

Abstract
Celiac disease (CeD) is an immune-mediated enteropathy with a strong genetic component where the main environmental trigger is dietary gluten, and currently a correct diagnosis of the disease is impossible if gluten-free diet (GFD) has already been started. We hypothesized that merging different levels of genomic information through Mendelian randomization (MR) could help discover genetic biomarkers useful for CeD diagnosis. MR was performed using public databases of expression quantitative trait loci (QTL) and methylation QTL as exposures and the largest CeD genome-wide association study conducted to date as the outcome, in order to identify potential causal genes. As a result, we identified UBE2L3, an ubiquitin ligase located in a CeD-associated region. We interrogated the expression of UBE2L3 in an independent data set of peripheral blood mononuclear cells (PBMCs) and found that its expression is altered in CeD patients on GFD when compared to non-celiac controls. The relative expression of UBE2L3 isoforms predicts CeD with 100% specificity and sensitivity and could be used as a diagnostic marker, especially in the absence of gluten consumption. This approach could be applicable to other diseases where diagnosis of asymptomatic patients can be complicated.
Introduction
In the last decade, genome-wide association studies (GWAS) have identified thousands of disease-associated susceptibility variants whose etiological role remains elusive (1). In this context, Mendelian randomization (MR) approaches that confront summary statistics from GWAS with results from expression quantitative trait loci (eQTL) studies have proven useful to detect pleiotropic associations between gene expression and particular traits and also to prioritize genes under GWAS peaks (2,3). More recently, methylation data have been used to propose a plausible mechanism whereby several single nucleotide polymorphism (SNP)–disease associations are mediated by transcription regulation through DNA methylation (4).
Celiac disease (CeD) is a common, immune-mediated enteropathy where alleles encoding human leukocyte antigen (HLA)-DQ2 and -DQ8 molecules account for 40% of disease heritability (5). The main environmental trigger of the disorder is dietary gluten, and being on a gluten-containing diet is necessary for a correct diagnosis of the disease. However, self-reported wheat sensitivity is a rising global phenomenon that is changing the consumption and eating habits and reaches 12.2% and 13% of the general population in Western countries like Italy and the United Kingdom, respectively (6). As a consequence, many will start gluten-free diet (GFD) before medical consultation, therefore hindering the diagnosis of CeD. In those cases, gluten challenge prior to duodenal endoscopy is the only available diagnostic protocol, albeit often non-effective and always invasive and troublesome (7). Other common disorders also present similar diagnostic challenges, like severe reactions during provocation with the causal compound in drug allergy (8) or difficulties to assess airflow limitation in childhood asthma (9). A genetic, constitutive biomarker present also when the disease-triggering insult is absent would be extremely useful for the diagnosis of these type of conditions.
We hypothesized that the overlapping results from methylation and expression-based MR analyses could not only clarify mechanisms underlying SNP–disease associations (4) but also provide genetic biomarkers that could be useful in the diagnosis of latent diseases. Here, we perform the first MR analysis in CeD using publicly available GWAS, eQTL and methylation QTL (mQTL) data and interrogate the resulting candidates in independent expression databases to test their diagnostic potential.
Results
We used MR to infer causal relationships between an exposure (e.g. altered gene expression or methylation) and an outcome (CeD). Since SNP alleles are randomly assigned to each individual, MR assumes that if an exposure is implicated in disease etiology, then the genetic variants associated with the exposure (eQTL/mQTL SNPs) will be associated with disease risk. Thus, confronting eQTL/mQTL (SNP–gene expression/methylation) and GWAS (SNP–disease) results, we can select those genes whose expression or methylation is altered by a SNP allele that in turn has been shown to increase the risk of developing the disease.
Therefore, in order to identify the causative genes driving GWAS associations, we first ran the summary-data-based MR (SMR) software (https://cnsgenomics.com/software/smr) (2) using results from the to-date largest CeD GWAS (9451 cases and 16 434 controls) (5) and from the Consortium for the Architecture of Gene Expression (CAGE) eQTL database (P < 1e-5) (10) (namely, genotype to transcription approach or G2T). We also ran SMR with the top blood mQTLs (P < 1e-5; n = 1366) available at the SMR website (11) to test the association of DNA methylation with CeD and detect possible effects of SNPs on methylation (G2M) or of methylation on the expression of nearby genes (M2T) and finally considered the intersection of these three analyses (G2T∩G2M∩M2T) (Fig. 1A).
Figure 1.
