

Abstract
Background
Salinity is a major abiotic stress seriously hindering crop yield. Development and utilization of tolerant varieties is the most economical way to address soil salinity. Upland cotton is a major fiber crop and pioneer plant on saline soil and thus its genetic architecture underlying salt tolerance should be extensively explored.
Results
In this study, genome-wide association analysis and RNA sequencing were employed to detect salt-tolerant qualitative-trait loci (QTLs) and candidate genes in 196 upland cotton genotypes at the germination stage. Using comprehensive evaluation values of salt tolerance in four environments, we identified 33 significant single-nucleotide polymorphisms (SNPs), including 17 and 7 SNPs under at least two and four environments, respectively. The 17 stable SNPs were located within or near 98 candidate genes in 13 QTLs, including 35 genes that were functionally annotated to be involved in salt stress responses. RNA-seq analysis indicated that among the 98 candidate genes, 13 were stably differentially expressed. Furthermore, 12 of the 13 candidate genes were verified by qRT-PCR. RNA-seq analysis detected 6640, 3878, and 6462 differentially expressed genes at three sampling time points, of which 869 were shared.
Conclusions
These results, including the elite cotton accessions with accurate salt tolerance evaluation, the significant SNP markers, the candidate genes, and the salt-tolerant pathways, could improve our understanding of the molecular regulatory mechanisms under salt stress tolerance and genetic manipulation for cotton improvement.
Electronic supplementary material
The online version of this article (10.1186/s12870-019-1989-2) contains supplementary material, which is available to authorized users.
Keywords: Salinity, Transcriptome, Genetic variation, Genome-wide association study, Germination stage, Comprehensive evaluation, Single-nucleotide polymorphisms (SNPs)
Background
Salinity is a significant abiotic stress that reduces crop productivity and quality throughout the world [1]. More than 6% of the world’s 800 million agricultural lands are affected by salinity [2]. A comprehensive understanding of the salt-responsive molecular mechanisms and exploring salt-tolerant genes will help increase crop tolerance to salinity [1, 3, 4]. Upland cotton (Gossypium spp.) is an important source of natural fiber, vegetable oil, and protein and is also a moderately salt-tolerant and pioneer crop that can be grown in saline-alkali land. Nevertheless, its yield will be drastically reduced as the soil salinization level increases [5]. Tolerance to salinity significantly varies among cotton germplasms. Thus, screening elite high salt-tolerant germplasms is key to breeding salt-tolerant cotton, as well as in identifying molecular mechanisms and key genes associated with salt tolerance.
Several methods have been developed for assessing germplasm. Factor analysis of principal component analysis (PCA) is commonly used in the evaluation of the status of each material in a group by analyzing a large number of samples and major correlation indicators [6–9]. Subordinate function analysis (SFA) is often used to evaluate stress tolerance [8, 10]. However, the evaluation may be one-sided when using only PCA or SFA [10, 11]. A comprehensive evaluation value combining PCA and SFA can convert each indicator into independent factors that can be compared with each other while maintaining the original information, thereby providing a more comprehensive assessment of plant tolerance [10]. In addition, this comprehensive evaluation value has higher accuracy and efficiency than grade evaluation with estimating intuitive withered area proportion. This integrated approach has been used to assess stress tolerance in sugarcane [10, 12], cucumber [13], tomato [11], alfalfa [8], wheat [14], and cotton [15].
Salinity tolerance is a multigene controlled trait and is susceptible to environmental factors. Association mapping based on linkage disequilibrium (LD) is a common and powerful technique for identifying genomic regions related to specific variants of phenotypic characteristics [16] based on its capability of dissecting a larger number of alleles than linkage mapping. However, studies on identifying salt-tolerant QTLs/genes in cotton using association mapping [7, 16, 17] or even linkage mapping [18, 19] are limited. In addition, the results of a single investigation on association mapping for salt tolerance in cotton using high-density single-nucleotide polymorphism (SNP) markers have been reported. A total of 23 significant SNPs and 280 possible candidate genes, of which most are involved with transcription factors, transporters, and enzymes, were found to be associated with two salt tolerance-related traits [20]. With the maturity and popularization of second-generation sequencing, RNA sequencing (RNA-seq) has become the major approach in excavating candidate genes, as well as in constructing molecular regulatory pathways and potential regulatory networks. Some salt-responsive mRNAs [21, 22], miRNAs [3], alternative splicing [23], or long non-coding RNAs (lncRNAs) [24] were detected in cotton using RNA-seq and the potential molecular regulatory pathways or regulatory networks of some genes were preliminarily explored. In cotton, several salt stress-inducible genes have been detected through association or linkage mapping and RNA-seq, including GhNHX1 [25], metallothionein (GhMT3a) [26], GhERF2-GhERF6 [27, 28], GhDREB1 [29], CCCH-type zinc finger (GhZFP1) [30], GhNAC1-GhNAC631 [31], GhMPK2 [32], GhMKK1 [33], GhSOD1 and GhCAT1 [34], GhWRKY17 [35], and GhAnn1 [36].
In this study, we combined comprehensive evaluation, association mapping, and RNA-seq to explore salt-tolerant candidate chromosomal regions/genes in cotton at the germination stage. This study provides candidate QTL (qualitative-trait locus) regions and genes for dissecting the genetic mechanisms of salt tolerance and variety breeding in cotton.
Results
Salt tolerance performance and evaluation
At germination stage, 10 traits were measured in 196 upland cotton genotypes, and the results are shown in Table 1. The results indicated that all 10 traits were significantly hindered by salt stress. All traits exhibited significantly lower means and extreme values under salt stress than the normal conditions. Comparatively, GP (germination potential) was the most affected trait, whereas SFM (shoot fresh mass) was least affected. The 0.3% (200 mmol/L) NaCl stress reduced not only the total germination rate but also the germination speed and subsequent growth. Under normal conditions, the CVs (coefficient variations) ranged from 7.1% in RDM (root dry mass) to 20.6% in VI (vigor index). The 0.3% (200 mmol/L) NaCl stress resulted in significantly increased CVs in nearly all 10 traits, except for VI and RFM (root fresh mass). SDM (shoot dry mass), GP, and GR (germination rate) ranked the top three CVs. These results indicated that most of the trait responses of this panel of upland cotton to salt stress are highly diverse.
Table 1.
Statistics and difference analysis of traits related to salt tolerance under salt stress and non-salt stress
| Trait | GR (%) | GP (%) | GI | VI | RL (cm) | RDM (mg) | RFM (mg) | PH (cm) | SDM (mg) | SFM (mg) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CK | Salt | CK | Salt | CK | Salt | CK | Salt | CK | Salt | CK | Salt | CK | Salt | CK | Salt | CK | Salt | CK | Salt | |
| Min | 90.0 | 23.0 | 75.0 | 0.0 | 58.5 | 9.3 | 92.6 | 3.9 | 7.8 | 3.0 | 201.0 | 10.0 | 412.0 | 40.0 | 7.9 | 2.9 | 231.0 | 20.0 | 1080.0 | 220.0 |
| Max | 100.0 | 87.0 | 100.0 | 74.0 | 63.5 | 53.0 | 103.3 | 60.5 | 9.7 | 7.1 | 240.0 | 120.1 | 450.0 | 230.0 | 12.6 | 8.6 | 260.0 | 160.0 | 1630.0 | 890.0 |
| Mean | 91.0 | 45.3 | 82.1 | 21.5 | 53.1 | 37.0 | 45.9 | 32.0 | 8.5 | 3.1 | 221.2 | 70.0 | 210.0 | 73.5 | 10.7 | 5.1 | 240.0 | 98.0 | 870.0 | 603.0 |
| SD | 7.7 | 13.0 | 9.9 | 6.8 | 6.7 | 8.8 | 9.5 | 5.7 | 0.9 | 0.4 | 15.8 | 9.3 | 0.0 | 10.3 | 0.9 | 0.8 | 0.0 | 35.3 | 0.1 | 90.6 |
| CV(%) | 8.5 | 28.8 | 12.0 | 27.9 | 12.7 | 23.7 | 20.6 | 17.8 | 11.0 | 12.2 | 7.1 | 13.3 | 15.2 | 14.0 | 8.7 | 15.1 | 13.6 | 36.0 | 13.2 | 15.0 |
| t-test | 56.248** | 30.778** | 37.452** | 43.032** | 22.774** | 1.439** | 4.716** | 51.944** | 33.782** | 38.912** | ||||||||||
**indicate significant difference at p < 0.01
To reduce the impact of environmental factors and to reach high evaluation accuracy, the BLUPed (BLUP: the best linear unbiased prediction) STI (salt tolerance index) was calculated based on the nine (three years × three replications) STIs in each trait, and the results were shown in Additional file 1: Table S1 [37, 38]. No significant differences among STIs among the 3 years and the BLUPed STIs of each trait were observed. However, the BLUPed STIs and their CVs significantly varied among traits. The GR, RL (root length), and PH (plant height) exhibited larger BLUPed STIs with smaller CVs, whereas VI showed the smallest BLUPed STIs with large CVs. GP and SFM had small BLUPed STIs with larger CVs. The traits that had high salt tolerance (high BLUPed STI) usually had low CVs and vice versa, except for SDM, which exhibited the second smallest BLUPed STI with medium CV value. To further reveal the relationship among the measured traits under salt stress, Pearson correlation coefficients were estimated and are listed in Additional file 2: Table S2. Significant positive correlations were detected among nearly all trait-pairs except for RFM-SDM and RFM-SFM, which exhibited very weak and insignificant correlations.
Comprehensive evaluation of salt tolerance
To comprehensively evaluate salt tolerance in this panel, PCA of STIs was performed. Before PCA, the Kaiser-Mayer-Olkin (KMO) measure and Bartlett’s test were adopted to check whether the STIs were suitable for factor analysis. The KMO values in the four data sets (2014: 0.825; 2015: 0.856; 2016: 0.852; and BLUP: 0.857) were all > 0.5, and the Bartlett’s test values all performed well (χ2 > χ20.01, 45, and Sig. = 0.000 < 0.05) [6], indicating that the STI data were suitable for PCA. The four STI data sets resulted in consistent results in PCA. For each set of STI data, two principal components that accounted for at least 86.85% of the total variance were obtained based on the Eigenvalues-more-than-1.0 rule (Additional file 3: Figure S1 and Additional file 4: Table S3). As shown in PCA plots (Additional file 5: Figure S2), Factor 1 represents all the salt-tolerance traits except for STI_RFM (salt tolerance index of root fresh mass), which was Factor 2.
Furthermore, the comprehensive evaluation value (D) of each genotype was obtained based on the subordinate function value (U) computed with the two principle factors (Additional file 6: Table S4). By K-mean cluster analysis, the 196 genotypes were divided into four groups based on the four sets of D values, which represented the salt tolerance of genotypes in 2014, 2015, 2016, and BLUPed data. Of the 196 genotypes, 27 accessions ranked as advanced salt-tolerant with D values for BLUPed STI within the range of 0.669–0.934, 35 accessions had medium salt tolerance, with D values ranging from 0.486 to 0.633), 67 were salt-sensitive with D values from 0.320 to 0.474, and 67 ranked as high salt-sensitive with D values from 0.053 to 0.312 (Additional file 6: Table S4).
Association of SNP markers and salt tolerance
In this GWAS (genome-wide association study), the comprehensive evaluation values (D) was used as phenotypic data to detect significant salt tolerance QTLs/genes. Thus, the covariant Q matrix, which can reduce the negative influence of group structure, was introduced into the mixed linear model (MLM) to reduce false positives. To test the quality of the mixed linear model combined structure Q matrix and kinship matrix (MLM [Q + K]) and association results, we employed a QQ (Quantile quantile) plot. Figure 1 showed that the MLM [Q + K] model was slightly too strict in detecting significant SNP markers, indicating that the probability of false positives was much lower. Consequently, a total of 33 SNPs that were significantly (p < 0.001) associated with salt tolerance were detected using MLM [Q + K] (Fig. 2 and Table 2). Among these, 17 SNPs were detected in at least two environments (3 years and BLUPed data) and 7 SNPs were identified in all four environments, explaining 5.98 to 10.76% phenotypic variation, with an average of 8.21%. These 17 SNPs were consequently used to identify candidate QTLs. When the distance between the first SNP and neighboring SNPs was less than the LD decay distance at pairwise r2 = 0.1 level on each chromosome (Additional file 7: Table S5) or r2 between the first SNP and the neighboring SNP was > 0.1, these SNP markers were regarded as a confidence interval for a candidate QTL. Finally, 13 QTLs were obtained on 10 chromosomes, including 4 QTLs on homologous chromosomes A07–D07.
Fig. 1.
