

Abstract
To maintain the lipid asymmetry of the cell envelope in Gram-negative bacteria, the MlaC protein serves as a lipid-transfer factor and delivers phospholipids from the outer to the inner membrane. A strategy of antibiotic discovery is to design a proper compound that can tightly bind to the MlaC protein and inhibit the MlaC function. In this study, we performed virtual screening on multiple MlaC structures obtained from molecular dynamics simulations to identify potential MlaC binders. Our results suggested that clorobiocin is a compound that could bind to the MlaC protein. Through the comparison of the bound geometry between clorobiocin and novobiocin, we pointed out that the methyl-pyrrole group of the noviose sugar in clorobiocin forms hydrophobic interactions with amino acids in the phospholipid binding pocket, which allows the compound to bind deep in the active site. This also explains why clorobiocin shows a tighter binding affinity than novobiocin. Our study highlights a practical path of antibiotic development against Gram-negative bacteria.
Keywords: antibiotic, Drug Design, Virtual Screening, MlaC protein
Graphical Abstract
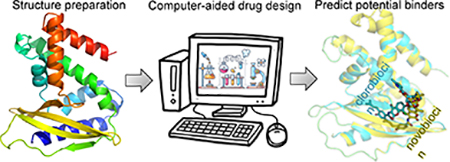
The MlaC protein serves as a lipid-transfer factor that can deliver phospholipids from the outer to the inner membrane to maintain the lipid asymmetry in Gram-negative bacteria. Through the design of a ligand binding to the MlaC protein, one could limit this bacteria’s capacity. Our virtual screening and docking simulations, combined with experimental measurements, suggest that clorobiocin is a compound that could inhibit the MlaC function.
Introduction
The outer membrane of Gram-negative bacteria has an asymmetric structure with lipopolysaccharides at the outer leaflet and phospholipids at the inner leaflet[1]. To maintain this lipid asymmetry, the Mla pathway, an ATP-binding cassette transport system, can transfer misplaced phospholipids from the outer to the inner membrane[2–4]. By inhibiting this fundamental mechanism, a strategy of antibiotic discovery is to design a small molecule that can tightly bind to the MlaC protein, a key element in the Mla pathway, and interrupt the phospholipid transport on the membranes further limiting the bacteria’s capacity to cause disease. This work aims to look for potential inhibitors of the MlaC protein through computational modeling.
A crystal structure of MlaC–phospholipid complex from Ralstonia solanacearum showed that the protein is folded into 9 alpha helices and 5 beta stands (Figure 1). The MlaC protein from Acinetobacter baumannii was built by homology modeling. Through performing molecular dynamic (MD) simulations, the dynamic properties of the MlaC protein in both apo and phospholipid-bound state from the two bacteria sources have been investigated[5]. The simulations revealed multiple protein conformations with different binding pocket volumes, which may directly contribute to the design of antibiotics of the MlaC protein[5].
Figure 1:

Structure of the MlaC protein.
Virtual screening has been widely applied in structure-based drug design. These approaches are now well established in a step-by-step process including refinements of protein structure, calculations of ligand binding thermodynamics, ranking compounds, and predicting binding poses[6]. The early docking studies were successful in antiviral discovery for HIV and influenza with a rigid protein-ligand model[7, 8]. Later on, the docking protocol allowed a flexible model of receptor and substrate, continuing to improve the success in drug discovery[9]. Thus, in this work, we performed both rigid and flexible docking to identify new inhibitors of the MlaC protein with potential antibiotic activity for Gram-negative bacteria.
Methods
MlaC protein structure preparation
The structure of the MlaC protein from Acinetobacter baumannii was built according to the MlaC–phospholipid complex from Ralstonia solanacearum (PDB ID: 2QGU) through the homology modeling tools on the i-Tasser server[10]. The sequence identity between the two MlaC proteins from different species has been explored in detail from the early MlaC dynamic study[5]. Following the MD protocol from the study[5], we performed 100-ns MD simulations on the MlaC protein from Acinetobacter baumannii using the Amber 14 package[11, 12]. The g-cluster program in Gromacs 4.5.5 package[13] was applied to cluster the MD trajectory into 30 clusters based on the root-mean-square-deviation (RMSD) of the protein backbone. The major conformations from the 30 clusters were used for the following screening simulations, following the relaxed complex scheme[14–16].