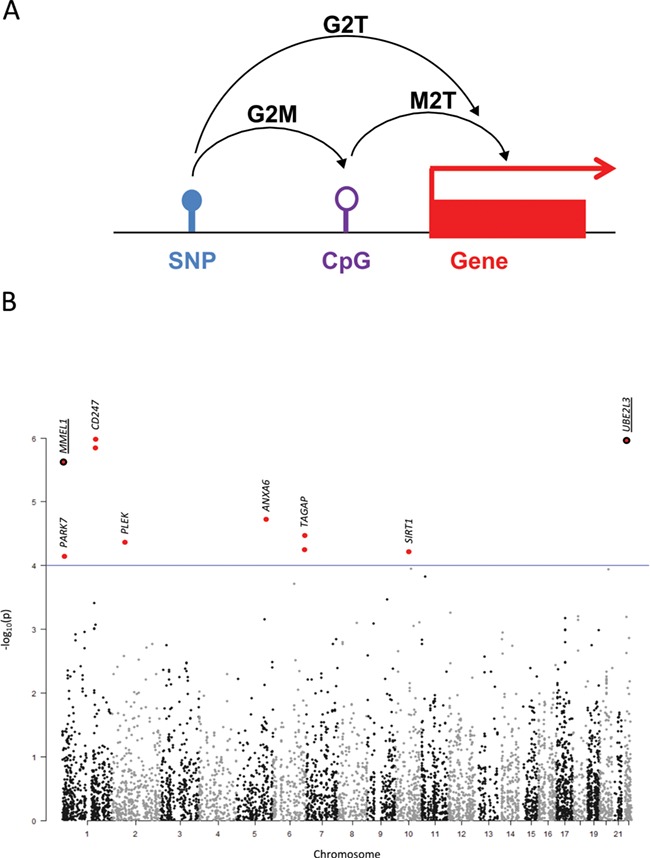
(A) Schematic representation of the SMR approach. (B) Manhattan plot representing the −log10P-values of the G2T-SMR analysis using the CAGE eQTL database. The genomic location of the resulting SNPs are shown in the x-axis while the −log10P-values of the G2T-SMR analysis are depicted in the y-axis. Those SNPs that do not pass the HEIDI cut-off have been excluded from the plot. The eight unique SNPs resulting from the G2T analysis are highlighted in red and marked with closest gene names. When two dots are plotted for the same genomic location, this means that two forms/probes of the same gene have been identified as mediators of the given SNP and CeD. When listed as G2T∩G2M∩M2T overlapping hits in the discovery data set, gene names and dots have been underlined.
The G2T analysis using the CAGE blood eQTL database identified 8 SNPs associated with the expression levels of 10 different Illumina probes representing 8 genes, namely CD247, UBE2L3, MMEL1, ANXA6, TAGAP, PLEK, SIRT1 and PARK7 (Table 1; Fig. 1B; Supplementary Material, Table 1.1). The G2M analysis resulted in 51 cytosine-phosphate-guanine (CpG) sites pleiotropically related to 27 CeD-associated SNPs (Supplementary Material, Table 1.2). On the other hand, the M2T analysis considered methylation of nearby CpG sites to mediate the association between a given SNP and the expression of a particular gene and therefore identified 22 824 CpG–SNP pairs (Supplementary Material, Table 2) whose pleiotropic association could drive the variation at expression level locally.
Table 1.
