

SUMMARY
Programmed death ligand 1 (PD-L1, also called B7-H1) is an immune checkpoint protein that inhibits immune function through its binding of the programmed cell death protein 1 (PD-1) receptor. Clinically approved antibodies block extracellular PD-1/PD-L1 binding, yet the role of intracellular PD-L1 in cancer remains poorly understood. Here, we discovered that intracellular PD-L1 acts as an RNA binding protein that regulates the mRNA stability of NBS1, BRCA1, and other DNA damage related genes. Through competition with the RNA exosome, intracellular PD-L1 protects targeted RNAs from degradation, thereby increasing cellular resistance to DNA damage. RNA immunoprecipitation and RNA-seq experiments demonstrated that PD-L1 regulates RNA stability genome-wide. Furthermore, we developed a PD-L1 antibody, H1A, which abrogates PD-L1’s interaction with CMTM6, thereby promoting PD-L1 degradation. Intracellular PD-L1 may be a potential therapeutic target to enhance the efficacy of radiotherapy and chemotherapy in cancer through inhibition of DNA damage response and repair.
INTRODUCTION
Programmed death ligand 1 (PD-L1, also called B7-H1) is an immune checkpoint protein that regulates the immune system through binding of the programmed cell death protein 1 (PD-1) receptor(Chen et al., 2012; Chen and Han, 2015; Dong et al., 1999; Freeman et al., 2000; Hamanishi et al., 2016; He et al., 2015; Nishimura et al., 1999; Ohaegbulam et al., 2015; Postow et al., 2015). PD-L1 is expressed on multiple types of immune cells and is also expressed in many cancers. By interacting with PD-1 on immune cells, PD-L1 helps tumor cells evade the immune system by inhibiting T-cell activity and proliferation, facilitating T cell anergy and exhaustion, and inducing activated T-cell apoptosis (Chen and Han, 2015; Dong et al., 2002; He et al., 2015). Therefore, abrogation of the PD-1/PD-L1 interaction has emerged as an effective therapeutic strategy to enhance antitumor immunity across multiple malignancies. Yet, despite high expression of PD-L1 many tumors do not respond to or eventually progress following clinically approved PD1/PD-L1 inhibitor therapy, suggesting there is more to understand regarding the function of PD-L1 in cancer (Hamanishi et al., 2016). Although the impact of extracellular PD-L1 on immune regulation is well established, the intracellular role of PD-L1 in tumor biology and cancer therapy has yet to be fully elucidated.
RESULTS
Knockdown of PD-L1 sensitizes cancer to radiotherapy
In order to examine the function of PD-L1 in response to cancer therapy independent of its role in immune cell regulation, we knocked down PD-L1 using two separate shRNAs in the HCT116 colorectal carcinoma and MDA-MB-231 breast cancer cell lines and assessed for sensitivity to chemotherapy (cisplatin) and ionizing radiation (IR) in vitro. Surprisingly, we observed that PD-L1 knockdown sensitized HCT116 and MDA-MB-231 cells to both cisplatin and IR (Fig. 1A–B). Meanwhile, restoration of PD-L1 expression in knockdown cells reversed the sensitivity to these treatments (Fig. 1A), suggesting that the observed impact on platinum and IR sensitivity was specific. Of note, the expression of PD-1 in HCT116 and MDA-MB-231 is not detectable (Supplementary Fig. 1A). Since platinum and IR kill cancer cells by inducing DNA damage, these results suggested that PD-L1 may have a function in the cellular response to DNA damage that is independent of its role in PD-1 binding and immune regulation.
Figure. 1, PD-L1 is required for the DNA damage response.

A-B, HCT116 cells (A) or MDA-MB-231 cells (B) transfected with the indicated constructs were treated with either ionizing radiation (IR) or cisplatin at the indicated doses and colony formation was assessed (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001) C, HeLa cells transfected with PD-L1 overexpression or control vectors were treated with either IR or cisplatin and colony formation was assessed (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001) D, Patient-derived breast cancer cells were isolated and infected with control or PD-L1 targeted shRNA lentivirus. Cells were seeded and treated with the indicated doses of irradiation and the viability of cell was assessed cell survival (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001 ) E, Control and PD-L1 knockdown U2OS cells were treated with 2 Gy and the formation and resolution of γH2AX foci were assessed using immunofluorescence. Representative images are shown with data quantification in Supplementary Figure 1B. F, Wild-type (+/+, n=16), PD-L1 heterozygous (+/−, n=24) and PD-L1 knockout (−/−, n=16) BALB/C mice were exposed to whole body irradiation (7 Gy) and survival was assessed. Comparisons between groups were made using the Log-rank test.
To further explore this possibility we assessed the impact of knocking down PD-L1 in HeLa and A549 cells, two extremely low PD-L1 expressing cell lines. Knockdown of PD-L1 resulted in either minor or no effect on sensitivity to DNA damage (Supplementary Fig. 1B–C, supplementary Fig 4A–B), suggesting the level of PD-L1 in those cell lines was insufficient to protect cells from DNA damage. We then examined the impact of exogenous expression of PD-L1 in HeLa cells and observed significantly increased resistance to both IR and cisplatin (Fig. 1C). In order to further establish the effect of PD-L1 on the cellular response to DNA damage in cancer, we assessed the radiosensitivity of patient-derived breast cancer spheroids following PD-L1 knock down(Yu et al., 2017). Notably, PD-L1 knockdown significantly sensitized these patient-derived cancer cells to IR (Fig. 1D).
Therefore, we next assessed whether DNA repair capacity could be compromised in cells depleted of PD-L1. First, we used immunofluorescence to examine the kinetics of IR-induced γH2AX foci, which are indicative of DNA double strand breaks, following PD-L1 knockdown. After PD-L1 knockdown, cells had higher basal levels of γH2X, likely as a result of decreased repair of spontaneous DNA damage (Fig 1E). Consistently, PD-L1 knockdown cells displayed delayed resolution of IR-induced γH2AX foci (Fig. 1E, and Supplementary Fig. 1D), indicative of compromised DNA repair function. We also evaluated the impact of PD-L1 on DNA damage sensitivity in vivo using PD-L1 knockout mice. PD-L1 knockout BALB/C mice were profoundly radiosensitive, exhibiting significantly decreased survival following whole body irradiation compared to wild-type controls (Fig. 1F, p=0.003). Notably, abrogating the PD-L1/PD-1 interaction with PD-L1 blocking antibody did not similarly increase the sensitivity of these mice to whole body irradiation (Supplementary Fig. 1E), suggesting again a function of PD-L1 independent of immune regulation. Taken together, these results provided strong evidence of a link between PD-L1 and the DNA damage response (DDR) independent of PD-L1/PD-1 binding.
Knockdown of PD-L1 decreased NBS1 and BRCA1 mRNA stability
We next sought to gain mechanistic insight into how PD-L1 was affecting the DDR. First, we assessed whether PD-L1 knockdown impacted the expression of important DDR pathway genes in cancer cells. Interestingly, we found that protein levels of all members of the MRN complex and BRCA1 decreased following knockdown of PD-L1 in both HCT116 and MDA-MB-231 cells, with a particularly robust effect on BRCA1 and NBS1 (Fig. 2A and Supplementary Fig. 2A). Therefore, we focused on NBS1 and BRCA1 expression in subsequent studies to understand how PD-L1 was regulating gene expression. First, we treated control and PD-L1 depleted MDA-MB-231 cells with the proteasome inhibitor, MG132. The level of NBS1 did not recover after MG132 treatment, implying that the reduced NBS1 level observed in PD-L1 depleted cells did not result from increased protein degradation through the proteasome (Supplementary Fig. 2B). We did observe, however, that NBS1 and BRCA1 mRNA levels were significantly decreased in PD-L1 depleted cells (Fig. 2B and Supplementary Fig. 2C).