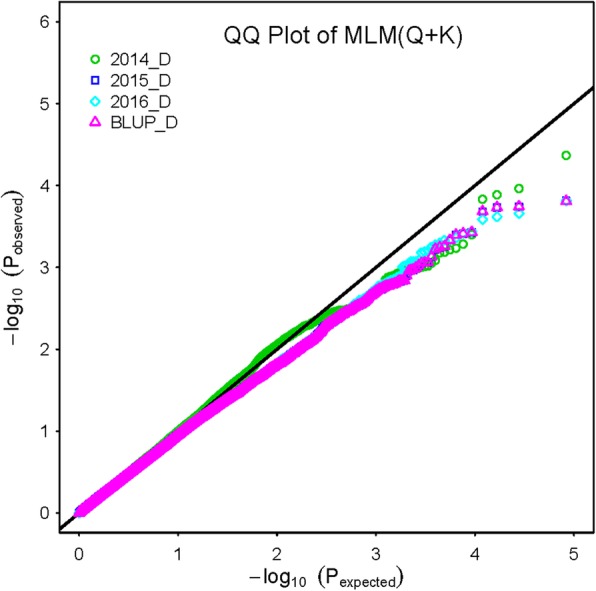
Q-Q plots for D values in 2014, 2015, 2016, and BLUPs using MLM (Q + K)
Fig. 2.

Circular-Manhattan plot for D values GWAS results. The threshold value was set at p < 0.001
Table 2.
Significant SNP markers, QTLs, and candidate genes related to salt tolerance
| Marker | Chr | Position (bp) | -lg(P) (BLUP) | MarkerR2 (BLUP) | Environments | QTL | Physical Chr | Genomic position | No. of genes |
|---|---|---|---|---|---|---|---|---|---|
| TM3300 | 1 | 92,501,634 | 3.41 | 0.084 | BLUP, 14, 15, 16 | qGhST-c1 | A01 | 92,402,641..92562754 | 7 |
| TM4974 | 2 | 56,676,599 | 3.05 | 0.079 | BLUP, 15 | qGhST-c2 | A02 | 56,176,476..56990091 | 6 |
| TM8361 | 3 | 98,536,836 | 3.81 | 0.108 | BLUP, 15, 16 | qGhST-c3 | A03 | 98,507,289..98618065 | 13 |
| TM18816 | 7 | 8,674,003 | 3.26 | 0.083 | BLUP, 15, 16 | qGhST-c7–1 | A07 | 8,654,507..8730789 | 5 |
| TM19026 | 7 | 11,292,696 | 3.22 | 0.090 | BLUP, 15, 16 | qGhST-c7–2 | A07 | 11,270,123..11494991 | 12 |
| TM19028 | 7 | 11,312,723 | 3.06 | 0.087 | BLUP, 15, 16 | A07 | |||
| TM19030 | 7 | 11,334,462 | 3.06 | 0.080 | BLUP, 15, 16 | A07 | |||
| TM19035 | 7 | 11,356,480 | 3.13 | 0.084 | BLUP, 15, 16 | A07 | |||
| TM41811 | 12 | 57,971,233 | 3.25 | 0.065 | BLUP, 15, 16 | qGhST-c12–1 | A12 | 57,740,976..58054684 | 11 |
| TM41814 | 12 | 58,100,057 | 3.33 | 0.069 | BLUP, 14, 15, 16 | qGhST-c12–2 | A12 | 58,097,661..58230406 | 3 |
| TM63245 | 16 | 2,688,021 | 3.00 | 0.060 | BLUP, 16 | qGhST-c16–1 | D07 | 2,555,918..2766294 | 27 |
| TM63387 | 16 | 5,545,380 | 3.73 | 0.099 | BLUP, 14, 15, 16 | qGhST-c16–2 | D07 | 5,518,153..5573449 | 3 |
| TM73567 | 20 | 6,921,040 | 3.43 | 0.068 | BLUP, 14, 15, 16 | qGhST-c20–1 | D10 | 6,887,955..6965236 | 7 |
| TM73573 | 20 | 6,959,800 | 3.02 | 0.077 | BLUP, 15 | D10 | |||
| TM73579 | 20 | 6,996,881 | 3.40 | 0.068 | BLUP, 14,15, 16 | qGhST-c20–2 | D10 | 6,991,910..7032029 | 2 |
| TM67763 | 24 | 16,771,670 | 3.74 | 0.099 | BLUP, 14, 15, 16 | qGhST-c24–1 | D08 | 16,766,258..16776082 | 0 |
| TM68258 | 24 | 19,887,759 | 3.68 | 0.096 | BLUP, 14, 15, 16 | qGhST-c24–2 | D08 | 19,884,918..19901251 | 2 |
| TM3510 | 1 | 96,629,867 | 3.24 | 0.092 | 14 | ||||
| TM776 | 1 | 14,254,094 | 3.40 | 0.098 | 14 | ||||
| TM4108 | 2 | 7,344,966 | 3.00 | 0.082 | 14 | ||||
| TM8353 | 3 | 98,481,949 | 3.01 | 0.079 | 16 | ||||
| TM18851 | 7 | 9,689,745 | 3.05 | 0.084 | 16 | ||||
| TM18989 | 7 | 11,034,281 | 3.07 | 0.081 | 16 | ||||
| TM19015 | 7 | 11,198,513 | 3.01 | 0.078 | 16 | ||||
| TM19017 | 7 | 11,226,724 | 3.09 | 0.080 | 16 | ||||
| TM19018 | 7 | 11,232,229 | 3.07 | 0.079 | 16 | ||||
| TM19023 | 7 | 11,276,537 | 3.20 | 0.082 | 16 | ||||
| TM53602 | 17 | 4,200,687 | 3.18 | 0.092 | 14 | ||||
| TM71972 | 23 | 33,356,265 | 3.02 | 0.064 | 14 | ||||
| TM71987 | 23 | 33,461,358 | 3.09 | 0.066 | 14 | ||||
| TM67849 | 24 | 17,321,002 | 3.89 | 0.112 | 14 | ||||
| TM68645 | 24 | 31,157,957 | 3.21 | 0.089 | 14 | ||||
| TM68879 | 24 | 44,114,822 | 3.03 | 0.060 | 16 |
Candidate genes detected with GWAS
According to the physical positions of the candidate QTLs referenced to the G. hirsutum (TM-1) genome [39], a total of 98 candidate genes were detected (Table 2), and all these genes were annotated in Arabidopsis thaliana (Additional file 8: Table S6). The candidate genes were retrieved in UniProtKB to annotate their biological processes (Additional file 8: Table S6). Functional annotation showed that 35 candidate genes were associated with salt tolerance (Table 3). Of these, 11 genes were involved in eight transcription factor (TF) families, including CO-like, MYB, bZIP, ERF, TALE, SBP, HD-ZIP, and ARR-B; 17 genes were related to “response to stress” or “defense response” such as the stress of acidic pH, heat, hydrogen peroxide, water deprivation, and hyperosmotic salinity; nine genes were associated with “signaling” or “response to signal factors” such as salicylic acid (SA), gibberellic acid (GA), jasmonic acid (JA), calcium-mediated signaling, cytokinin, and abscisic acid (ABA); six genes were involved in “ion homeostasis” or “ion transport”, one gene was associated with “fatty acid biosynthetic process”, and five genes were related to amino acid synthesis or transport. In addition, 12 of these 35 candidate genes were involved in two or three functional categories (Table 3).
Table 3.
Candidate genes belong to categories related to salt tolerance
| Category | Gene ID | Gene Name | Description | TF Family/Stress/Signal/Ion transport |
|---|---|---|---|---|
| Transcription factors | Gh_A01G1562 | COL16 | Zinc finger protein CONSTANS-LIKE 16 | CO-like |
| Gh_A01G1564 | MYB108 | Transcription factor MYB108 | MYB | |
| Gh_A03G1731 | BZIP53 | bZIP transcription factor 53 | bZIP | |
| Gh_A03G1738 | BLH6 | BEL1-like homeodomain protein 6 | TALE | |
| Gh_A12G0867 | SPL8 | Squamosa promoter-binding-like protein 8 | SBP | |
| Gh_A12G0870 | ERF034 | Ethylene-responsive transcription factor ERF034 | ERF | |
| Gh_A12G0871 | HAT3 | Homeobox-leucine zipper protein HAT3 | HD-ZIP | |
| Gh_A12G0875 | ERF13 | Ethylene-responsive transcription factor 13 | ERF | |
| Gh_A12G0876 | ERF13 | Ethylene-responsive transcription factor 13 | ERF | |
| Gh_A12G0877 | ERF2 | Ethylene-responsive transcription factor 2 | ERF | |
| Gh_D07G0251 | ARR12 | Two-component response regulator ARR12 | ARR-B | |
| Response to stress or defense response | Gh_A01G1563 | CUT1 | 3-ketoacyl-CoA synthase 6 | Cold |
| Gh_A03G1732 | AT2G16385 | CIF1:Proteincasparlan strip integrity factor 1 | Acidic pH | |
| Gh_A03G1733 | AT4G39130 | Late embryogenesis abundant protein | Stress, Water | |
| Gh_A07G0622 | CLPB1 | Chaperone protein ClpB1 | Heat, Hydrogen peroxide | |
| Gh_A07G0624 | DGK1 | Diacylglycerol kinase 1 | Defense response | |
| Gh_A12G0875 | ERF13 | Ethylene-responsive transcription factor 13 | Defense response | |
| Gh_A12G0876 | ERF13 | Ethylene-responsive transcription factor 13 | Defense response | |
| Gh_A12G0877 | ERF2 | Ethylene-responsive transcription factor 2 | Defense response | |
| Gh_D07G0240 | KAT3 | Potassium channel KAT3 | Nematode | |
| Gh_D07G0244 | EDR2L | Protein enhanced disease resistance 2-like | Defense response | |
| Gh_D07G0246 | CHIA | Acidic mammalian chitinase | Immune system process | |
| Gh_D07G0248 | CHIT1 | Chitotriosidase-1 | Immune response | |
| Gh_D07G0249 | CHIT1 | Chitotriosidase-1 | Immune response | |
| Gh_D07G0251 | ARR12 | Two-component response regulator ARR12 | Water deprivation | |
| Gh_D07G0252 | NKS1 | Ubiquitin-associated protein (DUF1068) | Hyperosmotic salinity | |
| Gh_D07G0256 | TIL | Temperature-induced lipocalin-1 | Hyperosmotic salinity, Cold, Freezing, Heat, Paraquat, Water deprivation | |
| Gh_D07G0500 | HVA22E | HVA22-like protein e | Hyperosmotic salinity, Cold, Water deprivation | |
| Signaling or response to signal factors | Gh_A01G1564 | MYB108 | Transcription factor MYB108 | Gibberellic acid, Jasmonic acid |
| Gh_A02G1099 | WAK2 | Wall-associated receptor kinase 2 | Salicylic acid | |
| Gh_A02G1100 | WAK2 | Wall-associated receptor kinase 2 | Salicylic acid | |
| Gh_A07G0624 | DGK1 | Diacylglycerol kinase 1 | Intracellular signal transduction | |
| Gh_A07G0729 | RALFL27 | Protein RALF-like 27 | Calcium mediated signaling, Cell-cell signaling | |
| Gh_D07G0251 | ARR12 | Two-component response regulator ARR12 | Cytokinin | |
| Gh_D07G0256 | TIL | Temperature-induced lipocalin-1 | Cytokinin | |
| Gh_D07G0500 | HVA22E | HVA22-like protein e | Abscisic acid | |
| Gh_D10G0643 | IAA27 | Auxin-responsive protein IAA27 | Auxin | |
| Ion homeostasis or ion transport | Gh_A03G1732 | AT2G16385 | CIF1:Proteincasparlan strip integrity factor 1 | Ion homeostasis |
| Gh_D07G0240 | KAT3 | Potassium channel KAT3 | Potassium ion transmembrane transport, Regulation of ion transmembrane transport, Regulation of membrane potential | |
| Gh_D07G0252 | NKS1 | Ubiquitin-associated protein (DUF1068) | Vacuolar sequestering of sodium ion | |
| Gh_D07G0254 | MOT1 | Molybdate transporter 1 | Molybdate ion transmembrane transporter activity, Molybdate ion transport | |
| Gh_D07G0255 | MOT1 | Molybdate transporter 1 | Molybdate ion transmembrane transporter activity, Molybdate ion transport | |
| Gh_D07G0256 | TIL | Temperature-induced lipocalin-1 | Cellular chloride ion homeostasis, Cellular sodium ion homeostasis | |
| Fatty acid | Gh_A01G1563 | CUT1 | 3-ketoacyl-CoA synthase 6 | Fatty acid biosynthetic process |
| Amino acid | Gh_D07G0242 | PROC | Pyrroline-5-carboxylate reductase | Proline biosynthetic process, Pyrroline-5-carboxylate reductase activity |
| Gh_A03G1734 | HISN7 | Bifunctional phosphatase IMPL2, chloroplastic | Histidine biosynthetic process, Inositol biosynthetic process, Phosphatidylinositol phosphorylation | |
| Gh_A03G1737 | SAE2 | SUMO-activating enzyme subunit 2 | Acid-amino acid ligase activity | |
| Gh_D08G0928 | GDU2 | Protein GLUTAMINE DUMPER 2 | Amino acid transport | |
| Gh_D08G0929 | GDU3 | Protein GLUTAMINE DUMPER 3 | Amino acid transport |
GO (Gene Ontology) enrichment analysis was conducted to further infer the functions of these 98 candidate genes. At a P-value < 0.05 and the number of genes > 3, the 98 candidate genes were categorized into eight GO terms (Fig. 3). Each of the two main categories, namely, biological process (BP) and molecular function (MF), contained four GO terms. The BP category in this study contained “chitin catabolic process”, “carbohydrate metabolic process”, and “metabolic process and regulation of transcription”, whereas the MF category contained “chitinase activity”, “hydrolase activity”, and “transcription factor activity and transporter activity”.