Summarized results of the G2T approach in whole-blood CAGE eQTLs. b refers to the beta-effect size or log (odds ratio) of the allele frequency/expression related to each SNP in each data set
| probeID | Gene | SNP | chr | bp | b_GWAS | p_GWAS | b_eQTL | p_eQTL | b_SMR | p_SMR |
|---|---|---|---|---|---|---|---|---|---|---|
| ILMN_1676924 | CD247 | rs864537 | 1 | 167 411 384 | −0.141 | 1.01E-07 | 0.344 | 6.22E-34 | −0.411 | 1.08E-06 |
| ILMN_1677877 | UBE2L3 | rs5754217 | 22 | 21 939 675 | 0.152 | 6.92E-07 | 0.917 | 6.88E-156 | 0.166 | 1.13E-06 |
| ILMN_2377669 | CD247 | rs864537 | 1 | 167 411 384 | −0.141 | 1.01E-07 | 0.320 | 1.96E-29 | −0.441 | 1.48E-06 |
| ILMN_1718488 | MMEL1 | rs3748816 | 1 | 2 526 746 | −0.136 | 4.93E-07 | −0.380 | 7.84E-41 | 0.359 | 2.51E-06 |
| ILMN_2326591 | ANXA6 | rs2303038 | 5 | 150 535 013 | 0.134 | 1.10E-05 | 0.530 | 4.47E-59 | 0.252 | 1.88E-05 |
| ILMN_2333774 | TAGAP | rs9295089 | 6 | 159 463 964 | 0.187 | 1.66E-06 | −0.338 | 2.47E-16 | −0.554 | 3.53E-05 |
| ILMN_1795762 | PLEK | rs3816281 | 2 | 68 607 947 | −0.124 | 2.85E-05 | 0.598 | 3.64E-77 | −0.207 | 4.49E-05 |
| ILMN_1676408 | TAGAP | rs9295089 | 6 | 159 463 964 | 0.187 | 1.66E-06 | −0.302 | 1.78E-13 | −0.620 | 5.92E-05 |
| ILMN_1739083 | SIRT1 | rs17712705 | 10 | 69 623 271 | 0.112 | 3.47E-05 | 0.456 | 4.24E-54 | 0.245 | 6.28E-05 |
| ILMN_1744713 | PARK7 | rs12727642 | 1 | 8 046 672 | 0.134 | 3.06E-05 | 0.428 | 2.45E-34 | 0.312 | 7.44E-05 |
We then considered the overlap between the three different studies (G2T∩G2M∩M2T) since it has been suggested that such an approach could provide information not only on the most relevant variation associated with complex traits but also on the putative mechanisms involved. As a result, we identified 10 SNP–gene pairs (Supplementary Material, Table 1.3), namely four different CpG sites mediating the association between rs3748816 on chromosome 1 and the metalloendopeptidase MMEL1 and six CpGs related to rs5754217 on chromosome 22 and the ubiquitin ligase UBE2L3. In particular, three out of the six later CpG sites were located close to the UBE2L3 promoter, while the rest spread across the 5′UTR of the gene (Fig. 2).
Figure 2.
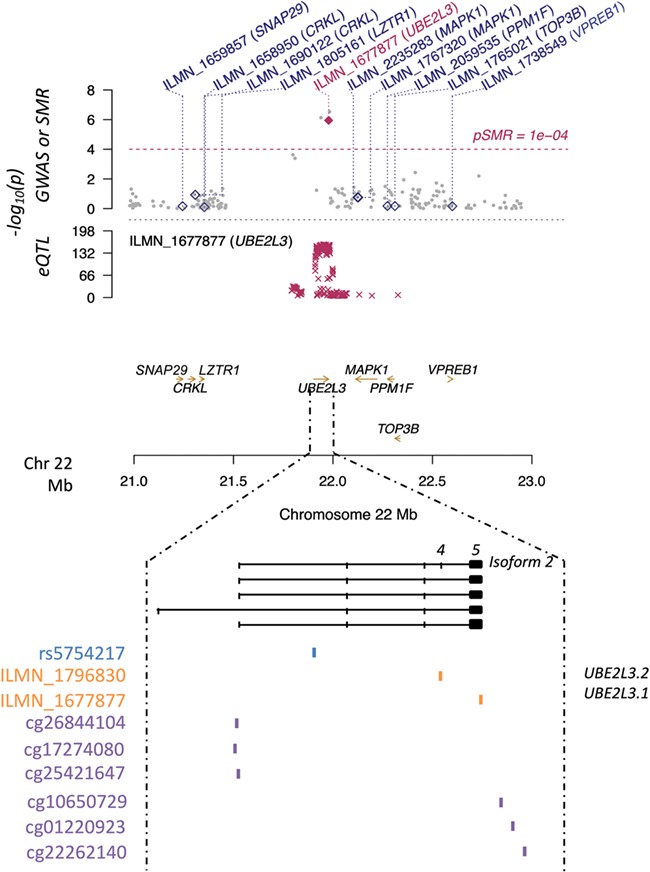
UBE2L3-rs5754217 region on Chr22. The y-axis in the upper panel shows the logarithmic P-values of both the eQTL and GWAS data sets, as well as the one resulting from the G2T-SMR analysis performed with the CAGE whole-blood data set. Diamonds and golden arrows represent genes. Particularly, empty diamonds represent genes in the region whose P-values in the G2T-SMR approach did not reach statistical significance, while the maroon diamond represents the statistically significant hit (UBE2L3). Numbers 4 and 5 above the isoform 2 indicate the UBE2L3 fourth and fifth exons, respectively.