Figure. 2, PD-L1 binds and stabilizes NBS1 and BRCA1 mRNA.
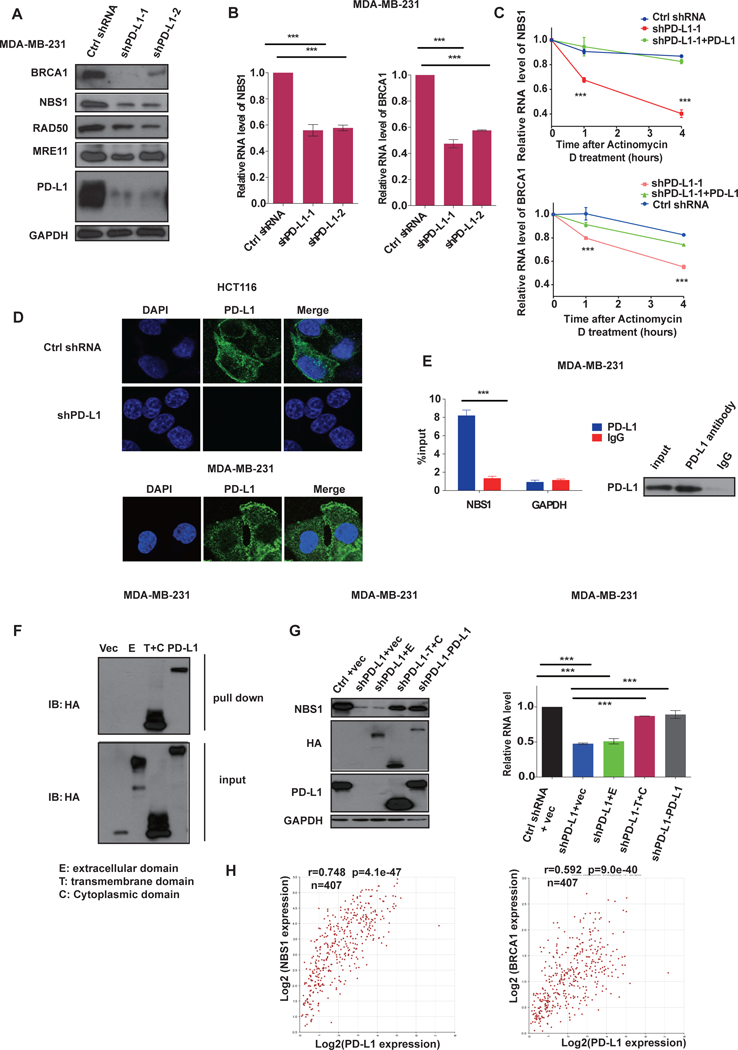
A, Western blot analysis of BRCA1, NBS1, RAD50, MRE11 and PD-L1 in MDA-MB-231 cells infected with lentiviruses encoding control shRNA or two different shRNAs targeting PD-L1. GAPDH was used as a loading control. B, Quantification of NBS1 and BRCA1 mRNA levels in MDA-MB-231 cells infected with lentiviruses encoding indicated shRNAs using quantitative real-time (qRT)-PCR (±s.e.m., n=3). GAPDH was used for normalization. (*p<0.05, **p<0.01, ***p<0.001) C, Control MDA-MB-231 cells, PD-L1 knockdown, and PD-L1 knockdown cells with PD-L1 re-expressed were treated with the transcription inhibitor actinomycin D (5 ug/ml). NBS1 and BRCA1 mRNA levels were quantified using qRT-PCR (±s.e.m., n=3). GAPDH was used for normalization. (*p<0.05, **p<0.01, ***p<0.001). D, Representative immunofluorescence images of the subcellular localization of PD-L1 in HCT116 cells and MDA-MB-231. PD-L1 was labeled with green fluorescence and the nucleus was labeled with DAPI. E, RNA immunoprecipitation (RIP) assay demonstrating significant enrichment of NBS1 mRNA by PD-L1 antibody compared to the negative control IgG. The result is shown as the percentage of input (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001). F, RNA pull down in MDA-MB-231 cells expressing different truncations of PD-L1 (vector control, extracellular domain [E], transmembrane plus cytoplasmic domain [T+C], and full length PD-L1) using in vitro biotin labeled 3’ NBS1 RNA. G, Following PD-L1 knockdown by shRNA in MDA-MB231 cells, various truncations of PD-L1 were overexpressed and the level of NBS1 protein (left), and NBS1 mRNA (right), was assessed using immunoblot or qRT-PCR, respectively. GAPDH was using as loading controls. For qRT-PCR the result is shown as the percentage of input (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001). H, The correlation of PD-L1 expression with NBS1 and BRCA1 was assessed using blood samples data from 407 healthy donors in The Cancer Genome Atlas (TCGA) database. The Pearson correlation coefficient (r) is shown with the associated p value.
In eukaryotic cells, mRNA homeostasis is achieved through a balance between mRNA synthesis and degradation. Therefore, we sought to investigate whether PD-L1 impacted NBS1 and BRCA1 mRNA stability or transcription. We first assessed the impact of PD-L1 on mRNA stability. To do so, we treated MDA-MB-231 cells with the transcriptional inhibitor, Actinomycin D, and assessed the mRNA levels of NBS1 and BRCA1 over time. The half-life of both NBS1 and BRCA1 mRNA were significantly shorter in PD-L1 depleted cells compared with control, while restoration of PD-L1 expression could restore NBS1 and BRCA1 mRNA stability (Fig. 2C and Supplementary Fig. 2E–2F). Similar findings were also observed in HCT116 (Supplementary Fig 2E) and U2OS cells (Supplementary Fig. 2H). We also used the nuclear run-on assay to assess whether the reduced NBS1 and BRCA1 mRNA expression in PD-L1 depleted cells were due to decreased gene transcription. However, the transcription of NBS1 and BRCA1 was either unchanged or increased in PD-L1 knockdown conditions (Supplementary Fig. 2G). Together, these data suggested that decreased NBS1 and BRCA1 mRNA levels in PD-L1-deficient cells was due to reduced mRNA stability, rather than reduced transcription.
Intracellular PD-L1 binds with NBS1 and BRCA1 mRNA
We next investigated how PD-L1 was regulating mRNA stability. First, we assessed the subcellular localization of PD-L1 using immunofluorescence and observed detectable but weak nuclear PD-L1 and strong PD-L1 staining in the membrane and cytoplasm of HCT116 and MDA-MB-231 cells (Fig. 2D). We confirmed a similar PD-L1 expression pattern in a number of other cancerous and non-cancerous cell lines (Supplementary Fig. 3A). These findings implied that rather than being expressed solely on cell membranes, PD-L1 is expressed ubiquitously in the cytoplasm of cells, and is consistent with other studies which have also reported the presence of intracellular PD-L1 (Chen et al., 2015; Satelli et al., 2016). Of note, we also detected the cell membrane expression levels of PD-L1 by flow cytometry. Cell membrane expression levels paralleled the whole cell expression levels of PD-L1, as assessed by immunofluorescence, in low (A549, HeLa) and high PD-L1 expressing (MDA-MB-231) lines (Supplementary Fig. 3A–B).
