

Abstract
Aim:
Pancreatic cancer is one of the worst malignant tumors in prognosis. Therefore, to reduce the mortality rate of pancreatic cancer, early diagnosis and prompt treatment are particularly important.
Results:
We put forward a new feature-selection method that was used to find clinical markers for pancreatic cancer by combination of Support Vector Machine Recursive Feature Elimination (SVM-RFE) and Large Margin Distribution Machine Recursive Feature Elimination (LDM-RFE) algorithms. As a result, seven differentially expressed genes were predicted as specific biomarkers for pancreatic cancer because of their highest accuracy of classification on cancer and normal samples.
Conclusion:
Three (MMP7, FOS and A2M) out of the seven predicted gene markers were found to encode proteins secreted into urine, providing potential diagnostic evidences for pancreatic cancer.
Keywords: : biomarker detection, LDM-RFE, pancreatic cancer, SVM-RFE, urinary excretory protein
According to the statistics from the American Cancer Association, cancer is the second leading cause of death after heart disease [1]. In China, cancer has also become the leading cause of death. According to statistics from the National Central Cancer Registry of China, there were 90,100 new cases of pancreatic cancer in 2015 and the death toll was as high as 79,400 [2]. Besides, among more than 60 cancers, the prognosis of pancreatic cancer is the worst and it has a very low 5-year survival rate of 8%. In contrast, the 5-year survival rate for breast cancer is as high as 90% [3]. Chemotherapy is the most commonly used adjunctive therapy for pancreatic ductal adenocarcinoma in patients with metaphase and metastasis; however, the development of drug resistance has dramatically reduced the effectiveness [4]. Therefore, to reduce the death toll of pancreatic cancer patients, early diagnosis and prompt treatment are particularly important.
With the improvement of genomic technology, we can understand cancer and its biological characteristics at the molecular level [5]. In the evolution of cancer, there is a significant difference in the amount of gene expression (or other components) between cancer tissues and normal tissues, and these components are generally genetically related. This difference between tissues can be detected through the comparison of relevant technologies. After determining these specific substances, quantitative measurement can be used to provide an essential basis for the early detection of cancer [6]. Due to the lack of adequate sensitivity and certain specificity, no final serum or urinary markers of cancer have been found until now. However, several cancer markers have been used in clinical and research fields, such as a prostate-specific antigen (PSA) which is a widely used biomarker for prostate cancer [7]. CA 19-9 is the most advanced serum tumor marker that can be used for pancreatic cancer. However, it has not been widely promoted due to some limitations [8]. In recent years, with the continuous development of machine learning, more and more researchers have begun to make predictions based on machine learning together with diverse feature representations [9]. At the same time, their research combined with clinical data accumulated by medical experts through traditional biological experiments and clinical methods. Fold-change (FC) [10] and t-test [11] are two common methods of feature selection. In addition, support vector machine recursive feature elimination (SVM-RFE) [12] is also a popular feature-selection method. In 2004, Zhang et al. performed a feature-based screening for cervical cancer detection based on support vector machine (SVM) [13]. In 2017, Qayyum conducted breast cancer research based on SVM and combined computer-aided diagnosis technology to detect breast cancer in mammograms automatically [14]. Large margin distribution machine (LDM) algorithm with better generalization performance was proposed in 2014 [15]. As we know, the SVM has better accuracy, and the LDM has better generalization performance. Considering the essential characteristics of the two methods above, we tried to merge the two algorithms to get better performance in this paper.
In the course of the experiment, we selected the optimal parameters of the 730 pancreatic cancer endemic genes first. Then the recursion feature was selected by the replacement test, and finally, we got a list of stable genes. Next, by comparing our method with the Random method, t-test, SVM-RFE and large margin distribution machine recursive feature elimination (LDM-RFE), we found that the classification accuracy of our method was higher and the result was better. What’s more, the top seven gene combinations (MMP7, MMP12, ANPEP, FOS, SFN, IL6 and A2M) had the best classification effect. The above experiments are based on the data set GSE15471 of GEO database [16]. Besides, we also validated our method on another dataset GSE28735 and the accuracy rate was well enough. To further understand the useful information of the genes obtained, we analyzed the pathways of the top 200 genes with the help of DAVID [17] and Cystoscope [18]. Through the algorithm proposed by Wang and Du [19] and the R2: Genomics Analysis and Visualization Platform [20], we proved that the genes selected were associated with the survival rate of patients with pancreatic cancer and their encoded proteins may enter urine and become useful urine markers. As a result, seven differentially expressed genes were predicted as specific biomarkers for pancreatic cancer because of their highest accuracy of classification on cancer and normal samples. Three (MMP7, FOS and A2M) out of the seven predicted gene markers were found to encode proteins secreted into urine, providing potential diagnostic evidences for pancreatic cancer.
Materials & methods
Datasets
During the experiment, the training data are 39 paired data from the GSE15471 dataset of the GEO database [16]. A total of 78 samples were selected from pancreatic ductal carcinoma (PDAC) and adjacent pancreatic tissues. Each sample contains more than 20,000 genes expression values. Most of the GEO cancer datasets are based on the GPL570 platform, which is one of the most commonly used platforms in the experiment. Therefore, to ensure the universality and generalization performance of the model, we finally chose GSE15471 dataset as training data. It also has excellent performance to be more widely applied to other cancers or more different datasets in the future.
Besides, we have processed other GEO datasets to find specific biomarkers for pancreatic cancer. Specially, these datasets include GSE15852 (breast cancer), GSE27342 (gastric cancer), GSE7670 (lung cancer), GSE14811 (liver cancer), GSE17951 (prostate cancer), GSE10810 (breast cancer), GSE8671 (colorectal cancer), GSE13911 (gastric cancer), GSE18842 (lung cancer) and GSE23878 (colorectal cancer). Also, we have further tested our method using the GSE28735 (pancreatic cancer) dataset with 45 paired data (90 samples), which is based on the platform GPL6244.