The rs5754217–UBE2L3 pleiotropic association and therefore the putative mediation of UBE2L3 expression levels between the SNP and CeD was further confirmed in the GTEx blood eQTL database (Supplementary Material, Table 3.1) and in the eQTL-Gen database (Supplementary Material, Table 3.2), with a P = 4.09e-05 and a P = 7.68e-07, respectively. Additionally, when the M2T analysis was rerun with each of these two eQTL databases, the G2T∩G2M∩M2T overlap revealed that the rs5754217–UBE2L3–CeD association was again apparently mediated by the methylation of adjacent CpGs. Interestingly, although other SNP–gene pairs were identified in the replication, UBE2L3 was the only hit resulting from the G2T∩G2M∩M2T analysis common to all three data sets studied (Supplementary Material, Table 3). In contrast, the G2T analysis performed in the small intestine eQTLs of the GTEx database did not reveal significant results, suggesting the cell type-specificity of our finding.
Regarding the expression analysis, two different probes interrogate UBE2L3 expression in the celiac peripheral blood mononuclear cell (PBMC) microarray. One is on an exon common to all isoforms (ILMN_1677877, here denoted as UBE2L3.1) and the other is exclusive of an alternative, non-coding transcript (ILMN_1796830 or UBE2L3.2) (Figure 2). Both probes showed a dramatic, albeit opposite alteration of UBE2L3 expression in CeD (P < 1e-4) (Fig. 3A). Interestingly, the minor allele of the rs5754217 SNP is the risk allele in CeD and is positively correlated with expression, a result that is consistent with the upregulation of the coding forms of UBE2L3. However, the expression of the coding isoforms shows strong negative correlation with the expression levels of the non-coding isoform 2, here represented as UBE2L3.2 (Fig. 3B).
Figure 3.
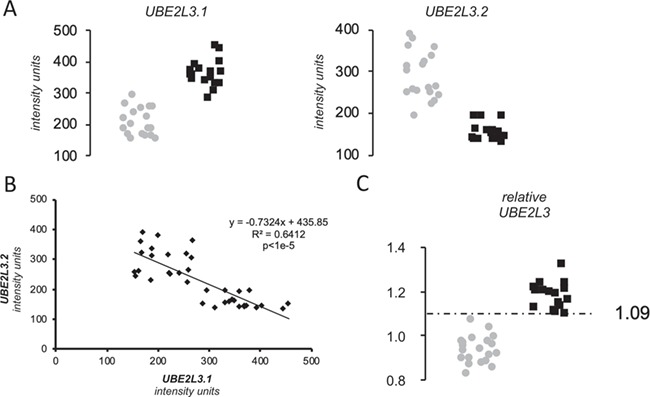
Results of the expression analysis. (A) Expression of UBE2L3 isoforms and ASHA2 in PBMCs of celiac patients on GFD. Gray dots and black squares represent non-celiac controls and CeD patients on GFD, respectively. (B) Pearson correlation across the entire sample set between the expression levels of the isoforms of UBE2L3 reported by the two different Illumina assays. (C) Relative expression score of UBE2L3. Gray dots and black squares represent non-celiac controls and CeD patients on GFD, respectively.
Additionally, the relative expression score of UBE2L3 was able to perfectly distinguish GFD–CeD patients from controls, at a relative expression threshold of 1.09 (Fig. 3C). This resulted in a receiver operating characteristic (ROC) curve with an area under the curve (AUC) value of 1 and therefore 100% specificity and sensitivity in the discrimination capacity of CeD patients in GFD, while each UBE2L3 exons independently showed less prediction potential (AUC of 0.997 and 0.997, respectively) (Fig. 4). Interestingly, none of the probes available to report the UBE2L3 expression in the Affymetrix array of the CD and UC samples was able to distinguish patients from controls, showing AUC values ranging between 0.528–0.699 and 0.452–0.752, respectively (Fig. 4).
Figure 4.
