

Abstract
Neonatal soft tissues sarcoma is a rare entity that comprises heterogeneous types of tumors. In this article we describe a neonatal case of round-cell sarcoma with an YWHAE-NUTM2B fusion gene. The patient was treated just after birth with neoadjuvant chemotherapy, then surgical resection, but evolution was quickly fatal. This fusion transcript has been reported in endometrial stromal sarcomas and clear cells renal sarcomas but its description in small round-cell sarcomas is recent. To our knowledge, this is the first case report describing this translocation in a newborn patient with soft tissues sarcoma and its clinical tumoral evolution.
Keywords: Sarcoma, Neonatal, Fusion transcript, Chemotherapy, Cancer biology
Introduction
Cancers in neonates comprises only 2% of childhood malignancies [1] and their presentation, clinical behavior, and treatment often differ from others neoplasms. They correspond to a varied group of tumors. The main are teratomas and neuroblastomas, followed by soft tissue sarcomas (STS), leukaemias, renal and then brain tumors. Their characteristics render the diagnosis challenging for the pathologists as normal developing tissues in fetal and neonatal period presents rapid cell proliferation and features similar to neoplasia [1]. In those situations, molecular analysis can be a real help for tumor diagnosis. Another difference in infants compared to children is the immaturity of their renal and hepatic metabolisms and this make them especially vulnerable to side effects of therapy [2]. Therefore, diagnostic and therapeutic issues require a multidisciplinary approach because of the particular immaturity and vulnerability of this population.
Among neonatal tumors, soft tissues sarcomas represent 8–10% of all neoplasms [1]. They are included in a heterogeneous group of malignancies, which originate in mesenchymal cells. Epidemiology is different for infants aged <1 year: rhabdomyosarcomas represent only one third of all STS (32.8%), followed by infantile fibrosarcomas (24.5%) and malignant rhabdoid tumours (14.2%) [3]. Other non-rhabdomyosarcoma soft tissue sarcomas (NRSTS) include a large number of different tumour types and still need to be described in this age group.
This clinical case reports the discovery and management of a new born presenting a small round-cell sarcoma with a YWHAE-NUTM2B fusion gene.
Case Report
We report a case occurring in a new born male which presented a left buttock mass. Parents were non-consanguineous, with no history of early onset malignancy in the family. The pregnancy was unremarkable with normal antenatal ultrasounds. The infant was born at term via normal vaginal delivery and was eutrophic (the youngest of four siblings). The adaptation to extra uterine life was good after birth but systematic clinical examination at 1 day of life revealed a 4cm soft and mobile mass in the left buttock, with a deflection of intergluteal sulcus and anal margin (Fig. 1).
Fig. 1.
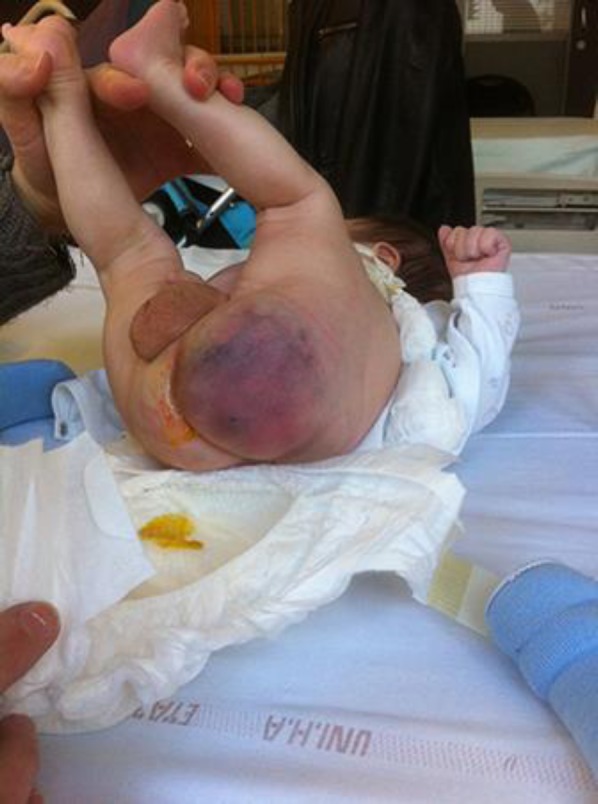
Diagnostic: Soft and mobile mass with bruise-like appearance measuring 4 cm in the newborn's left buttock, with a deflection of intergluteal sulcus and anal margin to the right.
