

Abstract
Background
Increasing seed oil content is one of the most important targets for rapeseed (Brassica napus) breeding. However, genetic mechanisms of mature seed oil content in Brassica napus (B. napus) remain little known. To identify oil content-related genes, a genome-wide association study (GWAS) was performed using 588 accessions.
Results
High-throughput genome resequencing resulted in 385,692 high-quality single nucleotide polymorphism (SNPs) with a minor allele frequency (MAF) > 0.05. We identified 17 loci that were significantly associated with seed oil content, among which 12 SNPs were distributed on the A3 (11 loci) and A1 (one loci) chromosomes, and five novel significant SNPs on the C5 (one loci) and C7 (four loci) chromosomes, respectively. Subsequently, we characterized differentially expressed genes (DEGs) between the seeds and silique pericarps on main florescences and primary branches of extremely high- and low-oil content accessions (HO and LO). A total of 64 lipid metabolism-related DEGs were identified, 14 of which are involved in triacylglycerols (TAGs) biosynthesis and assembly. Additionally, we analyzed differences in transcription levels of key genes involved in de novo fatty acid biosynthesis in the plastid, TAGs assembly and lipid droplet packaging in the endoplasmic reticulum (ER) between high- and low-oil content B. napus accessions.
Conclusions
The combination of GWAS and transcriptome analyses revealed seven candidate genes located within the confidence intervals of significant SNPs. Current findings provide valuable information for facilitating marker-based breeding for higher seed oil content in B. napus.
Keywords: Brassica napus, GWAS, Seed oil content, SNPs, Transcriptomics, Candidate genes
Background
Brassica napus (B. napus, AACC, 2n = 38) is now the second largest oil crop following soybean. Moreover, rapeseed oil is not only one of the major edible vegetable oil for human consumption but it is also important for biofuel and lubricant production for industry [43]. B. napus is also a valuable animal feed source and potential protein source for human nutrition owing to the high-quality protein and low glucosinolate content of the seed [29, 53]. Enhancing seed oil content (SOC) and oil production per unit area of land is of paramount importance to meet the growing demand in oilseed breeding programs [61]. Although a comprehensive overview of the biological and metabolic pathways for triacylglycerol (TAG) synthesis has been well recorded [3, 34], little is known about the genetic and complex molecular regulatory mechanisms underlying variations in SOC of B. napus.
Quantitative trait locus (QTL) mapping and genome-wide association study (GWAS) have been widely used to dissect the regulatory loci and genetic architecture of complex agronomical quantitative traits at the whole genome level. Previous studies have focused on QTL identification for oil content in B. napus and have identified numerous QTLs in all 19 linkage groups of B. napus [8, 10, 19, 24, 49, 51, 54, 60]. GWAS, as another alternative for identifying QTLs, is not restricted to the traditional biparental linkage mapping and offers a higher resolution. Additionally, GWAS has recently been widely used in the study of important complex traits in B. napus, such as seed germination and vigor [16], plant height and primary branch [32, 52], harvest index [38, 42], yield traits [39, 57]. In addition, GWAS has been reported in various plants, such as Arabidopsis [1], rice [21, 22, 65], soybean [23] and maize [28, 50] and so on.
Brassica napus is an allopolyploid species with a complex genome structure, which originated ~ 7500 years ago from a spontaneous hybridization between B. rapa (AA, 2n = 20) and B. oleracea (CC, 2n = 18) [7]. SOC is an important complex quantitative traits but its genetic and molecular mechanisms remain undefined. So far, there are relatively few reports on the study of SOC by GWAS. Liu et al. identified 50 loci that were significantly associated with SOC using 521 B. napus accessions genotyped with the Brassica 60 K SNP array by GWAS and validated a novel locus on chromosome A5 that could increase 1.5–1.7% of the seed oil content by linkage mapping [36]. Li et al. detected a QTL on chromosome A08 with a significant association with seed oil content using GWAS [30]. Wang et al. detected 17 loci associated with seed oil content by GWAS [56] using a total of 238 rapeseed cultivars. In this study, we selected 588 B. napus accessions for GWAS by high-throughput genome resequencing.
To understand the genetic control of SOC at the population level through the identification of associated loci with SNPs, we genotyped 588 B. napus accessions that were collected from Asia (466), Europe (102), North America (13) and Australia (7) using high-throughput genome resequencing, and carried out a GWAS with PCA + K statistical models. SNPs that were significantly associated with SOC were identified. In addition, we performed transcriptome sequencing of four tissues with extremely high-(HO) and low-oil content (LO) B. napus accessions. Among the genes identified in both the GWAS and transcriptome analysis, seven were identified as candidate genes involved in seed oil accumulation, which were verified by quantitative real-time PCR (qRT-PCR). The current study thus may contribute to marker-based breeding for higher seed oil content in B. napus.
Materials and methods
Plant materials and phenotyping
A total of 588 B. napus lines were collected from spring, winter and semi-winter accessions and cultivated in Southwest University of Beibei, Chongqing, China (29°45′N, 106°22′E, 238.57 m) for 3 consecutive years (2016–2018). All the field experiments followed a randomized complete block design with two biological replications. Each accession was planted in two rows of 10–12 plants per row, with 20 cm between plants within each row and 30 cm between rows. The trial management was performed in accordance with local standard breeding field protocols. At maturity, open-pollinated seeds including five representative plants in the middle of each plot were collected for the SOC measurements. The oil content of the desiccated seeds was measured by near-infrared reflectance spectroscopy (NIRS DS2500) using the parameters described by Gan et al. [14].
Genome-wide association analysis
The seed oil content with two biological replicates for 3 consecutive years (2016–2018) was evaluated by the method of the best linear unbiased prediction (BLUP) based on a linear model using an R script (http://www.eXtension.org/pages/61006). An association analysis was implemented in TASSEL5.2.1 software using the P + K model [6]. The population structure (Q), relative kinship (K) and SNP genotyping in the association panel has been well described in our previous report [41]. The uniform threshold of GWAS was P < 2.59 × 10−6 (1/valid SNPs used, − log10 (1/385,692) = 5.59) [56]. The quantile–quantile plot was shown with the expected P value and − log 10 (P) of each SNP, and the Manhattan plot was demonstrated using the R package qqman.
Transcriptome sequencing and identification of differentially expressed genes
Two extremely high-oil content (HO) lines and one extremely low-oil content (LO) line were selected from the GWAS population for transcriptome sequencing (RNA-Seq), respectively. The HO lines were SWU47 (CQ24) and Zhongshuang11 (CQ52), while the LO line was Ningyou12 (CQ46). Total RNA was extracted from four tissues of the HO and LO accessions, respectively. Tissues were harvested 30 days after flowering from seed and silique pericarps on the main inflorescence (30SM and 30SPM, respectively) and on the primary branch (30SB and 30SPB, respectively). For each sample, two biological replicates were performed, with each collected from three independent plants. All samples were immediately placed in liquid nitrogen and stored at − 80 °C for RNA sequencing (RNA-seq) and quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis.
Sequencing library preparation and sequencing reactions were conducted at the Biomarker Technologies Corporation (Beijing, China). Gene expression levels were estimated using FPKM (Fragments per kilobase of exon per million reads mapped). Differentially expressed genes (DEGs) between two samples were obtained with Cuffdiff, based on the criteria false discovery rate (FDR) < 0.05 and |log2fold change| > 2.0 [4].
Identification and expression analysis of acyl-lipid metabolism genes between extremely high- and low-oil content B. napus lines
To seek out genes associated with acyl-lipid metabolism (ALM), B. napus homologous gene sequences were analyzed against a list of genes involved in acyl-lipid metabolism obtained from the “Arabidopsis Acyl-Lipid Metabolism” website (ARALIP) (http://aralip.plantbiology.msu.edu/) [34]. The differentially expressed lipid metabolism-related genes were obtained on the basis of the RNA-Seq data of the B. napus HO and LO lines created in the present study. Similarly, to analyze the spatial expressions of differentially expressed acyl-lipid metabolism-related genes in the extremely HO and LO B. napus lines, the expression values of these genes in 30SM, 30SPM, 30SB and 30SPB were obtained from this RNA-Seq data as well. The expression heatmap of the differential expressed acyl-lipid metabolism genes was generated using HemI1.0 [58].
GO and KEGG enrichment analysis of DEGs
GO enrichment and KEGG pathway analysis of DEGs were performed using the online OmicShare tool (http://www.omicshare.com/tools/index.php/) [37, 45]. The threshold of significantly enriched GO terms was set to FDR < 0.05 [39].
Identification of potential candidate genes
To identify candidate genes associated with SOC, the 300-kb flanking regions on either side of the markers significantly associated with SOC were chosen as the confidence interval for in-depth analysis, as described previously [56]. The DEGs within the confidence interval of SNPs significantly associated with SOC were screened to identify differentially expressed (DE) candidate genes. Additionally, DEGs involved in acyl-lipid metabolism were also identified.
Validation of candidate genes by qRT-PCR analysis
Total RNA was extracted from all tested tissues with the EZ-10 DNAaway RNA Mini-prep Kit [Sangon Biotech (Shanghai), Co., Ltd], and then cDNA was synthesized from 1 µg RNA using the PrimeScript™ RT reagent kit with gDNA Eraser according to the manufacturer’s instructions (Perfect Real Time; TaKaRa Biotechnology, Dalian, China). The gene-specific primers for qRT-PCR of the candidate genes and reference gene are listed in Additional file 1: Table S7. The PCR consisted of 10 μL SYBR II (TakaRa), 2.0 μL cDNA, 1.6 μL primer, 0.4 μL ROX Reference Dye II and distilled water to a final volume of 20 μL. The PCR program was as follows: 95 °C for 30 s and 35 cycles of 95 °C for 5 s, followed by 56–60 °C (depending on the primers used) for 30 s. For each reaction, three biological replicates were performed, and relative expression levels were obtained using the 2−∆∆Ct method, with BnActin7 as internal controls.
Results
Phenotypic variation of SOC
The 588 B. napus accessions were planted in three environments (2016CQ, 2017CQ and 2018CQ) from 2016 to 2018, with two replications performed each year. Extensive phenotypic variations of SOC were observed (Table 1), and specific seed oil content (SOC, % of seed weight) phenotypes of 588 accessions for GWAS analysis are shown in Additional file 1: Table S1. In 2016CQ, SOCs ranged from 26.83 to 44.94, with an average of 35.05. And SOCs were from 30.21 to 48.41, with an average of 38.21 in 2017CQ, and from 29.46 to 49.13, with an average of 40.15 in 2018CQ. Among the three environments, the coefficient of variation (CV) of SOC was less than 10%, reflecting a relatively small variation of SOC within the entire GWAS panels. SOC among three environments in B. napus showed continuous variation and approximated a normal distribution (Fig. 1), suggesting SOC consisted of quantitative traits controlled by multiple genes. The broad sense heritability of SOC was 73.4%, which is less than previously reported by Liu et al. [36] (87.4%). These results suggested that most of the phenotypic variation in SOC was attributed to genetic effects despite being greatly affected by the environment in this study.