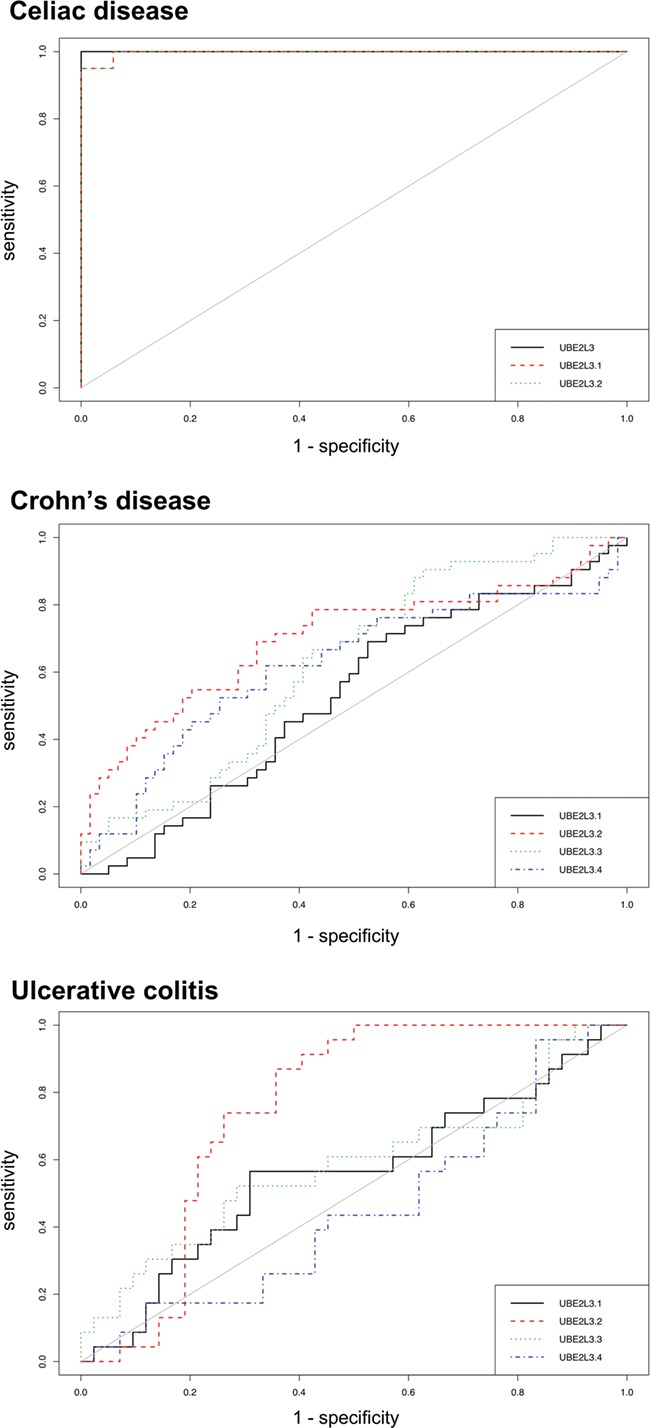
ROC curves with PBMC expression levels of the candidate genes as binary classifiers in CeD, CD and UC.
Finally, we interrogated the rest of the candidate genes resulting from the G2T∩G2M∩M2T approaches conducted separately in each of the discovery and replication eQTL data sets in the celiac PBMC expression study (Supplementary Material, File 1). In summary, CD247 and MMEL1 did not show any differential expression when PBMCs from CeD patients on GFD and from controls were compared. In contrast, some of the probes representing AHSA2, BACH2, CCDC116 and CDC73 were significantly altered in patients, especially the only probe covering AHSA2 (ILMN_1798308, P = 1.63e-05) and one of the BACH2 probes (ILMN_1670695, P = 8.15e-04), although an expression cutoff capable of distinguishing cases from controls could not be established in any of the cases (Supplementary Material, File 1).
Discussion
As far as we know, the number of published MR analyses aiming to discover clinically relevant gene expression signatures is limited. For example, a very interesting study proposed expression-SMR as a valuable tool for the discovery of functionally relevant genes in several diseases (12) but did not test the diagnostic potential of the identified candidates in independent gene expression data sets. Here, we demonstrate that the genetic and epigenetic dysregulation prioritized by MR can be of clinical interest. Furthermore, we propose that when gene expression and methylation of nearby CpG sites are related to the same disease-associated variants and also between each other, one is more likely to identify genes whose expression is selectively altered in a specific disease, since different information layers regulating gene expression are affected specifically in patients.
We present the first MR analysis performed in CeD, and we manage to identify a set of SNPs whose association with CeD could be mediated by the expression of genes in cis and by the methylation levels of nearby CpG positions. For example, CD247, although not altered in patients on GFD, participates in the T-helper 17 cell-signaling pathway and has been described to be diminished in the mucosa of active CeD individuals (13). On its side, AHSA2 is remarkably downregulated in PBMCs from CeD patients on GFD and has previously been shown to be subjected to CeD-associated SNP regulation in the thymus (14), and BACH2, an important immune regulator known to be repressed in CD4+ T cells in active CeD, remains partially silenced after GFD treatment (15). On the other hand, although MMEL1 is not altered in CeD patients on GFD, it is worthy to mention that in a pediatric CeD cohort, the same rs3748816-cg21209485 binomial in the MMEL1 gene body that is described here was one of the very few mQTLs found to be common to both the epithelial and immune fractions of the duodenal mucosa (16). Altogether, these results strongly support the mediation of DNA methylation in the GWAS-association peak on chromosome 1 that includes MMEL1.