Ultrasound showed a rather fibrous tissue mass of the gluteal region, without hypervascular character. Magnetic resonance imaging performed immediately showed a 49 mm tissue lesion focused on gluteal muscle under the fascia and in contact with the coccyx, with endo-pelvic extension (Fig. 2a). There was no invasion of the left sciatic nerve. The lesion was contiguous to the external sphincter with an aspect of retraction of the left perianal space. The mass was in contact with the rectum without apparent rectal invasion. MRI evoked first the hypothesis of a sacro-coccygeal teratoma, and a yolk sac component was eliminated by adequate alpha-fetoprotein level for this age, and decreased appropriately on repeated blood measurements.
Fig. 2.

Diagnostic and on-therapy MRI. (a) At diagnosis: Sagittal section in T2 sequence. Tumor mass measuring 49 × 37 × 48 mm. (b) After 2 courses of Vincristine Cyclophosphamide: Sagittal section in injected T1 sequence. Tumor mass measuring 72 × 74 × 56 mm. (c) After 4 courses of ICE: Axial section in T1 sequence with gadolinium injection. The white arrow shows the pelvic portion of the tumor in the left ischial fossa. The black arrow shows the intra muscular portion in the left gluteus maximus muscle. (d) After 4 courses of ICE: Coronal section in T2 sequence. The white arrow shows the pelvic portion of the tumor in the left rectal ischio fossa.
Core needle biopsy performed at day 6 showed a malignant tumor proliferation made of round cells, with a scanty eosinophilic cytoplasm and a hyperchromatic nucleus. These monomorphic cells were arranged on a myxoid background (Fig. 3a).
Fig. 3.

(a) Monomorphic round cells (HE ×20 and ×40). (b) Immunohistochemical staining for B-COR.
Immunostaining of the tumor cells was positive for CD56 (heterogeneous); and negative for CD45, desmin, myogenin, SALL4, CD99, EMA, HMB45, PS100, GFAP, KIT, ETV 4, Cyclin B3, FOX2B and NUT. BRG1 and INI1 staining were retained [4].
A multiplex reverse transcription polymerase chain reaction targeting the most common sarcomas including Ewing family sarcomas, synovial sarcomas, alveolar rhabdomyosarcomas and infantile fibrosarcomas transcripts remained negative [5, 6].
The mass presented rapid growth and a control scanner at day 9 showed an increase in the size of the tumor; the mass measured 63 mm of long axis with repression of the rectum.
Computed tomography (CT) of the chest and positron emission tomography-CT of the whole body showed no evidence of metastatic lesion. Cranio-spinal magnetic resonance imaging was normal. The myelogram and the lumbar puncture found no abnormality. Staging evaluation was therefore unremarkable.
After 2 courses of neoadjuvant chemotherapy with Vincristine and Cyclophosphamide (CO) (Vincristine 0.03 mg/kg given on day 1, and Cyclophosphamide 3.3 mg/kg day 1 to 5), the MRI showed a tumor progression (74 mm of maximal axe) (Fig. 2b).
Then, to go further into the characterization of this enigmatic infantile high-grade undifferentiated round-cell sarcoma, a whole transcriptome RNA-sequencing was performed and an YWHAE-NUTM2B fusion gene was identified. Immunostaining of the tumour cells was positive for BCOR (Fig. 3b), and a FISH analysis confirmed the YWHAE rearrangement in 90% of tumor cells. The final diagnosis obtained at day 20 was therefore a high grade round-cell sarcoma with YWHAE-NUTM2B fusion.
Guided by the fusion gene knowledge, the continuation of the treatment was switched to ICE chemotherapy (Carboplatine 15 mg/kg on day 1, Ifosfamide 65mg/kg and Etoposide 3.3 mg/kg day 1 to 3). The multidisciplinary board validated four courses of ICE: after an initial major tumor regression on MRI (56% of tumor volume regression after two courses), the benefits of more courses to reduce surgical sequelae was thought to be insufficient, as lesion measures were stable on the MRI (28 mm in the intra-muscular portion and 34 mm in its endopelvic portion - Fig. 2c, d).
The infant underwent surgery 4 months after the start of the neo-adjuvant treatment. A ventral decubitus allowed a large posterior sagittal incision. As the lesion was no longer palpable, an intraoperative ultrasound scan was necessary. This uncapsulated tumor was poorly limited and hardly differentiable from normal tissues. Limits of resection were the peritoneum, the sciatic nerve and the ischium, the gluteus maximus, the rectum, the anal canal and the external sphincter, and the coccyx which was removed with the operative specimen: the resection was complete but with millimeter margins (R1) because of an effraction of the tumor. A complementary resection beside this effraction showed healthy tissue on frozen examination. The pathological examination revealed a complete necrotic tumor.
Six days after surgery, suture disruption leaded to a colostomy and scar revision. Wound care necessitated 15 days of hospitalization.
Multidisciplinary board discussion advocated for two adjuvant courses of ICE. Local irradiation therapy was not retained due to the age of the patient and the good histologic response.