Table 1.
Phenotypic variation of seed oil content in three environments
| Trait | Mean | SD | Min | Max | CV (%) |
|---|---|---|---|---|---|
| 2016CQ | 35.05 | 2.88 | 26.83 | 44.94 | 8.22 |
| 2017CQ | 38.21 | 3.31 | 30.21 | 48.41 | 8.66 |
| 2018CQ | 40.15 | 3.18 | 29.46 | 49.13 | 7.92 |
CQ Chongqing, SD standard deviation, CV coefficient of variation
Fig. 1.

Frequency distribution of seed oil content (SOC) in the association panel of 588 accessions across three environments (2016CQ, 2017CQ and 2018CQ). CQ refers to Chongqing and the No. of Lines represents the number of accessions. CQ46 (Ningyou 12), CQ24 (SWU 47) and CQ52 (Zhongshuang 11) are accessions for RNA-Seq. LO and HO refers to extremely high- and low-oil content B. napus accessions, respectively
Genome-wide association analysis
GWAS for seed oil content (SOC) was performed using the P + K model [48]. The QQ plot is shown in Fig. 2a, and the results showed that this model could be used to identify association SNPs. A total of 17 significant SNPs for SOC (P < 2.59 × 10−6) were identified and these SNPs were unevenly distributed across four chromosomes (A1, A3, C5 and C7) (Fig. 2b and Table 2). Eleven significant SNPs were distributed on the A3 chromosome and for up to 64.71%, the significant correlation region ranged from 17.68 18.36 Mb, which was consistent with some previous QTL mapping results (Fig. 3) [10, 68]. Only one SNP was distributed on the A1 and C5 chromosomes, respectively. Additionally, the remaining four SNPs were distributed on the C7 chromosome. Individual significant SNPs for SOC explained 5.46–6.68% of the phenotypic variation (R2). The location and detailed information for all 17 SNPs are listed in Table 2. Among all the detected significant SNPs, twelve SNPs (70.59%) were within the previously identified significant QTL confidence intervals associated with seed oil content, suggesting the high reliability of SNPs identified in this study (Table 2) [10, 36, 54, 66, 68]. To find candidate genes associated with oil content, all these significant SNPs will be further analyzed.
Fig. 2.
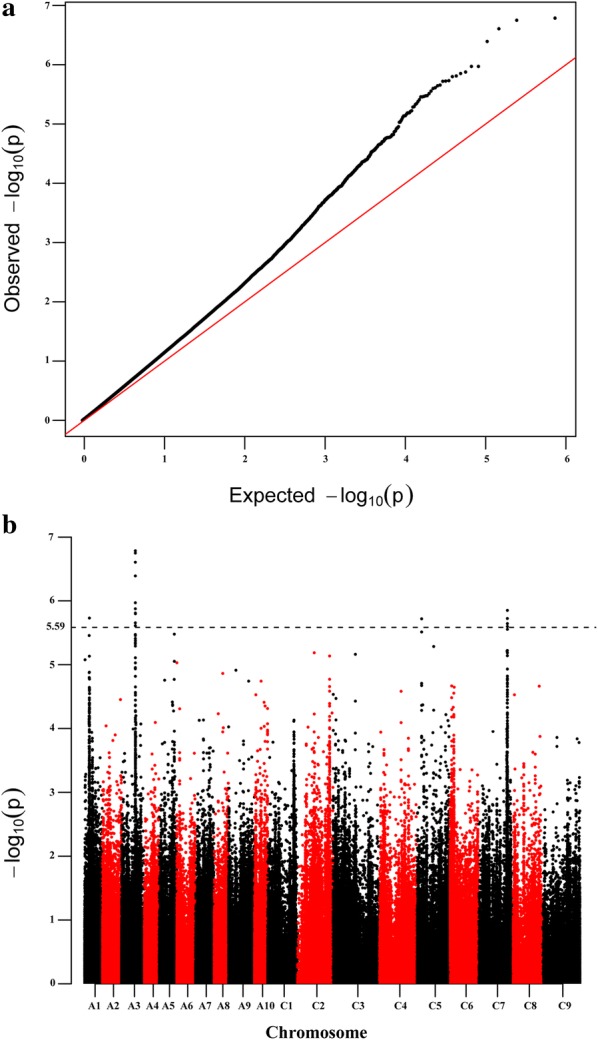
Quantile–quantile and Manhattan plots resulting from the GWAS results for seed oil content (SOC) in Brassica napus. a Quantile–quantile plot for SOC. b Manhattan plot for SOC. The dashed horizontal line indicates the Bonferroni-adjusted significance threshold (2.59 × 10−6)
Table 2.
Markers with significant association for SOC and genes in confidence interval
| Chromosome | Position | SNP | P value | R 2 | Confidence interval (300 kb up/downstream) | Genes in confidence interval | Detected in previous studies |
|---|---|---|---|---|---|---|---|
| A1 | 6244998 | S1_6244998 | 1.86E−06 | 0.06489 | 5944998–6,544,998 | BnaA01g11880D–BnaA01g13060D | SG-qOC-A1 [66]; qOC-A1-1 [54]; Bn-A01-p6560270 [36]; cqOC-A1 [8] |
| A3 | 17975486 | S3_17975486 | 1.64E−07 | 0.06679 | 17,675,486–18,275,486 | BnaA03g36150D–BnaA03g37050D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17981854 | S3_17981854 | 1.78E−07 | 0.06494 | 17,681,854–18,281,854 | BnaA03g36170D–BnaA03g37070D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17982022 | S3_17982022 | 2.48E−07 | 0.06503 | 17,682,022–18,282,022 | BnaA03g36170D–BnaA03g37070D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17981872 | S3_17981872 | 4.06E−07 | 0.06133 | 17,681,872–18,281,872 | BnaA03g36170D–BnaA03g37070D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17976024 | S3_17976024 | 1.07E−06 | 0.05832 | 17,676,024–18,276,024 | BnaA03g36160D–BnaA03g37050D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17982457 | S3_17982457 | 1.07E−06 | 0.0598 | 17,682,457–18,282,457 | BnaA03g36170D–BnaA03g37070D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17986408 | S3_17986408 | 1.33E−06 | 0.05819 | 17,686,408–18,286,408 | BnaA03g36180D–BnaA03g37080D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 18016213 | S3_18016213 | 1.55E−06 | 0.05585 | 17,716,213–18,316,213 | BnaA03g36240D–BnaA03g37110D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 18055410 | S3_18055410 | 1.59E−06 | 0.05956 | 17,755,410–18,355,410 | BnaA03g36320D–BnaA03g37130D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17986377 | S3_17986377 | 2.21E−06 | 0.05862 | 17,686,377–18,286,377 | BnaA03g36180D–BnaA03g37080D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| A3 | 17982838 | S3_17982838 | 2.45E−06 | 0.05525 | 17,682,838–18,282,838 | BnaA03g36170D–BnaA03g37070D | qOC-A3-DY, qOC-A3-4-TN [10, 68] |
| C5 | 5886965 | S15_5886965 | 1.91E−06 | 0.0638 | 5,586,965–6,186,965 | BnaC05g10000D–BnaC05g10760D | |
| C7 | 37164777 | S17_37164777 | 1.41E−06 | 0.05992 | 36,864,777–37,464,777 | BnaC07g33910D–BnaC07g34940D | |
| C7 | 37136168 | S17_37136168 | 1.89E−06 | 0.05761 | 36,836,168–37,436,168 | BnaC07g33820D–BnaC07g34890D | |
| C7 | 37142657 | S17_37142657 | 2.27E−06 | 0.05463 | 36,842,657–37,442,657 | BnaC07g33850D–BnaC07g34890D | |
| C7 | 37178443 | S17_37178443 | 2.52E−06 | 0.05577 | 36,878,443–37,478,443 | BnaC07g33910D–BnaC07g34970D |
Fig. 3.
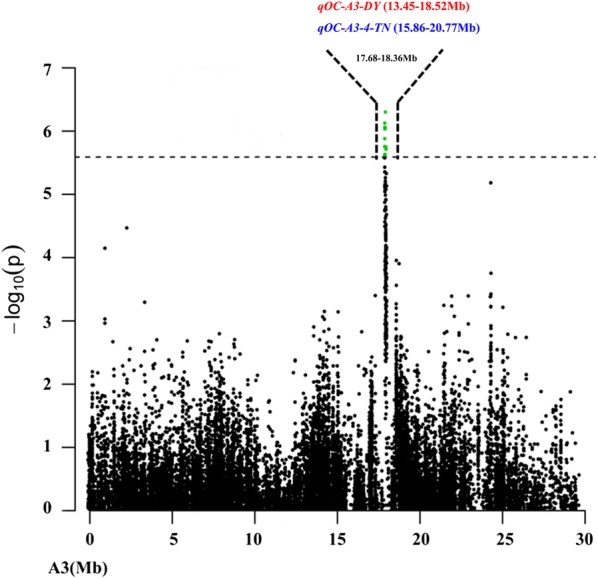
Comparison of the GWAS results in A3 with those of previous QTL mapping reported in Delourme et al. [10] and Zou et al. [68]. The markers that lie within the confidence interval of the seed oil content (SOC) QTL are in red for Delourme et al. [10] and blue for Zou et al. [68]
Comparative analysis of three Brassica napus lines using transcriptome sequencing
Three natural B. napus accessions with extremely significant differences in seed oil content (SOC) were selected from the genome-wide association analysis populations. The oil and protein contents of the desiccated seeds were determined by near-infrared reflectance spectroscopy (NIRS DS2500), and the results are shown in Fig. 4a. The average oil contents during the 3-year period for SWU47 (CQ24, high-oil) and Zhongshuang11 (CQ52, high-oil) were significantly higher than those of Ningyou12 (CQ46, low oil) and there is no significant difference in seed protein content and 1000-seed weight between the high- and low-oil lines (Fig. 4a). To determine the FA compositions of 30SM, 30SB and mature desiccated seeds of three accessions subjected to gas chromatography–mass spectrometry (GC–MS). In 30SM, 30SB (Fig. 4b) or mature desiccated seeds (Fig. 4c), the results showed significantly higher C18:1 in CQ24 and CQ52 (high-oil content), and significantly lower C16:0 and C18:2 in CQ24 and CQ52 compared to CQ46 (low-oil content).
Fig. 4.