Fig. 3.

Gene Ontology (GO) enrichment analysis of 98 salt-response genes detected in GWAS
Based on the physical positions, 21 of the 98 candidate genes detected in the GWAS were proximal to significant SNPs, and six (Gh_A07G0622-TM18816, Gh_A07G0729-TM19028, Gh_A12G0874-TM41811, Gh_D07G0251-TM63245, Gh_D10G0640-TM73567, and Gh_D10G0647-TM73579) of them had significant SNPs in their coding sequences (CDS) (Additional file 9: Table S7). Furthermore, based on their functional annotation or biological process (Table 3), three (Gh_A07G0622, Gh_A07G0729, and Gh_D07G0251) of the six genes were related to the functional categories of “response to stress or defense response”, “signaling or response to signal factors” or “transcription factors”, implying their involvements in salt tolerance responses.
Transcriptome sequencing
Root tissues of Han682 at the germination stage under 0.3% NaCl stress and normal conditions were harvested at 3, 24, and 72 h after treatment for RNA isolation. From the 12 RNA sequencing libraries of H3_1, H3_2, S3_1, S3_2, H24_1, H24_2, S24_1, S24_2, H72_1, H72_2, S72_1 and S72_2, a total of 50.5, 45.9, 46.6, 52.6, 46.8, 49.4, 45.1, 44.5, 43.6, 55.8, 45.3, and 40.6 million raw reads were obtained (Additional file 11: Table S8). At least 88% of the clean reads, which were raw reads without low-quality reads and adaptor sequences, were mapped to the G. hirsutum (TM-1) genome, in which uniquely mapped reads accounted for about 81% of the clean reads. A total of 6640, 3878, and 6462 DEGs (differentially expressed genes) (P-value < 0.05) were obtained between salt stress and the control at 3, 24, and 72 h post salt stress treatment, respectively, and 3956 (59.6%), 2238 (57.7%), and 3226 (49.9%) were upregulated (Fig. 4). Among the three sampling time points, we identified 869 shared DEGs, including 562 continuously upregulated and 307 continuously downregulated genes.
Fig. 4.

Statistical analysis of DEGs in the roots between salt stress and water control. a The number of upregulated and downregulated DEGs at different time points. b Venn diagram of DEGs at different time points after treatment
To verify our DEG results, qRT-PCR was adopted on 20 randomly selected DEGs, including 13 continuously upregulated and 7 downregulated genes (Additional file 10: Figure S3). Relative expression levels were calculated using the ΔΔCt method. The results of transcriptome sequencing coincided with our qRT-PCR findings.
To monitor salt tolerance gene expression, GO enrichment analysis (P-value corrected by FDR < 0.05) of 562 continuously upregulated genes and 307 continuously downregulated genes was conducted (Fig. 5). For continuously upregulated genes (Fig. 5a), the BP category consisted of “single-organism metabolic process”, “oxidation-reduction process”, “carbohydrate metabolic process”, “photosynthesis”, “organonitrogen compound catabolic process”, “oligosaccharide metabolic process” and “aromatic compound catabolic process”; the cellular component (CC) category was mainly related to “thylakoid” and “photosystem”; and the MF category comprised “oxidoreductase activity” and “O-methyltransferase activity”. For continuously downregulated genes (Fig. 5b), the BP category was mainly involved in “response to oxidative stress”, “obsolete GTP catabolic process”, and “polyol metabolic process”; the CC category was mainly involved in “viral capsid”, “integral component of organelle membrane”, and “intrinsic component of organelle membrane”; and the MF category was mainly involved in “transporter activity”, “peroxidase activity”, and “inorganic diphosphatase activity”. All the enriched GO terms of the 869 shared DEGs were associated with stress responses.
Fig. 5.
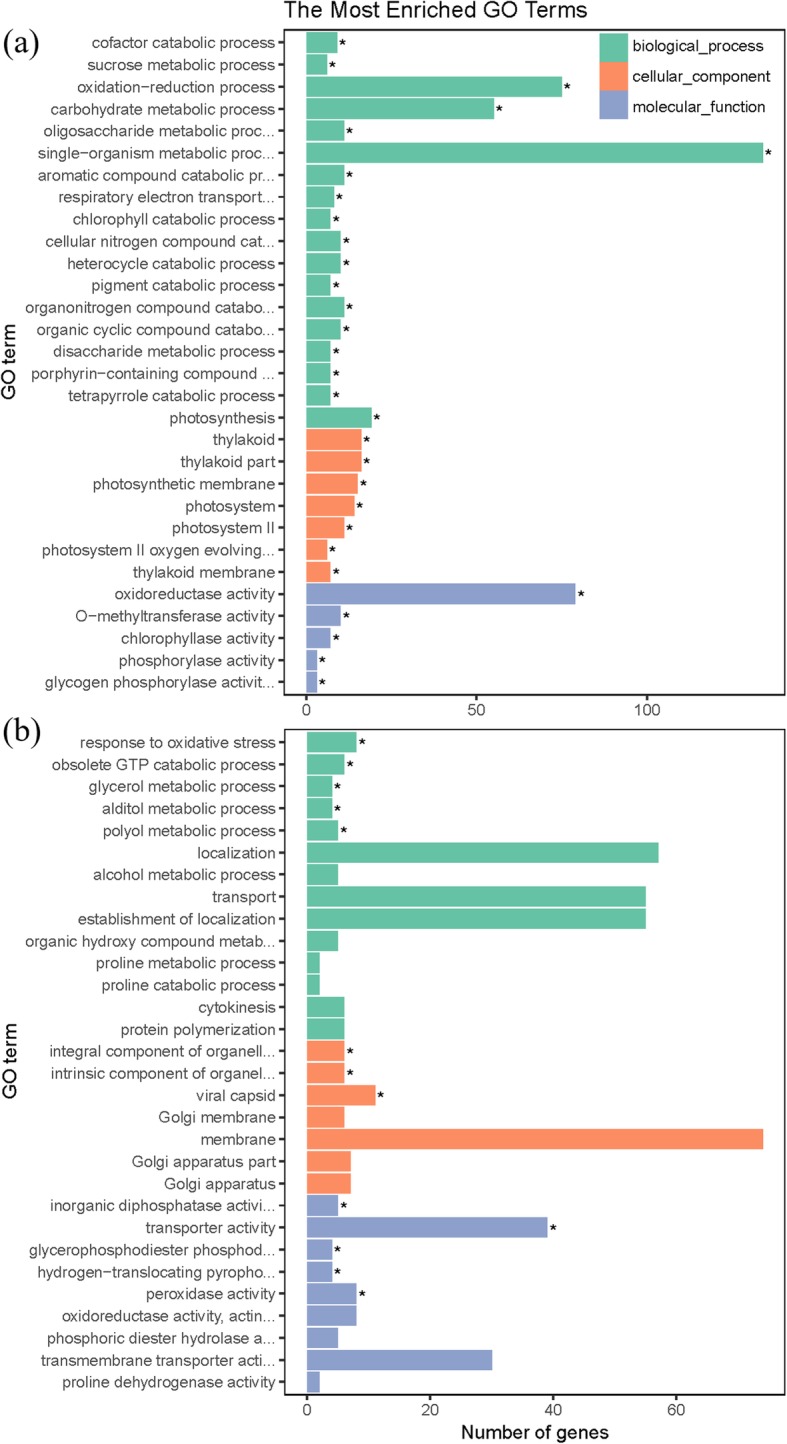
Gene Ontology (GO) functional enrichment analysis of salt-tolerance genes detected in RNA-seq. a GO functional classification of 562 continuously upregulated salt-tolerance genes at q-value < 0.05. b GO functional classification of 307 continuously downregulated salt tolerance genes at q-value < 0.05
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment of the 869 continuously up/downregulated DEGs was performed (Fig. 6). The continuously upregulated genes were significantly (P-value < 0.05) enriched in nine KEGG pathways (Fig. 6a), including “photosynthesis”, “flavonoid biosynthesis”, “starch and sucrose metabolism”, “photosynthesis-antenna proteins”, “pyruvate metabolism”, “plant hormone signal transduction”, “beta-alanine metabolism”, “galactose metabolism”, and “arachidonic acid metabolism”. The KEGG pathways of continuously upregulated genes correspond to the GO enrichment results and the functions of genes, which were associated with stress response. The continuously downregulated genes were mostly related to nine KEGG pathways (Fig. 6b), including phagosome, phenylalanine metabolism, phenylpropanoid biosynthesis, starch and sucrose metabolism, pentose and glucuronate interconversions, ascorbate and aldarate metabolism, glucosinolate biosynthesis, oxidative phosphorylation and alanine, and aspartate and glutamate metabolism.
Fig. 6.
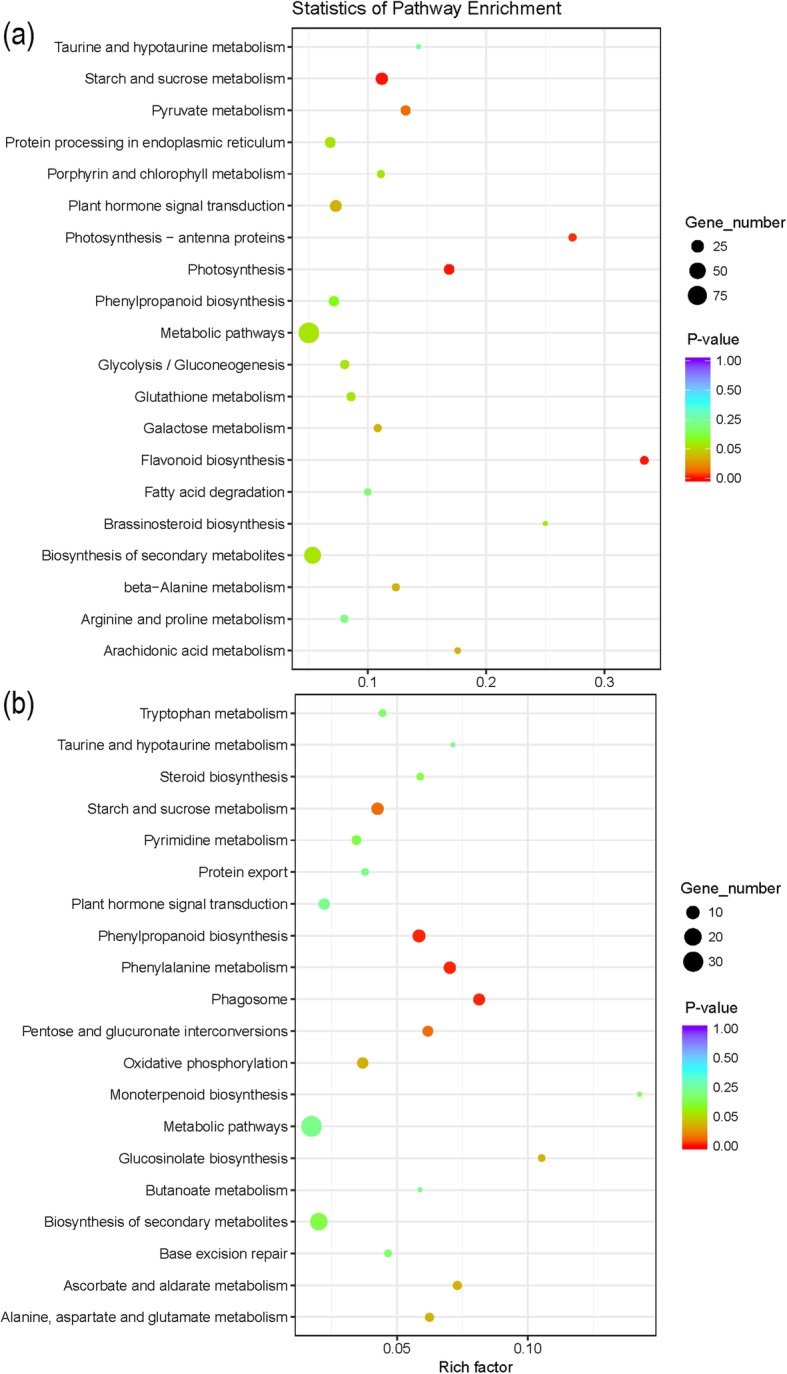
Statistics of pathway enrichment analysis of salt-tolerance genes detected in RNA-seq. a Pathway enrichment analysis of 562 continuously upregulated salt-tolerance genes at p-value < 0.05. b Pathway enrichment analysis of 307 continuously downregulated salt tolerance genes at p-value < 0.05
TFs play pivotal roles in plant stress responses [40]. In this study, 62 (7.13%) genes encoding for TFs were identified as shared DEGs, of which 41 were continuously upregulated and 21 continuously downregulated (Table 4). All the 62 genes were belonged to 19 TF families, including several key regulatory gene families responding to abiotic and biotic stresses such as ARF, bHLH, bZIP, C2H2, ERF, HD-ZIP, MYB, MYB_related, NAC, and WRKY.