Methods
In the feature extraction of cancer genes, the small number of samples and a large number of genes make the feature selection more challenging [21]. Therefore, data preprocessing is our primary task and first step, the same as in most experiments. The selection of these genes is also more reasonable as the differential expression (DE) is much more significant and the panel size might be more applicable in clinical practice. After detailed analysis, the final method we use is composed of three parts: identification of differentially expressed genes, feature selection and evaluation of features. The workflow of our combination algorithm can be seen in Supplementary Figure 1.
Identification of differentially expressed genes
First, we identified the differentially expressed genes unique to pancreatic cancer on the expression data. DE can be detected by conducting gene expression analysis, including two main methods: FC [10] and t-test [11]. Inspired by [22], FC can be expressed in Formula (1):
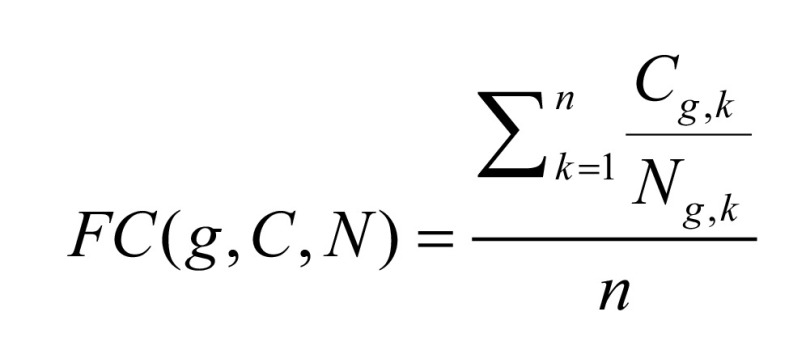 |
(1) |
where Cg,k represents the expression level of gene g in the kth cancer sample; Ng,k represents the expression level of gene g in the kth normal sample and n represents the number of sample pairs (the data in Cancer and Normal are corresponding).
A gene was identified as to be up- or downregulated if its FC > 2 (or <1/2), with a false discovery rate (FDR) value less than 0.01 measured by t-test. For details, FC > 2 means the expression value of a gene in the case C (cancer) is two-times larger than that in the control N (normal tissue), indicating the gene is upregulated; meanwhile FC < 1/2 means the expression value of a gene in the case N (normal tissue) is two-times larger than that in the control C (cancer), indicating the gene is downregulated.
The identification process of differentially expressed genes can be seen in Supplementary Figure 2.
Feature selection
SVM is a relatively common supervised machine learning algorithm and has many advantages that other common machine learning algorithms do not have, especially in dealing with high dimensions, small sample size and nonlinear problems [23].
The original SVM algorithm solves the two-class problem and its core goal is to obtain a classification surface. This classification plane can accurately separate the two types of samples. Since there are multiple such planes, the classification margin needs to be maximized to classify the prediction data better. The SVM problem can be expressed in Formula (2) [12].
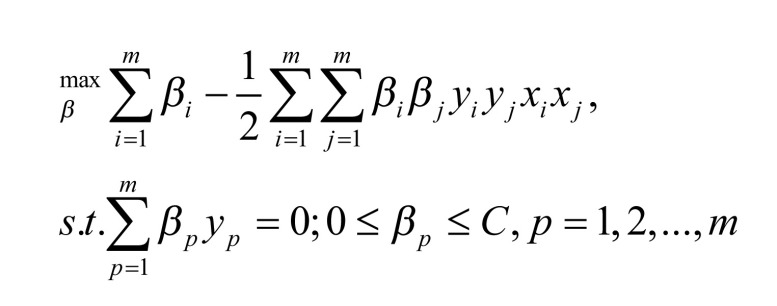 |
(2) |
where m represents the number of samples; xi and xj represents input data; yi and yj represents corresponding labels and βi and βj corresponds to Lagrange multiplier.
We implemented the SVM by using the LIBSVM toolkit. The user can select the optimal settings through crossvalidation methods to make the experimental results better [24]. The SVM-RFE method is one of the recursive feature elimination methods. Based on the SVM method, the gene is selected and deleted by using the weight vector of the hyperplane constructed from the sample on edge. Then, we use the remaining gene expression data to train the SVM classification model and repeat the process until the gene is eliminated. The final deleted gene is the most useful gene in the classifier [25], and we can select the required number of features according to the needs. To improve efficiency, we eliminated 1% of the genes in the list g in each iteration before the number of genes reached 400. When the number of genes in the list g reached 400, one gene is eliminated from the list g in each iteration. The following is a summary of the SVM-RFE algorithm in a linear case. The weight vector is a linear combination of training patterns and most of the weights are zero. The training samples of nonzero weights are support vectors (SV). The score of each gene is used in the Formula (3). (xp, yp) is the input data and the βp is the parameter in SVM train [12].
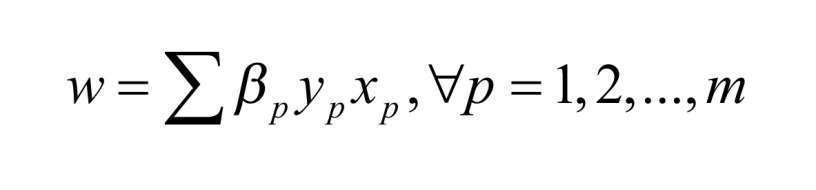 |
(3) |
Algorithm SVM-RFE:
 |
Unlike SVM, LDM tries to maximize the margin mean while minimizing the margin variance simultaneously. In the study [15], Zhang and Zhou found that LDM is more advantageous than SVM, and verified that maximizing the average margin of LDM can lead to better generalization performances than maximizing the minimum margin of SVM. It is worth noting that the margin theory is not only suitable for LDM, but also for many other learning algorithms. Such as AdaBoost [26], which is a representation of the integrated algorithms.
