

ABSTRACT
Osteoarthritis (OA) is one of the most common bone diseasesas it is reported that the impact of knee osteoarthritis symptomatic form is estimated at 240/100,000 people per year. The inflammation of articular cartilageis thought to be the pathologic drive for development of this disease. HMGB1(high mobility group box-1), a regulatory factor for gene transcription, could stimulate inflammation response. However, theexact regulatory role of HMGB1 in the inflammation of articular cartilage still need to be elucidated. In the current study, we used Quantitative Real-Time PCR(Q-PCR) to detect them RNA levels of Collagen Type II Alpha 1(Col2a1), Aggrecan, MMP3(Matrix Metallopeptidase 3), MMP13, ADAMTs4 and ADAMTs5; Enzyme-Linked Immunosorbent Assay(ELISA) was used to detect the content of IL-1β and calpain protein; Cell apoptosis was evaluated by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling(TUNEL) assay and flow cytometryanalysis; Western blot and immunofluorescence assays were applied to assess the expression of HMGB1; Lastly autophagic activity was mainly verified by monodansylcadaverine (MDC) staining. Our data revealed that in the early stage of chondrocyte inflammation(3 and 6 h of LPS stimulation), cytosolic HMGB1 attenuated inflammation response by facilitating cell autophagy and preventing cell apoptosis. While in the late stage (24 and 48 h of LPS stimulation), the extracellular HMGB1 stimulated inflammation reaction and contributed to the cartilage destruction in OA.
KEYWORDS: Osteoarthritis, HMGB1, inflammation, Col2a1, autophagy, apoptosis
Introduction
Osteoarthritis (OA) is one of the most prevalent skeletal diseases in the world [1]. It is estimated that 10% of men and 18% of women above the age of 60 are suffering from OA [2], due to the biomechanical and biochemical changes in the joint [3]. Articular cartilage mainly consists of chondrocytes and extracellular matrix. The later is riched in collagen type II and aggrecan [4]. There is a variety of factors that play important roles in the pathogenesis of OA, including proinflammatory mediators such as TNF-alpha IL-1, MMPs and ADAMTs [5]. Besides, cell apoptosis and autophagy are found to be crucial in the development of OA [6].
High mobility group box 1 (HMGB1) belongs to high mobility group and contains HMG-box domain [7]. Initially, HMGB1 was identified as a nuclear protein which can specifically recognize cruciform DNA and regulate chromatin remodeling [7]. Thereafter, HMGB1 was found to regulate gene expression and cell differentiation [8]. Later, HMGB1 was discovered as a secreted cytokine that regulates immunity. For example, it was reported that HMGB1 was probably a late mediator of endotoxin lethality in mice [9], secreting into the extracellular environment by necrotic cells and triggering inflammation response [10,11] and more extensively physiological or pathological activities [12,13]. Importantly, the extracellular HMGB1 was verified to play crucial roles in inflammation and sepsis [14,15], or even in other diseases such as neurocognitive dysfunction [16], ischemia-induced disruption [17], Alzheimer’s disease and trauma [18] and venous thrombosis [19]. Recently, HMGB1 was found to have close association with the pathogenesis of OA. Heinola T et al. discovered that HMGB1 could be translocated to the cytoplasm of chondrocytes or the extracellular matrix in osteoarthritic cartilage [20], and in synovial membranes and osteochondral fragments of OA patients, the levels of HMGB1 were high [21]. HMGB1 also played regulatory roles in the inflammation reaction of osteoarthritic synoviocytes and chondrocytes [22–24]. Although increasing evidence showed that HMGB1 potentiated the development of OA, the exact mechanism about how HMGB1 regulates the progression of OA, specifically its effects on chondrocytes and cartilage degradation are still uncertain.
In this study, we sought to investigate functions and molecular mechanism of HMGB1 using OA-like cell and animal models induced by LPS and monosodium iodoacetate (MIA) respectively. We provided the first evidence that HMGB1 regulated the inflammation reaction of chondrocytes in a location (or time)-dependent manner.
Materials and methods
Cell isolation and culturing
Mouse chondrocytes were isolated from the cartilage of knee joints of C57BL/6 mice (Hunan Slake Jingda Experimental Animal Co. Ltd., Changsha, China). The knee joint cartilage was cut and sectioned to small pieces, subsequently treated with 0.25% Trypsin (Invitrogen, USA) for 0.5 h and further treated with collagenase type-II for 5 h. The isolated chondrocytes were maintained with DMEM medium (Hyclone, USA), supplemented with 10% fetal bovine serum (FBS) (Hyclone, USA) and antibiotics (1% penicillin and streptomycin) (Invitrogen, USA). Chondrocytes were passed to next generation when the cells reached one another.
Animals
In total, 36 male C57BL/6 mice with the weight ranging from 20 to 30 g were purchased from Hunan Slake Jingda Experimental Animal Co. Ltd., China. The study was approved by Ethics Committee of the Second Xiangya Hospital of Central South University. The mice were then randomly assigned to six groups (six mice per group): 1) control (con); 2) monosodium iodoacetate (MIA)-3w (3 weeks after MIA injection, MIA-3w), 3)MIA-4w (4 weeks after MIA injection, MIA-4w), 4)MIA-6w (6 weeks after MIA injection, MIA-6w), 5) MIA+Glycyrrhizin (GL) (3w) and 6) MIA+GL (6w). The mouse model of OA was induced with MIA, as described previously [25,26]. Briefly, animals were anesthetized using 5% isoflurane in 100% O2(4.5 L/min) until the flexor reflex was abolished. The skin overlying the right knee joint was shaved and swabbed with 100% ethanol. A 27-gauge needle was introduced into the joint cavity through the patellar ligament and 3 mg of MIA, an irreversible NADPH inhibitor, diluted in 50 µL 0.9% saline was injected into the joint (intra-articular, i.a.) to induce OA-like lesions. As to the treatment of glycyrrhizic acid (Selleck, USA), an inhibitor of HMGB1, it was injected into the joints of the mice at a dose of 100 mg/kg/day.