Table 4.
Shared DEGs belonging to TF families
| TF family | Total | Up | Down |
|---|---|---|---|
| bZIP | 11 | 3 | 8 |
| HD-ZIP | 6 | 5 | 1 |
| MYB | 6 | 5 | 1 |
| NAC | 5 | 4 | 1 |
| bHLH | 4 | 2 | 2 |
| MYB_related | 3 | 2 | 1 |
| WRKY | 3 | 2 | 1 |
| G2-like | 2 | 1 | 1 |
| HSF | 4 | 4 | 0 |
| C3H | 3 | 3 | 0 |
| CO-like | 3 | 3 | 0 |
| LBD | 2 | 2 | 0 |
| M-type_MADS | 2 | 2 | 0 |
| NF-YB | 2 | 2 | 0 |
| NF-YA | 1 | 1 | 0 |
| ARF | 2 | 0 | 2 |
| C2H2 | 1 | 0 | 1 |
| ERF | 1 | 0 | 1 |
| TCP | 1 | 0 | 1 |
| Total | 62 | 41 | 21 |
Combination of association analysis and transcriptome sequencing
We combined the GWAS and RNA-Seq results to further screen salt tolerance candidate genes. Of the 98 candidate genes in GWAS, 13 exhibited significantly different expression levels (p < 0.005) at more than one sampling time point in RNA-seq analysis (Table 5). Of the 13 putative DEGs, eight exhibited significantly different expression at only one time point, four (Gh_A03G1740, Gh_A12G0877, Gh_D07G0263, and Gh_D07G0500) at two time points, and one, Gh_A07G0622, upregulated at all three time points. In addition, six of the 13 putative DEGs (Gh_A01G1563, Gh_A02G1100, Gh_A07G0622, Gh_A12G0877, Gh_D07G0251, and Gh_D07G0500) were proximal to significant SNPs in GWAS, and two (Gh_A07G0622 and Gh_D07G0251) of the six genes had significant SNPs within coding sequence regions (Additional file 9: Table S7). In addition, as shown in above, Gh_A07G0622 and Gh_D07G0251 were involved in “response to stress or defense response”, “signaling or response to signal factors”, and “transcription factors” (Table 3).
Table 5.
The differential expression of the putative genes detected in both GWAS and transcriptome sequencing
| Gene_id | readcount_H3 | readcount_S3 | log2FoldChange | p value | readcount_H24 | readcount_S24 | log2FoldChange | p value | readcount_H72 | readcount_S72 | log2FoldChange | p value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gh_A01G1563 | 40.232 | 78.450 | 0.963 | 0.261 | 4.001 | 51.024 | 3.673 | 0.000* | 0.000 | 5.109 | +∞ | 0.316 |
| Gh_A02G1100 | 4.161 | 26.612 | 2.677 | 0.001* | 2.994 | 2.954 | −0.020 | 1.000 | 5.117 | 1.099 | −2.219 | 0.297 |
| Gh_A03G1740 | 587.138 | 96.274 | −2.608 | 0.000* | 322.348 | 67.547 | −2.255 | 0.000* | 38.368 | 24.008 | −0.676 | 0.246 |
| Gh_A07G0622 | 180.915 | 912.736 | 2.335 | 0.000* | 185.769 | 1681.857 | 3.178 | 0.000* | 130.525 | 1972.522 | 3.918 | 0.000* |
| Gh_A07G0623 | 24.501 | 51.165 | 1.062 | 0.036 | 48.834 | 79.571 | 0.704 | 0.403 | 32.992 | 79.503 | 1.269 | 0.004* |
| Gh_A12G0877 | 10.051 | 4.262 | −1.238 | 0.306 | 27.855 | 0.484 | −5.848 | 0.000* | 25.105 | 3.719 | −2.755 | 0.002* |
| Gh_D07G0243 | 63.337 | 213.505 | 1.753 | 0.000* | 115.184 | 114.475 | − 0.009 | 0.990 | 88.638 | 69.300 | −0.355 | 0.441 |
| Gh_D07G0249 | 1.579 | 0.444 | −1.830 | 0.771 | 4.501 | 3.006 | −0.582 | 0.834 | 2.439 | 77.211 | 4.984 | 0.000* |
| Gh_D07G0250 | 3.196 | 39.993 | 3.645 | 0.004* | 0.501 | 4.404 | 3.137 | 0.205 | 7.333 | 4.395 | −0.738 | 0.621 |
| Gh_D07G0251 | 193.354 | 458.100 | 1.244 | 0.019 | 189.755 | 242.694 | 0.355 | 0.564 | 192.524 | 376.097 | 0.966 | 0.002* |
| Gh_D07G0258 | 220.517 | 323.412 | 0.552 | 0.060 | 141.005 | 164.415 | 0.222 | 0.586 | 95.896 | 206.969 | 1.110 | 0.002* |
| Gh_D07G0263 | 41.671 | 152.914 | 1.876 | 0.000* | 25.937 | 198.357 | 2.935 | 0.000* | 279.158 | 178.035 | −0.649 | 0.043 |
| Gh_D07G0500 | 37.958 | 426.805 | 3.491 | 0.000* | 37.418 | 163.250 | 2.125 | 0.000* | 262.034 | 249.357 | −0.072 | 0.846 |
*indicate significant at the p < 0.005 level
To further verify the putative genes, all the 13 putative DEGs except for Gh_A12G0877, which could not be distinguished from a homologous gene, were analyzed by qRT-PCR using a set of three replicates for each sample at 3, 12, 24, 48, and 72 h after salt or water treatment. Relative expression levels were calculated using the ΔΔCt method. Figure 7 showed that the putative genes Gh_A01G1563, Gh_A07G0622, Gh_D07G0243, and Gh_D07G0251 were continuously upregulated after salt treatment. The genes Gh_D07G0623, Gh_D07G0250, Gh_D07G0258, and Gh_D07G0500 responded to salt stress at the initial stage (3 h or 12 h post treatment). The gene Gh_A07G0623 was responsive to salt at 72 h post treatment. The candidate genes Gh_A03G1740, Gh_A02G1100, and Gh_D07G0249 were continuously downregulated under salt stress. The qRT-PCR results showed all the 12 DEGs might be involved in salt tolerance responses.
Fig. 7.

Quantitative real-time PCR validation of putative salt-responsive genes detected in combined analysis of GWAS and RNA-seq
Discussion
Comprehensive evaluation of salt tolerance
The salt tolerance traits of upland cotton are complex and vary with species, developmental stage, and tissue [41]. Thus, it is very important to evaluate germplasm salt tolerance accurately with a precise method. Several indexes have been used to assess salt tolerance in cotton, including indicators of seed germination (GR, GP, GI (germination index), and VI), plant morphological indexes (PH, SFM, SDM, and RL), physiological and biochemical indexes (Na+, K+, and betaine), and yield traits (boll number, boll weight, and lint yield) [7, 20, 42]. At present, it is generally believed that the comprehensive evaluation of salt tolerance combined with multi-indexes and multi-methods is more authentic and reliable such as PCA, SFA, and the comprehensive evaluation D value [6–11]. The comprehensive evaluation D value, which has the higher accuracy, has been widely used in evaluating stress tolerance in germplasms of different crops or vegetables [8, 10–15]. In this research, we investigated 10 traits related to salt tolerance and used their comprehensive evaluation D values in GWAS, which should help to improve salt tolerance evaluation of this panel and therefore the GWAS.
Candidate QTLs/genes detected with GWAS
Salt tolerance is a important and complex trait in cotton. Molecular tagging of salt tolerance has been investigated in several previous researches with different marker types and mapping populations [7, 16–19, 43, 44]. And, a meta analysis of salt tolerance QTLs in cotton was also reported [45]. Compared to the previous reports, there was no co-location QTL with the QTLs detected herein. This low consistency may be related to these factors, including the complexity of salt tolerance mechanisms in cotton, and differences in populations, salt tolerance assessment traits, marker types, and marker densities used in different studies. In this study, a high-density SNP array and four sets of D value (three years and BLUPed D value) were used in GWAS and 13 candidate QTLs controlling salt tolerance at germination stage were detected in no less than two environments. In addition, the QTLs regions harbored candidate genes whose functions were considered to be involved in salt tolerance and regulated by salt stress.
Many candidate genes from the 13 QTLs are very likely to be associated with salt tolerance, based on their annotation in Arabidopsis thaliana and functional annotation (Table 3 and Additional file 8: Table S6). Eleven genes are involved in seven TF families, namely, MYB, bZIP, ERF, TALE, SBP, HD-ZIP, and ARR-B. TFs play a major role in plant biotic and abiotic stress responses [46]. MYB [46–48], bZIP [49–51], and ERF [28, 52, 53] are also known to be involved in responses of biotic and abiotic stress. The CO-like gene (COL4) positively regulates abiotic (salt and osmotic) stress tolerance in Arabidopsis through an ABA-dependent method [54]. SBP, the plant-specific TF that participates in plant development, may enhance stress tolerance of plants by the growth regulation [55]. The ARR-B gene ARR12 (Gh_D07G0251 in this study) negatively regulates the responses of Arabidopsis to drought [56]. HD-ZIP TFs are involved in both development and stress (drought or salt) responses in A. thaliana [57, 58], rice [59], Manihot esculenta Crantz [60] and Craterostigma plantagineum [61]. Furthermore, the HD-ZIP gene (GhHB1) in G. hirsutum responds to root development, abscisic acid, and salt [62], suggesting that HD-ZIP genes play an important role in plant development and stress responses. As shown in Table 3, these 17 candidate genes detected in GWAS respond to acidic pH, heat, hydrogen peroxide, water deprivation, or hyperosmotic salinity stress or are involved in “defense response” and are highly likely to be related to salt tolerance. In addition, the gene Gh_A01G1564 responds to GA and JA, Gh_A02G1099 and Gh_A02G1100 responds to SA, Gh_D07G0251 and Gh_D07G0256 respond to cytokinin (CK), Gh_D07G0500 responds to ABA, and Gh_D10G0643 responds to auxin (IAA). Plant hormones such as GA, JA, SA, CK, ABA, and IAA play essential roles in plant adaptation to external stimuli and changes in the environment [3, 22, 63]. Those genes involved in “response to hormone stimulus” or “signaling” were concordant with the results of previous studies of expression profiling of plants under salt stress [5, 64, 65]. Gh_A07G0729 is also involved in calcium-mediated signaling, which plays an essential role in adapting to salt stress [3, 66, 67]. In addition, six genes were involved in “ion homeostasis” or “ion transport”, by which plant tolerance to salinity could be enhanced [68–70].
Furthermore, the GO terms enriched with candidate genes in this GWAS included carbohydrate metabolic process, metabolic process, hydrolase activity, regulation of transcription, transcription factor activity, and transporter activity, and were also mainly enriched in previous studies of responses to salt (NaCl) stress in cotton roots at the seedling stage [3, 21, 22, 65, 71]. Overexpression of chitinases in transgenic tobacco enhanced its tolerance to biotic (fungal and bacterial pathogens) and abiotic (salinity and heavy metals) stress [72], and accumulation of the chitinase isoforms was induced by heavy metal stress in plants [73]. In this GWAS, the enriched GO terms associated with salt stress included “chitin catabolic process” and “chitinase activity”.
Candidate genes and pathways associated with salt tolerance in transcriptome sequencing
To date, studies on the mechanism of salt tolerance in cotton are limited, with most investigations focusing on transcriptome sequencing of salt-responsive genes and pathways. Xu et al. (2013) [65] examined variations in gene expression of roots after exposing plants to 200 mM NaCl for 3, 12, 72, or 144 h and revealed that the enriched GO terms were related to cellular components, including “intrinsic to membrane”, “cytoplasmic vesicle”, and “membrane part”. Peng et al. (2014) [3] identified DEGs in cotton leaves at 4 and 24 h post-application of salt stress (200 mM NaCl) and revealed enriched GO terms such as “transcription factor activity”, “response to stress”, and “regulation of biological process”. Guo et al. (2015) [5] reported that the transcripts upregulated in both salt-tolerant and salt-sensitive cultivars under 150 mM NaCl treatment enriched GO terms related to “response to stimulus”, “transcription factor activity”, “peroxisome”, and “proline metabolic process”. Zhang et al. (2016) [22] identified DEGs in G. davidsonii roots and leaves at 12, 24, 48, 96, and 144 h post-salt stress (200 mM NaCl) and identified DEGs that enriched the salt-responsive GO categories, including “response to oxidative stress”, “responses to osmotic stress”, “ion transport”, “response to various hormone stimulus”, “response to sucrose stimulus”, and “metabolic processes”. Additionally, genes related to metabolic processes were involved in “carbohydrate”, “hormone”, “protein”, “lipid”, “amino”, “oxidation reduction”, and “organic substance”. Shu et al. (2017) [21] reported that the DEGs of NaCl/CK were associated with the GO terms of “oxidation-reduction”, “oligosaccharide metabolic process”, “photosynthesis”, “thylakoid”, and “oxidoreductase activity”.