Seed oil content, protein content and fatty acid (FA) composition of HO and LO Brassica napus accessions for RNA-Seq. a Three years (2016–2018) seed oil content (% of dry seed weight), protein content (% of dry seed weight) and weight/1000 dry seeds of three accessions. b Percentage of fatty acid species in 30SM and 30SB of three accessions (n = 4), 30SM and 30SB represent seeds on the main inflorescence and primary branch of 30 days after flowering, respectively. c FA composition in mature seeds of three accessions (mean ± SD, n = 4). CQ46 (Ningyou 12), CQ24 (SWU 47) and CQ52 (Zhongshuang 11) are accessions for RNA-Seq.*Denotes significance at P < 0.05, **P < 0.01, based on Student’s t test
Transcriptome analyses for differentially expressed genes
In the transcriptome analyses, RNA was obtained from four tissues of extremely high- (CQ24 and CQ52) and low-oil content (CQ46) B. napus lines at CQ including 12 independent samples and a total of 24 libraries (two biological replicates per sample) were constructed for transcriptome sequencing. After removing the low quality and contaminant reads, a total of 364.0 million clean reads were acquired, with an average of 23.05 million reads per sample. On average, 95.24% of the input reads mapped uniquely to the B. napus reference genome (Additional file 1: Table S2). The correlation coefficient between the two biological replicates of each sequencing sample exceeded 0.9 for all tested samples (Additional file 1: Table S2), suggesting a high reproducibility among the samples.
To identify differentially expressed genes (DEGs) between extremely high- and low-oil content B. napus lines, the following criterion were applied: |log2fold change| > 2.0 and FDR < 0.05. For CQ24/CQ46, 2185 and 4858 genes were differentially expressed in individual tissues. Among which, the number of DEGs was variable, and the number of upregulated genes exceeded that of the number of downregulated genes in all tissues tested. Under CQ52/CQ46, between 3347 and 4018 genes were differentially expressed in individual tissues, among which the number of differential genes was not very variable and the number of upregulated exceeded the number of downregulated genes in all tissues tested except 30SM (Additional file 1: Table S3, Additional file 2: Fig. S1a). These results suggested that positive regulatory genes might play a major role in the formation of high oil content in B. napus. To find conserved DEGs between high- and low-oil content lines, Venny analysis was conducted using CQ24/CQ46 and CQ52/CQ46 in all four tissues, respectively. We found 1628, 2658, 2146 and 1493 common DEGs in 30SM, 30SB, 30SPM and 30SPB, respectively, suggesting that transcriptomic variations are different in diverse tissues and variations in 30SB and 30SPM are greater than 30SM and 30SPB between high- oil and low-oil content B. napus (Fig. 5a).
Fig. 5.

Transcriptomic analysis of the four tissues among HO and LO B. napus cultivars. a Common differential genes (DEGs) were obtained in all four tissues under CQ24/CQ46 and CQ52/CQ46 by Venny analysis. b KEGG pathway analysis of common DEGs in the Fig. 5a
Functional classification of common DEGs between CQ24 (HO)/CQ46(LO) and CQ52(HO)/CQ46(LO)
To understand the functional classification of common DEGs in four tested tissues using CQ24(HO)/CQ46(LO) and CQ52(HO)/CQ46(LO) (hereinafter referred to as common DEGs), Gene Ontology (GO) enrichment analysis was performed. GO terms were divided into three main categories: biological process, cellular component and molecular function (Additional file 2: Fig. S1b, c). The results showed that common DEGs involved in cellular (GO:0009987), metabolism (GO:0008152) and single-organism processes (GO:0044699) were the most common in all four tissues between extremely high- and low-oil content B. napus lines. The most common DEGs were mainly enriched in the cell part (GO:0044464), cell (GO:0005623) and organelles (GO:0043226), and the dominant molecular function of the most common DEGs in all four tissues was binding (GO:0005488) and catalytic activity (GO:0003824) (Additional file 2: Fig. S1b, c).
To understand the functional distribution of these common DEGs, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was also conducted. The results showed that the most enriched pathway was involved in metabolism and the main pathways of common DEGs in all four tested tissues were carbohydrate metabolism, global and overview, amino acid metabolism, energy metabolism, lipid metabolism and biosynthesis of other secondary metabolites (Fig. 5b). In this study, we focused on common DEGs in lipid metabolism, and 64 differential expressed lipid metabolism-related genes were found in all four tissues, of which 4, 24, 10, and 6 were specific to 30SM, 30SB, 30SPM, and 30SPB, respectively. In addition, six common differential lipid metabolism genes were shared in 30SM and 30SB, two in 30SB, 30SM and 30SPB, two in 30SB, 30SM and 30SPM, three in 30SB, 30SPB and 30SPM, and seven among all four tissues (Additional file 1: Table S4).
To understand the expression patterns of 64 common differential expressed lipid metabolism-related genes between HO (CQ24, CQ52) and LO (CQ46) accessions, the heatmap of these genes was drawn based on the RNA-seq data which were normalized to the log2 FPKM using HemI1.0 (Additional file 1: Table S4, Additional file 2: Fig. S2a). Among all 64 common differential expressed lipid metabolism-related genes, 38 genes were upregulated in the HO (CQ24, CQ52) lines compared with the LO (CQ46) lines, and 26 genes showed opposite expression patterns. We found seven genes (BnaA07g31890D; BnaC02g06560D; BnaCnng62740D; BnaA03g60440D; BnaA07g35160D; BnaA07g20190D; BnaC08g44190D) that were differentially expressed in all tested tissues between HO and LO accessions, and only BnaC08g44190D was downregulated in HO compared with LO. Moreover, a total of 13 DEGs (BnaC03g16690D; BnaA07g31890D; BnaC02g00470D; BnaC03g04180D; BnaC02g04910D; BnaC04g40760D; BnaA03g13590D; BnaA04g17150D; BnaC08g12280D; BnaA01g15860D; BnaC01g18950D; BnaA10g09480D; BnaC09g31660D) involved in the TAG biosynthesis pathway and TAG assembly were upregulated in the HO compared with the LO lines. Therefore, we speculated that these upregulated lipid metabolism genes in HO lines may play an indispensable role in the formation of high oil content of B. napus.
TAG biosynthesis and assembly pathway gene expression in four tested tissues of the HO and LO accessions
To preliminarily elucidate the difference in seed oil content between transcriptome sequencing accessions at the transcription level, the genes involved in the TAG biosynthesis pathway were analyzed and presented using a Log2FPKM (calculation method) as relative transcript levels among the tissues and accessions, with a specific focus on FA synthesis in the plastid, and TAG accumulation/packaging pathways in the endoplasmic reticulum (Fig. 6) [2, 34].
Fig. 6.

Comparison of gene expression of TAG biosynthesis pathway and TAG assembly in all four tissues among HO and LO B. napus lines. The color boxes are the heatmap of gene expressions from RNA-seq which were normalized by the log2 FPKM (calculation method) (Additional file 1: Table S6). Abbreviation of genes that encode proteins (blue letters): PDHC pyruvate dehydrogenase complex, ACCase acetyl-CoA carboxylase, MCMT malonyl-CoA:ACP malonyltransferase, ACP acyl carrier protein, KASI/II/III 3-ketoacyl-ACP synthase I/II/III, KAR ketoacyl-ACP reductase, HAD hydroxyacyl-ACP dehydrase, ENR enoyl-ACP reductase, SAD stearoyl-acyl carrier protein desaturase, FATA/B fatty acyl-ACP thioesterase A/B, FAX1 plastid fatty acid export 1, LACS9 long-chain acyl-CoA synthetase 9, GPDH glycerol-3-phosphate dehydrogenase, GPAT9 glycerol-3-phosphate acyltransferase 9, LPAAT lysophosphatidic acid acyltransferase, PAP phosphatidic acid phosphatase, DGAT diacylglycerol acyltransferase, LPCAT lysophosphatidylcholine acyltransferase, FAD2/3 fatty acid desaturase 2/3, PLD phospholipase D, PDAT phospholipid:diacylglycerol acyltransferase, PDCT phosphatidylcholine:diacylglycerol cholinephosphotransferase, CPT CDP-choline:diacylglycerol cholinephosphotransferase, PLC phospholipase C, OBO oil body oleosin, CALO caleosin
In terms of FA synthesis in the plastid, most genes exhibited differential expression patterns among 30SM, 30SB, 30SPM, and 30SPB tissues in the same accession, especially in seeds (30SM, 30SB), and the expression level of most FA synthesis genes was significantly higher than in silique pericarps (30SPM and 30SPB). In addition, many genes showed differential expression levels in the same tissue among the HO (CQ24, CQ52) and LO (CQ46) accessions (Fig. 6 and Additional file 1: Table S6). For example, the transcript levels of the pyruvate dehydrogenase complex (PDHC), acetyl-CoA carboxylase (ACCase), malonyl-CoA:ACP malonyltransferase (MCMT), 3-ketoacyl-ACP synthase II/III (KASII/III), ketoacyl-ACP reductase (KAR), hydroxyacyl-ACP dehydrase (HAD), enoyl-ACP reductase (ENR), stearoyl-acyl carrier protein desaturase (SAD), and acyl-ACP thioesterase A (FATA) were generally lower in the seed tissues (30SM and 30SB) of the LO (CQ46) than the HO (CQ24 and CQ52) accessions. However, these genes showed no difference in transcription levels in silique pericarps (30SPM and 30SPB) among the HO and LO lines. Together, the differential expression of fatty acid synthesis genes in seeds is more likely to explain the difference in oil content among the three accessions than the silique pericarps; however, this does not preclude an effect of the silique pericarps in modulating the oil content difference.
Glycerol-3-phosphate (G-3-P) and acyl-CoA, which are precursors of glycerolipid assembly in the endoplasmic reticulum (ER), are produced by NAD-dependent glycerol-3-phosphate dehydrogenase (GPDH), and be involved in plastid fatty acid export 1(FAX1) and long-chain acyl-CoA synthetase 9 (LACS9) [5, 12, 31]. The transcript levels of GPDH and LACS9 were higher in the 30SM and 30SB of HO than in the LO lines, suggesting an increase in the acyl-CoA pool and glycerol-3-phosphate (G-3-P) substrate for HO lines in seeds. However, their expression in silique pericarps (30SPM and 30SPB) was not consistent. It is worth noting that the expression levels of GPDH and LACS9 in seeds were much higher than those in silique pericarps, but the expression of FAX1 was higher in silique pericarps than in seeds (Fig. 6). Thus, based from the transcript levels alone of the four transcriptome sequencing tissues, it remains vague whether these steps are important in regulating the oil content differences among the three accessions.
