres were loaded with 4 μm mag-fluo-4 for 20 min at room temperature followed by washout for 20 min in regular rodent Ringer supplemented with 25 μm benzyl-p-toluene sulfonamide, a skeletal muscle myosin inhibitor used to minimize movement. FDB fibres were stimulated with a series of electrically evoked twitch stimulations (1 Hz) or five consecutive tetani (500 ms at 100 Hz) using an extracellular electrode placed adjacent to the cell of interest. Mag-fluo-4 was excited at 480 ± 15 nm using an Excite epifluorescence illumination system (Nikon Instruments, Melville, NY), and fluorescence emission at 535 ± 30 nm was monitored with a 40× oil objective and a photomultiplier detection system (Photon Technologies Inc., Birmingham, NJ). Relative changes in mag-fluo-4 fluorescence from baseline (F/F0) were recorded using Clampex 9.0 software (Molecular Devices, San Jose, CA).
Temperature-dependent intracellular resting Ca2+ measurements
Resting intracellular Ca2+ concentration was measured in acutely isolated FDB fibres using fura-2 (Invitrogen, Carlsbad, CA) in a temperature-controlled chamber (17). Fura-2 was excited at 340 nm and 380 nm, and images of FDB fibres were collected using a charge-coupled device camera connected to a TILL monochromator (TILL Photonics Inc., Munich, Germany). The fibres were first imaged at room temperature (22°C), and then the temperature of the bathing solution was raised to 37°C. The same fibres were then imaged again at 37°C for paired statistical comparison. Ratio images of 340/380 emission were created using TILLvisION software (TILL Photonics Inc.) and analysed offline using ImageJ software. Ca2+ concentrations were calculated using a calibration curve for fura-2 as described previously (17).
Isoflurane exposure
Mice were exposed to isoflurane levels sufficient to induce stage 3 anaesthesia followed by continuous controlled delivery of isoflurane for the duration of the experiment. Following anaesthesia, a rectal thermometer was inserted, and mice were placed on pre-warmed ‘Hot Hands’ in order to bring their core temperature to 37°C at the beginning of the experiment. Core temperature was then continuously measured every minute for 30 min. If whole body contractures and death were observed, the rectal probe was immediately removed and the time of death recorded.
Statistics
All data are presented as mean ± standard error of the mean. Differences between two groups were assessed by paired or unpaired two-tailed student’s t-test as appropriate. Analysis of variance followed by Tukey post-hoc test was used for multiple comparisons.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to thank Dr Susan Hamilton for providing access to the Ryr1WT/YS mice used in this study.
Conflict of Interest statement. None declared.
Funding
RYR-1 Foundation (to R.T.D. and J.J.D.); Muscular Dystrophy Association (grant number 380397 to J.J.D. and R.T.D); Canadian Institutes for Health Research (363863 to J.J.D.); National Institutes of Health (AR053349 to R.T.D.).
References
- 1. Lawal T.A., Todd J.J. and Meilleur K.G. (2018) Ryanodine receptor 1-related myopathies: diagnostic and therapeutic approaches. Neurotherapeutics, 15, 885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gonorazky H.D., Bonnemann C.G. and Dowling J.J. (2018) The genetics of congenital myopathies. Handb. Clin. Neurol., 148, 549–564. [DOI] [PubMed] [Google Scholar]
- 3. Jungbluth H., Treves S., Zorzato F., Sarkozy A., Ochala J., Sewry C., Phadke R., Gautel M. and Muntoni F. (2018) Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat. Rev. Neurol., 14, 151–167. [DOI] [PubMed] [Google Scholar]
- 4. Santulli G., Lewis D.R. and Marks A.R. (2017) Physiology and pathophysiology of excitation–contraction coupling: the functional role of ryanodine receptor. J. Muscle Res. Cell Motil., 38, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jungbluth H., Dowling J.J., Ferreiro A., Muntoni F. and Consortium, R.Y.R.M (2016) 217th ENMC International Workshop: RYR1-related myopathies, Naarden, The Netherlands, 29–31 January 2016. Neuromuscul. Disord., 26, 624–633. [DOI] [PubMed] [Google Scholar]