UBE2L3 is an ubiquitin ligase that had been previously prioritized in CeD (17), although within a long list of candidate genes. Recently, UBE2L3 has been described to be relevant in lupus erythematosus (LE) after merging GWAS and methylome results (18). Interestingly, the CpG site that seems to mediate this LE association is an mQTL of rs5754217, the same SNP that results from our MR application in CeD, supporting the idea of a common regulatory mechanism in both autoimmune disorders. Additionally, it has been shown that the NF-κB pathway, an inflammatory route that is exacerbated in CeD (19), is very sensitive to the expression of UBE2L3, which in turn is very upregulated in LE plasma cells (20). Altogether, these facts suggest that UBE2L3 could be a relevant functional player in CeD as well as in other immune-mediated diseases.
Last but not least, the relative expression of UBE2L3 isoforms described here could be an important discovery for the diagnosis of CeD, mainly in individuals on self-imposed GFD. Moreover, the diagnostic potential of UBE2L3 seems to be CeD specific, since it does not work efficiently in other immune-mediated conditions of the gut. As previously stated, new eating habits are complicating enormously the diagnosis of CeD (6). This diagnostic challenge is of general interest among gastroenterologists and has encouraged relevant studies, such as a recent work that proposes HLA-DQ-gluten tetramers as blood biomarkers that could help distinguish CeD from non-CeD individuals in the absence of gluten consumption, with AUC values around 0.96 (21). The UBE2L3 expression score presented here has an AUC of 1 and is a potential diagnostic marker for CeD in PBMCs from individuals on GFD that, nevertheless, will have to be validated in prospective studies. Our approach is less time consuming and more easily adaptable to a clinical setting since it could be implemented through a simple polymerase chain reaction (PCR)-based expression analysis. However, in case UBE2L3 is confirmed as a marker of CeD in peripheral blood, it should always be used together with other determinations such as HLA genotyping.
In conclusion, our strategy could be applicable to other disorders in which diagnosis in the absence of the trigger driving the acute phase of the disease is challenging and is a good demonstration of the potential translation of the results of genomic studies, and particularly MR, to the routine clinical practice. However, the MR strategy itself is necessary but not sufficient to discover biomarkers, and follow-up expression analyses in fully independent data sets should be performed in order to fine-tune the pipeline.
Material and Methods
SMR analyses
In general, we used the default parameters suggested by the developers of the SMR software, including the application of the heterogeneity in dependent instruments (HEIDI) test, that resulted in filtering out those hits that arouse from significant linkage with pleiotropically associated variants (linkage disequilibrium cutoff of P = 0.05 in the HEIDI test, as suggested by SMR). More specifically, the software proposes a global test to account for linkage disequilibrium structure between SNPs by reading individual-level SNP genotype data from a reference sample. In our case, we uploaded not only the summary statistics of the celiac GWAS but also the complete data set in PLINK format.
We considered significant pleiotropic associations and kept for the subsequent analytical steps those hits that fulfilled the following criteria: an empirical P < 1e-4 and P > 0.05 in the SMR and HEIDI tests, respectively, in the G2T and G2M analyses and an empirical P < 1e-7 for M2T, given the high number of multiple comparisons performed in the latter case. If adjacent SNP information was not available to compute the HEIDI P-value, we kept the hit and proceeded to the next step, because since different genotyping technologies have been used to generate the different databases, excluding hits for which HEIDI cannot be computed in one of them often means removing from further consideration interesting SNPs that are replicated and pass the HEIDI cutoff elsewhere. Additionally, we plotted the UBE2L3 adjacent region on chromosome 22 by using the SMRLocusPlot function of the source code available at the SMR website, with a plot window of 1000 kb.