The assembly of TAGs in the ER occurs via two possible routes [9]. In the conventional Kennedy pathway, glycerol-3-phosphate (G-3-P) with acyl-CoA to yield TAG requires sequential enzymes; glycerol-3-phosphate acyltransferase 9 (GPAT9), 1-acylglycerol-3-phosphate acyltransferase (LPAAT), phosphatidic acid phosphatase (PAP) and diacylglycerol acyltransferase (DGAT) [5, 9, 12]. The transcript levels of PAP and DGAT (sum of DGAT1 and DGAT2) were higher in 30SM and 30SB of HO (CQ24, CQ52) than LO (CQ46), whereas GPAT9 and LPAAT exhibited similar transcript levels in 30SM and 30SB between HO (CQ24, CQ52) and LO (CQ46). In another TAG biosynthesis pathway, lysophosphatidylcholine acyltransferase (LPCAT) and phospholipid: diacylglycerol acyltransferase (PDAT) play an important role in forming TAG [9]. The transcript levels of LPCAT and PDAT were similar among the HO and LO accessions. Additionally, the transcript levels of FA desaturase 3 (FAD3) were higher in the seeds (30SM and 30SB) of the HO than the LO accessions, phosphatidylcholine: diacylglycerol cholinephosphotransferase (PDCT) and CDP-choline: diacylglycerol cholinephosphotransferase (CPT) could mediate the shuttling between PC-derived DAG and PC, and the transcript levels for PDCT were lower in seeds (30SB) of LO than HO accessions, while CPT transcript levels were higher in silique pericarps (30SPM and 30SPB) in the HO accessions. Phospholipase C (PLC) and phospholipase D (PLD) hydrolyze PC to produce DAG and PA, respectively, and their expression is increased in silique pericarps (30SPM and 30SPB) compared with seeds (30SM and 30SB). The oil body oleosin (OBO) and caleosin (CALO), are crucial for the stability of lipid droplets [44]. The RNA-Seq data showed that the expression level of OBO (sum of all isoforms) and CALO was lower in all tissues of the LO than HO accessions, and OBO had the highest expression in seeds (30SM and 30SB) of the HO accessions (Fig. 6).
Identification candidate genes by combining GWAS and transcriptome sequencing analysis
The candidate gene regions with SOC as determined by GWAS analysis were the 300-kb flanking regions on either side of the markers significantly associated with SOC, as described previously [56]. All genes within the confidence interval of all 17 significant SNPs with SOC are listed in Additional file 1: Table S5. By combination with transcriptome sequencing analysis, we found a total of 41 genes that were differentially expressed between high and low oil accessions, of which 16 and 32 were DEGs in CQ24/CQ46 (No. X1–X16) (Table 4) and CQ52/CQ46 (NO. Z1-Z32) (Table 5), respectively. Furthermore, seven common DEGs (X5/Z30, X7/Z31, X10/Z20, X12/Z13, X16/Z10, X15/Z9, and X14/Z1) were identified between CQ24/CQ46 and CQ52/CQ46. The biological functions of these differential expressed candidates were analyzed by applying Protein Basic Local Alignment Search Tool (BLASTP) searches against all Arabidopsis proteins. The results are listed in Tables 4 and 5 and the expression patterns of these genes between high (CQ24, CQ52) and low oil (CQ46) accessions are shown in Additional file 2: Fig. S3. Seven common DEGs (X5/Z30, X7/Z31, X10/Z20, X12/Z13, X16/Z10, X15/Z9, and X14/Z1) were considered to be important candidate genes related to seed oil content for the following study.
Table 4.
Candidate genes screened by combining GWAS and RNA sequencing (CQ24/46)
| Code | Candidate genes | At orthologs | Location | gDNA Length (bp) | CDS length (bp) | Exon number | Function description |
|---|---|---|---|---|---|---|---|
| X1 | BnaA01g11960D | AT4G22160 | chrA01:6001930–6002661 | 732 | 480 | 2 | Unknown protein |
| X2 | BnaA01g12400D | AT4G22730 | chrA01:6194675–6197498 | 2824 | 2064 | 3 | Leucine-rich repeat protein kinase family protein |
| X3 | BnaA01g12530D | AT4G22880 | chrA01:6294325–6295940 | 1616 | 1077 | 2 | Leucoanthocyanidin dioxygenase (LDOX) |
| X4 | BnaA01g12540D | AT4G22890 | chrA01:6297451–6299173 | 1723 | 960 | 9 | PGR5-LIKE A |
| X5 | BnaA01g13030D | AT4G23370 | chrA01:6518736–6520647 | 1912 | 1161 | 8 | Unknown protein |
| X6 | BnaA03g36210D | AT3G21320 | chrA03:17700381–17702480 | 2100 | 1473 | 5 | EARLY FLOWERING protein |
| X7 | BnaA03g36510D | AT3G29040 | chrA03:17886150–17887585 | 1436 | 744 | 3 | Domain of unknown function (DUF26) |
| X8 | BnaA03g36520D | AT3G22160 | chrA03:17918805–17921792 | 2988 | 264 | 2 | JASMONATE-ASSOCIATED VQ MOTIF GENE 1, JAV1 |
| X9 | BnaA03g36540D | AT4G11850 | chrA03:17925272–17933870 | 8599 | 4398 | 15 | Phospholipase D gamma 1 (PLDGAMMA1) |
| X10 | BnaA03g36620D | AT3G22370 | chrA03:17988398–17992403 | 4006 | 1065 | 6 | Alternative oxidase 1A (AOX1A) |
| X11 | BnaA03g37000D | AT3G23180 | chrA03:18246929–18248163 | 1235 | 564 | 6 | HR-like lesion-inducing protein-related |
| X12 | BnaA03g37060D | AT1G51400 | chrA03:18277785–18278767 | 983 | 438 | 3 | Photosystem II 5 kD protein |
| X13 | BnaC05g10400D | AT1G14130 | chrC05:5962733–5963804 | 1072 | 915 | 2 | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein |
| X14 | BnaC05g10690D | AT1G14450 | chrC05:6142756–6148159 | 5404 | 225 | 1 | NADH dehydrogenase (ubiquinone)s |
| X15 | BnaC05g10700D | AT1G51350 | chrC05:6148823–6151086 | 2264 | 312 | 7 | ARM repeat superfamily protein |
| X16 | BnaC05g10710D | AT5G07300 | chrC05:6151155–6152461 | 1307 | 357 | 8 | Encodes a copine-like protein, which is a member of a newly identified class of calcium-dependent, phospholipid binding proteins |
Table 5.
Candidate genes screened by combining GWAS and RNA sequencing (CQ52/46)
| Code | Genes in confidence interval | At orthologs | Location | gDNA length (bp) | CDS length (bp) | Exon number | Function description |
|---|---|---|---|---|---|---|---|
| Z1 | BnaC05g10690D | AT1G14450 | chrC05:6142756–6148159 | 5404 | 225 | 1 | NADH dehydrogenase (ubiquinone)s |
| Z2 | BnaA03g37070D | AT3G23410 | chrA03:18280209–18283825 | 3617 | 2166 | 3 | Fatty alcohol oxidase 3 (FAO3) |
| Z3 | BnaA01g12270D | AT4G22530 | chrA01:6118512–6120375 | 1864 | 786 | 2 | S-Adenosyl-l-methionine-dependent methyltransferases superfamily protein |
| Z4 | BnaA01g12520D | AT4G22860 | chrA01:6288006–6294204 | 6199 | 2358 | 23 | Cell cycle regulated microtubule-associated protein |
| Z5 | BnaA01g12870D | AT4G23060 | chrA01:6443916–6447139 | 3224 | 1467 | 5 | IQ-domain 22 (IQD22) |
| Z6 | BnaA03g36230D | AT3G21330 | chrA03:17713723–17714730 | 1008 | 1008 | 1 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
| Z7 | BnaA03g36910D | AT3G22960 | chrA03:18136808–18141526 | 4719 | 1728 | 7 | PKP-ALPHA |
| Z8 | BnaC07g34230D | AT4G17220 | chrC07:37116980–37119308 | 2329 | 1512 | 10 | Microtubule-associated proteins 70-5 (MAP70-5) |
| Z9 | BnaC05g10700D | AT1G51350 | chrC05:6148823–6151086 | 2264 | 312 | 7 | ARM repeat superfamily protein |
| Z10 | BnaC05g10710D | AT5G07300 | chrC05:6151155–6152461 | 1307 | 357 | 8 | Putative copine, regulates calcium signalling, BONZAI (AtBON2) |
| Z11 | BnaC07g34010D | AT4G16980 | chrC07:36973813–36974752 | 940 | 513 | 1 | Arabinogalactan-protein family |
| Z12 | BnaC07g34060D | AT4G17050 | chrC07:37001028–37003893 | 2866 | 903 | 13 | Ureidoglycine aminohydrolase (UGLYAH) |
| Z13 | BnaA03g37060D | AT1G51400 | chrA03:18277785–18278767 | 983 | 438 | 3 | Photosystem II 5 kD protein |
| Z14 | BnaA01g12040D | AT4G22220 | chrA01:6038697–6039866 | 1170 | 480 | 3 | ISU1 |
| Z15 | BnaA01g12370D | AT4G22680 | chrA01:6178215–6179541 | 1327 | 804 | 2 | Myb domain protein 85 (MYB85) |
| Z16 | BnaC07g34050D | AT4G17040 | chrC07:36997388–37000753 | 3366 | 921 | 7 | CLP protease R subunit 4 (CLPR4) |
| Z17 | BnaA01g12340D | AT4G22620 | chrA01:6165884–6166560 | 677 | 483 | 1 | SAUR-like auxin-responsive protein family |
| Z18 | BnaC05g10380D | AT1G14100 | chrC05:5938383–5940030 | 1648 | 1578 | 2 | Fucosyltransferase 8 (FUT8) |
| Z19 | BnaA03g36300D | AT3G21460 | chrA03:17739884–17740442 | 559 | 309 | 1 | Glutaredoxin family protein |
| Z20 | BnaA03g36620D | AT3G22370 | chrA03:17988398–17992403 | 4006 | 1065 | 6 | Alternative oxidase 1A (AOX1A) |
| Z21 | BnaA01g12320D | AT4G22590 | chrA01:6150232–6152082 | 1851 | 1128 | 7 | Haloacid dehalogenase-like hydrolase (HAD) superfamily protein |
| Z22 | BnaA01g12930D | AT4G23180 | chrA01:6474955–6477798 | 2844 | 2007 | 7 | Cysteine-rich RLK (RECEPTOR-like protein kinase) 10 (CRK10) |
| Z23 | BnaC05g10460D | AT1G14200 | chrC05:6006999–6010819 | 3821 | 1059 | 4 | RING/U-box superfamily protein |
| Z24 | BnaC07g34250D | AT4G17260 | chrC07:37125045–37126398 | 1354 | 1053 | 2 | Lactate/malate dehydrogenase family protein |
| Z25 | BnaC07g34450D | AT4G17615 | chrC07:37207760–37210093 | 2334 | 642 | 8 | Calcineurin B-like protein 1 (CBL1) |
| Z26 | BnaC07g34510D | AT4G17670 | chrC07:37221343–37222462 | 1120 | 507 | 2 | Protein of unknown function (DUF581) |
| Z27 | BnaA03g36680D | AT4G30090 | chrA03:18023844–18026170 | 2327 | 831 | 10 | Embryo defective 1353 (emb1353) |
| Z28 | BnaC05g10680D | AT1G14440 | chrC05:6131908–6132828 | 921 | 921 | 1 | Homeobox protein 31 (HB31) |
| Z29 | BnaA01g11920D | AT4G22120 | chrA01:5970386–5973641 | 3256 | 2088 | 8 | ERD (early-responsive to dehydration stress) family protein |
| Z30 | BnaA01g13030D | AT4G23370 | chrA01:6518736–6520647 | 1912 | 1161 | 8 | Unknown protein |
| Z31 | BnaA03g36510D | AT3G29040 | chrA03:17886150–17887585 | 1436 | 744 | 3 | Domain of unknown function (DUF26) |
| Z32 | BnaC05g10510D | AT1G14280 | chrC05:6028460–6029815 | 1356 | 1254 | 1 | Phytochrome kinase substrate 2 (PKS2) |
Verification of the transcriptome sequencing data by quantitative real-time polymerase chain reaction (qRT-PCR)
To confirm the accuracy of the RNA-Seq results, some genes were selected to perform qRT-PCR analysis, and the expression levels of these genes by qRT-PCR and transcriptome sequencing are shown in Fig. 7. Although the expression trend of individual genes, such as BnaA03g60440D, was not consistent with the RNA-Seq results between two high-oil content accessions, their expression trend was consistent between high- and low-oil content accessions. In short, the results of RNA-Seq are highly consistent with qRT-PCR. The results fully demonstrate the reliability and accuracy of the transcriptome sequencing data.