In the current study, we screened out 6640, 3878, and 6462 DEGs in the high salt-tolerance upland accession Han682 at 3, 24, and 72 h post salt stress respectively. Among all the DEGs, 562 were continuously upregulated and 307 were continuously downregulated. The GO terms enriched with continuously upregulated DEGs were related to “metabolic process”, “oxidation-reduction process”, “carbohydrate metabolic process”, “photosynthesis”, “oligosaccharide metabolic process”, “thylakoid”, and “oxidoreductase activity”, which were also identified in previous studies [5, 21, 22, 74]. The GO terms enriched with continuously downregulated DEGs included “intrinsic to membrane”, “cytoplasmic vesicle” and “membrane part”, “response to oxidative stress”, and “transporter activity”, were agree with the findings of previous studies [3, 5, 22, 65]. The GO term related to “oxidation-reduction” was enriched with both continuously up- and downregulated DEGs, suggesting that oxidation-reduction systems elicit more complex responses to stress. In addition, the GO terms related to “membrane”, “transporter activity”, and “thylakoid” were enriched with significant DEGs in both roots and leaves, suggesting that some mechanisms associated with salt tolerance may be shared in different plant tissues and organs [3, 5, 21, 22, 65].
The pathways related to salt tolerance, including flavonoid biosynthesis, starch and sucrose metabolism, plant hormone signal transduction, starch and sucrose metabolism, phenylpropanoid biosynthesis, phenylalanine metabolism, and phagosome, were also identified in previous studies on salt tolerance mechanism of roots of cotton seedlings [5, 21, 22, 74, 75]. These pathways may play an essential role in plant adaptation to stress. However, the pathways of pyruvate metabolism, galactose metabolism, and arachidonic acid metabolism are specific to the seedling stage. The pyruvate metabolism pathway was also founded in leaves with salt stress at the seedling stage [75]. The germination of cotton seeds under salt condition require a suitable physiological state such as salt ion homeostasis, sufficient energy supply, and the capacity to remove harmful substances. These specific pathways such as pyruvate metabolism may be able to improve seed survival rate at the seedling stage during salt stress, although the underlying mechanism is unclear. The pathway of flavonoid biosynthesis had the highest rich factor and was very highly significant (P-value < 0.0001) in the pathway enrichment of continuously upregulated DEGs. Flavonoids, which are polyphenolic secondary metabolic compounds, play an important role in growth, development, reproduction, and stress defense [76, 77]. Petrussa et al. (2013) [77] have shown that flavonoids constitute a secondary ROS-eliminating system in plants under severe or prolonged stress conditions. Petrussa et al. (2013) [77] and Fini et al. (2011) [76] suggest that the key role of vacuoles in ROS homeostasis might be mediated by flavonoids. Ma et al. (2014) [78] have investigated the expression of genes that are involved in the flavonoid pathway and the accumulation of flavonoids related to drought tolerance in wheat. Flavonoids also play key roles in defense responses against biotic stress [79].
Similar MYB-BHLH-WDR (MBW) complexes and a family of small MYB proteins (R3-MYB) have been shown to play a key role in the regulation of flavonoid biosynthesis [47, 80]. This confirmed that TFs play an essential role in the biotic and abiotic stress responses in plants. In the current study, 19 TF families that are always related to salt stress response [3, 5, 22], were continually differentially expressed (Table 4). Among these, three (ARF, ERF, and C2H2) were downregulated, and seven (C3H, CO-like, HSF, LBD, M-type_MADS, NF-YA, and NF-YB) were upregulated. The top six TFs were bZIP, MYB (MYB-related), HD-ZIP, NAC, bHLH, and HSF.
Putative salt-responsive genes
Based on the biological process in UniProtKB and the functions, most of the 13 putative DEGs detected from association mapping and transcriptome sequencing were likely to respond to salt stress (Table 6 and Additional file 8: Table S6). Of these, eight (Gh_A01G1563, Gh_A02G1100, Gh_A07G0622, Gh_A07G0623, Gh_A12G0877, Gh_D07G0249, Gh_D07G0251, and Gh_D07G0500) were involved in “response to stress or defense response”, “signaling or response to signal factors”, or “transcription factors” and very likely related to the salt stress response (Tables 3 and 5). The eight salt-regulated DEGs were homologous to genes that are related to salinity tolerance such as Gh_A01G1563(CUT1) involved in fatty acid biosynthetic process, Gh_A02G1100 (WAK2) encoding wall-associated receptor kinase 2 [81, 82], and responding to ABA, Gh_A07G0622 (CLPB1) and Gh_A07G0623 (HSP18.2) encoding heat shock protein [3], Gh_A12G0877 (ERF2) encoding ethylene-responsive transcription factor [53, 56], Gh_D07G0249 (CHIT1) encoding chitotriosidase 1 [5, 73], Gh_D07G0251 (RR23) involved in cytokinin-activated signaling pathway [3, 22, 64], and Gh_D07G0500 (HVA22E) responding to ABA and stress [83, 84]. In addition, Gh_A03G1740 (BGAL3) is involved in carbohydrate metabolic process which is always the mainly enriched GO term in plant under salt stress [3, 5, 22]; Gh_D07G0250 is related to methylation which plays a significant role in salt tolerance in cotton [46, 85]; and Gh_D07G0263 (GAPN) is involved in glyceraldehyde-3-phosphate dehydrogenase (NADP+) (non-phosphorylating) activity which is associated with salt stress [86, 87].
Table 6.
The biological process of the 13 putative genes related to salt stress in UniProtKB
| Gene ID | Gene Name | Description | GO - Biological process in UniProtKB |
|---|---|---|---|
| Gh_A01G1563 | CUT1 | 3-ketoacyl-CoA synthase 6 | Fatty acid biosynthetic process, response to cold, response to light stimulus, unidimensional cell growth, wax biosynthetic process |
| Gh_A02G1100 | WAK2 | Wall-associated receptor kinase 2 | Cell surface receptor signaling pathway, cellular water homeostasis, oligosaccharide metabolic process, response to salicylic acid, unidimensional cell growth |
| Gh_A03G1740 | BGAL3 | Beta-galactosidase 3 | Carbohydrate metabolic process |
| Gh_A07G0622 | CLPB1 | Chaperone protein ClpB1 | Positive regulation of translation, protein metabolic process, protein unfolding, response to heat, response to high light intensity,response to hydrogen peroxide |
| Gh_A07G0623 | HSP18.2 | 18.2 kDa class I heat shock protein | Stress response |
| Gh_A12G0877 | ERF2 | Ethylene-responsive transcription factor 2 | Cell division, ethylene-activated signaling pathway, induced systemic resistance, jasmonic acid mediated signaling pathway, phloem or xylem histogenesis, positive regulation of transcription, DNA-templated, response to chitin, transcription, DNA-templated |
| Gh_D07G0243 |
DDB _G0268948 |
Putative methyltransferase DDB_G0268948 | Methylation |
| Gh_D07G0249 | CHIT1 | Chitotriosidase-1 | Chitin catabolic process, immune response, neutrophil degranulation, polysaccharide catabolic process, polysaccharide digestion, response to bacterium |
| Gh_D07G0250 | abhd17c | Alpha/beta hydrolase domain-containing protein 17C | Palmitoyl-(protein) hydrolase activity |
| Gh_D07G0251 | RR23 | Two-component response regulator ORR23 | Cytokinin-activated signaling pathway, phosphorelay signal transduction system, transcription, DNA-templated |
| Gh_D07G0258 | At4g30993 | Calcineurin-like metallo-phosphoesterase superfamily protein | NA |
| Gh_D07G0263 | GAPN | NADP-dependent glyceraldehyde-3-phosphate dehydrogenase | Glyceraldehyde-3-phosphate dehydrogenase (NADP+) (non-phosphorylating) activity |
| Gh_D07G0500 | HVA22E | HVA22-like protein e | Flower development, hyperosmotic salinity response, negative regulation of autophagy, pollen development, response to abscisic acid, response to cold, response to water deprivation |
The qRT-PCR results also indicated the expression of these putative genes, Gh_A01G1563, Gh_A07G0622, Gh_D07G0243, Gh_D07G0251, Gh_D07G0623, Gh_D07G0250, Gh_D07G0258, Gh_D07G0500, Gh_A07G0623, Gh_A03G1740, Gh_A02G1100, and Gh_D07G0249, were regulated by salt stress at the germination stage. These results provide candidate genes for further research on salt tolerance mechanism of cotton. The specific functions and molecular regulation of these genes in salt tolerance of cotton need to be further studied [88].
Conclusions
In the current study, the salt tolerance of 196 accessions was comprehensively evaluated with the comprehensive D values of 10 salt-relevant traits. Based on this, a GWAS for salt tolerance was conducted. In GWAS, 98 candidate genes were obtained in the 13 candidate QTLs from 17 significant SNPs in at least two environments. Functional annotation revealed that 35 of the 98 candidate genes were involved in salt tolerance responses. Furthermore, transcriptome sequencing of a high salt resistant accession, Han682, at three time points after salt or control treatment were conducted to verify the results of GWAS. By combining the results of GWAS and RNA-seq, 13 putative genes were identified and the expressions of 12 of these genes were verified using qRT-PCR. These results will enhance our understanding of the molecular-genetic regulation of salt stress tolerance in cotton and aid in the the modification of salinity tolerance related traits.
Methods
Plant materials and SNP markers
A panel of 196 diverse upland cotton accessions, including 169 genotypes from five cotton-growing regions across China and 27 from other countries, were employed in association mapping of salt tolerance at the germination stage. All these 196 accessions were inbred for at least 3 years before use in this study. The detailed information on this panel is described in Additional file 12: Table S9 [89].
The 41,815 polymorphic SNP markers screened from 77,774 SNPs (CottonSNP80K, [90]) were applied in population structure analysis and GWAS. The 196 genotypes were divided into two subpopulations and confirmed using four methods (the UPGMA (unweighted pair group method with arithmetic means) phylogeny, PCA, STRUCTURE, and kinship matrix) (Additional file 13: Figure S4) [89]. The summary of SNPs, polymorphic information content (PIC), gene diversity, and LD decay were also calculated as described by Yuan et al. (2018) (Additional file 7: Table S5) [89].
Salt tolerance assessment at the germination stage
Salt tolerance evaluation at the germination stage, which is the most sensitive development stage to salt stress [91], was performed in a triplex randomized block experiment in 2017, with seeds from three calendar years (2014–2016). Cotton seeds delinted by sulfuric acid were surface-sterilized with 0.1% HgCl for 15 min, then uniform-sized seeds were selected for the germination test, which was performed in an incubator at 28 ± 1 °C and 80% ± 2% relative humidity. The seeds (100 seeds) were planted in a germination box (13 × 19 × 12 cm) containing 750 g dry sand and covered evenly with 250 g dry sand above the seeds, then 250 mL 200 mmol/L NaCl solution or distilled water (as control) was added. Every replicate (treatment or control) of each genotype consisted of 200 seeds in two germination boxes.
The number of germinated seeds was recorded from the 3rd day to 10th day after sowing. Then GR, GP, GI, and VI were calculated using the following formulas: GR = and GP = , where TS is the total number of seeds in each replicate (200 were used) and G3 or G7 is the number of total germinated seeds in two germination boxes from the first day to the third or seventh day after sowing; and GI = , VI = GI × S, where t is the number of days after planting, Gt is the number of germinated seeds at the tth day after sowing, Dt is the number of days after planting corresponding to Gt, and S is the fresh weight of a single plant seedling.
In addition to germination characteristics, several other salt tolerance-related traits were also investigated. On the 10th day, 10 plants from each germination box were randomly selected for determination of PH, SFM, SDM, RL, RFM, and RDM. The average value of two germination boxes of each replicate was used for further data analysis.
The STI for each trait was calculated using the following formula: , where Ynon-salt stress is the phenotypic value without salt stress, Ysalt stress is the phenotypic values under salt stress, and non-salt stress is the average phenotypic value without salt stress [92, 93].
The BLUPed STI were calculated with the nine STIs (three replications × three years) of each trait using the R lmer4 package. Then, four sets of STIs (STIs in three years and the BLUPed STIs) were used in the subsequent calculations. The weights of each principal component factor were calculated as Wi = Ri/ ∑ Ri (i = 1, 2, …, n), and Ri means the contribution rate of the ith principal component factor.