Inflammation of chondrocytes induced by LPS
Chondrocytes were pre-seeded in 6-well plate and maintained with growth medium for overnight. LPS (Sigma-Aldrich, USA, 20 μg/ml) was added to the medium to induce OA-like inflammation response, as described previously [27]. In order to produce an early stage of inflammation, the chondrocytes were treated with LPS for 3 to 6 h, and produce a late stage of inflammation using a treatment for 24 or 48 h. As to the Glycyrrhizin treatment, its working concentration was 25 μM.
RNA extraction and real-time PCR
Cells were harvested and animal tissues was freeze fractured in liquid nitrogen. Next, total RNA of chondrocyte cells or animal tissues was isolated by TriZol reagent (Thermo Fisher Scientific, USA), according to the manufacturers’ instructions. cDNA was synthesized from total RNA with High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, USA). Real-time PCR was performed using SYBR green Premix Ex Taq reagent kit (Takara, Japan). Primer sequences are Col2a1 (sense: TGGTGGAGCAGCAAGAGCA; antisense: TCAGTGGACAGTAGACGGAGGAA), Aggrecan (sense: TTCTGCTTCCGAGGTGTGTC; antisense: TCGAGTGACGATCCAGTCCT), MMP3 (sense: AGGTCTGGGAGGAGGTGA; antisense: GAGCAGCAACCAGGAATAG), MMP13 (sense: GCCAGAACTTCCCAACCA; antisense: ACCCTCCATAATGTCATACCC), ADAMTs4 (sense: GGAATGGTGGAAAGTATTGTGA; antisense: GAGGTCGGTTCGGTGGTT), ADAMTs5 (sense: AAGGTTACAGATGGGACAGAATG; antisense: GCTTTGAGCCGATGATGC), β-actin (sense: AGGCCCCTCTGAACCCTAAG; antisense: CCAGAGGCATACAGGGACAAC). The mRNA expression levels were calculated using 2−ΔΔCt method [28], in accordance with previous reports.
Western blot assay
The reagent RIPA lysis buffer (Thermo Fisher Scientific, USA) was used with protease inhibitor PMSF(selleck, USA) to extract the total protein of chondrocytes or cartilage tissue. Total protein concentration was determined by BCA kit (Thermo Fisher Scientific, USA). The lysis was then mixed with protein loading buffer (Thermo Fisher Scientific, USA) and loaded to the 12% SDS-PAGE gel for electrophoresis. Afterward, the gel was transferred to PVDF membrane (Thermo Fisher Scientific, USA). After incubation with blocking buffer, the membrane was incubated with primary antibody solution overnight at 4°C. Then, the membrane was washed with buffer three times and incubated with secondary antibody solution for another hour at room temperature. Finally, the signal was detected by ECL method. The antibodies for HMGB1, Caspase-1, Beclin-1, ATG5, p-JNK were purchased from ABCAM (UK) with a dilution around 1:1000 according to manufacturer’s instructions. p-ERK antibody was from Santa Cruz (USA).
RNA interference
The expression of HMGB1 in the chondrocytes was knocked down by the transfection of siRNAs, which were custom synthesized by Shanghai Genechem Co., LTD, China. The chondrocytes were maintained with growth medium, and then siRNAs were transfected to the chondrocyte using Lipofectamine 2000 (Life technology, CA, USA). For transfection, the cells were incubated with Lipofectamine 2000 and FBS-free medium for 4 h. Afterward, this medium was replaced by complete medium. The interference of HMGB1 was confirmed by Western blot.
Enzyme-linked immunosorbent assay (ELISA)
The content of extracellular HMGB1 and IL-1β was determined by ELISA kit (Biocompare, USA), according to the manufacturer’s instruction. Briefly, the supernatant was collected for the concentrations of cytokines. Measurement was conducted, followed by assay procedures. Absorbance value cytokines (optical density value) were measured at 450 nm and the concentrations of cytokines were calculated by using standard curve.
Immunofluorescence staining
The location of HMGB1 in the chondrocytes was traced by immunofluorescence staining. Cells were seeded on glass in 6-well plate. At the indicated time, the chondrocytes were fixed with 4% PFA(Paraformaldehyde) for 1 h and subsequently treated with 0.5% Triton X-100 for 10 min. Next, the cells were treated with 1% BSA (Bull Serum Albumin) to reduce the background. Afterward, it was then incubated with primary and secondary antibody solution, respectively, and the DAPI (4ʹ,6-diamidino-2-phenylindole) was used to detect cell nucleus. The samples were lastly observed using Leica microscopy.
Safranin O staining
The mice were sacrificed and the whole knee joints were dissected, and then fixed in 4% paraformaldehyde (PFA) buffered with PBS (phosphate buffer saline,pH 7.4) for 4 h at 4°C. The samples were decalcified for 2 weeks with 10% EDTA (Ethylene Diamine Tetraacetic Acid, pH 7.4) at 4°C. After dehydration with several concentrations of ethanol and embedded in paraffin, sections were cut from the whole medial compartment of the joints and were stained with Safranin O solution.
Toluidine blue (TB) staining
The cells were collected and fixed with 4% paraformaldehyde for 15 min at 25°C, followed by washing with PBS twice. Subsequently, the cells were stained with 0.5% (W/W) toluidine blue for 30 min at 25°C. The samples were detected with fluorescence microscope.
Monodansylcadaverine (MDC) staining
Autophagic vacuoles were detected with monodansylcadaverine (MDC) staining. Briefly, cells were digested with 0.25% trypsin for minutes and seeded on 6-well plate, and then the treated with LPS (lipopolysaccharides) for different time . The cells were collected, centrifugated, and then washed with PBS. About 80μL cell suspension was mixed with 20μL MDC staining solution for around 30 min at room temperature. Cells were then dropped onto slide glasses. The autophagic vacuoles were observed with fluorescence microscope.
Transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) staining
All steps were according to the manufacturer's instructions of TUNEL kit (KeygenBiotech, Nanjing, Jiangsu, China). In brief, chondrocytes grown on coverslips were exposed to LPS with different treatment times. TUNEL-positive cells were counted under fluorescence microscope.