In the process of the experiment, the LDM source provided by Zhang and Zhou is used for the realization of the LDM algorithm [15]. The steps are similar to those in the previous section SVM, and the Formula (4) and (5) are the definitions of the margin mean and variance: 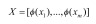 and
and 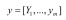 are input data. The parameter m represents the number of samples.
are input data. The parameter m represents the number of samples.
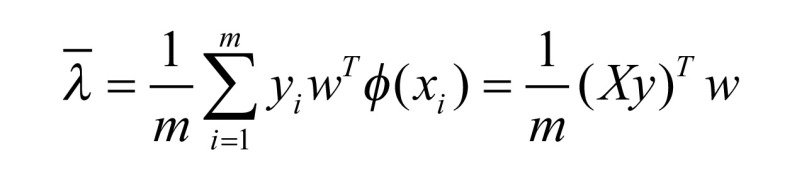 |
(4) |
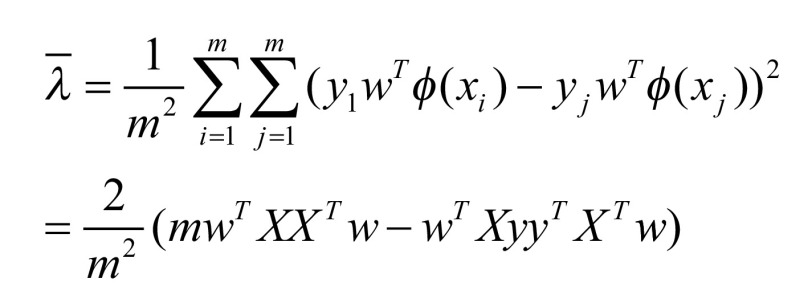 |
(5) |
Although SVM-RFE has better accuracy, it is easily affected by noise and outliers and leads to poor performance. For the small sample data with noise, the negative effects of the microarray data are especially obvious [27]. Good generalization performance is a prominent advantage of LDM-RFE.
To understand the LDM algorithm easier, we provided a detailed calculation process of the LDM-RFE algorithm. Similarly, we eliminated 1% of the genes in the list g in each iteration before the number of genes reached to 400. When the number of genes in the list g reached 400, one gene is eliminated from the list g in each iteration. The parameters of the algorithm LDM-RFE have the following meaning: the parameter g represents the list of original features of all the features; parameter l represents the list of final sorting features after the feature selection process; parameter s represents a subset with the same minimum value S(i) in each iteration [28].
Algorithm LDM-RFE:
 |
Although the most common feature-selection methods can obtain useful results, the results are not completely stable and reliable, due to certain limiting factors. Also, SVM is a well-known classification algorithm and has been widely used in the selection of cancer genes by the SVM-RFE. Therefore, in the RFE feature extraction, we tried to combine the two methods of SVM-RFE and LDM-RFE to expect our method to combine the advantages of them, so that the classification results are accurate and generalization performance is satisfied [22]. Formula (6) ≈SVM-RFE, and rank(g,LDM-RFE) is the ranking of gene g in LDM-RFE.
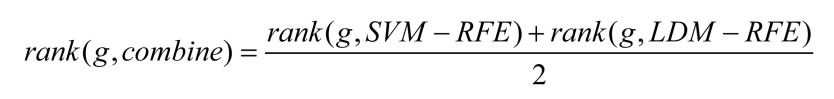 |
(6) |
After preprocessing the differentially expressed genes specific to pancreatic cancer, we use these data to select the optimal parameters. The detailed description of our method can be seen in Figure 1, which showed the combination process of SVM-RFE and LDM-RFE.
Figure 1. . The combination process of Support Vector Machine Recursive Feature Elimination and Large Margin Distribution Machine Recursive Feature Elimination.

LDM: Large Margin Distribution Machine; RFE: Recursive Feature Elimination; SVM: Support Vector Machine.
First of all, the prepared differentially expressed gene data (730) were imported. The genes in these samples were the unique genes of pancreatic cancer screened on the basis of 1585 DE of pancreatic cancer. In the test part, five methods (random method, t-test method, SVM-RFE method, LDM-RFE method and the combination of SVM-RFE and LDM-RFE method) were used to observe the accuracy rate;
This step was to obtain the optimal parameters. The 2/3 of the sample data were selected for training. This process also used fivefold crossvalidation, and the kernel function we used was the linear kernel. After obtaining the optimal parameters, the 3/4 training data were selected randomly for the next step;
Applied permutation test to random perturbation of data, then got a gene list. Once the model was trained, 3/4 of the disturbance data was selected, which makes the result more stable and excludes interference. Then, the recursive feature elimination (SVM-RFE and LDM-RFE) process was carried out using the parameters obtained by step 2). To speed up the process, each RFE process would remove 1% of the genes when the number of undeleted genes is greater than 400. When the remaining 400 genes were deleted, one gene was deleted each time. Repeated the above process 60-times and took average ranking;
To ensure that the final gene list was relatively stable and accurate, two lists from SVM-RFE and LDM-RFE were merged according to the Formula (6). We also tried to sort the genes by another formula, but the results were unsatisfactory. Finally, a stable gene list was accepted.
Figure 2 shows the score of 730 differentially expressed genes based on SVM-RFE and LDM-RFE. According to the picture, we found that all genes are roughly concentrated near the diagonal line, which indicates that there are some similarities between SVM-RFE and LDM-RFE for feature selection. That is to say, their fraction distribution is roughly the same, so we can see that the same gene has similar scores in the two methods. The emergence of this phenomenon provides an important basis for us to get a set of averages by combining two feature-selection methods.
Figure 2. . Gene scores calculated by Support Vector Machine Recursive Feature Elimination and Large Margin Distribution Machine Recursive Feature Elimination.

LDM: Large Margin Distribution Machine; RFE: Recursive Feature Elimination; SVM: Support Vector Machine.