Subordinate function analysis was performed as follow: U(Xi) = (Xi − Xmin)/(X max − Xmin) (i = 1, 2, …, n), where Xi is the value of the ith principal component factor, and Xmin and Xmax indicate the minimum and maximum value of the ith principal component factor [10, 11].
The comprehensive evaluation values (D) of salt tolerance were calculated as D = ∑ (Ui × Wi) (i = 1, 2, …, n) [10, 11]. Data processing was conducted with Excel (Office 365) and SPSS (version 23.0, RRID:SCR_002865).
Association mapping
Marker-trait association was performed using the software TASSEL version 5.2.40 (RRID:SCR_012837), and the threshold value was set at p < 0.001 for declaring a significant marker-trait association. For each chromosome, the LD decay distance was regarded as the confidence interval for the candidate QTL detected on it. With the G. hirsutum AD1 genome NAU-NBI assembly v1.1 [39], candidate genes were gained for each QTL.
RNA-seq and DEG
Based on the comprehensive evaluation of salt tolerance of the 196 genotypes, the high salt-resistant accession Han682 (namely H1) was selected for RNA-seq analysis under salt stress. After being surface-sterilized with 0.1% HgCl, seeds were sown in wet sand in germination boxes. When the radicle had grown to 2–3 mm long, seeds with uniform radicles were selected and planted in sand beds with 0.3% (weight percentage, approximately 200 mmol/L) or 0% NaCl (the control) in other germination boxes. Root tissues were harvested at 3, 24, and 72 h after planting and immediately stored in liquid nitrogen. The collected root samples were designated as S3, S24, and S72 for the salt-treated plants, and H3, H24, and H72 for the control plants, respectively. Each treatment was repeated twice. RNA-seq libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s recommendations. The library preparations were sequenced on an Illumina HiSeq platform in Novogene Bioinformatics Institute, Beijing, China.
An index of the reference genome (Gossypium hirsutum AD1 genome NAU-NBI assembly v1.1) [39] was built using Bowtie v2.2.3 [94], and clean reads were aligned to the reference genome using TopHat v2.0.12 [95]. HTSeq v0.6.1 was employed to estimate the gene expression levels using fragments per kilobase of exon per million mapped fragments [96]. DEGs in the two conditions were identified using the DESeq R package (1.18.0) based on an adjusted P-value < 0.05 [97].
GO and KEGG enrichment
The GOseq R package was employed to implement the GO enrichment analysis of DEGs [98]. GO terms with P-value corrected by FDR < 0.05 were regarded as significantly enriched. KOBAS software was used to test the statistical enrichment of DEGs in the KEGG pathways [99].
qRT-PCR verification
qRT-PCR was performed to validate the expression of DEGs. Total RNAs were extracted using an OmniPlant RNA kit (DNase I). The isolated RNA (1500 ng) was first reverse-transcribed to generate cDNAs using a HiFiScript cDNA synthesis kit. The cDNAs were then used as template for qRT-PCR on an Applied Biosystems® 7500 Real-Time PCR System using an UltraSYBR Mixture. In this process, all kits and mixtures were purchased from Beijing CoWin Bioscience Co. The specific primers for the target genes were designed using Primer-BLAST in NCBI and GSP (a web-based platform for designing genome-specific primers in polyploids) [100]. The GhUBQ7 was used as intro-reference gene [101]. All the specific primers (Additional file 14: Table S10) were synthesized by Sangon Biotech Shanghai Co., Ltd. (Shanghai, China).
Additional files
Table S1. Descriptive statistics of the salt tolerance index of 10 salt tolerance-related traits. (XLSX 11 kb)
Table S2. Correlation analysis among 10 traits related to salt tolerance. (XLSX 9 kb)
Figure S1. Screen plot of eigenvalues with component number for STIs in BLUP and 3 years. (DOCX 190 kb)
Table S3. Characteristics, contribution rates, and weights of two principal components at the germination stage. (XLSX 9 kb)
Figure S2. PCA plots for the STIs of 10 salt tolerance traits in BLUP and 3 years. a for 2014, b for 2015, c for 2016, and d for BLUP. (DOCX 79 kb)
Table S4. Comprehensive evaluation values (D) and K-mean cluster analysis of salt tolerance. (XLSX 20 kb)
Table S5. Summary of SNPs, PIC, gene diversity, and LD decay in Yuan et al. (2018) [89]. (XLSX 11 kb)
Table S6. Annotation of genes located within candidate QTL regions. (XLSX 25 kb)
Table S7. Summary of sequencing data quality. (XLSX 11 kb)
Figure S3. Validation of the 20 random genes in transcriptome sequencing with qRT-PCR. (XLSX 11 kb)
Table S8. Candidate genes near significant SNPs. (DOCX 7557 kb)
Table S9. Information for 196 upland cotton accessions [89]. (XLSX 23 kb)
Figure S4. Population structure of the 196 accessions in Yuan et al. (2018) [89]. (A) UPGMA tree based on Nei’s genetic distances. (B) Principal component analysis of 196 accessions based on genotype. (C) Population structure of the 196 accessions based on STRUCTURE when K = 2. (D) Kinship for this panel. (DOCX 693 kb)
Table S10. All the specific primers for the selected genes and the intro-reference gene UBQ7. (XLSX 12 kb)
Acknowledgments
We would like to thank the Germplasm Repository of Institute of Cotton Research, Chinese Academy of Agricultural Sciences (Anyang, Henan province, China) for providing the 27 exotic cotton accessions. We thank LetPub (www.letpub.com) for providing linguistic assistance during the preparation of this manuscript.
Abbreviations
- ABA
Abscisic acid
- BLUP
Best linear unbiased prediction
- BP
Biological process
- CC
Cellular component
- CDS
Coding sequences
- CV
Coefficient variation
- DEGs
Differentially expressed genes
- GA
Gibberellic acid
- GI
Germination index
- GO
Gene Ontology
- GP
Germination potential
- GR
Germination rate
- GWAS
Genome-wide association study
- JA
Jasmonic acid
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KMO
Kaiser-Mayer-Olkin
- LD
Linkage disequilibrium
- lncRNAs
Long non-coding RNAs
- MF
Molecular function
- MLM
Mixed linear model
- PCA
Factor analysis of principal component analysis
- PH
Plant height
- PIC
Polymorphic information content
Quantile quantile
- QTL
Qualitative-trait locus
- QTLs
Qualitative-trait loci
- RDM
Root dry mass
- RFM
Root fresh mass
- RL
Root length
- RNA-seq
RNA sequencing
- SA
Salicylic acid
- SDM
Shoot dry mass
- SFA
Subordinate function analysis
- SFM
Shoot fresh mass
- SNPs
Single-nucleotide polymorphisms
- STI
Salt tolerance index
- TF
Transcription factor
- UPGMA
Unweighted pair group method with arithmetic means
- VI
vVigor index
Authors’ contributions
XLS and XZS designed the experiments. YY and XLS wrote the manuscript. HX, WZ, JX, LM, LW, WF, JT, HW, HZ, QW, and GZ helped in collecting phenotype data. YY and HX analyzed the results. YY, and HX performed most of the experiments and contributed equally to this work. All authors have read and approved the final manuscript.
Funding
This research was financially supported by the National Key Research and Development Program (2018YFD0100303) and the Major Projects for Transgenic Breeding of China (2017ZX08005–004-006), the System of Modern Agriculture Industrial Technology (SDAIT-03-03/05), the Natural Science Foundation (ZR2017MC057) and the Agricultural Seed Project (cotton variety development, 2014–2017) of Shandong Province, and the Innovation Projects for improvement of Saline Land (2015) of Shandong Agricultural University. We thank all the foundation of economic support. The funding organizations provided the financial support to the research projects, but were not involved in the design of the study, data collection, analysis of the data, or the writing of the manuscript.
Availability of data and materials
Most of the data pertaining to the present study has been included in the Tables/Figures of the manuscript. The authors are pleased to share the rest of the raw data upon request.
Ethics approval and consent to participate
The collection of plant specimens used in our study complied with institutional and national guidelines. Field studies were conducted in accordance with local legislation. Ethical approval was not applicable for this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yanchao Yuan and Huixian Xing contributed equally to this work.
Contributor Information
Xianliang Song, Phone: +86 538 8242309, Email: songxl999@163.com.
Xue-Zhen Sun, Phone: +86 538 8242487, Email: sunxz@sdau.edu.cn.
References
- 1.Deinlein U, Stephan AB, Horie T, Luo W, Xu G, Schroeder JI. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014;19(6):371–379. doi: 10.1016/j.tplants.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munns R. Genes and salt tolerance: bringing them together. New Phytol. 2005;167(3):645–663. doi: 10.1111/j.1469-8137.2005.01487.x. [DOI] [PubMed] [Google Scholar]
- 3.Peng Z, He S, Gong W, Sun J, Pan Z, Xu F, Lu Y, Du X. Comprehensive analysis of differentially expressed genes and transcriptional regulation induced by salt stress in two contrasting cotton genotypes. BMC Genomics. 2014;15(1):760. doi: 10.1186/1471-2164-15-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saeed M, Dahab AHA, Guo WZ, Zhang TZ. Acascade of recently discovered molecular mechanisms involved in abiotic stress tolerance of plants. OMICS. 2012;16:188–199. doi: 10.1089/omi.2011.0109. [DOI] [PubMed] [Google Scholar]
- 5.Guo J, Shi G, Guo X, Zhang L, Xu W, Wang Y, Su Z, Hua J. Transcriptome analysis reveals that distinct metabolic pathways operate in salt-tolerant and salt-sensitive upland cotton varieties subjected to salinity stress. Plant Sci. 2015;238:33–45. doi: 10.1016/j.plantsci.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y. Analyzing blends of herbivore-induced volatile organic compounds with factor analysis: revisiting “cotton plant, Gossypium hirsutum L., defense in response to nitrogen fertilization”. J Econ Entomol. 2013;106:1053–1057. doi: 10.1603/EC12178. [DOI] [PubMed] [Google Scholar]
- 7.Du L, Cai C, Wu S, Zhang F, Hou S, Guo W. Evaluation and exploration of favorable QTL alleles for salt stress related traits in cotton cultivars (G. hirsutum L.) PloS One. 2016;11(3):e0151076. doi: 10.1371/journal.pone.0151076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han RH, Xinshi LU, Gao GJ, Yang XJ. Analysis of the principal components and the subordinate function of alfalfa drought resistance. Acta Agrestia Sinica. 2006;14(2):142–146. [Google Scholar]
- 9.Mancuso F, Horan WP, Kern RS, Green MF. Social cognition in psychosis: multidimensional structure, clinical correlates, and relationship with functional outcome. Schizophr Res. 2011;125(2–3):143–151. doi: 10.1016/j.schres.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su Y, Wang Z, Xu L, Peng Q, Liu F, Li Z, Que Y. Early selection for smut resistance in sugarcane using pathogen proliferation and changes in physiological and biochemical indices. Front Plant Sci. 2016;7:1133. doi: 10.3389/fpls.2016.01133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao Xue, Jiang Fangling, Wang Xu, Zang Yuwen, Wu Zhen. Comprehensive evaluation and screening for chilling-tolerance in tomato lines at the seedling stage. Euphytica. 2015;205(2):569–584. doi: 10.1007/s10681-015-1433-0. [DOI] [Google Scholar]
- 12.Zhang BQ, Yang LT, Li YR. Physiological and biochemical characteristics related to cold resistance in sugarcane. Sugar Tech. 2015;17(1):49–58. doi: 10.1007/s12355-014-0340-3. [DOI] [Google Scholar]
- 13.Mei Y, Ning Y, Cao YJ, Shen J, Pang X, Cui L, Cheng CY, Chen JF. Evaluation of cucumber's chilling tolerance at germination and seedling stages. J Appl Ecol. 2013;24(7):1914–1922. [PubMed] [Google Scholar]
- 14.Wang SG, Wang ZL, Wang P, et al. Evaluation of wheat freezing resistance based on the responses of the physiological indices to low temperature stress. Acta Ecol Sin. 2011;31(4):1064–1072. [Google Scholar]