Flow cytometry
Cell apoptosis was detected by Flow cytometry according to the instructions in the Annexin-V-FITC cell apoptosis detection kit (Sigma, USA). Briefly, after different treatment cells were collected, resuspended and washed with proper volume of PBS. Subsequently, Annexin-V-FITC binding buffer was used to resuspend the cell pellet, and then Annexin-V-FITC (20μg/mL) was added into the mixture, followed with 15-min incubation on ice. Lastly, propidium iodide(PI,50μg/mL) was added into the mixture for 2 min’s incubation on ice in the dark before it was measured using flow cytometry(CytoFLEX, Beckman Coulter, USA).
Calpain activity assay
Calpain activity was measured as the cleavage of fluorogenic substrate (Calbiochem, Germany) using fluorescence plate reader (Biotek Synergy 2, USA). Briefly, 10 mg of cell lysis was homogenized in lysis buffer and incubated on ice for 30 min. The samples were then centrifuged in prechilled tabletop centrifuge for 15 min. The protein concentration was determined by the bicinchoninic acid method. Fifty microliters of sample was used. The calpain activity in each sample was expressed as units per milligram of protein per min.
Hematoxylin-eosin (H&E) staining
The lateral and medial sides of the femoral condyle and tibial plateau were fixed with 10% neutral-buffered formalin (pH7.4) and decalcified with 20% EDTA solution. The decalcified femur and tibia were embedded in paraffin, and 5 mm microsections of them were prepared and stained with hematoxylin and eosin (H&E).
Statistical analysis
All results were expressed as the mean ± standard error of the mean (SEM). Statistical analyses was performed by GraphPad Prism 7 (GraphPad Software, La Jolla, CA). The differences between the mean values were analyzed for significance using a one-way analysis of variance (ANOVA). Values of p < 0.05 were considered to be statistically significant.
Results
Expression levels of HMGB1 in the LPS (lipopolysaccharides)-induced chondrocytes and MIA (monosodium iodoacetate)-induced mouse model of OA
We first created inflammatory response of primary chondrocyte cells (isolated from articular cartilage of C57BL/6 mice) using LPS (20 μg/ml) treatment. Consistent with previous reports [29], 48 h stimulation of LPS significantly suppressed the expression of cartilage markers, including Col2a1 and Aggrecan (Figure1a&b). Oppositely, the expression of cartilage matrix degradation-related enzymes such as MMP3, MMP13, ADAMTs4 and ADAMTs5, was notably increased (Figure 1c). Additionally, the expression of the critical inflammatory factor in OA, IL-1 beta, was also found to be stimulated by the treatment of LPS (Figure 1d). In addition to this cell model, we also produced an OA mouse model using MIA treatment (Figure 7d), as demonstrated by Safranin O staining result that the level of proteoglycan in OA mice femur tissue was reduced significantly, compared to the control group (Figure 1e).
Figure 1.
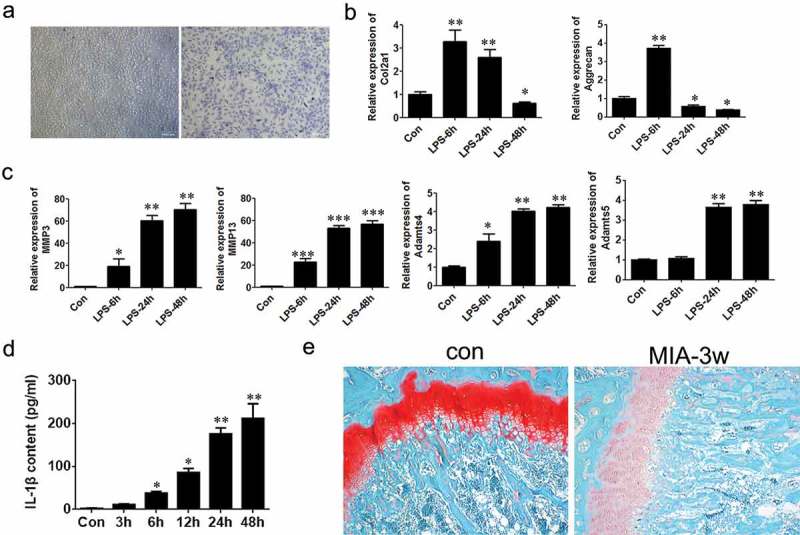
Establishment of osteoarthritis models using chondrocyte cells and mice.
(a) Toluidine blue staining of chondrocytes (×100, scale bars = 1000μm). (b) Relative mRNA levels of Col2a1 and Aggrecan in chondrocytes treated with LPS were detected by Q-PCR. (c) Relative mRNA levels of MMP3, MMP13, ADAMTs4, ADAMTs5 in chondrocytes treated with LPS. (d) Content of secreted IL-1β in medium was analyzed with ELISA assay. (e) The femur tissues of the mice were stained with Safranin O (×100). Data were expressed as mean ± SEM (n = 3). Values with p < 0.05 were considered to be statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group.
Figure 7.
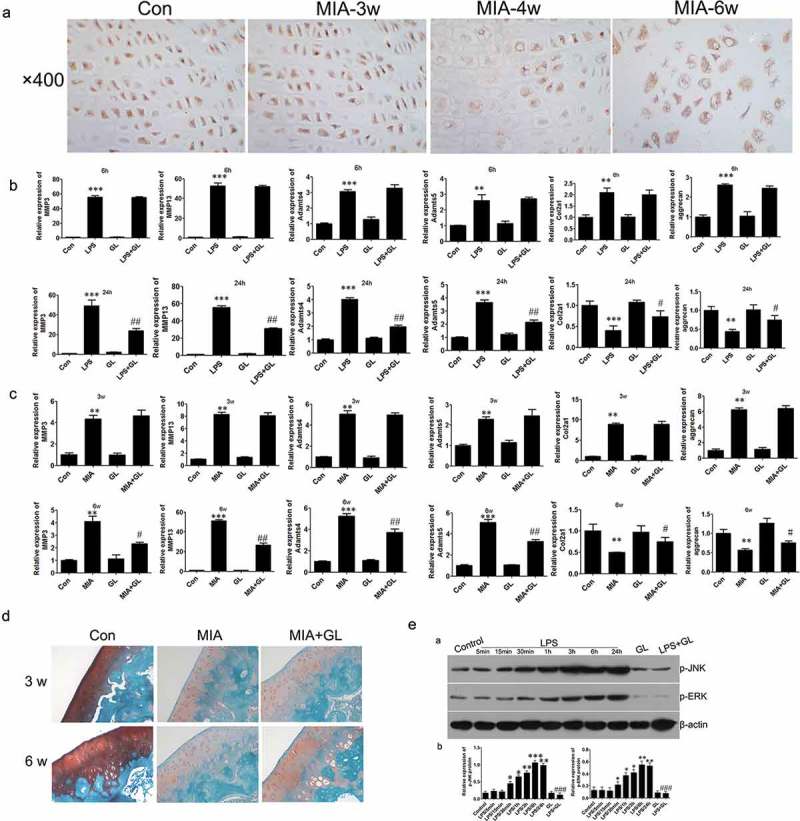
Regulation of autophagy in mice.