Evaluation
Algorithm evaluation
Accuracy is the most intuitive indicator for evaluating the outcome of a classification. The sample with the correct classification is divided into two parts. One represents that the positive sample is predicted to be positive and the other represents that the negative sample is predicted to be negative. The accuracy is used to evaluate the validity of the gene list that obtained from experiments, and it can be defined in Formula (7) [22].
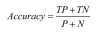 |
(7) |
Where TP represents the number of positive samples correctly identified as the positives; TN represents the number of negative samples correctly identified as the negative; P and N represent the total number of positive and negative samples, respectively.
In the algorithm evaluation part, we compare our method with the other four methods (random method, t-test method, SVM-RFE method and LDM-RFE method). Then the classification accuracy is tested by SVM, LDM and back propagation (BP) neural network classifier, respectively.
Gene list analysis
In this section, we performed functional analysis and pathway analysis of the candidate genes. The aim is to find out the biological significance of candidate genes that can better guide clinical and biological studies on the diagnosis of pancreatic cancer. Functional analysis reveals the biological functions of each gene. We conducted an indepth analysis by referring to the existing references and GeneCards [15]. Pathway analysis revealed the relationship between the top 200 genes and some biological pathways, which using Cystoscope [18] and DAVID [17].
In addition, survival analysis and urinary excretion protein prediction for pancreatic cancer were used in the top seven candidate genes. Survival analysis analyzed the relationship between genes and patient survival, which was carried out through the platform R2 (R2: Genomics Analysis and Visualization Platform) [20]. The predictive analysis of urinary protein excretion was analyzed by website developed by Wang and Du [19].
Results
After preprocessing the original GSE15471 dataset, we got 730 differentially expressed genes in pancreatic cancer finally. To verify the validity of the tagged genes obtained, we used SVM, LDM and BP neural network classifiers to classify them. At the same time, we compared our method with the common methods of feature selection: Random, t-test, SVM-RFE and LDM-RFE. In addition, to ensure that our tagged genes are still available on the datasets of other platforms, we also tested the candidate genes on another dataset GSE28735. In the process of the experiment, we implemented the implementation of SVM in the LIBSVM toolkit [29].
A total of 730 differentially expressed genes unique to pancreatic cancer were selected by our method that combined with SVM-RFE and LDM-RFE to obtain a new sorted gene list. The combined genes were ranked and used for classification. The results showed that the top seven genes (MMP7, MMP12, ANPEP, FOS, SFN, IL6 and A2M) had the highest classification accuracy. In the process of the experiment, the data in the GSE15471 were preprocessed to sort the genes and then the data were tested on the SVM, LDM and BP classifier, respectively.
Figure 3 shows that the average classification accuracy curve was obtained by using our method (red line) and Random method (black line), t-test method (blue line), SVM-RFE method (green line) and LDM-RFE method (purple line), respectively. It is clear that our method has a higher classification accuracy than the other methods, and SVM-RFE is suboptimal. It also can be seen from the figure that the accuracy of the top 50 genes combination can reach over 86%, of which the accuracy of the top seven genes is the highest. What’s more, the receiver operating characteristic curve (ROC) for SVM with top seven genes by five methods can be seen in Supplementary Figure 3 and the area under curve (AUC) for SVM with top 1–100 genes by five methods can be seen in Supplementary Figure 4.
Figure 3. . The performance of gene selection of five methods on Support Vector Machine, Large Margin Distribution Machine and back propagation classifiers.

LDM: Large Margin Distribution Machine; RFE: Recursive Feature Elimination; SVM: Support Vector Machine.
To show the experimental results more intuitively, we also carried out other cartographic analysis of the results. Figure 4 is a heatmap based on the set of the top seven genes obtained from the experiment on the GSE15471. According to the color and data comparison, it is easy to find that these genes’ expressions in pancreatic cancer and normal tissues are quite distinct.
Figure 4. . The expression heatmap of the top seven differentially expressed genes.

The expression levels of MMP7, MMP12, ANPEP, FOS, SFN, IL6 and A2M in cancer versus normal samples can be seen in Supplementary Figure 5. In this figure, it is evident that the expression of the matched normal pancreatic tissue gene is significantly lower than the expression of the gene in the pancreatic cancer tissue. In other words, we found that the expression of characteristic genes in pancreatic cancer was upregulated in cancer tissues. However, the expression of ANPEP gene in cancer tissue is significantly lower than that in normal tissues.
The above experiments were carried out on the dataset GSE15471. Also, to ensure that the feature genes obtained are reliable and meaningful, we used the GSE28735 dataset to test the identified differentially expressed genes by SVM, LDM and BP classifier. The results showed that the classification accuracy of the test data set reaches about 80% (see Supplementary Figure 6), which has a certain significance. It can be seen clearly that the classification accuracy is well enough.
Discussion
Functional analysis
Supplementary Table 1 lists the top seven differentially expressed genes (MMP7, MMP12, ANPEP, FOS, SFN, IL6 and A2M) and characteristic genes identified as markers of pancreatic cancer. Each column represents the name of their respective protein, the value of FC and the p-value of the t-test. To mine the information hidden behind the disease, we analyzed the genes selected from its biological function level. Supplementary Table 2 shows the function annotations of the top seven gene markers for pancreatic cancer, as mentioned in [30–36]. After consulting the literature, we found that they are all genes related to cancer, which may lead to various abnormalities.
MMP7, like other members of the matrix metalloproteinase family, has been known as an important factor involving the degradation of the extracellular matrix (ECM), which promotes the invasion and metastasis of PDAC since 1997 [37]. In 2011, Fukuda, providing another substantial evidence showed that Stat3 signaling enforces MMP7 expression in pancreatic ductal adenocarcinoma (PDA) cells and that MMP7 deletion limits tumor size and metastasis in mice model. They even demonstrated that serum MMP7 level in patients with PDA correlated with metastatic disease and survival [38].