Next, we used RNA immunoprecipitation (RIP) to explore whether PD-L1 might be regulating mRNA stability by interacting with mRNA. Indeed, we detected that PD-L1 interacted with NBS1 mRNA under both native and crosslinking conditions (Fig. 2E, and Supplementary Fig. 3C–D). PD-L1 contains three domains (Supplementary Fig. 3E): an extracellular domain, a transmembrane domain, and a cytoplasmic domain. The extracellular domain is the largest domain of the protein and the structure of which has been solved by X-ray crystallography (Lin et al., 2008). The transmembrane and domain and cytoplasmic domains are both very small and their structures have yet to be resolved. We had difficulty expressing the cytoplasmic domain alone and therefore we utilized extracellular (E) domain and transmembrane plus cytoplasmic (T+C) domain truncations (Supplementary Fig. 3E) for subsequent studies aimed at identifying which portion of PD-L1 interacted with RNA. First, we performed the RNA pull down assay using in vitro biotin labeling of the 3’ UTR of NBS1 RNAs. Interestingly, like full length PD-L1, we observed that the transmembrane plus cytoplasmic (T+C) domain had strong binding affinity with NBS1 mRNA, while the extracellular (E) domain alone could not interact with NBS1 mRNA (Fig. 2F). We also observed that expression of the PD-L1 transmembrane plus cytoplasmic domain rescued both NBS1 protein and mRNA levels in PD-L1-depleted cells (Fig. 2G), similar to the expression of full length PD-L1. However, expression of the PD-L1 extracellular domain alone had no effect (Fig. 2G). These results suggested that a motif in the PD-L1 transmembrane and/or cytoplasmic domain were required for its interaction and stabilizing effect on target RNAs. In order to identify the PD-L1 motif that interacts with NBS1 mRNA with greater precision, we constructed three additional PD-L1 truncations, each containing only the transmembrane domain plus distinct 10 amino acids sequences of the cytoplasmic domain (Supplementary Fig. 3F). We used these three PD-L1 truncations to pull down NBS1 mRNA and observed that amino acids 270–279 from the PD-L1 cytoplasmic domain are required for the PD-L1/RNA interaction (Supplementary Fig. 3G). Together, these data suggested that PD-L1 interacts with, and regulates NBS1 mRNA stability and that the cytoplasmic domain is required for this function.
To further explore the clinical relevance of these findings we calculated the expression correlation of PD-L1 with NBS1 and BRCA1 using blood samples data from 407 healthy donors in The Cancer Genome Atlas (TCGA) database. Consistently, we found that NBS1 and BRCA1 expression was significantly positively correlated with PD-L1 expression, with R values of 0.748 (p = 4.1 e−47) and 0.592 (p = 9.0 e−40), respectively (Fig. 2H).
PD-L1 protects targeted mRNA from degradation by RNA exosome
The RNA exosome complex is a multi-protein complex that plays a vital role in regulating RNA turnover. EXOSC10, which harbors 3’->5’ exoribonuclease activity, is an important catalytic component of the RNA exosome (Lejeune et al., 2003; Tomecki et al., 2010; van Dijk et al., 2007). EXOSC10 localizes to the nucleus in yeast but is ubiquitously expressed in human cells. Therefore, we sought to investigate the impact of PD-L1 on exosome-mediated mRNA turnover. To this end, we began by knocking down EXOSC10 in MDA-MB-231 cells. As expected, the level of NBS1 and BRCA1 mRNA increased after EXOSC10 knockdown (Fig. 3A). Interestingly, knockdown of PD-L1 increased the binding affinity of EXOSC10 with both NBS1 and BRCA1 mRNA, while knockdown of EXOSC10 increased the binding of PD-L1 with these two RNAs (Fig. 3B–C). In addition, knocking down EXOSC10 reversed the decrease in NBS1 and BRCA1 RNA levels and RNA half-lives caused by PD-L1 knockdown (Fig. 3D–E). These findings suggested that PD-L1 and EXOSC10 may be in competition to bind target RNAs. In order to rule out the possibility that Actinomycin D itself could be affecting BRCA1 and NBS1 mRNA stability we also measured RNA stability using the 5-Ethynyluridine (EU) labeling and releasing method. Consistently, we observed a significant decrease of EU labeled NBS1 and BRCA1 RNA (Figure. 3F) and shortened poly(A) tail of NBS1 mRNA after PD-L1 was knocked down (Supplementary Fig. 4A), consistent with an enhanced exosome activity. Furthermore, the decreased stability of NBS1 and BRCA1 mRNA could also be rescued by knockdown of EXOSC10 in this model (Fig.3F).
Figure. 3, PD-L1 protects NBS1 and BRCA1 mRNA from degradation by the RNA exosome.
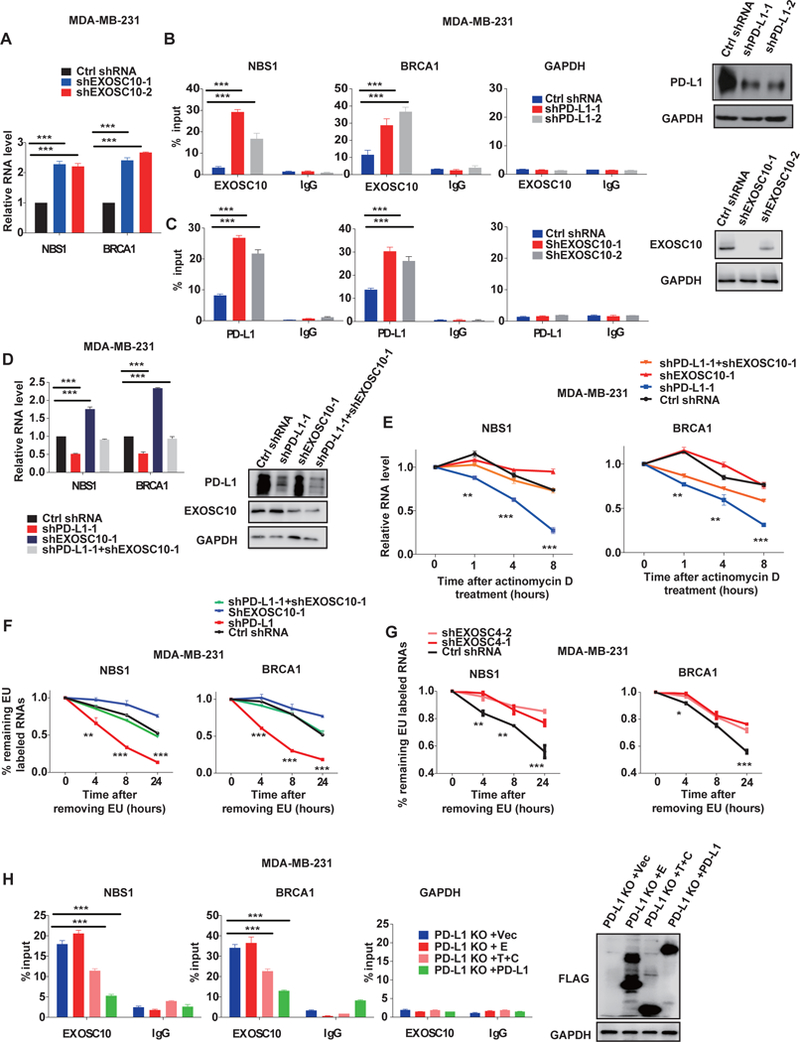
A, qRT-PCR analysis demonstrating NBS1 and BRCA1 mRNA levels following knockdown of EXOSC10 in MDA-MB-231 cells with two separate shRNA. GAPDH was used for normalization. (*p<0.05, **p<0.01, ***p<0.001). B, RIP assay in MDA-MB-231 cells demonstrating that knockdown of PD-L1 increases the binding affinity of EXOSC10 with NBS1 and BRCA1 mRNA. The result is shown as the percentage of input (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001). C, RIP assay in MDA-MB-231 cells demonstrating that knockdown of EXOSC10 increases the binding affinity of PD-L1 with NBS1 and BRCA1 mRNA. The result is shown as the percentage of input (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001). D, qRT-PCR analysis of NBS1 and BRCA1 mRNA levels in MDA-MB-231 cells infected with the indicated shRNAs (±s.e.m., n=3). GAPDH was used for normalization. (*p<0.05, **p<0.01, ***p<0.001). E, MDA-MB-231 cells infected with the indicated shRNAs were treated with the transcription inhibitor actinomycin D (5 ug/ml) and NBS1 (left) and BRCA1 (right) mRNA stability was assessed. (*p<0.05, **p<0.01, ***p<0.001). F and G, 5-Ethynyluridine (EU) labeling assay to determine NBS1 and BRCA1 mRNA stability under PD-L1 or EXOSC10 knockdown (F) and EXOSC4 knockdown (G) conditions. Cells were first treated with EU (0.1mM) for 24 hours, and the incorporation ratio of EU was determined. Then EU was removed and cells were cultured in fresh media for another 0, 4, 8, or 24 hours. The stability of BRCA1 and NBS1 mRNAs was determined as % of labeled mRNA after removal of EU divided by % of labeled mRNA before removal. H, RIP assay of PD-L1 knockout MDA-MB-231 cells stably transfected with indicated Flag-tagged PD-L1 truncations. The binding affinity of EXOSC10 with NBS1 (left) and BRCA1 (middle) mRNA was evaluated. GAPDH control is also displayed (right). The result is shown as the percentage of input (±s.e.m., n=3). (*p<0.05, **p<0.01, ***p<0.001).