(a) IHC assay was performed to assess HMGB1 expression level (×400). Relative mRNA levels were evaluated using Q-PCR in vitro(b) and in vivo(c) respectively (d) Safranin O staining of the joint tissue (×100). (e) Western blot analysis of the expression of p-JNK and p-ERK. Data are expressed as Mean±SEM (n = 3). Values with p < 0.05 were considered to be statistically significant. **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05 vs. LPS 24 h group.
Next, we evaluated the expression of HMGB1 in chondrocytes by Western blot assay. It was found that HMGB1 was suppressed after treatment with LPS for 6 h, whereas the expression was greatly promoted after 12 h(Figure 2a). These results demonstrated a changing expression pattern of HMGB1 between a short-time and a relative long-time of LPS stimulation. We then verified the localization of the HMGB1 using immunofluorescence assay, as shown in Figure 2b. After 3 h or 6 h of stimulation, more HMGB1 was located in the nucleus, While most of it translocated to cytoplasm after 12 to 48 h stimulation, compared with the control group. These results revealed that in the early stage of the inflammation (3–6 h of stimulation with LPS), HMGB1 accumulated in the nucleus, but it was translocated to the cytoplasm in the late stage of the inflammation (24–48 h of stimulation with LPS). Besides, HMGB1 was even secreted to the medium after 24 h stimulation with LPS, as shown from the ELISA assay result (Figure 2c), suggesting that HMGB1 was secreted to the extracellular matrix during the late stage of inflammation.
Figure 2.
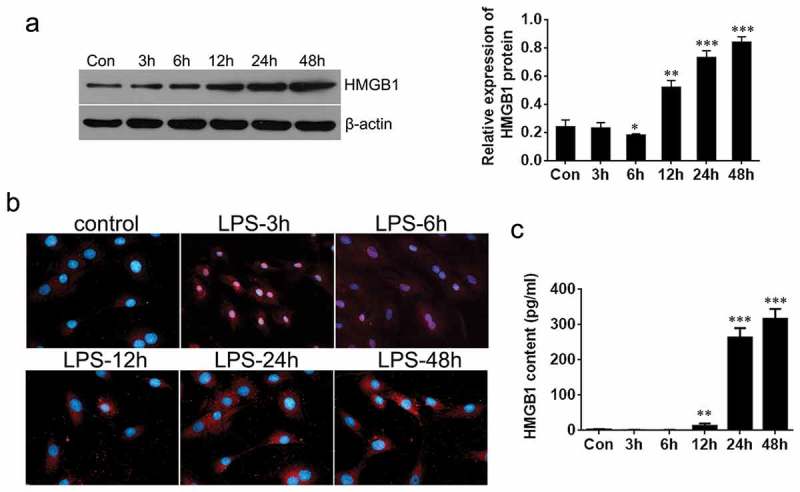
Expression of HMGB1 in chondrocytes treated by LPS. (a) Expression levels of HMGB1 after LPS treatment was detected by Western blot. (b) Immunofluorescence detection of HMGB1 at different time (×200). (c) The extracellular HMGB1 content was evaluated with ELISA assay. Data are expressed as Mean ± SEM (n = 3). Values with p < 0.05 was considered to be statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Autophagy and apoptosis in chondrocyte inflammation
Next, the autophagy and apoptosis of chondrocytes induced with LPS for different durations of time were investigated. The results from MDC staining indicated that there was strong autophagic activity in the chondrocytes at 3 h or 6 h. However, during the late stage of the inflammation, it was partly abolished (Figure 3a). Furthermore, the signal of autophagy marker LC-II in LPS-treated and RFP-GFP-LC3-transfected chondrocytes also showed that intense autophagy in the early stage of inflammation (3 and 6 h) reduced in the late stage of the inflammation (24 h)(Figure 3b). Lastly, we assessed the apoptosis of chondrocytes using flow cytometric analysis and Western blot. The data revealed that there was little apoptosis during the early inflammation (3 and 6 h). It was, however, the apoptosis of chondrocytes notably accelerated during the late stage of the inflammation as apoptosis marker caspase-1 level increased significantly (Figure 3c&d).
Figure 3.