Virtual screening
Virtual screening of the MlaC protein was performed with the virtual screening workflow in Schrodinger Suite 2016[17, 18]. The workflow includes ligand preparation using LigPrep, filtering using propfilter on QikProp properties, and Glide docking at the three accuracy levels, including the high-throughput virtual screening (HTVS), standard precision (SP) and extra precision (XP)[19]. The National Cancer Institute (NCI) diversity set IV, which contains 1596 compounds, was used as a screening library. First, the ligands were prepared using LigPrep with the OPLS2005 force field. Before running the workflow, we generated Glide grids for a receptor with the center located at the MlaC phospholipid-binding site. The inner and outer boxes for docking were set to 10 and 27 Å, respectively. Then, the HTVS, SP, and XP docking were carried out.
Induced-fit docking
We performed induced-fit docking using Glide in Schrodinger Suite 2016[17–19]. Two ligands, clorobiocin and novobiocin, were docked to 5 different MlaC structures, for which the protein conformations were generated by clustering the trajectory from the earlier molecular dynamic (MD) simulations[5]. We set the phospholipid-binding site of MlaC protein as the docking center. The inner docking box was set to10 Å, and the Glide default was used for the outer box value. The MlaC protein was rigid during the docking simulations except for the residues within 5 Å of the docking center.
Experimental methods
Checkerboards were performed in RPMI 1640 (Gibco) supplemented with 5% Luria-Bertani Broth (BD Difco). A. baumannii AB5075 was grown to mid log phase in LB broth, washed twice with PBS and suspended to a concentration of 5×106 cfu/mL in RPMI + 5% LB. Clorobiocin was prepared in a range of 500 – 7.8 μM and the antimicrobial peptide LL-37 in a range from 80 – 1.25 μM. 10 μL of diluted range LL-37 was added across 8 columns in a 96 well plate containing 70 μL of RPMI+5% LB followed by 10 μL of clorobiocin dilution range down the rows. 10 μL of bacteria was then added and placed at 37ºC overnight. Final concentrations were 50 – 0.78 μM clorobiocin, 8 – 0.125 μM LL-37, and 5×105 cfu/mL A. baumannii. Plates were read by eye and data analyzed. Fractional inhibitory concentration (FIC) was calculated using following formula: .
Results and discussion
In silico screen to select potential MlaC inhibitors
We performed a computational technique, virtual screening and docking simulation, to identify potential MlaC inhibitors. The methods have been broadly applied in drug discovery through searching libraries of small molecules to identify candidate compounds that bind to a target protein[20–22]. We first used the 30 protein conformations clustered from the MD simulations as receptor templates to perform virtual screening. For each conformational cluster, through the docking of ~1,600 compounds of the NCI library, we identified 120–150 compounds that show the lowest binding free energy score sorted by the screening. According to the docking score of each simulation, we listed the 20 compounds that have a preferred binding affinity to the MlaC protein in Table 1. Then, we performed experimental checkerboards to carefully examine these 20 compounds. One of the compounds, clorobiocin (NCI compound number 227186), was noticed to play an active role on the interference of Mla pathways. When clorobiocin was tested in an A. baumannii checkerboard with the human antimicrobial peptide LL-37, Figure 2 shows synergistic effects (FIC = 0.5 at concentration of 12.5 μM clorobiocin and 1 μM LL-37). This result implied potential interactions with the Mla pathway. Thus, we continued to learn molecular insights into the binding generated between the MlaC protein and clorobiocin.