In order to further elucidate the role of the RNA exosome in the RNA stabilizing function of PD-L1, we evaluated the impact of EXOSC4, an essential component of the core of exosome, on PD-L1 target RNAs. Indeed, knockdown of EXOSC4 significantly increased EU labeled NBS1 and BRCA1 RNA percentage (Fig. 3G and Supplementary Fig. 4B). Using the RIP assay, we confirmed that like EXOSC 10, EXOSC4 interacted with NBS1 and BRCA1 RNA (Supplementary Fig. 4C), and that knockdown of PD-L1 could increase the binding affinity of EXOSC4 with both NBS1 and BRCA1 mRNA (Supplementary Fig. 4D). Meanwhile, knockdown of EXOSC4 increased the binding of PD-L1 with these two RNAs (Supplementary Fig. 4D). Collectively, these results suggested that PD-L1 competes with the RNA exosome to bind targeted RNAs, including important DNA repair elements BRCA1 and NBS1, with PD-L1 promoting their stability by preventing RNA degradation. Finally, the competition between PD-L1 and EXOSC10 only exists when the T+C PD-L1 domain is present (Fig. 3H), demonstrating that the intracellular domain of PD-L1 binds RNAs to prevent their degradation by the RNA exosome.
PD-L1 regulates RNA stability genome-widely
All of these results on BRCA1 and NBS1 mRNA raised the question of whether PD-L1 might regulate mRNA stability genome-widely. Therefore, we performed crosslinked RIP-seq in MDA-MB-231 cells using PD-L1 antibody to identify RNA transcripts that interact with PD-L1. Notably, we identified 3152 RNAs that were significantly enriched by PD-L1 compared to control, including NBS1 and BRCA1 mRNA (Supplementary Table 1). In order to assess the impact of these interactions on the broader transcriptome we also depleted PD-L1 and performed RNA sequencing (RNA-seq). 6502 genes were identified that were downregulated under PD-L1 knockdown condition compared with control (counts>100, fold>1.5 and p-value<0.02), among which 1724 genes were identified for both RIP-seq and RNA-seq datasets (Fig. 4C, Supplementary Table 1). Through Gene Ontology (GO) pathway analysis, we confirmed that 408 genes from the RNA-seq analysis and 207 genes from the RIP-seq analysis were enriched in the ‘cellular response to DNA damage stimulus’ pathway (Fig. 4A–B), respectively, among which 135 genes were enriched in both datasets (Fig. 4C). Besides DDR and repair pathways, we also observed that other important pathways such as cell cycle, metabolism, transcription, and protein modification were also significantly enriched. These results suggest that PD-L1’s RNA stabilizing function may impact multiple other cellular processes (Fig. 4D). In order to confirm the validity of the sequencing data, we measured the level of several DNA damage related genes using quantitative reverse transcription PCR (qRT-PCR). As predicted by the sequencing data, ATM, POLQ, FANCL mRNA levels were all decreased to a similar extent as BRCA1 mRNA under PD-L1 knockdown condition (Fig. 4E).
Figure. 4, PD-L1 regulates RNA stability genome-wide.
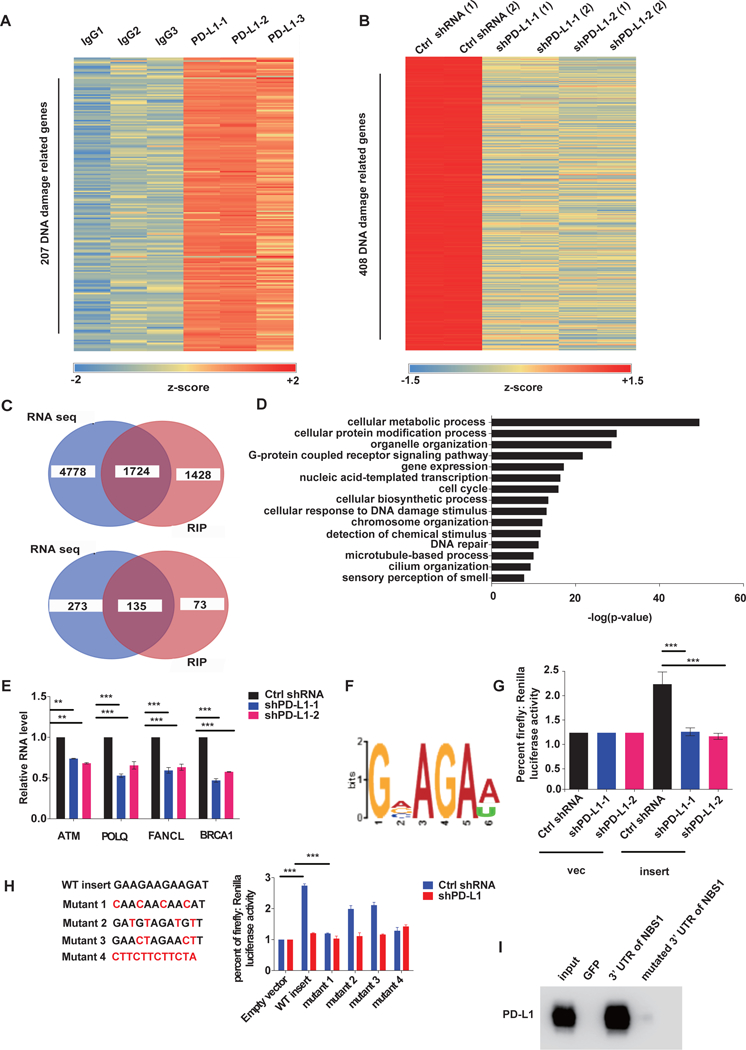
A, Cross-linked RIP was performed in MDA-MB-231 cells using PD-L1 antibody or control IgG followed by RNA sequencing on the extracted RNAs. Heatmap of DNA damage related genes enriched by PD-L1 through RIP-seq is shown (three independent replicates). The map was plotted according to the z-score of log (number of peaks). B, MDA-MB-231 cells were depleted of PD-L1 using control or two separate shRNAs and RNA sequencing was performed. Heatmap of DNA damage genes enriched by PD-L1 is shown. The map was plotted according to the z-score of log (normalized counts). C, Venny diagram of overlapping genes from the RIP-seq and RNA-seq analyses in A and B. The RNAs that were found to interact with PD-L1 through RIP-seq in A overlapped with the downregulated RNAs caused by PD-L1 knockdown in B. D, Gene Ontology (GO) analysis of RIP-seq and RNAseq data. RNAs that interacted with PD-L1 that were also downregulated by PD-L1 knockdown were significantly enriched in multiple important biological pathways including DNA damage response and repair related pathways. The data is shown as –log (p-value). E, qRT-PCR analysis of MDA-MB-231 cells infected with lentiviruses encoding indicated shRNAs to quantify several of the DNA damage response and repair related genes enriched by both RIP-Seq and RNA-Seq data (±s.e.m., n=3). GAPDH was used for normalization. (*p<0.05, **p<0.01, ***p<0.001). F, The top PD-L1 binding RNA motif identified using MEME ChIP software. G and H, The dual luciferase reporter assay using pmirGLO vector. Two copies of the candidate RNA motif (GAAGAAGAAGAT) and mutant sequences were inserted into the 3’UTR of the firefly luciferase gene, and then both empty vector and the vector with insert were transfected into control and PD-L1 knockdown cells. The signal of firefly luciferase was measured. Renilla luciferase was used as an internal control (±s.e.m., n=6, two tails t-test). (*p<0.05, **p<0.01, ***p<0.001). I, RNA pull down assay in MDA-MB-231 lysate using either the 3’ UTR of NBS1 or the 3’ UTR of NBS1 with binding motif mutations. GFP RNA was used as a negative control.