- 6. Colombo I., Scoto M., Manzur A.Y., Robb S.A., Maggi L., Gowda V., Cullup T., Yau M., Phadke R., Sewry C. et al. (2015) Congenital myopathies: natural history of a large pediatric cohort. Neurology, 84, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amburgey K., McNamara N., Bennett L.R., McCormick M.E., Acsadi G. and Dowling J.J. (2011) Prevalence of congenital myopathies in a representative pediatric United States population. Ann. Neurol., 70, 662–665. [DOI] [PubMed] [Google Scholar]
- 8. Dowling J.J., North K.N., Goebel H.H. and Beggs A.H. (2015) In Darras B.T., Jones H.R., Ryan M.M. and De Vivo D.C. (eds), Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician’s Approach. Elsevier, Butterwörth Heinemann, Philadelphia, PA. [Google Scholar]
- 9. Dowling J.J., Gonorazky H.D., Cohn R.D. and Campbell C. (2018) Treating pediatric neuromuscular disorders: the future is now. Am. J. Med. Genet. A, 176, 804–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maggi L., Scoto M., Cirak S., Robb S.A., Klein A., Lillis S., Cullup T., Feng L., Manzur A.Y., Sewry C.A. et al. (2013) Congenital myopathies—clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul. Disord., 23, 195–205. [DOI] [PubMed] [Google Scholar]
- 11. Klein A., Lillis S., Munteanu I., Scoto M., Zhou H., Quinlivan R., Straub V., Manzur A.Y., Roper H., Jeannet P.Y. et al. (2012) Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum. Mutat., 33, 981–988. [DOI] [PubMed] [Google Scholar]
- 12. Jungbluth H., Sewry C.A. and Muntoni F. (2011) Core myopathies. Semin. Pediatr. Neurol., 18, 239–249. [DOI] [PubMed] [Google Scholar]
- 13. Zhou H., Rokach O., Feng L., Munteanu I., Mamchaoui K., Wilmshurst J.M., Sewry C., Manzur A.Y., Pillay K., Mouly V. et al. (2013) RyR1 deficiency in congenital myopathies disrupts excitation-contraction coupling. Hum. Mutat., 34, 986–996. [DOI] [PubMed] [Google Scholar]
- 14. Amburgey K., Bailey A., Hwang J.H., Tarnopolsky M.A., Bonnemann C.G., Medne L., Mathews K.D., Collins J., Daube J.R., Wellman G.P. et al. (2013) Genotype–phenotype correlations in recessive RYR1-related myopathies. Orphanet J. Rare Dis., 8, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Durham W.J., Aracena-Parks P., Long C., Rossi A.E., Goonasekera S.A., Boncompagni S., Galvan D.L., Gilman C.P., Baker M.R., Shirokova N. et al. (2008) RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell, 133, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dowling J.J., Arbogast S., Hur J., Nelson D.D., McEvoy A., Waugh T., Marty I., Lunardi J., Brooks S.V., Kuwada J.Y. et al. (2012) Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain, 135, 1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lanner J.T., Georgiou D.K., Dagnino-Acosta A., Ainbinder A., Cheng Q., Joshi A.D., Chen Z., Yarotskyy V., Oakes J.M., Lee C.S. et al. (2012) AICAR prevents heat-induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat. Med., 18, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee C.S., Hanna A.D., Wang H., Dagnino-Acosta A., Joshi A.D., Knoblauch M., Xia Y., Georgiou D.K., Xu J., Long C. et al. (2017) A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat. Commun., 8, 14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takekura H., Nishi M., Noda T., Takeshima H. and Franzini-Armstrong C. (1995) Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc. Natl. Acad. Sci. USA, 92, 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zvaritch E., Depreux F., Kraeva N., Loy R.E., Goonasekera S.A., Boncompagni S., Kraev A., Gramolini A.O., Dirksen R.T., Franzini-Armstrong C. et al. (2007) An Ryr1I4895T mutation abolishes Ca2+ release channel function and delays development in homozygous offspring of a mutant mouse line. Proc. Natl. Acad. Sci. U. S. A., 104, 18537–18542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang T., Riehl J., Esteve E., Matthaei K.I., Goth S., Allen P.D., Pessah I.N. and Lopez J.R. (2006) Pharmacologic and functional characterization of malignant hyperthermia in the R163C RyR1 knock-in mouse. Anesthesiology, 105, 1164–1175. [DOI] [PubMed] [Google Scholar]