MMP12, also known as human macrophage metalloelastase (HME), has verified its biological function through Balaz’s work: they used Northern blot analysis, reverse transcriptase-PCR, Western blot analysis and immunohistochemistry in 39 pancreatic cancer tissues and 13 normal controls. Combined with clinicopathologic parameters and patient survival analysis they concluded that HME participates in pancreatic cancer progression and that its presence worsens the prognosis [39].
ANPEP, aminopeptidase N/cluster of differentiation antigen 13 (APN/CD13), has been implicated in the tumor invasion. Researchers analyzed the APN/CD13 gene expression status by reverse transcriptase-PCR and immunohistochemistry in PDAC tumor samples and suggested that APN/CD13 may be a prognostic marker for patients [40].
FOS encodes leucine zipper protein, which is closely related to cell proliferation, differentiation and apoptosis. By comparing with c-fos protein in normal pancreatic tissues and chronic pancreatitis tissues, the protein is expressed more in pancreatic cancer tissues [33].
SFN, together with S100P, S100A6, AGR2 and other genes belong to a set of 33 genes differentially expressed in common between primary PDAC and liver metastases. This study has been validated in primary PDACs and matched metastatic liver lesions [41].
IL-6 and its receptors are involved in the induction of VEGF in pancreatic cancer and the expression of VEGF is also associated with microvessel density in pancreatic cancer [35].
A2M, the encoded protein has been verified through mass spectrometry approaches using pancreatic adenocarcinoma patient sera. Hanas, published the result, that three large-mass proteins were found to be elevated in pancreatic cancer sera versus normal sera, α-2 macroglobulin, ceruloplasmin and complement 3C [42].
Pathway analysis
Many studies have shown that when compared with any specific single gene or gene product, changes in pathways or networks are more related to complex diseases [43]. Figure 5 shows the pathway and network analysis of the top 200 genes. The graph is based on a software tool called Reactome FIViz [44], which is a plugin of Cystoscope. In Figure 5, nodes represent genes and connections between nodes represent pairs of interactions between genes. The real line represents the connection between genes identified by the experiment and the dotted line represents the predictive relationship through the algorithm.
Figure 5. . Pathway and network analysis of the top 200 genes by combination of Support Vector Machine Recursive Feature Elimination and Large Margin Distribution Machine Recursive Feature Elimination.

From the Supplementary Table 3, we can see the following facts: genes FOS and IL6 are divided into module 0 (containing 27 genes). Interestingly, chemokines in this pathway can attract leukocytes to the site of infection and play a greater role in inflammatory response. We found that there were many Fc receptors (FCGR) in module 0, and FCGR2A and FCGR3A polymorphisms were associated with rheumatoid arthritis in Europe [45]. The top five pathways most significantly enriched in each of the ten modules can be seen in Supplementary Table 4.
Furthermore, A2M was divided into module 3. This module enriches with fibrin clot (coagulation cascade) formation, complement and coagulation cascade, platelet cytoplasmic Ca2+ reaction, endogenous prothrombin activation pathway and so on. MMP7 and MMP12 are divided into module 4. This module enriches the ECM, the interaction of integrin cells, the interaction of integrin ligand, the prion pathway and the role of GRAIN in postsynaptic differentiation. Studies have shown that the matrix metalloproteinase family is the most important protease that degrades ECM.
The above data indicate some vital cellular mechanisms related to pancreatic cancer, which may provide new clues for further research on pancreatic cancer or new drug development.
Table 1A shows that most genes are mapped to p-value as the keyword ‘disulfide bond’ of 6.40E-20 and p-value as the keyword ‘signal’ of 1.50E-22. This may indicate that there might be some important changes in the disulfide bond function and signaling pathway if a pancreatic tissue is cancerous. Also, the ‘extracellular matrix’, ‘secreted’ and ‘glycoprotein’ also have significant statistical significance. ECM is a dynamic niche for cancer progression. It is clear that ECM abnormalities can affect the behavior of the release of stromal cells, promote tumor-related angiogenesis and inflammation, and lead to the production of in the tumorigenic microenvironment [46]. The deregulation of ECM components can drive cell cycle progression, possibly through their interaction with cytoskeletal structure [47]. The analysis of DAVIDUP_SEQ_FEATURE shown in Table 1B also supports the above conclusions.
Table 1A. . Results of the top 200 genes analyzed by DAVID SP_PIR_KEYWORDS.
| Term | Count | % | p-value |
|---|---|---|---|
| Disulfide bond | 83 | 41.7 | 6.40E-20 |
| Signal | 92 | 46.2 | 1.50E-22 |
| Extracellular matrix | 16 | 8 | 2.40E-08 |
| Secreted | 55 | 27.6 | 8.40E-15 |
| Glycoprotein | 96 | 48.2 | 3.60E-16 |
| Duplication | 16 | 8 | 1.10E-08 |
| Calcium binding | 7 | 3.5 | 6.20E-04 |
| Heterodimer | 10 | 5 | 9.90E-07 |
| Homotrimer | 2 | 1 | 6.90E-02 |
| Pyroglutamic acid | 3 | 1.5 | 7.80E-02 |
| Transmembrane protein | 27 | 13.6 | 1.70E-09 |
Table 1B. . Results of the top 200 genes analyzed by DAVID UP_SEQ_FEATURE.
| Term | Count | % | p-value |
|---|---|---|---|
| Signal peptide | 91 | 47.2 | 1.80E-23 |
| Disulfide bond | 79 | 40.9 | 1.30E-19 |
| Glycosylation | 91 | 47.2 | 2.80E-16 |
In addition, we also observed the enrichment results of gene ontology biological process and found that the genes were enriched in some important biological processes. Gene ontology biological process enrichment analysis can be seen in Supplementary Figure 7. All these findings are consistent with the observed phenotype of pancreatic cancer, which involves excessive proliferation of ECM and inflammation. It supports the notion that cancer is an abnormal response to biological processes [48].