We next sought to assess the RNA sequence preference of PD-L1 binding to RNA in the available RIP-seq data set using MEME-ChIP (Machanick and Bailey, 2011). The sequence ‘GVAGAW’ was on the top of the list of predicted preferred PD-L1 binding sequence motifs (Fig. 4F). To validate that this sequence was recognized by PD-L1, we inserted two copies of the sequence (GAAGAAGAAGAT) into the dual-luciferase reporter vector, pmirGLO, and performed the dual luciferase reporter assay, detecting the level of RNAs under control or PD-L1 knockdown conditions. As showed in Fig. 4G and H, insertion of the sequence increased the level of firefly luciferase mRNA level, which was significantly decreased after PD-L1 knock down. Insertion with antisense sequence had no effect, indicating that the ‘GVAGAW’ motif is important for PD-L1 binding and regulation of RNA. Partial mutation of this motif decreased PD-L1’s function in RNA protection, with the base 1 and 4 having the most robust effect (Fig. 4H). Meanwhile, in order to further confirm GVAGAW as the binding region, we searched the 3’ UTR of NBS1 and 3 binding motifs were found (Supplementary Table. 2). We mutated all three regions to see if they would affect binding efficiency. Notably, through the RNA pull down assay, we found that mutation of all three regions significantly decreased binding of NBS1 with PD-L1 (Fig. 4I).
PD-L1 antibody H1A destabilizes PD-L1 and sensitizes cancer to radiotherapy
Our findings that PD-L1 regulates the stability of RNAs involved in DDR suggested that intracellular PD-L1 may be a target in cancer therapy beyond its established role as an immune checkpoint protein in collaboration with PD-1. The clinically available PD-L1 antibodies durvalumab and atezolizumab bind PD-L1 extracellularly, abrogating the PD-L1/PD-1 checkpoint. However, neither durvalumab (Supplementary Fig 5A) nor atezolizumab (not shown) significantly impacted PD-L1 or NBS1 expression. Therefore, we developed several human PD-L1 targeted mouse antibodies in BALB/C mice, as previously described (Liu et al., 2016), and screened for their ability to downregulate PD-L1 and NBS1 expression. Notably, one of the clones (H1A targeting epitope 20–32 aa, Fig. 5A), which has previously been shown to disrupt PD-L1 function in activated T cells (Liu et al. 2016(Liu et al., 2016)), was found to not only cause downregulation of PD-L1, but also decrease NBS1 mRNA and protein levels (Fig. 5B–C). Previously, CMTM6 has been reported to protect PD-L1 from lysosomal degradation (Burr et al., 2017; Mezzadra et al., 2017). Therefore, in order to investigate the mechanism of how H1A was regulating PD-L1 levels, we first treated MDA-MB-231 cells with the lysosomal inhibitor chloroquine. The reduction in PD-L1 expression following treatment with H1A was reversed after chloroquine treatment, suggesting that the lysosome is involved in H1A mediated PD-L1 downregulation (Fig. 5D).
Figure. 5. The anti-PD-L1 antibody, H1A, destabilizes PD-L1 and sensitizes cancer to radio-therapy.
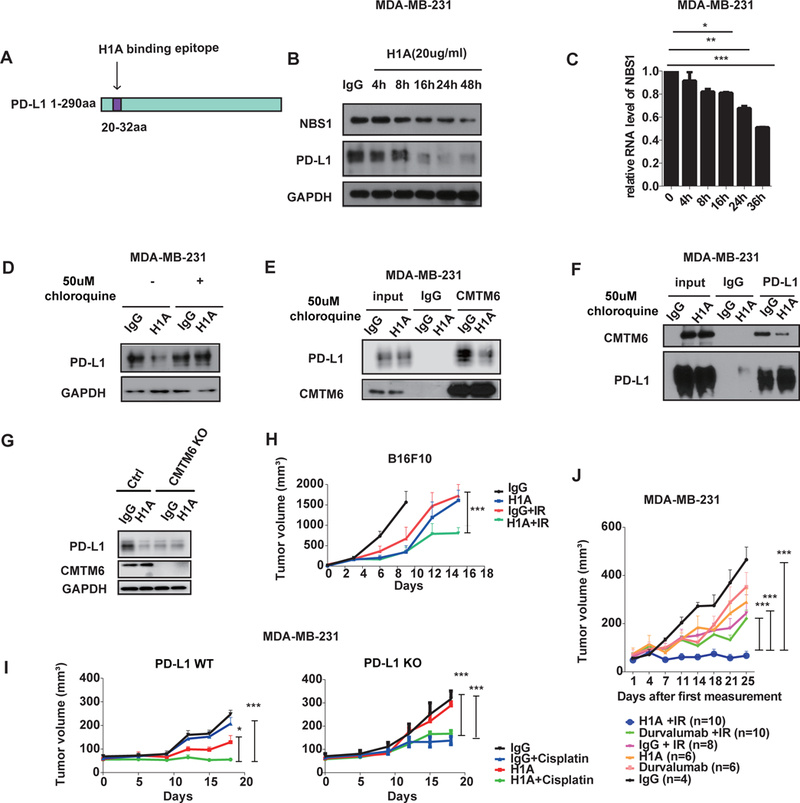
A, Diagram of the H1A binding epitope on PD-L1 (20–32 aa). B-C, MDA-MB-231 cells were plated one day before treatment with either H1A (20 ug/ml) or control IgG. Cells were lysed at the indicated time points and NBS1 and PD-L1 expression was assessed using immunoblot (B) or qRT-PCR (C). GAPDH was used as a loading control. (*p<0.05, **p<0.01, ***p<0.001 ) D, MDA-MB-231 cells were plated one day before either H1A (20 ug/ml) or control IgG treatment, with or without the lysosome inhibitor, chloroquine, and expression of PD-L1 was assessed using immunoblot. GAPDH was used as a loading control. E-F, MDA-MB-231 cells were treated with H1A (20 ug/ml) and chloroquine (50 uM). 16 hours later, cells were harvested and immunoprecipitation was performed using CMTM6 (E) or PD-L1 (F) antibodies and CMTM6 and PD-L1 was detected using immunoblot. G, Control and CMTM6 knockout cells were treated either IgG or H1A (20ug/ml) antibody and PD-L1 level was determined by Western blot. H, human PD-L1 expressing murine melanoma B16F10 cells with endogenous PD-L1 knocked out were subcutaneously injected into the flank of C57BL/6 mice. Animals were randomized into four groups (n=5) and treated with either IgG, H1A, IgG + IR or H1A + IR. Bars represent SEM. The p value for the primary comparison of IgG + IR vs H1A + IR was calculated using a t test. (*p<0.05, **p<0.01, ***p<0.001). I, Wild-type (WT, left) or PD-L1 knock out (KO, right) MDA-MB-231 cells were subcutaneously injected into the flank of NOD-SCID mice. Animals were randomized into four groups (n=5) and treated with either control IgG, H1A, IgG + cisplatin or H1a + cisplatin. Bars represent SEM. The p value for the primary comparison of IgG + cisplatin vs H1A + cisplatin was calculated using a t test. (*p<0.05, **p<0.01, ***p<0.001 ) J, MDA-MB-231 cells were subcutaneously injected into the flank of NOD-SCID mice and mice were randomized and treated with either IgG, H1A, Durvalumab, IgG + IR, H1A + IR or Durvalumab + IR. Bars represent SEM. The p value for the comparisons of H1A + IR vs durvalumab + IR, H1A+ IR vs IgG + IR, and H1A + IR vs IgG are shown and were calculated using a t test.(*p<0.05, **p<0.01, ***p<0.001 )
Therefore, we sought to investigate whether H1A could be destabilizing PD-L1 by abrogating CMTM6’s function in shielding PD-L1 from lysosomal degradation. We treated MDA-MB231 cells with H1A or control IgG and observed significantly decreased interaction between PD-L1 and CMTM6 following H1A treatment (Fig. 5E–F), suggesting that H1A induces PD-L1 degradation by blocking the PD-L1/CMTM6 interaction. In contrast, durvalumab did not impact the interaction between PD-L1 and CMTM6, or sensitize MDA-MB-231 cells to IR (Supplementary Fig. 5B–C). To further confirm the role of H1A in PD-L1 regulation, we knocked out CMTM6 in MDA-MB-231 cells and assessed PD-L1 levels following treatment with H1A or control IgG. As anticipated, PD-L1 level was significantly decreased in CMTM6 knockout cells, but PD-L1 level could not be further decreased when these cells were treated with H1A (Fig. 5G). Collectively, these results indicated that H1A acts to destabilize PD-L1 in a CMTM6 and lysosome dependent manner. In turn, PD-L1 is no longer available to bind and stabilize target RNAs including NBS1, and would lead to greater sensitivity to DNA damage.