- 15.Wu H, Hou L, Zhou Y, Fan Z, Shi J, Zhang J. Analysis and evaluation indicator selection of chilling tolerance of different cotton genotypes. Agric Sci Technol. 2012;13(11):2338. [Google Scholar]
- 16.Saeed M, Guo WZ, Zhang TZ. Association mapping for salinity tolerance in cotton (Gossypium hirsutum L.) germplasm from US and diverse regions of China. Aust J Crop Sci. 2014;8(3):338–346. [Google Scholar]
- 17.Jia YH, Sun JL, Wang XW, Zhou ZL, Pan ZE, He SP, Pang BY, Wang LR, Du XM. Molecular diversity and association analysis of drought and salt tolerance in Gossypium hirsutum L. germplasm. J Integr Agric. 2014;13(9):1845–1853. doi: 10.1016/S2095-3119(13)60668-1. [DOI] [Google Scholar]
- 18.Diouf Latyr, Pan Zhaoe, He Shou-Pu, Gong Wen-Fang, Jia Yin, Magwanga Richard, Romy Kimbembe, or Rashid Harun, Kirungu Joy, Du Xiongming. High-Density Linkage Map Construction and Mapping of Salt-Tolerant QTLs at Seedling Stage in Upland Cotton Using Genotyping by Sequencing (GBS) International Journal of Molecular Sciences. 2017;18(12):2622. doi: 10.3390/ijms18122622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oluoch G, Zheng J, Wang X, et al. QTL mapping for salt tolerance at seedling stage in the interspecific cross of Gossypium tomentosum with Gossypium hirsutum. Euphytica. 2016;209(1):223–235. doi: 10.1007/s10681-016-1674-6. [DOI] [Google Scholar]
- 20.Sun Z, Li H, Zhang Y, Li Z, Ke H, Wu L, Zhang G, Wang X, Ma Z. Identification of SNPs and candidate genes associated with salt tolerance at the seedling stage in cotton (Gossypium hirsutum L.) Front Plant Sci. 2018;9:1011. doi: 10.3389/fpls.2018.01011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shu H. M., Guo S. Q., Gong Y. Y., Jiang L., Zhu J. W., Ni W. C. RNA-seq analysis reveals a key role of brassinolide-regulated pathways in NaCl-stressed cotton. Biologia Plantarum. 2017;61(4):667–674. doi: 10.1007/s10535-017-0736-5. [DOI] [Google Scholar]
- 22.Zhang F, Zhu G, Du L, Shang X, Cheng C, Yang B, Hu Y, Cai C, Guo W. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci Rep. 2016;6:20582. doi: 10.1038/srep20582. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Zhu G, Li W, Zhang F, Guo W. RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii. BMC Genomics. 2018;19(1):73. doi: 10.1186/s12864-018-4449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng F, Zhang X, Wang W, Yuan R, Shen F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018;18(1):23. doi: 10.1186/s12870-018-1238-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu CA, Yang GD, Meng QW, Zheng CC. The cotton GhNHX1 gene encoding a novel putative tonoplast Na+/H+ antiporter plays an important role in salt stress. Plant Cell Physiol. 2004;45(5):600–607. doi: 10.1093/pcp/pch071. [DOI] [PubMed] [Google Scholar]
- 26.Xue T, Li X, Zhu W, Wu C, Yang G, Zheng C. Cotton metallothionein GhMT3a, a reactive oxygen species scavenger, increased tolerance against abiotic stress in transgenic tobacco and yeast. J Exp Bot. 2008;60(1):339–349. doi: 10.1093/jxb/ern291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Champion A, Hébrard E, Parra B, Bournaud C, Marmey P, Tranchant C, Nicole M. Molecular diversity and gene expression of cotton ERF transcription factors reveal that group IXa members are responsive to jasmonate, ethylene and xanthomonas. Mol Plant Pathol. 2009;10(4):471–485. doi: 10.1111/j.1364-3703.2009.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin LG, Li H, Liu JY. Molecular characterization of three ethylene responsive element binding factor genes from cotton. J Integr Plant Biol. 2010;52(5):485–495. doi: 10.1111/j.1744-7909.2010.00914.x. [DOI] [PubMed] [Google Scholar]
- 29.Gao SQ, Chen M, Xia LQ, Xiu HJ, Xu ZS, Li LC, Zhao CP, Cheng XG, Ma YZ. A cotton (Gossypium hirsutum) DRE-binding transcription factor gene, GhDREB, confers enhanced tolerance to drought, high salt, and freezing stresses in transgenic wheat. Plant Cell Rep. 2009;28(2):301–311. doi: 10.1007/s00299-008-0623-9. [DOI] [PubMed] [Google Scholar]
- 30.Guo YH, Yu YP, Wang D, Wu CA, Yang GD, Huang JG, Zheng CC. GhZFP1, a novel CCCH-type zinc finger protein from cotton, enhances salt stress tolerance and fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5. New Phytol. 2009;183(1):62–75. doi: 10.1111/j.1469-8137.2009.02838.x. [DOI] [PubMed] [Google Scholar]
- 31.Meng C, Cai C, Zhang T, Guo W. Characterization of six novel NAC genes and their responses to abiotic stresses in Gossypium hirsutum L. Plant Sci. 2009;176(3):352–359. doi: 10.1016/j.plantsci.2008.12.003. [DOI] [Google Scholar]
- 32.Zhang L, Xi D, Li S, Gao Z, Zhao S, Shi J, Wu C, Guo X. A cotton group C MAP kinase gene, GhMPK2, positively regulates salt and drought tolerance in tobacco. Plant Mol Biol. 2011;77(1–2):17–31. doi: 10.1007/s11103-011-9788-7. [DOI] [PubMed] [Google Scholar]
- 33.Lu W, Chu X, Li Y, Wang C, Guo X. Cotton GhMKK1 induces the tolerance of salt and drought stress, and mediates defence responses to pathogen infection in transgenic Nicotiana benthamiana. PLoS One. 2013;8(7):e68503. doi: 10.1371/journal.pone.0068503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo X, Wu J, Li Y, Nan Z, Guo X, Wang Y, Zhang A, Wang Z, Xia G, Tian Y. Synergistic effects of GhSOD1 and GhCAT1 overexpression in cotton chloroplasts on enhancing tolerance to methyl viologen and salt stresses. PLoS One. 2013;8(1):e54002. doi: 10.1371/journal.pone.0054002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan H, Jia H, Chen X, Hao L, An H, Guo X. The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production. Plant Cell Physiol. 2014;55(12):2060–2076. doi: 10.1093/pcp/pcu133. [DOI] [PubMed] [Google Scholar]
- 36.Zhang F, Li S, Yang S, Wang L, Guo W. Overexpression of a cotton annexin gene, GhAnn1, enhances drought and salt stress tolerance in transgenic cotton. Plant Mol Biol. 2015;87(1–2):47–67. doi: 10.1007/s11103-014-0260-3. [DOI] [PubMed] [Google Scholar]
- 37.de los Campos G, Vazquez AI, Fernando R, Klimentidis YC, Sorensen D. Prediction of complex human traits using the genomic best linear unbiased predictor. PLoS Genet. 2013;9(7):e1003608. doi: 10.1371/journal.pgen.1003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang C, Nie X, Shen C, You C, Li W, Zhao W, Zhang X, Lin Z. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnol J. 2017;15(11):1374–1386. doi: 10.1111/pbi.12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang T, Hu Y, Jiang W, et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol. 2015;33(5):531–537. doi: 10.1038/nbt.3207. [DOI] [PubMed] [Google Scholar]
- 40.Singh KB, Foley RC, Oñate-Sánchez L. Transcription factors in plant defense and stress responses. Curr Opin Plant Biol. 2002;5(5):430–436. doi: 10.1016/S1369-5266(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 41.Wang N, Qi H, Su G, Yang J, Zhou H, Xu Q, Huang Q, Yan G. Genotypic variations in ion homeostasis, photochemical efficiency and antioxidant capacity adjustment to salinity in cotton (Gossypium hirsutum L.) Soil Sci Plant Nutr. 2016;62(3):240–246. doi: 10.1080/00380768.2016.1172022. [DOI] [Google Scholar]
- 42.Higbie Sarah M., Wang Fei, Stewart J. McD., Sterling Tracy M., Lindemann William C., Hughs E., Zhang Jinfa. Physiological Response to Salt (NaCl) Stress in Selected Cultivated Tetraploid Cottons. International Journal of Agronomy. 2010;2010:1–12. doi: 10.1155/2010/643475. [DOI] [Google Scholar]
- 43.Abdelraheem A, Fang DD, Zhang J. Quantitative trait locus mapping of drought and salt tolerance in an introgressed recombinant inbred line population of upland cotton under the greenhouse and field conditions. Euphytica. 2018;214(1):8. doi: 10.1007/s10681-017-2095-x. [DOI] [Google Scholar]
- 44.Dilnur Tussipkan, Peng Zhen, Pan Zhaoe, Palanga Koffi, Jia Yinhua, Gong Wenfang, Du Xiongming. Association Analysis of Salt Tolerance in Asiatic cotton (Gossypium arboretum) with SNP Markers. International Journal of Molecular Sciences. 2019;20(9):2168. doi: 10.3390/ijms20092168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdelraheem A, Liu F, Song M, Zhang JF. A meta-analysis of quantitative trait loci for abiotic and biotic stress resistance in tetraploid cotton. Mol Gen Genomics. 2017;292(6):1221–1235. doi: 10.1007/s00438-017-1342-0. [DOI] [PubMed] [Google Scholar]
- 46.He Q, Jones DC, Li W, Xie F, Ma J, Sun R, Wang Q, Zhu S, Zhang B. Genome-wide identification of R2R3-MYB genes and expression analyses during abiotic stress in Gossypium raimondii. Sci Rep. 2016;6:22980. doi: 10.1038/srep22980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu W, Dubos C, Lepiniec L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015;20(3):176–185. doi: 10.1016/j.tplants.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Xu FC, Liu HL, Xu YY, Zhao JR, Guo YW, Long L, Gao W, Song CP. Heterogeneous expression of the cotton R2R3-MYB transcription factor GbMYB60 increases salt sensitivity in transgenic Arabidopsis. Plant Cell Tissue Organ Cult. 2018;133(1):15–25. doi: 10.1007/s11240-017-1357-6. [DOI] [Google Scholar]
- 49.Liang C, Meng Z, Meng Z, Malik W, Yan R, Lwin KM, Lin F, Wang Y, Sun G, Zhou T, Zhu T, Li J, Jin S, Guo S, Zhang R. GhABF2, a bZIP transcription factor, confers drought and salinity tolerance in cotton (Gossypium hirsutum L.) Sci Rep. 2016;6:35040. doi: 10.1038/srep35040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang C, Lu G, Hao Y, Guo H, Guo Y, Zhao J, Cheng H. ABP9, a maize bZIP transcription factor, enhances tolerance to salt and drought in transgenic cotton. Planta. 2017;246(3):453–469. doi: 10.1007/s00425-017-2704-x. [DOI] [PubMed] [Google Scholar]
- 51.Zou M, Guan Y, Ren H, Zhang F, Chen F. A bZIP transcription factor, OsABI5, is involved in rice fertility and stress tolerance. Plant Mol Biol. 2008;66(6):675–683. doi: 10.1007/s11103-008-9298-4. [DOI] [PubMed] [Google Scholar]
- 52.Ma L, Hu L, Fan J, Amombo E, Khaldun ABM, Zheng Y, Chen L. Cotton GhERF38 gene is involved in plant response to salt/drought and ABA. Ecotoxicology. 2017;26(6):841–854. doi: 10.1007/s10646-017-1815-2. [DOI] [PubMed] [Google Scholar]
- 53.Guo W, Jin L, Miao Y, He X, Hu Q, Guo K, Zhu L, Zhang X. An ethylene response-related factor, GbERF1-like, from Gossypium barbadense improves resistance to Verticillium dahliae via activating lignin synthesis. Plant Mol Biol. 2016;91(3):305–318. doi: 10.1007/s11103-016-0467-6. [DOI] [PubMed] [Google Scholar]
- 54.Min JH, Chung JS, Lee KH, Kim CS. The b-box zinc finger family constans-like 4 transcription factor, AtCOL4, positively regulates abiotic stress tolerance through an abscisic acid-dependent manner in Arabidopsis. J Integr Plant Biol. 2015;57(3):313–324. doi: 10.1111/jipb.12246. [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, Dou L, Pang C, Song M, Wei H, Fan S, Wang C, Yu S. Genomic organization, differential expression, and functional analysis of the SPL gene family in Gossypium hirsutum. Mol Gen Genomics. 2015;290(1):115–126. doi: 10.1007/s00438-014-0901-x. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen KH, Ha CV, Nishiyama R, et al. Arabidopsis type B cytokinin response regulators ARR1, ARR10, and ARR12 negatively regulate plant responses to drought. Proc Natl Acad Sci. 2016;113(11):3090–3095. doi: 10.1073/pnas.1600399113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hjellström M. Drought stress signal transduction by the HD-ZIP transcription factors ATHB6 and ATHB7. J Appl Phys. 2002;92(6):3124–3129. doi: 10.1063/1.1500786. [DOI] [Google Scholar]
- 58.Söderman E, Hjellström M, Fahleson J, Engström P. The HD-zip gene ATHB6 in Arabidopsis is expressed in developing leaves, roots and carpels and up-regulated by water deficit conditions. Plant Mol Biol. 1999;40(6):1073–1083. doi: 10.1023/A:1006267013170. [DOI] [PubMed] [Google Scholar]