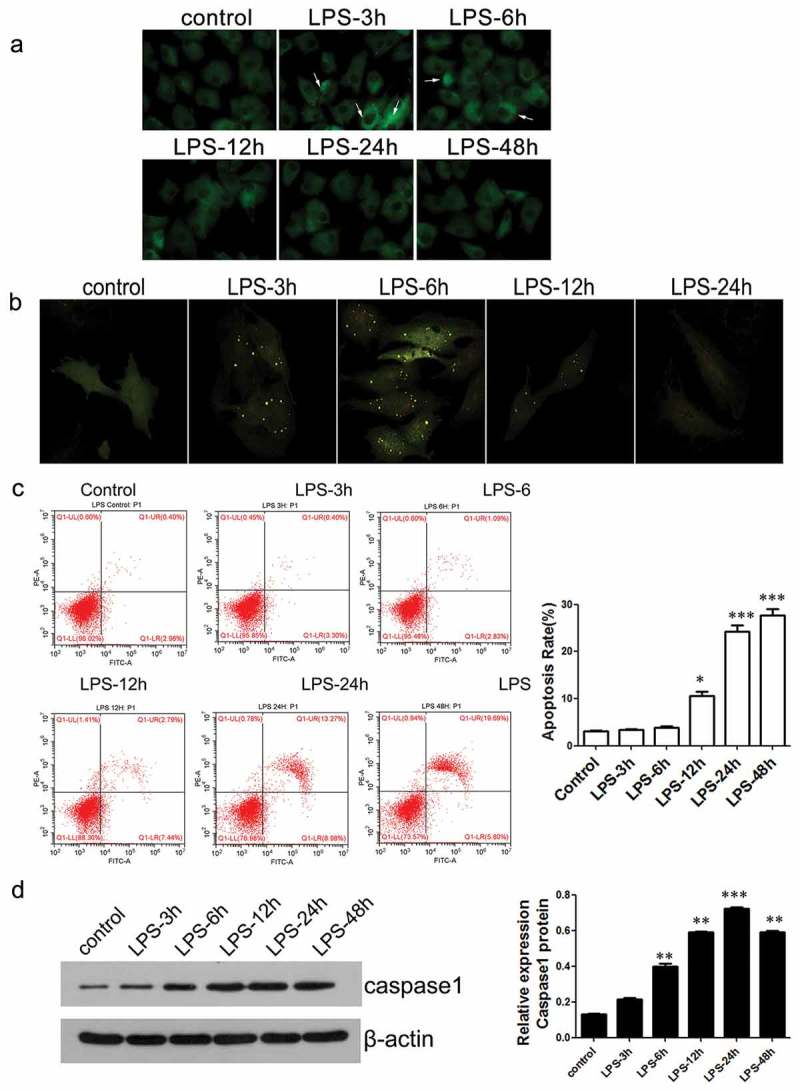
The levels of autophagic activity and cell apoptosis in chondrocytes after different treatment time with LPS.
(a) Chondrocyte autophagic activity was detected by MDC staining (×400). (b) Expression of intracellular RFP- GFP-LC3 was observed using fluorescence microscope (×200). Detection of apoptosis of chondrocytes by flow cytometry. (d) The expression level of caspase-1 was detected using Western blot. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control
Cytosolic HMGB1 affected chondrocyte inflammation by regulating the balance of autophagy and apoptosis
In order to ascertain the regulatory function of HMGB1 in OA. We used specific siRNA (we got three siRNA duplexes, named randomly siRNA-1, siRNA-2 and siRNA-3, respectively) to supress HMGB1 in mouse chondrocytes and chose siRNA-3 for the next study (Figure 4a). Although si-HMGB1 led to moderate suppression of osteoarthritis-related genes in the early stage of the inflammation (6 h), including MMP3, MMP13, ADAMTs4 and ADAMTs5 (Figure 4b), the expression of these factors significantly increased in the late stage of the inflammation (24 h) in the LPS-induced OA phenotype of chondrocytes (Figure 4c). Further studies revealed that in the early stage of the inflammation (6 h), si-HMGB1 promoted autophagic activity in chondrocytes. However, in the late stage of the inflammation (24 h), no autophagic activity could be observed in neither the siRNA group or the scramble (Figure 5a&b). Moreover, it was seen that si-HMGB1 group presented a moderate inhibition of cell apoptosis at 6 h stimulation of LPS. Apoptosis of chondrocytes increased when LPS treated for 24 h and knockdown of HMGB1 led to a more lower apoptotic activity (Figure 5c&d). All these evidence indicated that in the late stage of the inflammation (24 h),cytosolic HMGB1 played roles in regulating autophagy and apoptosis of chondrocytes.
Figure 4.
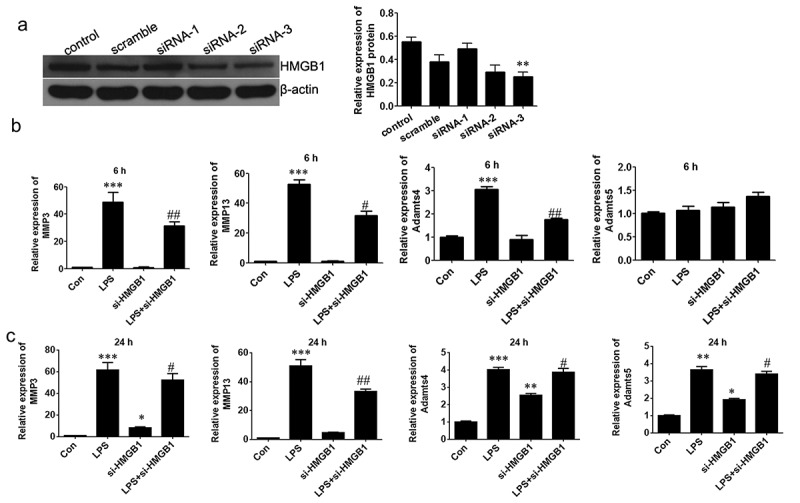
HMGB1 regulates the expression levels of several osteoarthritis-related genes in chondrocytes.
(a) Interference of siRNAs for HMGB1 was confirmed by Western blot. Relative mRNA levels were evaluated at 6 h (b) and 24 h (c) respectively. Data are expressed as Mean ± SEM (n = 3). Values with p < 0.05 were considered to be statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05, ##P < 0.01 vs. LPS group.
Figure 5.
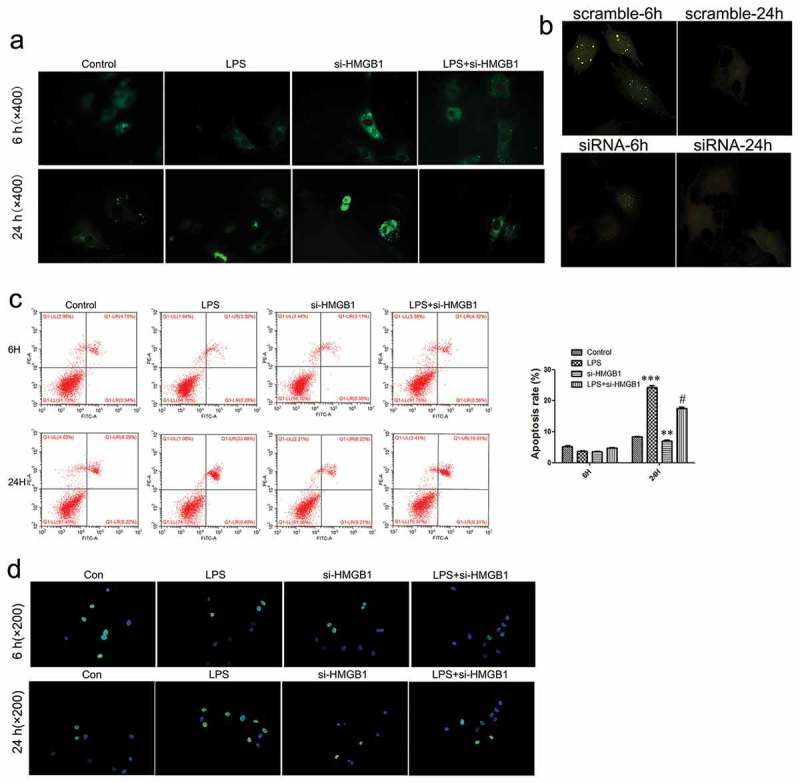
HMGB1-mediated autophagic activity and apoptosis of chondrocytes.