In order to further examine how H1A impacts cancer therapy we constructed human PD-L1 expressing murine melanoma B16F10 cells with endogenous PD-L1 knocked out. The substitution of PD-L1 did not affect the growth of B16F10 cells in mice (Supplementary Fig 5D–E). These human PD-L1 expressing murine melanoma cells were subcutaneously injected into the right flank of C57BL/6 normal mice and tumors were treated with IR, H1A, or the combination. While H1A or IR alone had modest therapeutic effects on tumor growth, the combination of H1A and IR significantly delayed tumor growth in the immune competent mice (Fig. 5H).
Next, we sought to examine the therapeutic effects of H1A on tumor response to DNA damaging therapy in immune deficient NOD-SCID mice. First, we injected PD-L1 wild-type and PD-L1 knockout MDA-MB-231 cells into immune deficient NOD SCID mice and treated them with H1A, cisplatin, or the combination. Cisplatin alone did not significantly delay tumor growth of PD-L1 wild-type MDA-MB-231 tumors whereas H1A alone resulted in a modest tumor growth delay. Notably, H1A markedly sensitized these PD-L1 positive tumors to treatment of cisplatin (Fig. 5I). Since PD-L1 knockout tumor cells were already more sensitive to cisplatin, the combination of H1A to cisplatin did not provide additional benefit over cisplatin alone (Fig. 5I). In order to further compare the impact of H1A antibody vs FDA approved PD-L1 antibody in radiation therapy in the absence of significant immune effects, we injected PD-L1 wild-type MDA-MB-231 cells into NOD SCID mice and treated these animals with either H1A, an FDA approved PD-L1 antibody (Durvalmab), or control IgG in combination with irradiation. In contrast with Durvalumab and control IgG, H1A significantly delayed tumor growth in combination with irradiation (Fig. 5J supplementary Fig. 5E), supporting an inhibition of intracellular function by H1A.
In summary, these data suggest that PD-L1 antibody H1A sensitizes cancer to DNA damaging therapy by downregulating PD-L1 and inhibiting its role in the DDR. Furthermore, this distinct mechanism of action of PD-L1 antibody (H1A) is distinct from the effect of currently approved PD-L1 inhibitors that mainly regulate anti-tumor immune response by abrogating PD-1/PD-L1 binding.
DISCUSSION
The emergence of immune checkpoint therapy into the clinic, including the approval of monoclonal antibodies targeting PD-L1, has altered the treatment paradigms of many malignancies. However, primary and acquired resistance to these agents remains an important challenge. Here, we have identified a different function of PD-L1 in cancer that is independent of its role in immune regulation. In this report, we establish that PD-L1 acts as a RNA binding protein. By competing with key components of the RNA exosome including an important catalytic component, EXOSC10, and the essential core component, EXOSC4, PD-L1 protects target RNAs from degradation. Multiple PD-L1 target RNAs were identified as being involved in DDR and repair, and their binding and shielding from degradation by PD-L1 promoted resistance to DNA damaging therapy. Importantly, we developed a PD-L1 antibody, H1A, could target intracellular PD-L1’s regulation of the DDR. H1A destabilizes PD-L1 by disrupting its binding with the PD-L1 stabilizer CMTM6, resulting in greater PD-L1 degradation through the lysosome and sensitivity to radiotherapy and cisplatin. The mechanism of action H1A is unique from anti-PD-L1 antibodies that are currently available in the clinic which were unable to mimic the therapeutic effect observed when cancer cells and tumors were treated with H1A. There is considerable interest in investigating combinations of PD-L1 inhibitors with DNA damaging therapies in an effort to optimally prime the anti-tumor immune response (Antonia et al., 2017). Our data suggests that targeting both intracellular and extracellular PD-L1 may be a potential therapeutic approach that not only unleashes the antitumor activity of the patient’s immune system, but also enhances the effects of DNA damaging cancer therapy through inhibition of DDR and repair.
The stability of RNA is regulated by many RNA binding proteins, which in turn affect the functions of one or several RNA-degrading enzymes such as ribonucleases or RNases (Houseley and Tollervey, 2009). The RNA exosome was discovered almost two decades ago and is responsible for regulation of RNAs turnover (Zinder and Lima, 2017). In eukaryotic cells, the RNA exosome core contains a barrel-like structure (constituted by EXOSC4, EXOSC5, EXOSC6, EXOSC7, EXOSC8 and EXOSC9) and a cap (constituted by EXOSC1, EXOSC2 and EXOSC3)(Chlebowski et al., 2013). Besides functioning in RNA turnover, the RNA exosome has also been reported to be involved in cellular processes such as ribosome biogenesis (Knight et al., 2016) and transcription(Fox and Mosley, 2016). The discovery of the relationship between the RNA exosome and PD-L1 in the context of DDR and repair indicates the RNA exosome can also be used as a therapeutic target for cancer therapy.
Besides DDR and repair, pathway analyses revealed that PD-L1 regulates target RNAs involved in a number of cellular processes important to cancer such as metabolism, cell cycle, and protein modification. Further investigation into these other non-immune PD-L1 functions may provide additional clues into the role of PD-L1 in cancer pathogenesis and potential opportunities for therapeutic targeting.
Taken together, our study revealed an intracellular function of PD-L1 in stabilization of RNAs of coding genes required for DDR and repair. PD-L1 has previously been reported to be upregulated following DNA damage(Sato et al., 2017). Our results suggest that increased expression of PD-L1 following DNA damage may not only be important in assisting tumors in avoiding immune attack, but also in promoting resistance to DNA damaging therapy. Therefore, targeting the intracellular function of PD-L1 using a PD-L1 antibody (H1A) that can induce PD-L1 degradation may be a promising way to increase the efficacy of cancer radiation and chemotherapy. Furthermore, this strategy may be tumor specific because PD-L1 is overexpressed in cancer relative to surrounding normal tissues.
STAR ★ METHOD
METHORD DETAILS
Cell lines, PD-L1 knockout mice, antibodies and plasmid
MDA-MB-231 was cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% Fetal Bovine Serum (FBS). HCT116 was cultured in McCoy’s 5A with 10% FBS. PD-L1 WT/heterozygous/knockout mice were obtained from Dr. Haidong Dong’ lab, which were originally created from BALB/C mice.