Replication using GTEx blood and intestine eQTLs and the eQTL-Gen database
We also ran the SMR software using results from the celiac GWAS and from the GTEx eQTL (P < 1e-5) database (22). In particular, we used the whole blood eQTLs (n = 122) for the replication of the findings of the discovery set and the small intestine eQTLs (n = 369) to conduct a sensitivity analysis with the final aim of assessing heterogeneity of effects across the main tissues relevant to CeD. Additionally, we rerun the analysis in the eQTL-Gen database, where blood of 31 684 individuals has recently been studied (23). For both GTEx and eQTL-Gen, we kept the same criteria used for the CAGE eQTL analysis, namely an empirical SMR threshold of P < 1e-4 for the G2T analysis, an empirical SMR threshold of P < 1e-7 for the M2T analysis, as well as P > 0.05 in the HEIDI test in both cases.
Expression analysis
We interrogated UBE2L3, the only candidate gene resulting from the G2T∩G2M∩M2T approach and replicated in the two additional eQTL data sets, in two fully independent expression microarray experiments performed in PBMCs available in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/): an Illumina HumanHT-12 V4.0 expression Beadchip analysis of 17 CeD patients on GFD and 20 controls (GSE113469) (24) and an Affymetrix Human Genome U133A Array study of 42 controls and 59 Crohn’s disease and 26 ulcerative colitis patients (GSE3365) (25). Comparisons were performed using Mann–Whitney U tests, and ROC curves were constructed using the expression levels of the candidates as binary classifiers. The UBE2L3 relative expression score was calculated from the intensity units of the two UBE2L3 probes available at the celiac PBMC study and the GAPDH housekeeping gene with this formula: 2^((UBE2L3.1 − UBE2L3.2)/GAPDH), where UBE2L3.1 = ILMN_1677877 Illumina probe (mapping to exon 5, common to all UBE2L3 variants); UBE2L3.2 = ILMN_1796830 Illumina probe (mapping to exon 4, exclusive for the UBE2L3 non-coding variant 2) and GAPDH = ILMN_1343295 Illumina probe.
We decided to test the full set of genes resulting from the G2T∩G2M∩M2T approach performed separately in each of the discovery and replication eQTL data sets, with the final aim of ascertaining whether there was any other biomarker among them. In particular, we interrogated all the probes available for AHSA2 (ILMN_1798308), BACH2 (ILMN_1659943, ILMN_2058468 and ILMN_1670695), CCDC116 (ILMN_1795085), CD247 (ILMN_2377669 and ILMN_1676924), CDC73 (ILMN_1730745 and ILMN_1701614) and MMEL1 (ILMN_2151241 and ILMN_1718488) in the same PBMC expression data set of celiac patients on GFD and controls (GSE113469). We performed unpaired, two-tailed T-tests in Microsoft Excel version 14.0.7232.5000 (32 bits) to test for differential expression.
Funding
Project SAN2018111086 from the Basque Department of Health (to J.R.B.); ISCIII Research Projects PI16/00258 and PI18/01142, co-financed by the Spanish Ministry of Economy and Competitiveness (http://www.mineco.gob.es/) and by the European Union ERDF/ESF ‘A way to make Europe’ (to J.R.B. and N.F.-J., respectively).
Conflict of Interest statement
None declared.
Supplementary Material
References
- 1. Gallagher M.D. and Chen-Plotkin A.S. (2018) The post-GWAS era: from association to function. Am. J. Hum. Genet., 102, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M. et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
- 3. Porcu E., Rüeger S., eQTLGen Consortium, Santoni F.A., Reymond A. and Kutalik Z. (2018) Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. 10.1101/377267. [DOI] [PMC free article] [PubMed]
- 4. Wu Y., Zeng J., Zhang F., Zhu Z., Qi T., Zheng Z., Lloyd-Jones L.R., Marioni R.E., Martin N.G., Montgomery G.W. et al. (2018) Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun., 9, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dubois P.C., Trynka G., Franke L., Hunt K.A., Romanos J., Curtotti A., Zhernakova A., Heap G.A., Adány R., Aromaa A. et al. (2010) Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet., 42, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aziz I. (2018) The global phenomenon of self-reported wheat sensitivity. Am. J. Gastroenterol., 113, 945–948. [DOI] [PubMed] [Google Scholar]
- 7. Bruins M.J. (2013) The clinical response to gluten challenge: a review of the literature. Nutrients, 5, 4614–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mayorga C., Ebo D.G., Lang D.M., Pichler W.J., Sabato V., Park M.A., Makowska J., Atanaskovic-Markovic M., Bonadonna P. and Jares E. (2019) Controversies in drug allergy: in vitro testing. J. Allergy Clin. Immunol., 143, 56–65. [DOI] [PubMed] [Google Scholar]