- 59.Zhang S, Haider I, Kohlen W, Jiang L, Bouwmeester H, Meijer AH, Schluepmann H, Liu CM, Ouwerkerk PB. Function of the HD-zip I gene Oshox22 in ABA-mediated drought and salt tolerances in rice. Plant Mol Biol. 2012;80(6):571–585. doi: 10.1007/s11103-012-9967-1. [DOI] [PubMed] [Google Scholar]
- 60.Lokko Y, Anderson JV, Rudd S, et al. Characterization of an 18,166 EST dataset for cassava (Manihot esculenta Crantz) enriched for drought-responsive genes. Plant Cell Rep. 2007;26(9):1605–1618. doi: 10.1007/s00299-007-0378-8. [DOI] [PubMed] [Google Scholar]
- 61.Deng X, Phillips J, Meijer AH, Salamini F, Bartels D. Characterization of five novel dehydration-responsive homeodomain leucine zipper genes from the resurrection plant Craterostigma plantagineum. Plant Mol Biol. 2002;49(6):601–610. doi: 10.1023/A:1015501205303. [DOI] [PubMed] [Google Scholar]
- 62.Ni Y, Wang X, Li D, Wu Y, Xu W, Li X. Novel cotton homeobox gene and its expression profiling in root development and in response to stresses and phytohormones. Acta Biochim Biophys Sin. 2008;40(1):78–84. doi: 10.1111/j.1745-7270.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 63.Peleg Z, Blumwald E. Hormone balance and abiotic stress tolerance in crop plants. Curr Opin Plant Biol. 2011;14(3):290–295. doi: 10.1016/j.pbi.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 64.Yao D, Zhang X, Zhao X, Liu C, Wang C, Zhang Z, Zhang C, Wei Q, Wang Q, Yan H, Li F, Su Z. Transcriptome analysis reveals salt-stress-regulated biological processes and key pathways in roots of cotton (Gossypium hirsutum L.) Genomics. 2011;98(1):47–55. doi: 10.1016/j.ygeno.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 65.Xu P, Liu Z, Fan X, Gao J, Zhang X, Zhang X, Shen X. De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene. 2013;525(1):26–34. doi: 10.1016/j.gene.2013.04.066. [DOI] [PubMed] [Google Scholar]
- 66.Gao W, Xu FC, Guo DD, et al. Calcium-dependent protein kinases in cotton: insights into early plant responses to salt stress. BMC Plant Biol. 2018;18(1):15. doi: 10.1186/s12870-018-1230-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilkins KA, Matthus E, Swarbreck SM, Davies JM. Calcium-mediated abiotic stress signaling in roots. Front Plant Sci. 2016;7:1296. doi: 10.3389/fpls.2016.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang N, Wang X, Shi J, Liu X, Xu Q, Zhou H, Song M, Yan G. Mepiquat chloride-priming induced salt tolerance during seed germination of cotton (Gossypium hirsutum L.) through regulating water transport and K+/Na+ homeostasis. Environ Exp Bot. 2019;159:168–178. doi: 10.1016/j.envexpbot.2018.12.024. [DOI] [Google Scholar]
- 69.Peng Z, He S, Sun J, Pan Z, Gong W, Lu Y, Du X. Na+ compartmentalization related to salinity stress tolerance in upland cotton (Gossypium hirsutum) seedlings. Sci Rep. 2016;6:34548. doi: 10.1038/srep34548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen Y, Shen L, Shen Z, Jing W, Ge H, Zhao J, Zhang W. The potassium transporter OsHAK21 functions in the maintenance of ion homeostasis and tolerance to salt stress in rice. Plant Cell Environ. 2015;38(12):2766–2779. doi: 10.1111/pce.12586. [DOI] [PubMed] [Google Scholar]
- 71.Zhang X, Zhen J, Li Z, Kang D, Yang Y, Kong J, Hua J. Expression profile of early responsive genes under salt stress in upland cotton (Gossypium hirsutum L.) Plant Mol Biol Report. 2011;29(3):626–637. doi: 10.1007/s11105-010-0269-y. [DOI] [Google Scholar]
- 72.de las Mercedes Dana M, Pintor-Toro JA, Cubero B. Transgenic tobacco plants overexpressing chitinases of fungal origin show enhanced resistance to biotic and abiotic stress agents. Plant Physiol. 2006;142(2):722–730. doi: 10.1104/pp.106.086140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Békésiová B, Hraška Š, Libantová J, Moravčíková J, Matušíková I. Heavy-metal stress induced accumulation chitinase isoforms in plants. Mol Biol Rep. 2008;35(4):579–588. doi: 10.1007/s11033-007-9127-x. [DOI] [PubMed] [Google Scholar]
- 74.Shi G, Guo X, Guo J, Liu L, Hua J. Analyzing serial cDNA libraries revealed reactive oxygen species and gibberellins signaling pathways in the salt response of upland cotton (Gossypium hirsutum L.) Plant Cell Rep. 2015;34(6):1005–1023. doi: 10.1007/s00299-015-1761-5. [DOI] [PubMed] [Google Scholar]
- 75.Chen T, Zhang L, Shang H, et al. iTRAQ-based quantitative proteomic analysis of cotton roots and leaves reveals pathways associated with salt stress. PLoS One. 2016;11(2):e0148487. doi: 10.1371/journal.pone.0148487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fini A, Brunetti C, Di Ferdinando M, Ferrini F, Tattini M. Stress-induced flavonoid biosynthesis and the antioxidant machinery of plants. Plant Signal Behav. 2011;6(5):709–711. doi: 10.4161/psb.6.5.15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petrussa E, Braidot E, Zancani M, Peresson C, Bertolini A, Patui S, Vianello A. Plant flavonoids-biosynthesis, transport and involvement in stress responses. Int J Mol Sci. 2013;14(7):14950–14973. doi: 10.3390/ijms140714950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma D, Sun D, Wang C, Li Y, Guo T. Expression of flavonoid biosynthesis genes and accumulation of flavonoid in wheat leaves in response to drought stress. Plant Physiol Biochem. 2014;80:60–66. doi: 10.1016/j.plaphy.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 79.Koskimäki JJ, Hokkanen J, Jaakola L, Suorsa M, Tolonen A, Mattila S, Pirttilä AM, Hohtola A. Flavonoid biosynthesis and degradation play a role in early defence responses of bilberry (Vaccinium myrtillus) against biotic stress. Eur J Plant Pathol. 2009;125(4):629. doi: 10.1007/s10658-009-9511-6. [DOI] [Google Scholar]
- 80.Dubos C, Le Gourrierec J, Baudry A, Huep G, Lanet E, Debeaujon I, Routaboul JM, Alboresi A, Weisshaar B, Lepiniec L. MYBL2 is a new regulator of flavonoid biosynthesis in Arabidopsis thaliana. Plant J. 2008;55(6):940–953. doi: 10.1111/j.1365-313X.2008.03564.x. [DOI] [PubMed] [Google Scholar]
- 81.He ZH, He D, Kohorn BD. Requirement for the induced expression of a cell wall associated receptor kinase for survival during the pathogen response. Plant J. 1998;14(1):55–63. doi: 10.1046/j.1365-313X.1998.00092.x. [DOI] [PubMed] [Google Scholar]
- 82.Li H, Zhou SY, Zhao WS, Su SC, Peng YL. A novel wall-associated receptor-like protein kinase gene, OsWAK1, plays important roles in rice blast disease resistance. Plant Mol Biol. 2009;69(3):337–346. doi: 10.1007/s11103-008-9430-5. [DOI] [PubMed] [Google Scholar]
- 83.Brands A, Ho THD. Function of a plant stress-induced gene, HVA22. Synthetic enhancement screen with its yeast homolog reveals its role in vesicular traffic. Plant Physiol. 2002;130(3):1121–1131. doi: 10.1104/pp.007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shen Q, Chen CN, Brands A, Pan SM, Ho TH. The stress-and abscisic acid-induced barley gene HVA22: developmental regulation and homologues in diverse organisms. Plant Mol Biol. 2001;45(3):327–340. doi: 10.1023/A:1006460231978. [DOI] [PubMed] [Google Scholar]
- 85.Wang B, Zhang M, Fu R, et al. Epigenetic mechanisms of salt tolerance and heterosis in upland cotton (Gossypium hirsutum L.) revealed by methylation-sensitive amplified polymorphism analysis. Euphytica. 2016;208(3):477–491. doi: 10.1007/s10681-015-1586-x. [DOI] [Google Scholar]
- 86.Wood AJ, Reski R, Frank W. Isolation and characterization of ALDH11A5, a novel non-phosphorylating GAPDH cDNA from Physcomitrella patens. Bryologist. 2004;107(3):385–387. doi: 10.1639/0007-2745(2004)107[0385:IACOAA]2.0.CO;2. [DOI] [Google Scholar]
- 87.Zeng L, Deng R, Guo Z, Yang S, Deng X. Genome-wide identification and characterization of glyceraldehyde-3-phosphate dehydrogenase genes family in wheat (Triticum aestivum) BMC Genomics. 2016;17(1):240. doi: 10.1186/s12864-016-2527-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Hai-Jun, Yan Jianbing. Crop genome-wide association study: a harvest of biological relevance. The Plant Journal. 2018;97(1):8–18. doi: 10.1111/tpj.14139. [DOI] [PubMed] [Google Scholar]
- 89.Yuan Y, Wang X, Wang L, et al. Genome-wide association study identifies candidate genes related to seed oil composition and protein content in Gossypium hirsutum L. Front Plant Sci. 2018;9:1359. doi: 10.3389/fpls.2018.01359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cai C, Zhu G, Zhang T, Guo W. High-density 80 K SNP array is a powerful tool for genotyping G. hirsutum accessions and genome analysis. BMC Genomics. 2017;18(1):654. doi: 10.1186/s12864-017-4062-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khan AH, Ashraf MY, Azmi AR. Effect of sodium chloride on growth and nitrogen metabolism of sorghum. Acta Physiol Plant. 1990;12(12):233–238. [Google Scholar]
- 92.Ravelombola W, Shi A, Weng Y, et al. Association analysis of salt tolerance in cowpea (Vigna unguiculata (L.) Walp) at germination and seedling stages. Theor Appl Genet. 2018;131(1):79–91. doi: 10.1007/s00122-017-2987-0. [DOI] [PubMed] [Google Scholar]
- 93.Saad FF, El-Mohsen AAA, El-Shafi MAA, Al-Soudan IH. Effective selection criteria for evaluating some barley crosses for water stress tolerance. Egypt J Plant Breed. 2014;203(1893):1–40. [Google Scholar]
- 94.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2009;26(1):136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 98.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, Tiwari VK, Rawat N, Gill BS, Huo N, You FM, Coleman-Derr D, Gu YQ. GSP: a web-based platform for designing genome-specific primers in polyploids. Bioinformatics. 2016;32(15):2382–2383. doi: 10.1093/bioinformatics/btw134. [DOI] [PubMed] [Google Scholar]
- 101.Wang M, Wang Q, Zhang B. Evaluation and selection of reliable reference genes for gene expression under abiotic stress in cotton (Gossypium hirsutum L.) Gene. 2013;530(1):44–50. doi: 10.1016/j.gene.2013.07.084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Descriptive statistics of the salt tolerance index of 10 salt tolerance-related traits. (XLSX 11 kb)
Table S2. Correlation analysis among 10 traits related to salt tolerance. (XLSX 9 kb)
Figure S1. Screen plot of eigenvalues with component number for STIs in BLUP and 3 years. (DOCX 190 kb)
Table S3. Characteristics, contribution rates, and weights of two principal components at the germination stage. (XLSX 9 kb)
Figure S2. PCA plots for the STIs of 10 salt tolerance traits in BLUP and 3 years. a for 2014, b for 2015, c for 2016, and d for BLUP. (DOCX 79 kb)
Table S4. Comprehensive evaluation values (D) and K-mean cluster analysis of salt tolerance. (XLSX 20 kb)
Table S5. Summary of SNPs, PIC, gene diversity, and LD decay in Yuan et al. (2018) [89]. (XLSX 11 kb)
Table S6. Annotation of genes located within candidate QTL regions. (XLSX 25 kb)
Table S7. Summary of sequencing data quality. (XLSX 11 kb)
Figure S3. Validation of the 20 random genes in transcriptome sequencing with qRT-PCR. (XLSX 11 kb)
Table S8. Candidate genes near significant SNPs. (DOCX 7557 kb)
Table S9. Information for 196 upland cotton accessions [89]. (XLSX 23 kb)
Figure S4. Population structure of the 196 accessions in Yuan et al. (2018) [89]. (A) UPGMA tree based on Nei’s genetic distances. (B) Principal component analysis of 196 accessions based on genotype. (C) Population structure of the 196 accessions based on STRUCTURE when K = 2. (D) Kinship for this panel. (DOCX 693 kb)
Table S10. All the specific primers for the selected genes and the intro-reference gene UBQ7. (XLSX 12 kb)
Data Availability Statement
Most of the data pertaining to the present study has been included in the Tables/Figures of the manuscript. The authors are pleased to share the rest of the raw data upon request.