GAPDH antibody was purchased from Sigma-Aldrich, PD-L1 antibody was purchased from cell signaling Technology (CST). EXOSC10 and EXOSC4 antibodies were purchased from abcam. NBS1 antibody was purchased from Bethyl Lab.
The shRNAs targeting PD-L1, EXOSC10 and EXOSC4 were purchased from Mayo Clinic RNA interference shared resource (RISR) which were inserted into pLKO.1- puro vector. The following is the sequence information of shRNAs for PD-L1 and EXOSC10.
For shRNA knockdown, we used a second generation lentiviral system. Briefly, we co-transfected 293T cells with package plasmids and shRNA (with puro resistance). The cells were then cultured for 48 hours to produce virus. The media containing viruses were harvested to perform infection. Infected cells were than screened with puromycin after 72 hours of culture.
For CMTM6 Knockout, we got CMTM6 Knockout gRNAs as a friendly gift from Dr. Dawson MA’ s lab, and then CMTM6 knockout was performed on MDA-MB-231 according to standard protocol(Burr et al., 2017). Briefly, cells were first transfected with Cas9 expression vector with Blasticidin resistance and then screened by blasticidin. Then cells were transduced with a lentiviral sgRNA expression vector followed by isolation of clones by single-cell dilution. Finally, the knockout efficiency was confirmed by western blot.
The NBS1 RNA sequence was cloned into pGEMT-easy (Promega) vector which was used for in vitro transcription. The RNA motif for the dual-fluorescent reporter was cloned into pmirGLO (Promega). The full length and truncations of PD-L1 was cloned into plvx3-puro and pCMV-HA vectors, respectively.
Dual reporter luciferase assay
Dual reporter luciferase assay was performed using Dual-Luciferase® Reporter Assay System according to manufacturer’s instructions (promega).
RNA extraction, reverse transcription and quantitative RT-PCR
RNA extraction was performed using quick-RNA™ miniPrep kit (zymo) according to the manufacturer’s instructions. Reverse transcription was performed with PrimeScript RT-PCR Kit (Takara). Quantitative RT-PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems).
RNA immunoprecipitation and RNA pull down
Native RNA immunoprecipitation (RIP) was performed using Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to the manufacturer’s instructions. Crosslinked RIP was performed as previously described with minor modification(Gilbert and Svejstrup, 2006). Briefly, cells were first fixed with 0.3% formaldehyde for 10min followed by stopping crosslinking reaction with 0.2M glycine. Then the cells were lysed with FA lysis buffer (50 mM This.HCl, pH=7.5, 140 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 0.1% (w/v) sodium deoxycholate) followed by sonication (10% AMP, 5s sonication, 5s pause, 10 cycles). The supernatant of the cell lysate was then mixed with antibody and protein A/G magnetic beads followed by incubation overnight at 4°C. Then the beads were washed two times of FA lysis buffer, FA500 buffer (50 mM This.HCl, pH=7.5, 500 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 0.1% (w/v) sodium deoxycholate), LiCl buffer (10 mM Tris HCl, pH 8, 250 mM LiCl, 0.5% (v/v) NP-40, 0.1% (w/v) sodium deoxycholate, 1 mM EDTA), and TE/100 mM NaCl buffer (10 mM Tris HCl, pH 8, 1 mM EDTA, 100 mM NaCl). The RNA was then eluted by elution buffer (100 mM Tris⋅Cl, pH 8, 10 mM EDTA, 1% (w/v) SDS) and extracted using Trizol (invitrogen).
RNA pull down was performed as previously described with minor modification (Tsai et al., 2010). In short, In vitro biotin labeled RNAs were mixed with MDA-MB-231 lysate, followed by targeting RNAs-protein complex with streptavidin beads. Western blot analysis was then performed on the co-precipitated proteins.
RNA stability determined by 5-ethynyl Uridine (EU) labeling assay.
5-ethynyl Uridine (EU) labeling assay was performed using Click-iT® Nascent RNA Capture Kit as manufacturer’s instructions. Briefly, cells were first treated with EU (0.1mM) for 24 hours, then EU was removed and cells were continued to be cultured for another 24 hours. Both samples before or after EU removal were harvested to extract RNAs. Then EU labeled RNAs were Biotinylated by click reaction and isolated by streptavidin beads. Then the stability of mRNAs were determined as % of labeled mRNA after removal of EU at different time points divided by % of labeled mRNA before removal.
Poly (A) Tail-length assay
Poly (A) Tail-length assay was performed using the Poly(A) Tail-Length Assay Kit permanufacturer’s instructions. Briefly, total RNAs were first added poly(G/I) and reverse transcribed. Then the poly (A) length was detected by PCR using both gene specific and Universal PCR Reverse Primer.
Immunofluorescence
Immunofluorescence was performed as previously described with minor modification(Huang et al., 2006). Briefly, cells were first fixed by 3% paraformaldehyde then permeabilized with 0.5% Triton X-100, then cells were blocked by 5% goat serum followed by incubating with primary antibody. Fluorescent secondary antibody and DAPI were then incubated with cells to stain the targeted proteins and nucleus. The data was then analyzed by fluorescent microscopy.
Co-immunoprecipitation
Cells were first lysed by NETN buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM, EDTA, 0.5% Nonidet P-40). 3ug antibody and protein A/G beads were then added to the cell lysate. After overnight of incubation, the beads were washed 3–5 times with NETN. The co-immunoprecipitated protein was then analyzed by Western blot.
Nuclear run on
Nuclear run on was performed as previously described (Patrone et al., 2000), with minor modification. Briefly, nuclei of cells were extracted using nuclear extraction buffer (10 mM Tris-HCl, pH 7.4, 3 mM, MgCl2, 10 mM NaCl, 150 mM sucrose and 0.5% NP-40) followed by suspension in freezing buffer (50 mM Tris-HCl, pH 8.3, 40% glycerol, 5 mM MgCl2 and 0.1 mM EDTA). The nuclei were then mixed with the same volume of 2x transcription buffer (200 mM KCl, 20 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 4 mM dithiothreitol (DTT), 4 mM each of ATP, GTP and CTP, 1mM biotin-16-UTP, 200 mM sucrose and 20% glycerol). After 30 min incubation at 30°C, the de novo synthesized transcripts were purified by Dynabeads M-280 (invitrogen). Reverse transcription and quantification was then performed on the purified RNAs.
RIP-seq and RNA-seq
RIP seq was performed using an Illumina HiSeq 2000 sequencer at the Genomic Core facility of the Mayo Clinic, Rochester, MN. Paired-end sequencing libraries were prepared using the TruSeq Stranded Total Sample Preparation kit (Illumina) by the Mayo Clinic sequencing core facilities followed by quality control, cluster generation and sequencing on the Illumina HiSeq 2000 platform. Sequence alignment was performed using Tophat 2.0.14(Kim et al., 2013)against the hg19 human reference genome. Peak calling and annotation were performed using HOMER 4.8.3 (Heinz et al., 2010). The PD-L1 binding RNAs were defined as RNAs that were at least 5-folds higher in the PD-L1 group compared to IgG. Motif scan was performed on peaks called by HOMER for each sample using MEME-ChIP (Machanick and Bailey, 2011) against the HOCOMOCO transcription factor binding site database(Neer, 1973).
RNA seq was performed using an Illumina HiSeq 2000 sequencer at the Genomic Core facility of the Mayo Clinic, Rochester, MN. Paired-end sequencing libraries were prepared using the TruSeq Stranded Total Sample Preparation kit (Illumina) by the Mayo Clinic sequencing core facilities followed by quality control, cluster generation and sequencing on the Illumina HiSeq 2000 platform. RNA-Seq results were delivered as BAM files which were converted to FASTQ format using Picard. FASTQ files were aligned to the hg19 human reference genome using Tophat 2.0.14(Kim et al., 2013). Expression values were calculated with featureCounts v1.4.6-p2(Liao et al., 2014), and differential expression analysis was determined by DESeq2(Love et al., 2014). The downregulated genes for RNA seq were then defined by counts>100, folds>1.5 and p<0.02.