Fig. 7.

qRT-PCR analysis confirmed the accuracy of transcriptome sequencing by identifying the expression patterns of nine genes in all four tested tissues. The blue line represents the RNA-Seq results and the orange line represents the qRT-PCR results. *Denotes significance at P < 0.05, **P < 0.01, based on Student’s t test
Discussion
The identification of significant SNPs for seed oil content (SOC)
Genome-wide association analysis (GWAS) based on millions of markers is currently widely used in analysis of complex agronomic traits for crops such as rice [22], maize [50], soybean [23] and sorghum [46]. Many reports have addressed QTL mapping of seed oil content in Brassica napus [8, 10, 13, 24, 49, 54, 55, 59, 68]. However, there have been relatively few reports on the identification of significant SNPs associated with seed oil content by GWAS compared with the QTL mapping method. Liu et al. identified 50 SNPs that were significantly associated with seed oil content using 521 B. napus accessions combined with the Brassica 60K SNP array [36]. Seven stable QTLs for seed oil content were identified by Sun et al. by combined linkage and association mapping methods in Brassica napus [51]. Four significant SNPs for seed oil content were located on chromosomes A1, C3, and C5 [27]. Seventeen loci associated with seed oil content were identified by combining high-throughput genome resequencing and phenotyping using GWAS [56]. Although previous studies have identified many SNPs that were significantly associated with SOC in Brassica napus, none of them have combined the analysis with transcriptome sequencing to obtain candidate genes contributing to SOC. In our previous study, the population structure and linkage disequilibrium of 588 Brassica napus accessions were evaluated, and provided a high-resolution genomic variation map consisting of 616053 SNPs by the resequencing method. In the present study, we obtained 17 SNPs that were significantly associated with seed oil content (SOC) in Brassica napus, 12 of which were overlapped with previous studies (Table 2), supporting the high reliability of loci detected in current study. In addition, we also found five new SNPs significantly associated with SOC in Brassica napus (Table 2), and all the significant SNPs detected in current study were further analyzed by combination with the RNA-Seq data.
Comparative analysis of three Brassica napus lines using transcriptome sequencing from phenotype to gene expression
Identification of differentially expressed (DE) candidate genes in combination with GWAS and RNA-seq has been proven to be more successful than each separate strategy [38, 64]. In this study, an extremely low-oil (CQ46) and two extremely high-oil content accessions (CQ24, CQ52) were selected from the GWAS analysis population to perform transcriptome sequencing (Fig. 4). A difference of approximately 10% SOC was observed between the HO and LO accessions (Fig. 4a), without a difference between the seed protein content and 1000-grain weight. Additionally, C18:1 was higher in HO (CQ24 and CQ52) than LO (CQ46) accessions, and C16:0 and C18:2 showed the opposite pattern either in seeds at 30 days after flowering or at maturity, which are consistent with the higher expression levels of KASIII, SAD and FAD3 in seeds (30SM and 30SB) of high-oil content than low-oil content accessions (Fig. 6). Moreover, a previous study has shown that fatty acid accumulation is accelerated from 26 to 34 days after flowering [40]. Therefore, seeds and silique pericarps at 30 days after flowering were selected for transcriptome analysis among typical high- and low-oil content accessions. In addition, we analyzed the transcriptional levels of major genes in the TAG biosynthesis and assembly pathway in four tissues of HO (CQ24, CQ52) and LO (CQ46) accessions. As shown in Fig. 6, transcripts of the genes involved in the TAG biosynthesis and assembly pathway were generally higher in seeds (30SM and 30SB) than silique pericarps (30SPM and 30SPB), which suggested that the seeds were more active in TAG biosynthesis and assembly than silique pericarps. Exceptionally, we found that FAX1, PLD, CPT, PLC, and PDAT were expressed at higher levels in silique pericarps than seeds, indicating that silique pericarps may also play a non-negligible role in TAG biosynthesis, which is consistent with a previous study examining the importance of the silique wall in the regulation of seed oil content [18]. The expression level of most of genes involved in fatty acid (FA) and TAG biosynthesis in seeds (30SM and 30SB) was significantly enhanced in HO (CQ24 and CQ52) compared with LO, which including PDHC, ACCase, MCMT, KAR, HAD, KASIII, FATA, KASII, SAD, LACS9, GPDH, PAP, FAD3, DGAT and so on (Fig. 6, Additional file 1: Table S6). To our surprise, the transcription levels of OBO and CALO were significantly higher in all tested tissues of HO (CQ24 and CQ52) than LO (CQ46) (Fig. 6, Additional file 1: Table S6), which may suggest an important role for these OBO and CALO in high seed oil formation. This result is consistent with the findings of Liu et al. showing that the overexpression of soybean oleosin can increase the seed lipid content in transgenic rice [35]. These results strongly suggested that it is feasible to select these accessions as extremely high- (CQ24 and CQ52) and low-oil content (CQ46) lines for RNA-Seq.
The identification of SOC-related candidate genes
Brassica napus is one of the most important oil crops in the world along with soybean and palm. The typical SOC of B. napus germplasm varies from 35 to 50% [36]. Recently, through the efforts of breeders, ultra-high oil content germplasm materials with 55–65% oil content have been produced [20]. Hu et al. predicted that the B. napus seed oil content could be increased to 75% [17]. Therefore, there is still a great potential to increase the B. napus seed oil content, especially in the main Chinese rapeseed producing areas such as Chongqing of the Yangtze River Basin. Previous studies examining the seed oil content of B. napus have focused on many QTL mappings or a small number of GWAS, but in this study, the combination of GWAS and transcriptome sequencing was implemented for this purpose. According to the 300-kb flanking regions on either side of the 17 significantly associated SNPs with SOC [56], we obtained a total of 411 genes. These genes were annotated by applying the BLASTP program against the Arabidopsis proteome (TAIR10) with an E-value threshold of 1E−5 [38] (Additional file 1: Table S5), and a total of 14 acyl-lipid metabolism (ALM)-related genes were found (Table 3). Although these 14 genes did not differ significantly in the four detested tissues of high- and low-oil accessions, and their expression patterns are shown in Additional file 2: Fig. S2b, we found BnaC05g10520, BnaA01g12060 and BnaA01g12140D were expressed at higher levels in all tested tissues of high- and low-oil accessions. In previous reports, BnaA01g12140D which is homologous to AT4G22330 (ATCES1), encodes a nuclear and endoplasmic reticulum localized Acyl-CoA-independent ceramide synthase that is involved in sphingolipid metabolism, disease resistance, nutrient limitation, and response to salt stress [62, 67]. Therefore, we speculate that these genes (BnaC05g10520, BnaA01g12060 and BnaA01g12140D) play an important role in lipid metabolism of B. napus.
Table 3.