Twenty-two days after the surgery, and before the chemotherapy was resumed, the infant was hospitalized for sudden onset of intracranial hypertension symptoms with lethargy and vomiting. The cerebral MRI revealed multiple intra-parenchymal brain lesions with triventricular dilation requiring urgent CSF diversion. Massive bleeding at the location of the metastases in the posterior brain fossa led to the death of the patient at the age of 5 months, only 2 days after the beginning of the neurological symptomatology.
Discussion
Soft tissue sarcomas (STS) are rare tumors but occur with an overall annual incidence of 1.6/100,000 in children aged <1 year, which is higher than in older pediatric and adolescent patients (1–4 years, 0.94/100,000, 5–9 years 0.8/100,000, 10–14 years 1.06/100,000) [2]. Among NRSTS, some tumour entities like undifferentiated round cell sarcoma (URCS) remain unclassified [7].
In the literature, these infant URCS are localized in pelvis, trunk, retroperitoneum, head and neck, with a large morphological spectrum. Histologically, these round cell sarcomas share significant overlap with clear cell sarcomas of the kidney (CCSK) and primitive myxoid mesenchymal tumours of infancy (a fuzzy entity initially considered as a more aggressive variant of infantile fibrosarcomas without ETV6-NTRK3 transcript) [8].
As these three entities showing clinical and histological similarities, the team of Kao YC investigated the existence of possible common genetic abnormalities. Among 22 so called infant URCS and 7 primitive myxoid mesenchymal tumors of infancy, they individualized a group sharing the same genetic profiles as the CCSK, but harbouring either an YWHAE-NUTM2B fusion (2 cases) or a tandem internal duplication of BCOR (15 cases) [9]. Remarkably, in CCSK, these somatic genetic alterations are mutually exclusive and found only in infants [10].
Likewise, the majority of biologically tested lesions classified as primitive myxoid mesenchymal tumor of infancy has an internal BCOR duplication [8]. Both BCOR internal duplication and YWHAE-NUTM2B fusion harbor an overexpression of BCOR that might be the oncogenesis starting point. The exact mechanism remains to be understood but internal tandem duplication located at the C terminal of BCOR may affect the conformation of BCOR protein and interfere with PCGF1 binding [11]. This BCOR complex modification could affect gene transcription through an epigenetic mechanism [8, 12]. Based on these data, primitive myxoid mesenchymal tumours of infancy, some round-cell sarcomas in infants, and CCSK are very likely one and the same lesion group. We do not know yet if we can talk about a new biologically defined tumoral entity or simply border forms of tumors, but we may refer to them as “BCOR rearranged sarcomas” [13].
YWHAE-NUTM2A/B fusions were otherwise described in some low grade endometrial stromal sarcoma, with more often aggressive behavior and high-grade transformation [14].
Should we rely on histology or genetics to develop therapeutic strategies? In the above clinical case, of the age and initial lack of knowledge of the fusion transcript, the infant was first treated with CO chemotherapy. This first line chemotherapy failed to reduce the tumor, but after reception of the RNA sequencing, the multidisciplinary team decided to perform ICE chemotherapy. This second line treatment regimen was biologically driven as similar to chemotherapy used in pediatric forms of CCSK [15], and after only 2 courses, the tumor decreased dramatically. However, despite this chemosensitivity, this neonatal sarcoma with YWHAE-NUTM2B fusion was very aggressive and lead to the death of the infant due to cerebral metastases dissemination only 3 weeks after chemotherapy interrupted in order to perform surgical resection of the primary tumor.
Difficulties in therapeutic management together with the aggressiveness of this tumor lead to the death of the patient before 5 months of life.
This clinical report leads us not only to consider age and histology in infant round cell sarcomas management, but also the molecular analyses to prefigure the therapeutic strategy.
Conclusions
In front of this complex new tumoral entity mimicking neonatal immature sacro-coccygeal teratoma, we underline its original presentation (pelvic primary tumor, aggressive behavior despite early chemotherapy, and early development of brain metastasis) and the difficulty of therapy adjustment. As this YWHAE-NUTM2B fusion-positive tumor was very aggressive despite its chemosensitivity, we advocate for a systematic search for somatic translocation in all cases of undifferentiated neonatal round-cell tumors.
More clinical and biological studies are needed to learn about this entity and find a possible curable treatment.
Statements of Ethics
Written informed consent was obtained from the parents of the patient for publication of this case report and any accompanying images.
The authors have no ethical conflicts to report.
Compliant with CNIL declaration MR004 no 2211365 (data collection file).
Disclosure Statement
The authors have no conflicts of interest to report.
Funding Sources
There is no funding to report.