Go analysis was performed using Gene Ontology Consortium website (http://www.geneontology.org/)
Development of anti-PD-L1 antibody
Human 624mel cells were transfected with full-length human B7-H1 (PD-L1) and injected (5 × 106 cells per injection) intraperitoneally into Balb/c mice weekly for 6 weeks. Splenocytes from immunized mice were isolated and fused with A38 cells to form a hybridoma by standard techniques. Then screens were performed by ELISA for reactivity against a recombinant human protein B7-H1, the extracellular domain of B7-H1 (amino acids 19–239) fused with human IgG Fc domain (R&D Systems), and to exclude their cross reactivity with irrelevant recombinant Fc fusion proteins of other B7 family members (R&D Systems).
Animal models
PD-L1 WT and KO MDA-MB-231 cells were subcutaneously injected into the flank of NOD SCID mice (6–8 weeks age female) at 2 × 106 cells per mouse. Animals were randomized when tumors reached 60 mm3. Cisplatin was administrated intraperitoneally (i.p.) at 5 mg/kg for 2 doses on days 1 and 6. Anti-human PD-L1 monoclonal antibody (clone H1A) or isotype control (mouse IgG) was injected at 200 μg/mouse i.p. on days 0, 3, 6, 9 and 12.
For the xenograft model comparing the anti-tumor effects of H1A and Durvalumab, we subcutaneously injected 2 × 106 MDA-MB-231 cells into the flank of NOD SCID mice (6–8 weeks age, female). After tumor formation, IgG, H1A or Durvalumab (200ug per mice) were injected at 200 μg/mouse i.p. 24 hours and again one hour prior to irradiation with 2 Gy administered on day 1 and day 7.
For the B16F10 (humanized PD-L1) xenograft tumor model (6–8 weeks age female), 5 × 105 cells were injected into C57BL/6 mice. Animals were randomized when tumors reached 30 mm3 Anti-human PD-L1 monoclonal antibody (clone H1A) and isotype control (mouse IgG) was injected at 200 ug/mouse i.p. on day 1, 3, 5, 7, 9, while IR (3 Gy per fraction) or mock treatment was applied on day 2, 4, 6, 8 and 10.
Patient derived triple negative breast cancer cell line irradiation
When triple negative breast cancer PDX tumors grew to 1cm in diameter, tumors were dissected, minced and then incubated with the enzyme mix and from a human Tumor Dissociation Kit (Miltenyi Biotech). Human breast cancer tumor cells were isolated using the Mouse Cell Depletion Purification Kit (Miltenyi Biotech) following the manufacturer’s protocol. The purified breast cancer cells were transduced with control or PD-L1 shRNA lentivirus for 2 hours. The transduced cells were seed in 96 well plate and irradiated with indicated dose of ionizing radiation at the following day. Cell spheroids were counted using a Nikon Eclipse Ts100 (Nikon Instruments Inc.), and the susceptibility of the cells to IR was investigated using the percentage of viability.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by the American Society for Radiation Oncology, the National Cancer Institute of the National Institutes of Health under Award Number CA203561, P50CA116201, K12 HD065987, HALT Cancer at X, NCI R21 CA197878 (HD), R01-AI095239 and NIAID R01 AI095239.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Yokoi T, Chiappori A, Lee KH, de Wit M, et al. (2017). Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med 377, 1919–1929. [DOI] [PubMed] [Google Scholar]
- Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, et al. (2017). CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, Irving BA, and Hodi FS (2012). Molecular pathways: next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res 18, 6580–6587. [DOI] [PubMed] [Google Scholar]
- Chen L, and Han X (2015). Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 125, 3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, et al. (2015). Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol 10, 910–923. [DOI] [PubMed] [Google Scholar]
- Chlebowski A, Lubas M, Jensen TH, and Dziembowski A (2013). RNA decay machines: the exosome. Biochim Biophys Acta 1829, 552–560. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8, 793–800. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, and Chen L (1999). B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5, 1365–1369. [DOI] [PubMed] [Google Scholar]
- Fox MJ, and Mosley AL (2016). Rrp6: Integrated roles in nuclear RNA metabolism and transcription termination. Wiley Interdiscip Rev RNA 7, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, and Svejstrup JQ (2006). RNA immunoprecipitation for determining RNA-protein associations in vivo. Curr Protoc Mol Biol Chapter 27, Unit 27 24. [DOI] [PubMed] [Google Scholar]
- Hamanishi J, Mandai M, Matsumura N, Abiko K, Baba T, and Konishi I (2016). PD-1/PD-L1 blockade in cancer treatment: perspectives and issues. Int J Clin Oncol 21, 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Hu Y, Hu M, and Li B (2015). Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci Rep 5, 13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J, and Tollervey D (2009). The many pathways of RNA degradation. Cell 136, 763–776. [DOI] [PubMed] [Google Scholar]
- Huang H, Regan KM, Lou Z, Chen J, and Tindall DJ (2006). CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 314, 294–297. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JR, Bastide A, Peretti D, Roobol A, Roobol J, Mallucci GR, Smales CM, and Willis AE (2016). Cooling-induced SUMOylation of EXOSC10 down-regulates ribosome biogenesis. RNA 22, 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Li X, and Maquat LE (2003). Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 12, 675–687. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N, and Garboczi DN (2008). The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A 105, 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu X, Cao S, Harrington SM, Yin P, Mansfield AS, and Dong H (2016). B7-H1 antibodies lose antitumor activity due to activation of p38 MAPK that leads to apoptosis of tumor-reactive CD8(+) T cells. Sci Rep 6, 36722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, et al. (2017). Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neer EJ (1973). Vasopressin-responsive, soluble adenylate cyclase from the rat renal medulla. J Biol Chem 248, 3742–3744. [PubMed] [Google Scholar]
- Nishimura H, Nose M, Hiai H, Minato N, and Honjo T (1999). Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151. [DOI] [PubMed] [Google Scholar]
- Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, and Zang X (2015). Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med 21, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrone G, Puppo F, Cusano R, Scaranari M, Ceccherini I, Puliti A, and Ravazzolo R (2000). Nuclear run-on assay using biotin labeling, magnetic bead capture and analysis by fluorescence-based RT-PCR. Biotechniques 29, 1012–1014, 1016–1017. [DOI] [PubMed] [Google Scholar]
- Postow MA, Callahan MK, and Wolchok JD (2015). Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 33, 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A, Batth IS, Brownlee Z, Rojas C, Meng QH, Kopetz S, and Li S (2016). Potential role of nuclear PD-L1 expression in cell-surface vimentin positive circulating tumor cells as a prognostic marker in cancer patients. Sci Rep 6, 28910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, et al. (2017). DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 8, 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomecki R, Kristiansen MS, Lykke-Andersen S, Chlebowski A, Larsen KM, Szczesny RJ, Drazkowska K, Pastula A, Andersen JS, Stepien PP, et al. (2010). The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J 29, 2342–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, and Chang HY (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk EL, Schilders G, and Pruijn GJ (2007). Human cell growth requires a functional cytoplasmic exosome, which is involved in various mRNA decay pathways. RNA 13, 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Qin B, Moyer AM, Sinnwell JP, Thompson KJ, Copland JA 3rd, Marlow LA, Miller JL, Yin P, Gao B, et al. (2017). Establishing and characterizing patient-derived xenografts using pre-chemotherapy percutaneous biopsy and post-chemotherapy surgical samples from a prospective neoadjuvant breast cancer study. Breast Cancer Res 19, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinder JC, and Lima CD (2017). Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev 31, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.