(a) Autophagic activity was detected by MDC staining (×400). (b) Expression of intracellular GFP-LC3 was observed under the fluorescence microscope (×200). (c) Flow cytometry analysis of apoptosis in chondrocytes. (d) Detection of apoptosis in chondrocytes using TUNEL staining (×200). Data are expressed as Mean ± SEM (n = 3). Values with p < 0.05 were considered to be statistically significant. **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05 vs. LPS group.
HMGB1-regulated autophagy and apoptosis by modulating calpain activity
To confirm the effects of HMGB1 on regulating autophagy of chondrocytes, we performed an enzyme assay to assess the activity of calpain protein. Interestingly, si-HMGB1 could decrease the activity of calpain, as we expected(Figure 6a). Additionally, to ascertain the role of calpain in HMGB1-regulated autophagic activity we used MDL28170, a specific calpain inhibitor, to block calpain activity. The result from enzyme activity assay showed that the calpain activity was stimulated by LPS, while neutralized by MDL28170 treatment and the knockdown of HMGB1 (Figure 6b). Furthermore, results from immunofluorescence-confocal assay revealed that inhibition of calpain significantly elevated autophagic activity (Figure 6c), reduced LPS-induced cell apoptosis and significantly suppressed the si-HMGB1-induced apoptosis of chondrocytes (Figure 6d). Finally, inhibition of calpain activity blocked the LPS-induced cleavage of Beclin1 and ATG5 (Figure 6e).
Figure 6.
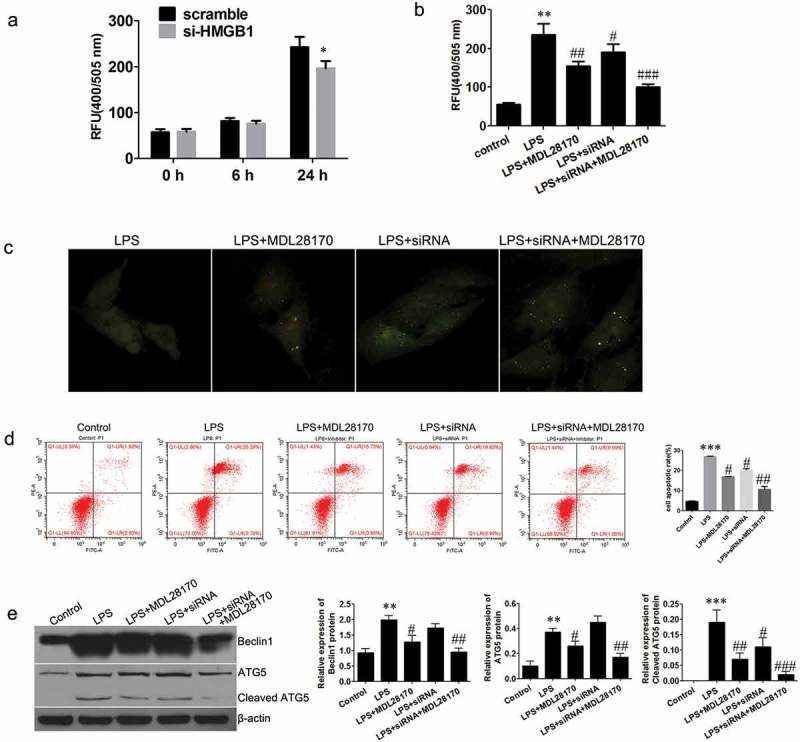
Regulation of autophagy in chondrocytes.
(a) & (b)Detection of calpain activity. (c) Expression of intracellular GFP-LC3 was observed under the fluorescence microscope (×200). (d) Apoptosis levels of chondrocytes were detected using flow cytometry. (e) The expression of Beclin1, ATG5, and cleaved ATG5. Data are expressed as Mean±SEM (n = 3). Data are expressed as Mean ± SEM (n = 3). Values with p < 0.05 were considered to be statistically significant. **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05 vs. LPS group.
Secreted HMGB1-stimulated inflammation in chondrocytes
To clarify the regulatory function of the secreted HMGB1 in the pathogenesis of OA, we firstly detected the level of secreted HMGB1 in the culture medium and the cartilage matrix of OA. HMGB1cytokine was seen in the medium of chondrocytes after 24 and 48 h stimulation of LPS (Figure2c). In the OA animal model, the secreted HMGB1 can be detected at 4 to 6 weeks (Figure 7a). These results suggested that the chondrocytes secrets HMGB1 to the extracellular matrix at the late stage of inflammation. Next, we used Glycyrrhizin (GL), a potent inhibitor of HMGB1, to counteract the cytokine activity of HMGB1. As shown in Figure7b, in the early stage of inflammation of chondrocytes (6 h), Glycyrrhizin had little effect on the expression of the osteoarthritis-related genes. However, in the late stage of inflammation (24 h), MMP3, MMP13, ADAMTs4 and ADAMTs5 were significantly blocked by Glycyrrhizin treatment. Furthermore, the expression of Col2a1 and Aggrecan was rescued by the treatment with Glycyrrhizin.