- 22. Boncompagni S., Rossi A.E., Micaroni M., Hamilton S.L., Dirksen R.T., Franzini-Armstrong C. and Protasi F. (2009) Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc. Natl. Acad. Sci. U. S. A., 106, 21996–22001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lopez J.R., Kaura V., Diggle C.P., Hopkins P.M. and Allen P.D. (2018) Malignant hyperthermia, environmental heat stress, and intracellular calcium dysregulation in a mouse model expressing the p.G2435R variant of RYR1. Br. J. Anaesth., 121, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou H., Brockington M., Jungbluth H., Monk D., Stanier P., Sewry C.A., Moore G.E. and Muntoni F. (2006) Epigenetic allele silencing unveils recessive RYR1 mutations in core myopathies. Am. J. Hum. Genet., 79, 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bevilacqua J.A., Monnier N., Bitoun M., Eymard B., Ferreiro A., Monges S., Lubieniecki F., Taratuto A.L., Laquerriere A., Claeys K.G. et al. (2011) Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol. Appl. Neurobiol., 37, 271–284. [DOI] [PubMed] [Google Scholar]
- 26. Nakai J., Dirksen R.T., Nguyen H.T., Pessah I.N., Beam K.G. and Allen P.D. (1996) Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature, 380, 72–75. [DOI] [PubMed] [Google Scholar]
- 27. Takeshima H., Iino M., Takekura H., Nishi M., Kuno J., Minowa O., Takano H. and Noda T. (1994) Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature, 369, 556–559. [DOI] [PubMed] [Google Scholar]
- 28. Capote J., Bolanos P., Schuhmeier R.P., Melzer W. and Caputo C. (2005) Calcium transients in developing mouse skeletal muscle fibres. J. Physiol., 564, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Litman R.S., Griggs S.M., Dowling J.J. and Riazi S. (2018) Malignant hyperthermia susceptibility and related diseases. Anesthesiology, 128, 159–167. [DOI] [PubMed] [Google Scholar]
- 30. Chelu M.G., Goonasekera S.A., Durham W.J., Tang W., Lueck J.D., Riehl J., Pessah I.N., Zhang P., Bhattacharjee M.B., Dirksen R.T. et al. (2006) Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J., 20, 329–330. [DOI] [PubMed] [Google Scholar]
- 31. Yuen B., Boncompagni S., Feng W., Yang T., Lopez J.R., Matthaei K.I., Goth S.R., Protasi F., Franzini-Armstrong C., Allen P.D. et al. (2012) Mice expressing T4826I-RYR1 are viable but exhibit sex- and genotype-dependent susceptibility to malignant hyperthermia and muscle damage. FASEB J., 26, 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cacheux M., Blum A., Sebastien M., Wozny A.S., Brocard J., Mamchaoui K., Mouly V., Roux-Buisson N., Rendu J., Monnier N. et al. (2015) Functional characterization of a central core disease RyR1 mutation (p.Y4864H) associated with quantitative defect in RyR1 protein. J. Neuromuscul. Dis., 2, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dowling J.J., Lillis S., Amburgey K., Zhou H., Al-Sarraj S., Buk S.J., Wraige E., Chow G., Abbs S., Leber S. et al. (2011) King–Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul. Disord., 21, 420–427. [DOI] [PubMed] [Google Scholar]
- 34. Wilmshurst J.M., Lillis S., Zhou H., Pillay K., Henderson H., Kress W., Muller C.R., Ndondo A., Cloke V., Cullup T. et al. (2010) RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol., 68, 717–726. [DOI] [PubMed] [Google Scholar]
- 35. Jungbluth H., Sewry C., Brown S.C., Manzur A.Y., Mercuri E., Bushby K., Rowe P., Johnson M.A., Hughes I., Kelsey A. et al. (2000) Minicore myopathy in children: a clinical and histopathological study of 19 cases. Neuromuscul. Disord., 10, 264–273. [DOI] [PubMed] [Google Scholar]
- 36. Lynch P.J., Tong J., Lehane M., Mallet A., Giblin L., Heffron J.J., Vaughan P., Zafra G., MacLennan D.H. and McCarthy T.V. (1999) A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc. Natl. Acad. Sci. U. S. A., 96, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hernandez-Lain A., Husson I., Monnier N., Farnoux C., Brochier G., Lacene E., Beuvin M., Viou M., Manere L., Claeys K.G. et al. (2011) De novo RYR1 heterozygous mutation (I4898T) causing lethal core-rod myopathy in twins. Eur. J. Med. Genet., 54, 29–33. [DOI] [PubMed] [Google Scholar]