- 9. Herzog R. and Cunningham-Rundles S. (2011) Pediatric asthma: natural history, assessment, and treatment. Mt. Sinai J. Med., 78, 645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lloyd-Jones L.R., Holloway A., McRae A., Yang J., Small K., Zhao J., Zeng B., Bakshi A., Metspalu A., Dermitzakis M. et al. (2017) The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet., 100, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McRae A.F., Marioni R.E., Shah S., Yang J., Powell J.E., Harris S.E., Gibson J., Henders A.K., Bowdler L., Painter J.N. et al. (2018) Identification of 55,000 replicated DNA methylation QTL. Sci. Rep., 8, 17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pavlides J.M., Zhu Z., Gratten J., McRae A.F., Wray N.R. and Yang J. (2016) Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome Med., 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lahdenperä A.I., Fälth-Magnusson K., Högberg L., Ludvigsson J. and Vaarala O. (2014) Expression pattern of T-helper 17 cell signaling pathway and mucosal inflammation in celiac disease. Scand. J. Gastroenterol., 49, 145–156. [DOI] [PubMed] [Google Scholar]
- 14. Amundsen S.S., Viken M.K., Sollid L.M. and Lie B.A. (2014) Coeliac disease-associated polymorphisms influence thymic gene expression. Genes Immun., 15, 355–360. [DOI] [PubMed] [Google Scholar]
- 15. Quinn E.M., Coleman C., Molloy B., Dominguez Castro P., Cormican P., Trimble V., Mahmud N. and McManus R. (2015) Transcriptome analysis of CD4+ T cells in coeliac disease reveals imprint of BACH2 and IFNγ regulation. PLoS One, 10, e0140049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandez-Jimenez N., Garcia-Etxebarria K., Plaza-Izurieta L., Romero-Garmendia I., Jauregi-Miguel A., Legarda M., Ecsedi S., Castellanos-Rubio A., Cahais V., Cuenin C. et al. (2019) The methylome of the celiac intestinal epithelium harbours genotype-independent alterations in the HLA region. Sci. Rep., 9, 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar V., Gutierrez-Achury J., Kanduri K., Almeida R., Hrdlickova B., Zhernakova D.V., Westra H.J., Karjalainen J., Ricaño-Ponce I., Li Y. et al. (2015) Systematic annotation of celiac disease loci refines pathological pathways and suggests a genetic explanation for increased interferon-gamma levels. Hum. Mol. Genet., 24, 397–409. [DOI] [PubMed] [Google Scholar]
- 18. Imgenberg-Kreuz J., Carlsson Almlöf J., Leonard D., Alexsson A., Nordmark G., Eloranta M.L., Rantapää-Dahlqvist S., Bengtsson A.A., Jönsen A., Padyukov L. et al. (2018) DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis., 77, 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandez-Jimenez N., Castellanos-Rubio A., Plaza-Izurieta L., Irastorza I., Elcoroaristizabal X., Jauregi-Miguel A., Lopez-Euba T., Tutau C., de Pancorbo M.M., Vitoria J.C. et al. (2014) Coregulation and modulation of NFκB-related genes in celiac disease: uncovered aspects of gut mucosal inflammation. Hum. Mol. Genet., 23, 1298–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lewis M.J., Vyse S., Shields A.M., Boeltz S., Gordon P.A., Spector T.D., Lehner P.J., Walczak H. and Vyse T.J. (2015) UBE2L3 polymorphism amplifies NF-κB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am. J. Hum. Genet., 96, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarna V.K., Lundin K.E.A., Mørkrid L., Qiao S.W., Sollid L.M. and Christophersen A. (2018) HLA-DQ-gluten tetramer blood test accurately identifies patients with and without celiac disease in absence of gluten consumption. Gastroenterology, 154, 886–896. [DOI] [PubMed] [Google Scholar]
- 22. Consortium G.T.E. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Võsa U., Claringbould A., Westra H.J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Kasela K. et al. (2018) Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 447367, doi: 10.1101/447367. [DOI]
- 24. Sangineto M., Graziano G., D'Amore S., Salvia R., Palasciano G., Sabbà C., Vacca M. and Cariello M. (2018) Identification of peculiar gene expression profile in peripheral blood mononuclear cells (PBMC) of celiac patients on gluten free diet. PLoS One, 13, e0197915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burczynski M.E., Peterson R.L., Twine N.C., Zuberek K.A., Brodeur B.J., Casciotti L., Maganti V., Reddy P.S., Strahs A., Immermann F. et al. (2006) Molecular classification of Crohn’s disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J. Mol. Diagn., 8, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.