Subsistence analysis
Furthermore, we analyzed the relationship between genes and the survival rate of pancreatic cancer through the R2: Genomics Analysis and Visualization Platform [20] and found that there was a significant negative correlation between the expression of genes and the survival rate of patients with diseases. Its statistical significance is very clear.
The p-values of seven gene markers calculated by R2 Kaplan–Meier can be seen in Supplementary Table 5. It can be seen that the p-values of MMP7, FOS and A2M are lower than 0.05. That means that the genes we found are related to the survival rate of patients with pancreatic cancer and their abnormal expression is closely associated with the survival rate of patients. Figure 6 shows the survival curves of our selected genes.
Figure 6. . Survival curves of MMP7, MMP12, ANPEP, FOS, SFN, IL6 and A2M obtained by R2.

Literature shows that MMP7 is associated with metastasis of pancreatic cancer and colorectal cancer, which has been involved in the UPA-UPAR pathway and WNT signaling pathway [38]. MMP12 can decompose almost all the extracellular mechanisms and vascular wall components, although there is no reliable record of how it functions. However, it can be confirmed that with the indepth study of MMP12, targeted therapy is beneficial to cancer.
ANPEP is commonly found in small intestinal and renal microbubble membranes, although it is also found in other keratinocytes. It is known that this gene is closely related to upper respiratory tract infection. The FOS gene family encodes leucine zipper, which is closely associated with cell proliferation, differentiation and apoptosis. SFN is a gene that can encode proteins and the known diseases related to its encoded proteins are benign breast adenocarcinoma. IL-6 and its receptors are involved in the induction of VEGF in pancreatic cancer, and the expression of VEGF is also associated with microvessel density in pancreatic cancer. By combining A2M with trypsin to form giant globulin trypsin complex (MTLS), the expression of this complex in pancreatic disease samples is significantly higher than that of without the disease sample [49]. Based on the findings, we can infer that the lesions of pancreatic cancer are closely related to the above genes, which may be the breakthrough point for the study of biomarkers for pancreatic cancer.
Prediction of urinary excretory protein
As described in the introduction, serum and urine tumor markers are important for tumor diagnosis, which prompts us to predict and analyze these genes. Table 2 shows the predicted results of urinary excretion of proteins carried out by the website developed, by Wang and Du [19]. The top seven differentially expressed genes in pancreatic cancer were analyzed to predict whether their protein products were urinary secretory proteins. In Table 2, the higher the value of excretory is, the more significant the result is. It means that the protein is more likely to discharge urine protein.
Table 2. . Prediction results of urinary excretory protein.
| Query name | Value of excretory | Value of no urological origin | Property of secreted | Property of membrane |
|---|---|---|---|---|
| MMP7 | 0.206 | 2.291 | Secreted | Nonmembrane proteins |
| MMP12 | -1.29857 | 0.97 | Secreted | Nonmembrane proteins |
| ANPEP | 0.169 | -0.2 | Released | Membrane proteins |
| FOS | -0.096 | 0.651 | Nonsecreted | Nonmembrane proteins |
| SFN | 1.602 | -1.071 | Nonsecreted | Nonmembrane proteins |
| IL6 | 0.048 | 1.578 | Secreted | Nonmembrane proteins |
| A2M | 0.204 | 0.488 | Secreted | Nonmembrane proteins |
Conclusion
As the 5-year survival rate of pancreatic cancer is lower than all types of malignant tumor [50], the need for early diagnosis and prediction of biomarkers for faster treatment to this deadly disease is urgent. Because most pancreatic cancer patients are accompanied by other diseases at the time of diagnosis, surgical and medical interventions are relatively ineffective. Studies have shown that Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation [51]. Therefore, early prediction of pancreatic cancer is particularly important. By analyzing the methods of SVM-RFE and LDM-RFE, we put forward a new method, which combines the above two feature-selection methods.
The experiments are based on the dataset GSE15471 of GEO database [16]. In addition, we conducted data preprocessing on ten other GSE datasets. The characteristics of pancreatic cancer were then compared with one by one, and the same genes were deleted from the list of pancreatic cancer features. Finally, we screened 730 differentially expressed genes specific for pancreatic cancer. In the end, seven differentially expressed genes (MMP7, MMP12, ANPEP, FOS, SFN, IL6 and A2M) were obtained as the specific biomarkers for pancreatic cancer. To explore the relationship between the survival rate of pancreatic cancer patients and the marker genes, we used the R2 platform to analyze three meaningful genes (MMP7, FOS and A2M) and found they were closely related to the survival rate of the patients. Finally, we made a prediction analysis of urinary protein in pancreatic cancer and found that all three genes were urinary excretion proteins. From the experimental results, we could find multiple feature sets are more likely to improve the classification accuracy of PDAC than a single feature. Compared with the previously reported pancreatic cancer marker CA19-9, the combination of the seven genes we selected had higher classification accuracy and generalization performance [52]. In conclusion, the results of our experiments may help the biomedical experts to have a better basis for the diagnosis of pancreatic cancer.
To sum up, although the method we used in this paper has extracted the related characteristic genes for the identification of pancreatic cancer markers, it is still not perfect in the design of algorithms and experiments. And it should be improved to help the identification of pancreatic cancer better in the future.
Summary points.
Materials & methods
Experimental datasets are all from the GEO database. The training data are 39 paired data from the GSE15471 dataset.
We put forward a new feature-selection method that can be used for pancreatic cancer by merging the results of Support Vector Machine Recursive Feature Elimination and Large Margin Distribution Machine Recursive Feature Elimination algorithms.
Results
Seven differentially expressed genes were obtained as a specific biomarker for pancreatic cancer.
Three genes (MMP7, FOS and A2M) were found to closely related to the survival rate of the patients.