- 38. Michelucci A., Boncompagni S., Canato M., Reggiani C. and Protasi F. (2017) Estrogens protect calsequestrin-1 knockout mice from lethal hyperthermic episodes by reducing oxidative stress in muscle. Oxid. Med. Cell. Longev., 2017, 6936897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murayama T., Kurebayashi N., Ogawa H., Yamazawa T., Oyamada H., Suzuki J., Kanemaru K., Oguchi K., Iino M. and Sakurai T. (2016) Genotype–phenotype correlations of malignant hyperthermia and central core disease mutations in the central region of the RYR1 channel. Hum. Mutat., 37, 1231–1241. [DOI] [PubMed] [Google Scholar]
- 40. Wu S., Ibarra M.C., Malicdan M.C., Murayama K., Ichihara Y., Kikuchi H., Nonaka I., Noguchi S., Hayashi Y.K. and Nishino I. (2006) Central core disease is due to RYR1 mutations in more than 90% of patients. Brain, 129, 1470–1480. [DOI] [PubMed] [Google Scholar]
- 41. Clarke N.F. (2011) Congenital fiber-type disproportion. Semin. Pediatr. Neurol., 18, 264–271. [DOI] [PubMed] [Google Scholar]
- 42. Clarke N.F., Waddell L.B., Cooper S.T., Perry M., Smith R.L., Kornberg A.J., Muntoni F., Lillis S., Straub V., Bushby K. et al. (2010) Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum. Mutat., 31, E1544–E1550. [DOI] [PubMed] [Google Scholar]
- 43. Bachmann C., Noreen F., Voermans N.C., Schar P.L., Vissing J., Fock J.M., Bulk S., Kusters B., Moore S.A., Beggs A.H. et al. (2019) Aberrant regulation of epigenetic modifiers contributes to the pathogenesis in patients with selenoprotein N-related myopathies. Hum. Mutat., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rokach O., Sekulic-Jablanovic M., Voermans N., Wilmshurst J., Pillay K., Heytens L., Zhou H., Muntoni F., Gautel M., Nevo Y. et al. (2015) Epigenetic changes as a common trigger of muscle weakness in congenital myopathies. Hum. Mol. Genet., 24, 4636–4647. [DOI] [PubMed] [Google Scholar]
- 45. Klatt Shaw D., Gunther D., Jurynec M.J., Chagovetz A.A., Ritchie E. and Grunwald D.J. (2018) Intracellular calcium mobilization is required for sonic hedgehog signaling. Dev. Cell, 45, 512–525 e515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tinklenberg J.A., Siebers E.M., Beatka M.J., Fickau B.A., Ayres S., Meng H., Yang L., Simpson P., Granzier H.L. and Lawlor M.W. (2019) Myostatin inhibition using ActRIIB-mFc does not produce weight gain or strength in the nebulin conditional KO mouse. J. Neuropathol. Exp. Neurol., 78, 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tinklenberg J., Meng H., Yang L., Liu F., Hoffmann R.G., Dasgupta M., Allen K.P., Beggs A.H., Hardeman E.C., Pearsall R.S. et al. (2016) Treatment with ActRIIB-mFc produces myofiber growth and improves lifespan in the Acta1 H40Y murine model of nemaline myopathy. Am. J. Pathol., 186, 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lawlor M.W., Read B.P., Edelstein R., Yang N., Pierson C.R., Stein M.J., Wermer-Colan A., Buj-Bello A., Lachey J.L., Seehra J.S. et al. (2011) Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am. J. Pathol., 178, 784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maani N., Sabha N., Rezai K., Ramani A., Groom L., Eltayeb N., Mavandadnejad F., Pang A., Russo G., Brudno M. et al. (2018) Tamoxifen therapy in a murine model of myotubular myopathy. Nat. Commun., 9, 4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wei-Lapierre L., Carrell E.M., Boncompagni S., Protasi F. and Dirksen R.T. (2013) Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun., 4, 2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sabha N., Volpatti J.R., Gonorazky H., Reifler A., Davidson A.E., Li X., Eltayeb N.M., Dall'Armi C., Di Paolo G., Brooks S.V. et al. (2016) PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J. Clin. Invest., 126, 3613–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reifler A., Li X., Archambeau A.J., McDade J.R., Sabha N., Michele D.E. and Dowling J.J. (2014) Conditional knockout of pik3c3 causes a murine muscular dystrophy. Am. J. Pathol., 184, 1819–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.