Table 1:
List of the 20 compounds from the NCI library with the best docking scores to the MlaC protein.
| ranking | NCI compound number | binding energy (kcal/mol) |
|---|---|---|
| 1 | 268251 | −13.53 |
| 2 | 354844 | −13.50 |
| 3 | 227186 | −13.14 |
| 4 | 345647 | −13.12 |
| 5 | 37553 | −12.90 |
| 6 | 122819 | −12.48 |
| 7 | 89821 | −12.37 |
| 8 | 91397 | −12.08 |
| 9 | 111210 | −12.08 |
| 10 | 128606 | −12.05 |
| 11 | 84100 | −11.96 |
| 12 | 335504 | −11.83 |
| 13 | 275266 | −11.79 |
| 14 | 309892 | −11.75 |
| 15 | 9037 | −11.74 |
| 16 | 163443 | −11.69 |
| 17 | 80997 | −11.69 |
| 18 | 654260 | −11.64 |
| 19 | 186200 | −11.63 |
| 20 | 156565 | −11.61 |
Figure 2:

Experimental measurements of FIC change between clorobiocin and LL-37 concentration.
Comparison between clorobiocin and novobiocin
Novobiocin is structurally similar to clorobiocin. It has been known as an agent for the treatment of resistant bacterial infections since 1950s[23]. Both novobiocin and clorobiocin are composed of three chemical segments: noviose sugar, coumarin and benzamide group (see Figure 3A). The only two structural differences between novobiocin and clorobiocin are 1) the functional group at noviose sugar and 2) the replacement of chloride by a methyl group at coumarin. Although earlier studies showed that novobiocin is effective in antibacterial therapy, our experimental investigation does not show it alters the bacterial activity involving Mla mechanisms; however, clorobiocin does. Thus, this prompted us to study why the slight structural differences between novobiocin and clorobiocin could result in different behaviors in antibiotics.
Figure 3:

(A) Chemical structures of novobiocin and clorobiocin. (B) Structural alignment of novobiocin (yellow) and clorobiocin (cyan) complex reported from induced-fit docking simulations. (C) Structure of MlaC-novobiocin complex reported from induced-fit docking calculations. Bond representations of cyan, magenta and purple color indicate that the protein residues form interactions with the isobutylene functional group on benzamide, the benzamide group and the amine group of noviose sugar, respectively. (D) Structure of MlaC-clorobiocin complex reported from induced-fit docking simulations. Bond representations of yellow, magenta and purple color indicate that the residues form interactions with the isobutylene group of clorobiocin, the center coumarin group and the methyl-pyrrole group of noviose sugar, respectively.
We performed induced-fit docking of novobiocin and clorobiocin to 5 different MlaC conformations to explore ligand-protein binding geometries of the MlaC systems. Compared to conventional docking tools with a rigid protein coordinate, the induced-fit protocol here allows conformational changes of the receptor binding site induced by a bound ligand, which enables us to quickly predict active site geometries with minimal expense. Our results show that the best docking scores of novobiocin and clorobiocin binding to the MlaC protein are −12.38 and −12.83 kcal/mol, respectively (Table 2). Both bound poses of novobiocin and clorobiocin from docking simulations demonstrate that the benzamide group points toward the active site, while the noviose sugar is exposed to solvent and tends to interact with the protein residues near the entrance of the lipid-binding site (Figure 3B). The structural alignment of novobiocin and clorobiocin shows that clorobiocin could be placed deeper in the binding pocket, and the ligand geometry of clorobiocin fits better to MlaC protein conformation than novobiocin (Figure 3B), which may explain why clorobiocin could bind tighter than novobiocin to MlaC.
Table 2:
The docking scores reported from Schrodinger Suite of clorobiocin and novobiocin binding to five different conformations of MlaC protein.
| MlaC protein conformation | novobiocin (kcal/mol) | clorobiocin (kcal/mol) |
|---|---|---|
| conformation 1 | −12.375 | −12.826 |
| conformation 2 | −10.229 | −11.539 |
| conformation 3 | −10.328 | −11.218 |
| conformation 4 | −11.648 | −11.522 |
| conformation 5 | −10.076 | −10.855 |
We then examined closely the detailed interactions between the ligands and MlaC protein. Three major contacts are formed between novobiocin and the MlaC. First, the isobutylene functional group on benzamide interacts with the hydrophobic residues, Val43, Leu73 and Phe158 (Figure 3C benzamide interactions). Second, the benzamide group displays pi-pi stacking with Tyr123 and Tyr128, which allows binding of the compound in the protein pocket (Figure 3C coumarin interactions). Third, the amine group of noviose sugar forms polar interactions with Tyr116 and Gln182 (Figure 3C noviose sugar interactions). Compared to novobiocin, the isobutylene group of clorobiocin binds deeper in the binding pocket, interacting with Tyr76, Val77 and Ile169 (Figure 3D benzamide interactions). The center coumarin group is clamped between Tyr116 and Val141 by non-polar interactions (Figure 3D coumarin interactions). Importantly, the methyl-pyrrole group of noviose sugar in clorobiocin binds nicely at the hydrophobic pocket formed by Leu112, Asn115, Gln182 and Phe183 (Figure 3D noviose sugar interactions). Apparently, the replacement of amide from novobiocin to methyl-pyrrole group in clorobiocin enables the ligand to form more contacts with the MlaC protein and further increases the binding affinity.