Conclusion
The proteins encoded by the genes (MMP7, FOS and A2M) can be secreted into urine, which may provide a basis for the clinical diagnosis of pancreatic cancer.
Supplementary Material
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: https://www.futuremedicine.com/doi/suppl/10.2217/bmm-2018-0273
Financial & competing interests disclosure
This research was funded by the National Natural Science Foundation of China (Nos. 61572227, 61772227), and Development Project of Jilin Province of China (Nos. 20170203002GX, 20180414012GH, 20190201293JC). This work was also supported by Jilin Provincial Key Laboratory of Big Date Intelligent Computing (No. 20180622002JC). Support for this project was also provided by an RO1 award #1R01GM131399-01 from the National Institute of General Medical Sciences of the NIH. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation grant number ACI-1548562. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct
The authors state that experimental datasets are all downloaded from the public database (GEO database), not self-measured. All work has been cited following the GEO guidelines.
Author’s contributions
Y Wang, Y Tian and W Du conceived and designed the experiments. K Liu, Y Tan and Y Lv performed the experiments. Q Ma and H Wang analyzed the data. Y Wang, Y Tian, K Liu and Y Lv wrote the paper.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Siegel RL, Miller KD, Ahmedin Jemal., DVM Cancer statistics, 2017. CA. Cancer J. Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA. Cancer J. Clin. 2016;66(2):115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Warshaw AL, Fernándezdel CC. Pancreatic carcinoma. N. Engl. J. Med. 1992;326(7):455–465. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 4.Zhao YP, Chen G, Feng B, et al. Microarray analysis of gene expression profile of multidrug resistance in pancreatic cancer. Chin. Med. J. 2007;120(20):1743–1752. [PubMed] [Google Scholar]
- 5.Sawyers CL. The cancer biomarker problem. Nature. 2008;452(7187):548–552. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]; • Discusses the biological indicators or biomarkers of cancer.
- 6.Chen YL, Ge GJ, Qi C, et al. A five-gene signature may predict sunitinib sensitivity and serve as prognostic biomarkers for renal cell carcinoma. J. Cell. Physiol. 2018;233(10):6649–6660. doi: 10.1002/jcp.26441. [DOI] [PubMed] [Google Scholar]
- 7.Stephan C, Ralla B, Jung K. Prostate-specific antigen and other serum and urine markers in prostate cancer. Biochim. Biophys. Acta. 2014;1846(1):99–112. doi: 10.1016/j.bbcan.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Steinberg W. The clinical utility to the CA 19–9 tumor-associated antigeen. Am. J. Gastroenterol. 1990;85:350–355. [PubMed] [Google Scholar]
- 9.Wei L, Ding Y, Su R, Tang J, Zou Q. Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 2018;117:212–217. [Google Scholar]; • Proposes DeepPSL by using SAE network for protein subcellular localization prediction.
- 10.Grishin NV. Fold change in evolution of protein structures. J. Struct. Biol. 2001;134(2–3):167–185. doi: 10.1006/jsbi.2001.4335. [DOI] [PubMed] [Google Scholar]
- 11.Zhou N, Wang L. A modified t-test feature selection method and its application on the HapMap genotype data. Genomics Proteomics Bioinf. 2007;5(3–4):242–249. doi: 10.1016/S1672-0229(08)60011-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002;46(1–3):389–422. [Google Scholar]
- 13.Zhang J, Liu Y. Medical Image Computing and Computer-Assisted Intervention, MICCAI 2004 – 7th International Conference. Saint-Malo, France: 2004. Cervical cancer detection using SVM based feature screening. Presented at. 26–29 September. [Google Scholar]
- 14.Qayyum A, Basit A. 12th International Conference on Emerging Technologies. Islamabad, Pakistan: 2016. Automatic breast segmentation and cancer detection via SVM in mammograms. Presented at. 18–19 October. [Google Scholar]
- 15.Zhang T, Zhou Z-H. 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. New York, NY, USA: 2014. Large margin distribution machine. Presented at. 24–27 August. [Google Scholar]; • Presents the Large Margin Distribution Machine (LDM).
- 16.Barrett T, Suzek TO, Troup DB, et al. NCBI GEO: mining millions of expression profiles – database and tools. Nucleic Acids Res. 2005;33:D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis G, Sherman BT, Hosack DA, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(9):11. [PubMed] [Google Scholar]
- 18.Cline MS, Smoot M, Cerami E. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2(10):2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Du W, Liang Y, et al. 12th International Conference on Advanced Data Mining and Applications. Gold Coast, Queensland, Australia: 2016. PUEPro: a computational pipeline for prediction of urine excretory proteins. Presented at. 12–15 December. [Google Scholar]; •• Discusses how to predict urine excretory protein.
- 20.Koster J, Molenaar JJ, Versteeg R. R2: accessible web-based genomics analysis and visualization platform for biomedical researchers. Cancer Res. 2015;75(22):2. [Google Scholar]
- 21.Duan K, Rajapakse JC. Proceedings of the 2004 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology. La Jolla, CA, USA: 2004. A variant of SVM-RFE for gene selection in cancer classification with expression data. Presented at. 7–8 October. [Google Scholar]
- 22.Lv Y, Wang Y, Tan Y, Du W, Liu K, Wang H. 4th International Conference on Systems and Informatics. Hangzhou, China: 2017. Pancreatic cancer biomarker detection using recursive feature elimination based on Support Vector Machine and large margin distribution machine. Presented at. 11–13 November. [Google Scholar]
- 23.Ukil A. Support vector machine. Comput. Sci. 2002;1(4):1–28. [Google Scholar]
- 24.Chen HL, Yang B, Liu J, Liu DY. A support vector machine classifier with rough set-based feature selection for breast cancer diagnosis. Expert Syst. Appl. 2011;38(7):9014–9022. [Google Scholar]
- 25.Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002;46(1–3):389–422. [Google Scholar]; • Presents a method of gene selection to eliminates gene redundancy automatically and yields better and more compact gene subsets by using Recursive Feature Elimination.