We next confirmed this using an animal model. Local injection of Glycyrrhizin did not impact the progression of OA in the early stage of OA (3 weeks after MIA injection), as real-time PCR results revealed that the expression levels of OA-related biomarkers did not change notably. However, in the late stage of OA (6 weeks after MIA injection), Glycyrrhizin treatment abolished the promotion of OA-related biomarkers like MMP3, MMP13, ADAMTs4 and ADAMTs5. Importantly, the dysregulation of proteoglycans could be partly blocked by Glycyrrhizin as Safranin O staining result showed (Figure 7d). Collectively, these results demonstrated that blockage of HMGB1 by Glycyrrhizin in the late stage of inflammation could retard the progression of OA in vivo.
Extracellular HMGB1 regulated the inflammation of chondrocyte through activation of ERK and JNK pathway
To investigate the exact mechanism of HMGB1, we analyzed the activity of ERK and JNK pathway as they were involved in inflammation response. Interestingly, the level of p-ERK1/2 in chondrocytes was significantly increased by LPS treatment for 24 h or more, and this activation was partly blocked by Glycyrrhizin treatment (Figure7e). These results suggested that HMGB1 exerts inflammatory effects on chondrocytes by activating ERK signaling. Further, we continued to evaluate the activation of JNK signaling also. Similar to the ERK signaling, further activation of JNK pathway and an inhibition of it by Glycyrrhizin treatment were verified by Western blot assay. These results indicated that extracellular HMGB1 could regulate inflammation response of chondrocytes in the late stage of inflammation, at least, partly through ERK and JNK pathway.
Discussion
HMGB1 was initially identified as a nuclear protein which has chromatin regulatory function [30]. Later discoveries revealed that HMGB1 can be secreted to the extracellular environment, and acts as a cytokine regulating the cellular activity [31]. In addition to its roles in the pathogenesis of various inflammatory diseases such as sepsis [32] and rheumatoid arthritis [33]. HMGB1 was reported to be stimulated in OA tissues, and HMGB1 A-box, an antagonist of HMGB1, could reduce the inflammation response of chondrocytes [24]. However, the exact roles of HMGB1 in the pathogenesis of OA are still to be elucidated. In this study, we performed systematic investigations to shed light on the regulatory functions of HMGB1 in OA. Based on the well-established OA cell model and OA animal model, we confirmed that the expression of HMGB1 was up-regulated during the whole stage of inflammation and partly secreted to extracellular matrix in the late stage of inflammation. These results also implied that HMGB1 may regulate chondrocyte activity in both endogenous and extracellular ways [34].
To clarify the regulatory function of HMGB1 in chondrocyte inflammation, the expression of HMGB1 was disrupted by RNA interference. We observed a stimulation of OA-related genes in the early stage of inflammation, suggesting that endogenous HMGB1 might suppress OA progression in the early stage. In addition, HMGB1 knockdown led to autophagic activity inhibition and cell apoptosis promotion in this stage [35]. In the animal model, MIA induced the expression of HMGB1 in cartilage in the early stage, promoted autophagy and reduced cell apoptosis. These results suggested that the endogenous HMGB1 facilitated chondrocyte autophagy and attenuated chondrocyte apoptosis. Importantly, our results demonstrated that HMGB1 could regulate the balance between autophagic activity and apoptosis of chondrocytes. This was consistent with previous reports [36,37] which demonstrated that the endogenous HMGB1 controls the cellular autophagy/apoptosis checkpoint in inflammatory bowel disease.
Present study also showed that cytosolic HMGB1 potentiate the autophagy-associated proteins like Beclin1 and ATG5, by counteracting the effects of calpain protein. Additionally, the cleavage of these proteins can be promoted by knockdown of HMGB1 expression, suggesting that HMGB1 inhibits the degradation of Beclin1 and ATG5. Furthermore, it was found that the calpain activity was stimulated by inflammation stimuli, and then was promoted further by HMGB1 knockdown. All these evidence demonstrated that HMGB1 inhibits calpain activity preventing the degradation of Beclin1 and ATG5, enhancing autophagic activity and maintaining cell viability [37]. It was reasonable to suppose that this might be a common mechanism in inflammation-associated diseases. Due to the fact that HMGB1 can be secreted to the extracellular matrix in the late stage of inflammation, we sought to determine the role of the extracellular HMGB1 in the pathogenesis of OA. Glycyrrhizin, an inhibitor which specifically blocks the cytokine activity of HMGB1 [38], was used and verified that it could suppress the LPS-induced expression of OA-related markers and neutralized the cartilage degradation in OA animal model.
As a highly prevalent disease, osteoarthritis is a considerable burden to public health. Despite several decades of investigation, there is still no effective therapeutic agents for OA treatment [39]. Our study demonstrated that HMGB1 can exert dual effects on inflammatory reaction based on its locations in chondrocytes. In one way, during the early stage of inflammation, the endogenous cytosolic HMGB1 protects chondrocytes through inducing autophagic activity and preventing cell apoptosis, protecting cartilage integrity from inflammation-mediated damage. In the other way, during the late stage of inflammation, HMGB1 is secreted into the extracellular matrix of chondrocytes and acts as a pro-inflammation cytokine inducing OA-like phenotype in cartilage and promoting inflammation response.