Acknowledgments
This work was supported by the NIH, NBCR, and NSF supercomputer centers.
References
- [1].Kamio Y, Nikaido H, Biochemistry 1976, 15, 2561. [DOI] [PubMed] [Google Scholar]
- [2].Narita S.-i., Biosci Biotechnol Biochem 2011, 75, 1044. [DOI] [PubMed] [Google Scholar]
- [3].Malinverni JC, Silhavy TJ, Proc Natl Acad Sci U S A 2009, 106, 8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Munguia J, LaRock DL, Tsunemoto H, Olson J, Cornax I, Pogliano J, et al. , Journal of Molecular Medicine-Jmm 2017, 95, 1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Huang Y.-m. M., Miao YL, Munguia J, Lin L, Nizet V, McCammon JA, Protein Sci 2016, 25, 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Amaro RE, Baudry J, Chodera J, Demir O, McCammon JA, Miao YL, et al. , Biophys J 2018, 114, 2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kaldor SW, Kalish VJ, Davies JF, Shetty BV, Fritz JE, Appelt K, et al. , J Med Chem 1997, 40, 3979. [DOI] [PubMed] [Google Scholar]
- [8].Vonitzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, et al. , Nature 1993, 363, 418. [DOI] [PubMed] [Google Scholar]
- [9].Rosenfeld R, Vajda S, Delisi C, Annu Rev Biophys Biomol Struct 1995, 24, 677. [DOI] [PubMed] [Google Scholar]
- [10].Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y, Nat Methods 2015, 12, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, et al. , J Comput Chem 2005, 26, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Salomon-Ferrer R, Case DA, Walker RC, WIREs Comput Mol Sci 2013, 3, 198. [Google Scholar]
- [13].Lange OF, Grubmuller H, Proteins: Struct, Funct, Bioinf 2006, 62, 1053. [DOI] [PubMed] [Google Scholar]
- [14].Amaro RE, Baron R, McCammon JA, J Comput-Aided Mol Des 2008, 22, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lin JH, Perryman AL, Schames JR, McCammon JA, Biopolymers 2003, 68, 47. [DOI] [PubMed] [Google Scholar]
- [16].Lin JH, Perryman AL, Schames JR, McCammon JA, J Am Chem Soc 2002, 124, 5632. [DOI] [PubMed] [Google Scholar]
- [17].Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. , J Med Chem 2004, 47, 1739. [DOI] [PubMed] [Google Scholar]
- [18].Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. , J Med Chem 2004, 47, 1750. [DOI] [PubMed] [Google Scholar]
- [19].Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. , J Med Chem 2006, 49, 6177. [DOI] [PubMed] [Google Scholar]
- [20].Miao YL, Goldfeld DA, Von Moo E, Sexton PM, Christopoulos A, McCammon JA, et al. , Proc Natl Acad Sci U S A 2016, 113, E5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Joshi P, McCann GJP, Sonawane VR, Vishwakarma RA, Chaudhuri B, Bharate SB, J Chem Inf Model 2017, 57, 1309. [DOI] [PubMed] [Google Scholar]
- [22].Singh VK, Coumar MS, J Mol Model 2017, 23, 1309. [DOI] [PubMed] [Google Scholar]
- [23].Bisacchi GS, Manchester JI, Acs Infectious Diseases 2015, 1, 4. [DOI] [PubMed] [Google Scholar]