Author Contributions
MG and NC wrote the manuscript. CFC diagnosed this case and reviewed the manuscript. MK, FD and FT performed the histologic interpretation as well as RNA sequencing, immunostaining and FISH analysis. AB interpreted the MRI and selected the images. FH operated the patient and wrote the paragraph concerning the surgery. All authors conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
References
- 1.Orbach D, Sarnacki S, Brisse HJ, Gauthier-Villars M, Jarreau PH, Tsatsaris V, et al. Neonatal cancer. Lancet Oncol. 2013 Dec;14((13)):e609–20. doi: 10.1016/S1470-2045(13)70236-5. [DOI] [PubMed] [Google Scholar]
- 2.Sultan I, Casanova M, Al-Jumaily U, Meazza C, Rodriguez-Galindo C, Ferrari A. Soft tissue sarcomas in the first year of life. Eur J Cancer. 2010 Sep;46((13)):2449–56. doi: 10.1016/j.ejca.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Ferrari A, Orbach D, Sultan I, Casanova M, Bisogno G. Neonatal soft tissue sarcomas. Semin Fetal Neonatal Med. 2012 Aug;17((4)):231–8. doi: 10.1016/j.siny.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Boudjemaa S, Petit A, Dainese L, Bourdeaut F, Lipsett J, Coulomb A. Congenital disseminated extrarenal malignant rhabdoid tumor. Pediatr Dev Pathol. 2015 Sep-Oct;18((5)):401–4. doi: 10.2350/14-07-1533-CR.1. [DOI] [PubMed] [Google Scholar]
- 5.Qadir MA, Zhan SH, Kwok B, Bruestle J, Drees B, Popescu OE, et al. ChildSeq-RNA: A next-generation sequencing-based diagnostic assay to identify known fusion transcripts in childhood sarcomas. J Mol Diagn. 2014 May;16((3)):361–70. doi: 10.1016/j.jmoldx.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Machado I, Navarro L, Pellin A, Navarro S, Agaimy A, Tardío JC, et al. Defining Ewing and Ewing-like small round cell tumors (SRCT): the need for molecular techniques in their categorization and differential diagnosis. A study of 200 cases. Ann Diagn Pathol. 2016 Jun;22:25–32. doi: 10.1016/j.anndiagpath.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher CD. The evolving classification of soft tissue tumours - an update based on the new 2013 WHO classification. Histopathology. 2014 Jan;64((1)):2–11. doi: 10.1111/his.12267. [DOI] [PubMed] [Google Scholar]
- 8.Kao YC, Sung YS, Zhang L, Huang SC, Argani P, Chung CT, et al. Recurrent BCOR internal tandem duplication and YWHAE-NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy. Overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol. 2016 Aug;40((8)):1009–20. doi: 10.1097/PAS.0000000000000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy A, Kumar V, Zorman B, Fang E, Haines KM, Doddapaneni H, et al. Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun. 2015 Nov;6((6)):8891. doi: 10.1038/ncomms9891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karlsson J, Valind A, Gisselsson D. BCOR internal tandem duplication and YWHAE-NUTM2B/E fusion are mutually exclusive events in clear cell sarcoma of the kidney. Genes Chromosomes Cancer. 2016 Feb;55((2)):120–3. doi: 10.1002/gcc.22316. [DOI] [PubMed] [Google Scholar]
- 11.Wong SJ, Gearhart MD, Taylor AB, Nanyes DR, Ha DJ, Robinson AK, et al. KDM2B recruitment of the polycomb group complex, PRC1.1, requires cooperation between PCGF1 and BCORL1. Structure. 2016 Oct;24((10)):1795–801. doi: 10.1016/j.str.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gearhart MD, Corcoran CM, Wamstad JA, Bardwell VJ. Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol Cell Biol. 2006 Sep;26((18)):6880–9. doi: 10.1128/MCB.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson S, Perrin V, Guillemot D, Reynaud S, Coindre JM, Karanian M, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. 2018 May;245((1)):29–40. doi: 10.1002/path.5053. [DOI] [PubMed] [Google Scholar]
- 14.Aisagbonhi O, Harrison B, Zhao L, Osgood R, Chebib I, Oliva E. YWHAE Rearrangement in a purely conventional low-grade endometrial stromal sarcoma that transformed over time to high-grade sarcoma: importance of molecular testing. Int J Gynecol Pathol. 2018 Sep;37((5)):441–7. doi: 10.1097/PGP.0000000000000451. [DOI] [PubMed] [Google Scholar]
- 15.Sudour-Bonnange H, Dijoud F, Leclair MD, Rocourt N, Bergeron C. [Clear cell sarcoma of kidney in children] Bull Cancer. 2016 Apr;103((4)):402–11. doi: 10.1016/j.bulcan.2016.01.017. [DOI] [PubMed] [Google Scholar]