Funding Statement
This study was supported by the Natural Science Foundation of Hunan Province [grant S2017JJMSXM1134].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Vina ER, Kwoh CK.. Epidemiology of osteoarthritis: literature update. Curr Opin Rheumatol. 2018;30:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Woolf AD, Pfleger B.. Burden of major musculoskeletal conditions. Bull World Health Organ. 2003;81:646–656. [PMC free article] [PubMed] [Google Scholar]
- [3].Goldring MB. Articular cartilage degradation in osteoarthritis. Hss J. 2012;8:7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Asanbaeva A, Tam J, Schumacher BL, et al. Articular cartilage tensile integrity: modulation by matrix depletion is maturation-dependent. Arch Biochem Biophys. 2008;474:175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage. 2013;21:16–21. [DOI] [PubMed] [Google Scholar]
- [6].Wang Z, Hu J, Pan Y, et al. Mir-140-5p/mir-149 affects chondrocyte proliferation, apoptosis, and autophagy by targeting fut1 in osteoarthritis. Inflammation. 2018;41:959–971. [DOI] [PubMed] [Google Scholar]
- [7].Bianchi ME, Beltrame M, Paonessa G. Specific recognition of cruciform DNA by nuclear protein hmg1. Science. 1989;243:1056–1059. [DOI] [PubMed] [Google Scholar]
- [8].Loick J, Schulz HG, Rupp KD, et al. Is the thyroidectomy a surgical standard procedure for therapy of graves’ disease?. Zentralbl Chir. 2005;130:368–371. [DOI] [PubMed] [Google Scholar]
- [9].Wang H, Bloom O, Zhang M, et al. Hmg-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. [DOI] [PubMed] [Google Scholar]
- [10].Lotze MT, Tracey KJ. High-mobility group box 1 protein (hmgb1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. [DOI] [PubMed] [Google Scholar]
- [11].Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein hmgb1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. [DOI] [PubMed] [Google Scholar]
- [12].Palumbo R, Sampaolesi M, De Marchis F, et al. Extracellular hmgb1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164:441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schiraldi M, Raucci A, Munoz LM, et al. Hmgb1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with cxcl12 and signaling via cxcr4. J Exp Med. 2012;209:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang H, Yang H, Tracey KJ. Extracellular role of hmgb1 in inflammation and sepsis. J Intern Med. 2004;255:320–331. [DOI] [PubMed] [Google Scholar]
- [15].Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Terrando N, Yang T, Wang X, et al. Systemic hmgb1 neutralization prevents postoperative neurocognitive dysfunction in aged rats. Front Immunol. 2016;7:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang J, Takahashi HK, Liu K, et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke. 2011;42:1420–1428. [DOI] [PubMed] [Google Scholar]
- [18].Fujita K, Motoki K, Tagawa K, et al. Hmgb1, a pathogenic molecule that induces neurite degeneration via tlr4-marcks, is a potential therapeutic target for Alzheimer’s disease. Sci Rep. 2016;6:31895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stark K, Philippi V, Stockhausen S, et al. Disulfide hmgb1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Heinola T, Kouri VP, Clarijs P, et al. High mobility group box-1 (hmgb-1) in osteoarthritic cartilage. Clin Exp Rheumatol. 2010;28:511–518. [PubMed] [Google Scholar]
- [21].Ley C, Ekman S, Roneus B, et al. Interleukin-6 and high mobility group box protein-1 in synovial membranes and osteochondral fragments in equine osteoarthritis. Res Vet Sci. 2009;86:490–497. [DOI] [PubMed] [Google Scholar]
- [22].Garcia-Arnandis I, Guillen MI, Gomar F, et al. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1beta in osteoarthritic synoviocytes. Arthritis Res Ther. 2010;12:R165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sun XH, Liu Y, Han Y, et al. Expression and significance of high-mobility group protein b1 (hmgb1) and the receptor for advanced glycation end-product (rage) in knee osteoarthritis. Med Sci Monit. 2016;22:2105–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fu Y, Lei J, Zhuang Y, et al. Overexpression of hmgb1 a-box reduced il-1beta-induced mmp expression and the production of inflammatory mediators in human chondrocytes. Exp Cell Res. 2016;349:184–190. [DOI] [PubMed] [Google Scholar]
- [25].Mlost J, Kostrzewa M, Malek N, et al. Molecular understanding of the activation of cb1 and blockade of trpv1 receptors: implications for novel treatment strategies in osteoarthritis. Int J Mol Sci. 2018;19:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Park JW, Yun YP, Park K, et al. Ibuprofen-loaded porous microspheres suppressed the progression of monosodium iodoacetate-induced osteoarthritis in a rat model. Colloids Surf B Biointerfaces. 2016;147:265–273. [DOI] [PubMed] [Google Scholar]
- [27].Jin H, Zhang H, Ma T, et al. Resveratrol protects murine chondrogenic atdc5 cells against lps-induced inflammatory injury through up-regulating mir-146b. Cell Physiol Biochem. 2018;47:972–980. [DOI] [PubMed] [Google Scholar]
- [28].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- [29].Sun T, Li X, Song H, et al. Mir-146a aggravates lps-induced inflammatory injury by targeting cxcr4 in the articular chondrocytes. Cell Physiol Biochem. 2017;44:1282–1294. [DOI] [PubMed] [Google Scholar]
- [30].Muller S, Scaffidi P, Degryse B, et al. New embo members’ review: the double life of hmgb1 chromatin protein: architectural factor and extracellular signal. Embo J. 2001;20:4337–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gardella S, Andrei C, Ferrera D, et al. The nuclear protein hmgb1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Utsuno H, Kageyama T, Uchida K, et al. Facial soft tissue thickness in japanese children. Forensic Sci Int. 2010;199(109):e101–106. [DOI] [PubMed] [Google Scholar]
- [33].Park SY, Lee SW, Kim HY, et al. Hmgb1 induces angiogenesis in rheumatoid arthritis via hif-1alpha activation. Eur J Immunol. 2015;45:1216–1227. [DOI] [PubMed] [Google Scholar]
- [34].Lee WJ, Song SY, Roh H, et al. Profibrogenic effect of high-mobility group box protein-1 in human dermal fibroblasts and its excess in keloid tissues. Sci Rep. 2018;8:8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Luo P, Zhu Y, Chen M, et al. Hmgb1 contributes to adriamycin-induced cardiotoxicity via up-regulating autophagy. Toxicol Lett. 2018;292:115–122. [DOI] [PubMed] [Google Scholar]
- [36].Tang D, Kang R, Livesey KM, et al. Endogenous hmgb1 regulates autophagy. J Cell Biol. 2010;190:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhu X, Messer JS, Wang Y, et al. Cytosolic hmgb1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest. 2015;125:1098–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang Y, Zhang Y, Peng G, et al. Glycyrrhizin ameliorates atopic dermatitis-like symptoms through inhibition of hmgb1. Int Immunopharmacol. 2018;60:9–17. [DOI] [PubMed] [Google Scholar]
- [39].Ji Q, Qi D, Xu X, et al. Cryptotanshinone protects cartilage against developing osteoarthritis through the mir-106a-5p/glis3 axis. Mol Ther Nucleic Acids. 2018;11:170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]