- 26.Zhu J, Zou H, Rosset S, Hastie T. Multi-class adaboost. Stat. Interface. 2009;2(3):349–360. [Google Scholar]
- 27.Niijima S, Kuhara S. Recursive gene selection based on maximum margin criterion: a comparison with SVM-RFE. BMC Bioinformatics. 2006;7(1):543. doi: 10.1186/1471-2105-7-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ou G, Wang Y, Pang W, Coghill GM. 4th International Conference on Systems and Informatics. Hangzhou, China: 2017. Large margin distribution machine recursive feature elimination. Presented at. 11–13 November. [Google Scholar]; •• Presents Large Margin Distribution Machine Recursive Feature Elimination to eliminate irrelevant features for classification.
- 29.Chang CC, Lin CJ. LIBSVM: a Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011;2(3):27. [Google Scholar]
- 30.Grindel BJ, Martinez JR, Pennington CL, et al. Matrilysin/matrix metalloproteinase-7(MMP7) cleavage of perlecan/HSPG2 creates a molecular switch to alter prostate cancer cell behavior. Matrix Biol. 2014;36:64–76. doi: 10.1016/j.matbio.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christoph K, Maria F, Gabriel M, et al. Prognostic impact of a compartment-specific angiogenic marker profile in patients with pancreatic cancer. Oncotarget. 2014;5(24):12978–12989. doi: 10.18632/oncotarget.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore BD. Graduate School of Arts and Sciences, Washington University. St Louis, MO, USA: 2015. Transcriptional regulation of the endoplasmic reticulum in dedicated secretory cells [PhD Thesis] . [Google Scholar]
- 33.Lee CS, Charalambous D. Immunohistochemical localisation of the c-fos oncoprotein in pancreatic cancers. Zentralbl. Pathol. 1994;140(3):271–275. [PubMed] [Google Scholar]
- 34.Li SH, Fu J, Watkins DN, Srivastava RK, Shankar S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of sonic hedgehog–gli pathway. Mol. Cell. Biochem. 2013;373(1-2):217–227. doi: 10.1007/s11010-012-1493-6. [DOI] [PubMed] [Google Scholar]
- 35.Masui T, Hosotani R, Doi R, et al. Expression of IL-6 receptor in pancreatic cancer: involvement in VEGFinduction. Anticancer Res. 2002;22(6C):4093–4100. [PubMed] [Google Scholar]
- 36.Craigbarnes HA, Doumouras BS, Palaniyar N. Surfactant protein d interacts with alpha2-macroglobulin and increases its innate immune potential. J. Biol. Chem. 2010;285(18):13461–13470. doi: 10.1074/jbc.M110.108837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bramhall SR, Neoptolemos JP, Stamp GWH, Lemoine NR. Imbalance of expression of matrix metalloproteinases (MMPs) and tissue inhibitors of the matrix metalloproteinases (TIMPs) in human pancreatic carcinoma. J. Pathol. 1997;182(3):347–355. doi: 10.1002/(SICI)1096-9896(199707)182:3<347::AID-PATH848>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 38.Fukuda A, Wang SC, Th MJ, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19(4):456–469. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balaz P, Friess H, Kondo Y, Zhu Z, Zimmermann A, Büchler MW. Human macrophage metalloelastase worsens the prognosis of pancreatic cancer. Ann. Surg. 2002;235(4):519–527. doi: 10.1097/00000658-200204000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikeda N, Nakajima Y, Tokuhara T, et al. Clinical significance of aminopeptidase n/cd13 expression in human pancreatic carcinoma. Clin. Cancer Res. 2003;9(4):1503–1508. [PubMed] [Google Scholar]
- 41.Shimojo Y, Akimoto M, Hisanaga T, et al. Attenuation of reactive oxygen species by antioxidants suppresses hypoxia-induced epithelial–mesenchymal transition and metastasis of pancreatic cancer cells. Clin. Exp. Metastasis. 2013;30(2):143–154. doi: 10.1007/s10585-012-9519-8. [DOI] [PubMed] [Google Scholar]
- 42.Hanas JS, Hocker JR, Cheung JY, et al. Biomarker identification in human pancreatic cancer sera. Pancreas. 2008;36(1):61. doi: 10.1097/mpa.0b013e3180d0a738. [DOI] [PubMed] [Google Scholar]
- 43.Barabási A, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vidal M, Cusick ME, Barabási AL. Interactome networks and human disease. Cell. 2011;144(6):986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee YH, Bae SC, Song GG. FCGR2A, FCGR3A, FCGR3B polymorphisms and susceptibility to rheumatoid arthritis: a meta-analysis. Clin. Exp. Rheumatol. 2015;33(5):647–654. [PubMed] [Google Scholar]
- 46.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 2012;196(4):395. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91(4):439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 48.Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato-Gastroenterology. 2008;55(88):2016–2027. [PubMed] [Google Scholar]; • Discusses the genes that are specifically overexpressed in pancreatic ductal carcinoma tumor epithelia.
- 49.Kato M, Hayakawa S, Naruse S, Kitagawa M, Nakae Y, Hayakawa T. Plasma alpha 2-macroglobulin-trypsin complexlike substance (MTLS) in pancreatic disease. J. Clin. Lab. Anal. 1996;10(6):399. doi: 10.1002/(SICI)1098-2825(1996)10:6<399::AID-JCLA14>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 50.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J. Clin. 2010;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 51.Størling J, Pociot F. Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes. 2017;8(2):72. doi: 10.3390/genes8020072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goggins M. Molecular markers of early pancreatic cancer. J. Clin. Oncol. 2005;23(20):4524–4531. doi: 10.1200/JCO.2005.19.711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.