All genes within the confidence interval of significant related SNPs with SOC belong to ALM genes
| ALM genes in confidence interval | At orthologs | Pathways | Function description |
|---|---|---|---|
| BnaC05g10520D | AT1G14290 | Sphingolipid biosynthesis 1 | Sphingoid base hydroxylase 2 (SBH2) |
| BnaA01g12830D | AT1G73550 | Fatty acid elongation and wax biosynthesis | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| BnaC07g34330D | AT3G10550 | Phospholipid signaling | Myotubularin-like phosphatases II superfamily |
| BnaA03g36710D | AT3G22620 | Fatty acid elongation and wax biosynthesis | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| BnaA03g37090D | AT3G23530 | Unknown | Cyclopropane-fatty-acyl-phospholipid synthase |
| BnaA03g36540D | AT4G11850 | Phospholipid signaling | Phospholipase D gamma 1 (PLDGAMMA1) |
| BnaC07g33970D | AT4G16820 | Prokaryotic galactolipid, sulfolipid, and phospholipid synthesis 2 | Alpha/beta-hydrolases superfamily protein |
| BnaC07g34360D | AT4G17483 | Unknown | Alpha/beta-hydrolases superfamily protein |
| BnaA01g12060D | AT4G22240 | Unknown | Plastid-lipid associated protein PAP/fibrillin family protein |
| BnaA01g12140D | AT4G22330 | Sphingolipid biosynthesis 2 | ATCES1 |
| BnaA01g12150D | AT4G22340 | Prokaryotic galactolipid, sulfolipid, and phospholipid synthesis 1 | Cytidinediphosphate diacylglycerol synthase 2 (CDS2) |
| BnaA01g12260D | AT4G22520 | Fatty acid elongation and wax biosynthesis | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| BnaA01g12290D | AT4G22550 | Prokaryotic galactolipid, sulfolipid, and phospholipid synthesis 1 | Phosphatidic acid phosphatase (PAP2) family protein |
| BnaA01g12350D | AT4G22640 | Fatty acid elongation and wax biosynthesis | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
A combination of our GWAS and the DEGs of the transcriptome sequencing results between HO and LO accessions revealed 16 genes (X1–X16) under CQ24/CQ46 (Table 4) and 32 genes (Z1–Z32) under CQ52/CQ46 (Table 5). The expression patterns of all the genes using HemI1.0 are shown in Additional file 2: Fig. S3 [58]. Interestingly, seven common genes were obtained under CQ24/CQ46 and CQ52/CQ46, and they were considered important candidate genes associated with seed oil content in B. napus. BnaA01g13030D (X5/Z30) and BnaA03g36510D (X7/Z31) represented two genes of unknown function, and their expression levels were significantly lower in seeds of HO (CQ24 and CQ52) than LO (CQ46) (Additional file 2: Fig. S3c). The gene (BnaA03g36620D, X10/Z20) encodes AOX1a, an isoform of alternative oxidase. The absence of AOX1a in Arabidopsis leads to acute sensitivity to combined light and drought stress [15]. AOX1a has been implicated in the modulation of metabolic homeostasis in cadmium (Cd)-exposed Arabidopsis plant and displays a differential role in roots and leaves in response to sublethal cadmium exposure [25, 26]. The maize AOX1a gene plays an essential role under oxidative stress [47]. In Brassica napus roots, selenite can activate the alternative oxidase pathway and alter primary metabolism and may ultimately improve selenium tolerance [11]. BnaA03g37060D (X12/Z13) was a putative component of photosystem II complex and may be involved in response to UV-B, ozone and wounding. In all tested tissues, BnaA03g37060D is less expressed in HO and is not expressed in LO accessions. BnaC05g10690D (X14/Z1) exhibited extremely high expression levels in LO compared with HO lines, and this finding was verified by qRT-PCR (Fig. 7, Additional file 2: Fig. S3c). Therefore, we speculate that interfering with the expression of this gene may contribute to increase seed oil content in Brassica napus. BnaC05g10700D (X15/Z9) was an ARM repeat superfamily protein and its specific function is unknown, and its expression level in HO is higher than that of LO accessions. Increasing the expression level of BnaC05g10700D may help to increase seed oil content. BON2 (BnaC05g10710D, X16/Z10) encodes a copine-like protein. In Arabidopsis, the BON gene family can promote cell growth and development in addition to repressing cell death [63]. Additionally, Li et al. [33] have validated that plasma membrane-localized calcium pumps and copines coordinately regulate pollen germination and fertility in Arabidopsis. Furthermore, BnaC05g10710D exhibited a significantly higher expression level in HO compared with LO accessions, and this result also further verified by qRT-PCR (Fig. 7). This gene (BnaC05g10710D) may play a positive regulatory role in the formation of high oil content in B. napus. Of course, the role of the seven candidate genes mentioned above in the formation of seed oil content in B. napus must be further confirmed, which will be an important task for our next studies.
Conclusion
In the present study, 17 loci significantly associated with seed oil content in B. napus were successfully obtained, and 12 significant SNPs were found to overlap with QTLs from previous studies, which proved the reliability of this study. In addition, five novel significant SNPs distributed on the C5 and C7 chromosomes were identified, which provided valuable information for further exploration of genes that contribute to increase seed oil content in B. napus. Subsequently, the combination of GWAS and transcriptome analyses revealed seven functional candidate genes located within the confidence intervals of significant SNPs associated with seed oil content in B. napus. These results may facilitate marker-based breeding for higher seed oil content in B. napus.
Supplementary information
Additional file 1: Table S1. Seed oil content (SOC, % of seed weight) phenotypes of 588 accessions for GWAS analysis. 2016CQ, 2017CQ and 2018CQ refer to the three environments, CQ refers to Chongqing. BLUP represents the SOC phenotypic value obtained by the best linear unbiased prediction in three environments. Missing data are replaced by ‘− 999’. Table S2. The quality statistics of RNA sequencing data. Table S3. The number of differential genes is counted under HO/LO accessions. Table S4. Common differential lipid metabolism genes in four tested tissues under CQ24/CQ46 and CQ52/CQ46 based on KEGG pathway analysis. Table S5. All genes within the confidence interval of significant related SNPs with SOC. Table S6. Summary of gene expression of TAG biosynthesis and assembly pathway in all four tissues among LO (CQ46) and HO (CQ24, CQ52) Brassica napus lines. Table S7. Primer sequences for qRT-PCR verification in this study.
Additional file 2: Fig. S1. (a) Statistics on the number of differential genes in different tissues with different seed oil content (SOC) Brassica napus lines. (b) Gene ontology (GO) enrichment analysis of common DEGs in 30SM and 30SB in Fig. 5a. (c) Gene ontology (GO) enrichment analysis of common DEGs in 30SPM and 30SPB in Fig. 5a. Fig. S2. Expression patterns of identified ALM genes within the confidence interval of significant related SNPs with SOC. (a) Heatmap of identified common differential ALM genes was derived from KEGG pathway analysis in all tested tissues under CQ24/CQ46 and CQ52/CQ46. (b) Heat map of identified all ALM genes within the confidence interval significantly associated with SOC was derived from transcriptome sequencing among HO (CQ24, CQ52) and LO (CQ46) lines. Fig. S3. Expression patterns of candidate genes identified by GWAS and transcriptome sequencing. Heatmap of identified candidate genes was derived from RNA sequencing data in all tested tissues between CQ24/CQ46 (a) and CQ52/46 (b) and common candidate genes (c).
Abbreviations
- GWAS
Genome-wide association study
- SNP
Single nucleotide polymorphism
- MAF
Minor allele frequency
- DEGs
Differentially expressed genes
- HO
High-oil content accessions
- LO
Low-oil content accessions
- TAG
Triacylglycerol
- ER
Endoplasmic reticulum
- SOC
Seed oil content
- QTL
Quantitative trait locus
- qRT-PCR
Quantitative real-time PCR
- BLUP
Best linear unbiased prediction
- 30SM
Seeds on the main inflorescence after flowering 30 days
- 30SPM
Silique pericarps on the main inflorescence after flowering 30 days
- 30SB
Seed on the primary branch after flowering 30 days
- 30SPB
Silique pericarps on the primary branch after flowering 30 days
- RNA-seq
RNA sequencing
- FPKM
Fragments per kilobase of exon per million reads mapped
- FDR
False discovery rate
- ALM
Acyl-lipid metabolism
- CV
Coefficient of variation
- GC–MS
Gas chromatography–mass spectrometry
- KEGG
Kyoto encyclopedia of genes and genomes
- FA
Fatty acid
- PDHC
Pyruvate dehydrogenase complex
- ACCase
Acetyl-CoA carboxylase
- MCMT
Malonyl-CoA:ACP malonyltransferase
- ACP
Acyl carrier protein
- KASI/II/III
3-Ketoacyl-ACP synthase I/II/III
- KAR
Ketoacyl-ACP reductase
- HAD
Hydroxyacyl-ACP dehydrase
- ENR
Enoyl-ACP reductase
- SAD
Stearoyl-acyl carrier protein desaturase
- FATA/B
Acyl-ACP thioesterase A/B
- G-3-P
Glycerol-3-phosphate
- GPDH
Glycerol-3-phosphate dehydrogenase
- FAX1
Fatty acid export 1
- LACS9
Long-chain acyl-CoA synthetase 9
- GPAT9
Glycerol-3-phosphate acyltransferase 9
- LPAAT
Lysophosphatidic acid acyltransferase
- PAP
Phosphatidic acid phosphatase
- DGAT
Diacylglycerol acyltransferase
- LPCAT
Lysophosphatidylcholine acyltransferase
- PDAT
Phospholipid:diacylglycerol acyltransferase
- FAD2
FA desaturase 2
- FAD3
FA desaturase 3
- PDCT
Phosphatidylcholine:diacylglycerol cholinephosphotransferase
- CPT
CDP-choline:diacylglycerol cholinephosphotransferase
- PLC
Phospholipase C
- PLD
Phospholipase D
- OBO
Oil body oleosin
- CALO
Caleosin
Authors’ contributions
JNL and NNL conceived and designed the experiments. ZCX performed the experiments. ZCX, CZ, FT, BY, LYZ, JSL, QH, SFW, STL, LJW, HD, CMQ and KL analyzed the data. ZCX and NNL wrote the paper. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Funding and acknowledgments
This study was funded by National Key R & D Program of China (2018YFD0200903; 2018YFD0100500), the “111” Project (B12006), Modern Agro-industry Technology Research System (CARS-13), the National Natural Science Foundation of China (31870587; 31400063; 31571701, U1302266), Chongqing Min sheng Technology Funds (cstc2016shms-ztzx80020), Fundamental Research Funds for the Central Universities (XDJK2018C095; XDJK2017B030). We extend our thanks to the reviewers for their careful reading and helpful comments on this manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Zhongchun Xiao, Chao Zhang and Fang Tang contributed equally to this work
Contributor Information
Jiana Li, Phone: +86-023-68250701, Email: ljn1950@swu.edu.cn.
Nannan Li, Email: linannan2013@swu.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13068-019-1557-x.
References
- 1.Atwell S, Huang YS, Vilhjalmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JD, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bates PD, Stymne S, Ohlrogge J. Biochemical pathways in seed oil synthesis. Curr Opin Plant Biol. 2013;16:358–364. doi: 10.1016/j.pbi.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Baud S, Lepiniec L. Physiological and developmental regulation of seed oil production. Prog Lipid Res. 2010;49:235–249. doi: 10.1016/j.plipres.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol. 1995;57:289–300. [Google Scholar]
- 5.Bourgis F, Kilaru A, Cao X, Ngando-Ebongue GF, Drira N, Ohlrogge JB, Arondel V. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc Natl Acad Sci USA. 2011;108:18186. doi: 10.1073/pnas.1106502108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 7.Chalhoub B, Denoeud F, Liu SY, Parkin IAP, Tang HB, Wang XY, Chiquet J, Belcram H, Tong CB, Samans B, Correa M, Da Silva C, Just J, Falentin C, Koh CS, Le Clainche I, Bernard M, Bento P, Noel B, Labadie K, Alberti A, Charles M, Arnaud D, Guo H, Daviaud C, Alamery S, Jabbari K, Zhao MX, Edger PP, Chelaifa H, Tack D, Lassalle G, Mestiri I, Schnel N, Le Paslier MC, Fan GY, Renault V, Bayer PE, Golicz AA, Manoli S, Lee TH, Thi VHD, Chalabi S, Hu Q, Fan CC, Tollenaere R, Lu YH, Battail C, Shen JX, Sidebottom CHD, Wang XF, Canaguier A, Chauveau A, Berard A, Deniot G, Guan M, Liu ZS, Sun FM, Lim YP, Lyons E, Town CD, Bancroft I, Wang XW, Meng JL, Ma JX, Pires JC, King GJ, Brunel D, Delourme R, Renard M, Aury JM, Adams KL, Batley J, Snowdon RJ, Tost J, Edwards D, Zhou YM, Hua W, Sharpe AG, Paterson AH, Guan CY, Wincker P. Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science. 2014;345:950–953. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- 8.Chao H, Wang H, Wang X, Guo L, Gu J, Zhao W, Li B, Chen D, Raboanatahiry N, Li M. Genetic dissection of seed oil and protein content and identification of networks associated with oil content in Brassica napus. Sci Rep. 2017;7:46295. doi: 10.1038/srep46295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman KD, Ohlrogge JB. Compartmentation of triacylglycerol accumulation in plants. J Biol Chem. 2012;287:2288–2294. doi: 10.1074/jbc.R111.290072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delourme R, Falentin C, Huteau V, Clouet V, Horvais R, Gandon B, Specel S, Hanneton L, Dheu JE, Deschamps M, Margale E, Vincourt P, Renard M. Genetic control of oil content in oilseed rape (Brassica napus L.) Theor Appl Genet. 2006;113:1331–1345. doi: 10.1007/s00122-006-0386-z. [DOI] [PubMed] [Google Scholar]
- 11.Dimkovikj A, Van Hoewyk D. Selenite activates the alternative oxidase pathway and alters primary metabolism in Brassica napus roots: evidence of a mitochondrial stress response. BMC Plant Biol. 2014;14:259. doi: 10.1186/s12870-014-0259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dussert S, Guerin C, Andersson M, Joet T, Tranbarger TJ, Pizot M, Sarah G, Omore A, Durand-Gasselin T, Morcillo F. Comparative transcriptome analysis of three oil palm fruit and seed tissues that differ in oil content and fatty acid composition. Plant Physiol. 2013;162:1337–1358. doi: 10.1104/pp.113.220525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Y, Zhang DQ, Gleeson M, Zhang YF, Lin BG, Hua SJ, Ding HD, Frauen M, Li JN, Qian W, Yu HS. Analysis of QTL for seed oil content in Brassica napus by association mapping and QTL mapping. Euphytica. 2017;213:17. doi: 10.1007/s10681-016-1817-9. [DOI] [Google Scholar]
- 14.Gan L, Sun X, Jin L, Wang G, Xiu J, Wei Z, Fu T. Establishment of math models of NIRS analysis for oil and protein contents in seed of Brassica napus. Sci Agric Sin. 2003;23613:1609–1613. [Google Scholar]
- 15.Giraud E, Ho LHM, Clifton R, Carroll A, Estavillo G, Tan YF, Howell KA, Ivanova A, Pogson BJ, Millar AH, Whelan J. The absence of alternative oxidase1a in Arabidopsis results in acute sensitivity to combined light and drought stress. Plant Physiol. 2008;147:595–610. doi: 10.1104/pp.107.115121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatzig SV, Frisch M, Breuer F, Nesi N, Ducournau S, Wagner MH, Leckband G, Abbadi A, Snowdon RJ. Genome-wide association mapping unravels the genetic control of seed germination and vigor in Brassica napus. Front Plant Sci. 2015;6:221. doi: 10.3389/fpls.2015.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu ZY, Hua W, Zhang L, Deng LB, Wang XF, Liu GH, Hao WJ, Wang HZ. Seed structure characteristics to form ultrahigh oil content in rapeseed. PLoS ONE. 2013;8:e62099. doi: 10.1371/journal.pone.0062099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hua W, Li RJ, Zhan GM, Liu J, Li J, Wang XF, Liu GH, Wang HZ. Maternal control of seed oil content in Brassica napus: the role of silique wall photosynthesis. Plant J. 2012;69:432–444. doi: 10.1111/j.1365-313X.2011.04802.x. [DOI] [PubMed] [Google Scholar]
- 19.Hua JF, Zhang S, Cui JJ, Wang DJ, Wang CY, Luo JY, Lv LM, Ma Y. Functional characterizations of one odorant binding protein and three chemosensory proteins from Apolygus lucorum (Meyer-Dur) (Hemiptera: Miridae) legs. J Insect Physiol. 2013;59:690–696. doi: 10.1016/j.jinsphys.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Hua W, Liu J, Wang HZ. Molecular regulation and genetic improvement of seed oil content in Brassica napus L. Front Agric Sci Eng. 2016;3:186–194. doi: 10.15302/J-FASE-2016107. [DOI] [Google Scholar]
- 21.Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Li W, Lin Z, Buckler ES, Qian Q, Zhang QF, Li J, Han B. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42:961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 22.Huang XH, Zhao Y, Wei XH, Li CY, Wang A, Zhao Q, Li WJ, Guo YL, Deng LW, Zhu CR, Fan DL, Lu YQ, Weng QJ, Liu KY, Zhou TY, Jing YF, Si LZ, Dong GJ, Huang T, Lu TT, Feng Q, Qian Q, Li JY, Han B. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet. 2012;44:32–U53. doi: 10.1038/ng.1018. [DOI] [PubMed] [Google Scholar]
- 23.Hwang EY, Song QJ, Jia GF, Specht JE, Hyten DL, Costa J, Cregan PB. A genome-wide association study of seed protein and oil content in soybean. BMC Genomics. 2014;15:1. doi: 10.1186/1471-2164-15-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang CC, Shi JQ, Li RY, Long Y, Wang H, Li DR, Zhao JY, Meng JL. Quantitative trait loci that control the oil content variation of rapeseed (Brassica napus L.) Theor Appl Genet. 2014;127:957–968. doi: 10.1007/s00122-014-2271-5. [DOI] [PubMed] [Google Scholar]
- 25.Keunen E, Schellingen K, Van Der Straeten D, Remans T, Colpaert J, Vangronsveld J, Cuypers A. ALTERNATIVE OXIDASE1a modulates the oxidative challenge during moderate Cd exposure in Arabidopsis thaliana leaves. J Exp Bot. 2015;66:2967–2977. doi: 10.1093/jxb/erv035. [DOI] [PubMed] [Google Scholar]
- 26.Keunen E, Florez-Sarasa I, Obata T, Jozefczak M, Remans T, Vangronsveld J, Fernie AR, Cuypers A. Metabolic responses of Arabidopsis thaliana roots and leaves to sublethal cadmium exposure are differentially influenced by ALTERNATIVE OXIDASE1a. Environ Exp Bot. 2016;124:64–78. doi: 10.1016/j.envexpbot.2015.11.015. [DOI] [Google Scholar]
- 27.Korber N, Bus A, Li J, Parkin IA, Wittkop B, Snowdon RJ, Stich B. Agronomic and seed quality traits dissected by genome-wide association mapping in Brassica napus. Front Plant Sci. 2016;7:386. doi: 10.3389/fpls.2016.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kump KL, Bradbury PJ, Wisser RJ, Buckler ES, Belcher AR, Oropeza-Rosas MA, Zwonitzer JC, Kresovich S, McMullen MD, Ware D, Balint-Kurti PJ, Holland JB. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet. 2011;43:163-U120. doi: 10.1038/ng.747. [DOI] [PubMed] [Google Scholar]
- 29.Leckband G, Frauen M, Friedt W. NAPUS 2000. rapeseed (Brassica napus) breeding for improved human nutrition. Food Res Int. 2002;35:273–278. doi: 10.1016/S0963-9969(01)00196-X. [DOI] [Google Scholar]
- 30.Li F, Chen B, Xu K, Wu J, Song W, Bancroft I, Harper AL, Trick M, Liu S, Gao G, Wang N, Yan G, Qiao J, Li J, Li H, Xiao X, Zhang T, Wu X. Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.) DNA Res. 2014;21:355–367. doi: 10.1093/dnares/dsu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li N, Gugel IL, Giavalisco P, Zeisler V, Schreiber L, Soll J, Philippar K. FAX1, a novel membrane protein mediating plastid fatty acid export. PLoS Biol. 2015;13:e1002053. doi: 10.1371/journal.pbio.1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F, Chen BY, Xu K, Gao GZ, Yan GX, Qiao JW, Li J, Li H, Li LX, Xiao X, Zhang TY, Nishio T, Wu XM. A genome-wide association study of plant height and primary branch number in rapeseed (Brassica napus) Plant Sci. 2016;242:169–177. doi: 10.1016/j.plantsci.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Guo JP, Yang ZY, Yang DL. Plasma membrane-localized calcium pumps and copines coordinately regulate pollen germination and fertility in Arabidopsis. Int J Mol Sci. 2018;19:1774. doi: 10.3390/ijms19061774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li-Beisson Y, Shorrosh B, Beisson F, Andersson MX, Arondel V, Bates PD, Baud S, Bird D, Debono A, Durrett TP, Franke RB, Graham IA, Katayama K, Kelly AA, Larson T, Markham JE, Miquel M, Molina I, Nishida I, Rowland O, Samuels L, Schmid KM, Wada H, Welti R, Xu C, Zallot R, Ohlrogge J. Acyl-lipid metabolism. Arabidopsis Book. 2013;11:e0161. doi: 10.1199/tab.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu WX, Liu HL, le Qu Q. Embryo-specific expression of soybean oleosin altered oil body morphogenesis and increased lipid content in transgenic rice seeds. Theor Appl Genet. 2013;126:2289–2297. doi: 10.1007/s00122-013-2135-4. [DOI] [PubMed] [Google Scholar]
- 36.Liu S, Fan C, Li J, Cai G, Yang Q, Wu J, Yi X, Zhang C, Zhou Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor Appl Genet. 2016;129:1203–1215. doi: 10.1007/s00122-016-2697-z. [DOI] [PubMed] [Google Scholar]
- 37.Liu YM, Zhao ZF, Wei G, Zhang P, Lan H, Zhang SZ, Li C, Cao MJ. Characterization of the ZmbHLH122 transcription factor and its potential collaborators in maize male reproduction. Plant Growth Regul. 2018;85:113–122. doi: 10.1007/s10725-018-0377-5. [DOI] [Google Scholar]
- 38.Lu K, Xiao Z, Jian H, Peng L, Qu C, Fu M, He B, Tie L, Liang Y, Xu X, Li J. A combination of genome-wide association and transcriptome analysis reveals candidate genes controlling harvest index-related traits in Brassica napus. Sci Rep. 2016;6:36452. doi: 10.1038/srep36452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu K, Peng L, Zhang C, Lu J, Yang B, Xiao Z, Liang Y, Xu X, Qu C, Zhang K, Liu L, Zhu Q, Fu M, Yuan X, Li J. Genome-wide association and transcriptome analyses reveal candidate genes underlying yield-determining traits in Brassica napus. Front Plant Sci. 2017;8:206. doi: 10.3389/fpls.2017.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu S, Sturtevant D, Aziz M, Jin C, Li Q, Chapman KD, Guo L. Spatial analysis of lipid metabolites and expressed genes reveals tissue-specific heterogeneity of lipid metabolism in high- and low-oil Brassica napus L. seeds. Plant J. 2018;94:915–932. doi: 10.1111/tpj.13959. [DOI] [PubMed] [Google Scholar]
- 41.Lu K, Wei L, Li X, Wang Y, Wu J, Liu M, Zhang C, Chen Z, Xiao Z, Jian H, Cheng F, Zhang K, Du H, Cheng X, Qu C, Qian W, Liu L, Wang R, Zou Q, Ying J, Xu X, Mei J, Liang Y, Chai YR, Tang Z, Wan H, Ni Y, He Y, Lin N, Fan Y, Sun W, Li NN, Zhou G, Zheng H, Wang X, Paterson AH, Li J. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat Commun. 2019;10:1154. doi: 10.1038/s41467-019-09134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo X, Ma C, Yue Y, Hu K, Li Y, Duan Z, Wu M, Tu J, Shen J, Yi B, Fu T. Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genomics. 2015;16:379. doi: 10.1186/s12864-015-1607-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer M. Rapeseed oil fuel—the crisis-proof home-made eco-fuel. Agrarforschung. 2009;16:262–267. [Google Scholar]
- 44.Miquel M, Trigui G, d’Andrea S, Kelemen Z, Baud S, Berger A, Deruyffelaere C, Trubuil A, Lepiniec L, Dubreucq B. Specialization of oleosins in oil body dynamics during seed development in Arabidopsis seeds([W][OPEN]) Plant Physiol. 2014;164:1866–1878. doi: 10.1104/pp.113.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mo YD, Yang SX, Zhao JY, Jin PY, Hong XY. Comparative transcriptomes and reciprocal best hit analysis revealed potential pigment genes in two color forms of Tetranychus urticae. Exp Appl Acarol. 2017;73:159–176. doi: 10.1007/s10493-017-0188-9. [DOI] [PubMed] [Google Scholar]
- 46.Morris GP, Ramu P, Deshpande SP, Hash CT, Shah T, Upadhyaya HD, Riera-Lizarazu O, Brown PJ, Acharya CB, Mitchell SE, Harriman J, Glaubitz JC, Buckler ES, Kresovich S. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci USA. 2013;110:453–458. doi: 10.1073/pnas.1215985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polidoros AN, Mylona PV, Pasentsis K, Scandalios JG, Tsaftaris AS. The maize alternative oxidase 1a (Aox1a) gene is regulated by signals related to oxidative stress. Redox Rep. 2005;10:71–78. doi: 10.1179/135100005X21688. [DOI] [PubMed] [Google Scholar]
- 48.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 49.Qiu D, Morgan C, Shi J, Long Y, Liu J, Li R, Zhuang X, Wang Y, Tan X, Dietrich E, Weihmann T, Everett C, Vanstraelen S, Beckett P, Fraser F, Trick M, Barnes S, Wilmer J, Schmidt R, Li J, Li D, Meng J, Bancroft I. A comparative linkage map of oilseed rape and its use for QTL analysis of seed oil and erucic acid content. Theor Appl Genet. 2006;114:67–80. doi: 10.1007/s00122-006-0411-2. [DOI] [PubMed] [Google Scholar]
- 50.Riedelsheimer C, Lisec J, Czedik-Eysenberg A, Sulpice R, Flis A, Grieder C, Altmann T, Stitt M, Willmitzer L, Melchinger AE. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc Natl Acad Sci USA. 2012;109:8872–8877. doi: 10.1073/pnas.1120813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun F, Liu J, Hua W, Sun X, Wang X, Wang H. Identification of stable QTLs for seed oil content by combined linkage and association mapping in Brassica napus. Plant Sci. 2016;252:388–399. doi: 10.1016/j.plantsci.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Sun CM, Wang BQ, Yan L, Hu KN, Liu S, Zhou YM, Guan CY, Zhang ZQ, Li JN, Zhang JF, Chen S, Wen J, Ma CZ, Tu JX, Shen JX, Fu TD, Yi B. Genome-wide association study provides insight into the genetic control of plant height in rapeseed (Brassica napus L.) Front Plant Sci. 2016;7:1102. doi: 10.3389/fpls.2016.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wanasundara JPD. Proteins of Brassicaceae oilseeds and their potential as a plant protein source. Crit Rev Food Sci Nutr. 2011;51:635–677. doi: 10.1080/10408391003749942. [DOI] [PubMed] [Google Scholar]
- 54.Wang XD, Wang H, Long Y, Li DR, Yin YT, Tian JH, Chen L, Liu LZ, Zhao WG, Zhao YJ, Yu LJ, Li MT. Identification of QTLs associated with oil content in a high-oil Brassica napus cultivar and construction of a high-density consensus map for QTLs comparison in B. napus. PLoS ONE. 2013;8:e80569. doi: 10.1371/journal.pone.0080569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang D, Yang C, Dong L, Zhu J, Wang J, Zhang S. Comparative transcriptome analyses of drought-resistant and—susceptible Brassica napus L. and development of EST-SSR markers by RNA-Seq. J Plant Biol. 2015;58:259–269. doi: 10.1007/s12374-015-0113-x. [DOI] [Google Scholar]
- 56.Wang B, Wu Z, Li Z, Zhang Q, Hu J, Xiao Y, Cai D, Wu J, King GJ, Li H, Liu K. Dissection of the genetic architecture of three seed-quality traits and consequences for breeding in Brassica napus. Plant Biotechnol J. 2017;16:1336–1348. doi: 10.1111/pbi.12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang P, Yang C, Chen H, Song C, Zhang X, Wang D. Transcriptomic basis for drought-resistance in Brassica napus L. Sci Rep. 2017;7:40532. doi: 10.1038/srep40532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang T, Song Z, Wei L, Li LB. Molecular characterization and expression analysis of WRKY family genes in Dendrobium officinale. Genes Genomics. 2018;40:265–279. doi: 10.1007/s13258-017-0602-z. [DOI] [PubMed] [Google Scholar]
- 59.Wang P, Yang C, Chen H, Luo L, Leng Q, Li S, Han Z, Li X, Song C, Zhang X, Wang D. Exploring transcription factors reveals crucial members and regulatory networks involved in different abiotic stresses in Brassica napus L. BMC Plant Biol. 2018;18:202. doi: 10.1186/s12870-018-1417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z, Yang C, Chen H, Wang P, Wang P, Song C, Zhang X, Wang D. Multi-gene co-expression can improve comprehensive resistance to multiple abiotic stresses in Brassica napus L. Plant Sci. 2018;274:410–419. doi: 10.1016/j.plantsci.2018.06.014. [DOI] [PubMed] [Google Scholar]
- 61.Weselake RJ, Taylor DC, Rahman MH, Shah S, Laroche A, McVetty PBE, Harwood JL. Increasing the flow of carbon into seed oil. Biotechnol Adv. 2009;27:866–878. doi: 10.1016/j.biotechadv.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 62.Wu JX, Li J, Liu Z, Yin J, Chang ZY, Rong C, Wu JL, Bi FC, Yao N. The Arabidopsis ceramidase AtACER functions in disease resistance and salt tolerance. Plant J. 2015;81:767–780. doi: 10.1111/tpj.12769. [DOI] [PubMed] [Google Scholar]
- 63.Yang S, Yang H, Grisafi P, Sanchatjate S, Fink GR, Sun Q, Hua J. The BON/CPN gene family represses cell death and promotes cell growth in Arabidopsis. Plant J. 2006;45:166–179. doi: 10.1111/j.1365-313X.2005.02585.x. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J, Mason AS, Wu J, Liu S, Zhang XC, Luo T, Redden R, Batley J, Hu LY, Yan GJ. Identification of putative candidate genes for water stress tolerance in canola (Brassica napus) Front Plant Sci. 2015;6:1058. doi: 10.3389/fpls.2015.01058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, McClung AM, Bustamante CD, McCouch SR. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun. 2011;2:467. doi: 10.1038/ncomms1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao J, Huang J, Chen F, Xu F, Ni X, Xu H, Wang Y, Jiang C, Wang H, Xu A, Huang R, Li D, Meng J. Molecular mapping of Arabidopsis thaliana lipid-related orthologous genes in Brassica napus. Theor Appl Genet. 2012;124:407–421. doi: 10.1007/s00122-011-1716-3. [DOI] [PubMed] [Google Scholar]
- 67.Zheng P, Wu JX, Sahu SK, Zeng HY, Huang LQ, Liu Z, Xiao S, Yao N. Loss of alkaline ceramidase inhibits autophagy in Arabidopsis and plays an important role during environmental stress response. Plant Cell Environ. 2018;41:837–849. doi: 10.1111/pce.13148. [DOI] [PubMed] [Google Scholar]
- 68.Zou J, Jiang C, Cao Z, Li R, Long Y, Chen S, Meng J. Association mapping of seed oil content in Brassica napus and comparison with quantitative trait loci identified from linkage mapping. Genome. 2010;53:908–916. doi: 10.1139/G10-075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Seed oil content (SOC, % of seed weight) phenotypes of 588 accessions for GWAS analysis. 2016CQ, 2017CQ and 2018CQ refer to the three environments, CQ refers to Chongqing. BLUP represents the SOC phenotypic value obtained by the best linear unbiased prediction in three environments. Missing data are replaced by ‘− 999’. Table S2. The quality statistics of RNA sequencing data. Table S3. The number of differential genes is counted under HO/LO accessions. Table S4. Common differential lipid metabolism genes in four tested tissues under CQ24/CQ46 and CQ52/CQ46 based on KEGG pathway analysis. Table S5. All genes within the confidence interval of significant related SNPs with SOC. Table S6. Summary of gene expression of TAG biosynthesis and assembly pathway in all four tissues among LO (CQ46) and HO (CQ24, CQ52) Brassica napus lines. Table S7. Primer sequences for qRT-PCR verification in this study.
Additional file 2: Fig. S1. (a) Statistics on the number of differential genes in different tissues with different seed oil content (SOC) Brassica napus lines. (b) Gene ontology (GO) enrichment analysis of common DEGs in 30SM and 30SB in Fig. 5a. (c) Gene ontology (GO) enrichment analysis of common DEGs in 30SPM and 30SPB in Fig. 5a. Fig. S2. Expression patterns of identified ALM genes within the confidence interval of significant related SNPs with SOC. (a) Heatmap of identified common differential ALM genes was derived from KEGG pathway analysis in all tested tissues under CQ24/CQ46 and CQ52/CQ46. (b) Heat map of identified all ALM genes within the confidence interval significantly associated with SOC was derived from transcriptome sequencing among HO (CQ24, CQ52) and LO (CQ46) lines. Fig. S3. Expression patterns of candidate genes identified by GWAS and transcriptome sequencing. Heatmap of identified candidate genes was derived from RNA sequencing data in all tested tissues between CQ24/CQ46 (a) and CQ52/46 (b) and common candidate genes (c).
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional files