

Abstract
Klebsiella pneumoniae is an important cause of Gram-negative pneumonia and sepsis. Mice deficient for TIR-domain-containing adaptor-inducing interferon-β (TRIF) demonstrate enhanced bacterial growth and dissemination during Klebsiella pneumonia. We show here that the impaired antibacterial defense of TRIF mutant mice is associated with absent interferon (IFN)-γ production in the lungs. IFN-γ production by splenocytes in response to K. pneumoniae in vitro was critically dependent on Toll-like receptor 4 (TLR4), the common TLR adaptor myeloid differentiation primary response gene (MyD88) and TRIF. Reconstitution of TRIF mutant mice with recombinant IFN-γ via the airways reduced bacterial loads in lungs and distant body sites to levels measured in wild-type mice, and partially restored pulmonary cytokine levels. The IFN-γ-induced, improved, enhanced antibacterial response in TRIF mutant mice occurred at the expense of increased hepatocellular injury. These data indicate that TRIF mediates antibacterial defense during Gram-negative pneumonia, at least in part, by inducing IFN-γ at the primary site of infection.
Key Words: Pneumonia, Toll-like receptors, Gram-negative sepsis
Introduction
Globally, pneumonia is a common cause of morbidity and mortality and the most common cause of sepsis [1, 2, 3]. The emerging antibiotic resistance among Gram-negative pathogens, including Enterobacteriaceae such as Klebsiella pneumoniae, is an issue of major concern, since therapeutic options are limited and infections with these pathogens are associated with an unfavorable outcome [3, 4]. K. pneumoniae is a common sepsis pathogen in humans, in particular in the context of lower respiratory tract infection [2].
Pathogens entering the lower airways are detected by innate immune cells via pattern recognition receptors, among which the family of Toll-like receptors (TLRs) features prominently; this interaction initiates the early immune response [5]. TLR signaling can proceed via two different routes that are dependent on the myeloid differentiation primary response gene 88 (MyD88) and TIR-domain-containing adaptor-inducing interferon-β (TRIF), respectively [6]. MyD88 is the universal adaptor for all TLRs except TLR3 and leads to NF-κB and MAP kinase activation and the induction of inflammatory cytokines. TRIF is the sole adaptor for TLR3 and also contributes to TLR4 signaling, leading to the activation of NF-κB and interferon regulatory factor 3 (IRF3) and the induction of type I interferon (IFN) and inflammatory cytokine production [6]. Notably, TLR4, which recognizes lipopolysaccharide (LPS), activates the MyD88-dependent pathway before it initiates downstream signaling via the TRIF-dependent pathway once the TLR4 complex has been transported to the endosome for degradation [7]. However, activation of both pathways is necessary for the induction of inflammatory cytokines via TLR4 [7].
We previously reported the crucial role of the TLR adaptors MyD88 and TRIF during K. pneumoniae infection and their differential contribution to the host response in different body compartments [8, 9]. In these studies, we noted that mice deficient for TRIF were incapable of IFN-γ production at the primary site of infection (unpubl. data). IFN-γ is an important cytokine for innate and adaptive immunity that influences a wide array of immunologically relevant cellular programs, such as the enhancement of leukocyte attraction, the upregulation of pathogen recognition, the processing and presentation of antigens and microbicidal effector cell functions [10]. A previous report, making use of IFN-γ gene deficient mice, demonstrated the importance of IFN-γ for antibacterial defense and survival during K. pneumoniae infection [11, 12]. In several models of experimental respiratory tract infection, it has been found that IFN-γ deficient mice are more susceptible to airway infection with Legionella pneumophila and Burkholderia pseudomallei [13, 14] and that therapeutic administration of recombinant (r)IFN-γ is beneficial [15, 16]. rIFN-γ also demonstrated a beneficial effect in several human studies when used as an adjunctive therapy for opportunistic pathogens [17, 18, 19, 20, 21].
We report here the impact of TRIF deficiency on pulmonary IFN-γ production during Klebsiella pneumonia. We explored to what extent the absence of local IFN-γ production during K. pneumoniae pneumonia in TRIF-deficient mice contributes to their susceptible phenotype. We demonstrate that TRIF-dependent signaling is crucial for IFN-γ production in vivo and in vitro and that reconstitution of IFN-γ levels in the airways improves antibacterial defense in TRIF-deficient but not in wild-type (WT) mice.
Materials and Methods
Animals
TRIF mutant mice, generated on a C57Bl/6 genetic background [22], were provided by Dr. B. Beutler (Center for the Genetics of Host Defense, University of Texas Southwestern Medical Center, Tex., USA). MyD88-deficient (Myd88-/-)[23] and Tlr4-/- mice [24] were provided by Dr. S. Akira (Research Institute for Microbial Diseases, Osaka, Japan) and backcrossed >8 times to a C57Bl/6 genetic background. All gene-deficient mice were bred at the animal facility of the Academic Medical Center (Amsterdam, the Netherlands). Age- and sex-matched WT C57Bl/6 control mice were obtained from Harlan Nederland (Horst, The Netherlands). The mice were infected at 10–12 weeks of age. The Animal Care and Use Committee of the University of Amsterdam approved all experiments.
Induction of Pneumonia and Sampling of Organs
Pneumonia was induced by intranasal inoculation with about 1 × 104 CFU of K. pneumoniae serotype 2 (ATCC 43816; American Type Culture Collection, Manassas, Va., USA) [8, 9]. The mice were sacrificed at the indicated time points after infection and their organs were harvested and processed exactly as described [8, 25]. In the reconstitution experiment, the mice were administered 50 ng of rIFN-γ (R&D Systems, Abbington, UK) or vehicle (0.1% human serum albumin in sterile saline) intranasally 30 min before and 24 h after inoculation; they were euthanized after 48 h of infection.
Quantitative RT-PCR
RNA was isolated from lung homogenates using the Nucleospin RNA II kit (Machery-Nagel, Duren, Germany). Total RNA was reverse-transcribed using oligo (dT) primer and Moloney murine leukemia virus reverse transcriptase (Invitrogen, Breda, The Netherlands). Quantitative (q)PCR of the Ifng gene product was performed as described [26]. Data were analyzed using the LinRegPCR program. Results were normalized to the β2m transcript.
In vitro Studies
Splenocytes were obtained, seeded at a density of 500,000 cells per well and cultured exactly as described [9]. Cells were stimulated for 48 h in at least quadruplicate with the indicated concentrations of mitomycin C-treated (0.05 mg/ml) (Sigma-Aldrich) growth-arrested K. pneumoniae diluted in RPMI medium without antibiotics, LPS derived from Klebsiella pneumoniae (100 ng/ml; Sigma) or ultrapure Escherichia coli O111 B4 LPS (100 ng/ml; Invivogen) diluted in RPMI medium with antibiotics in a final volume of 200 μl. Supernatants were stored and analyzed for cytokine concentrations by ELISA.
Assays
IFN-γ levels in cell supernatants and lung levels of IL-1β, CXCL1, CXCL2 and CCL2 were measured by ELISA (R&D Systems, Minneapolis, Minn., USA and Invitrogen). Lung levels of IFN-γ, TNF-α, IL-6 and IL-10 were measured by using a cytometric bead array multiplex assay (BD Biosciences). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using kits from Sigma and a Hitachi analyzer (Boehringer Mannheim).
Histopathology
Histologic examination of lungs and liver was performed exactly as described [25, 27]. Granulocyte immunohistochemical staining was prepared using a FITC-labeled anti-mouse Ly6-C/G mAb (BD Biosciences, San Jose, Calif., USA) exactly as previously described [9].
Statistical Analysis
Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation (in vivo experiments) or as means ± standard error (SE) of the mean (tables, cell stimulation experiments). Bacterial loads are expressed as scatter plots, each symbol representing an individual mouse, with horizontal lines indicating medians. For experiments with 2 groups, the Mann-Whitney U test was used to determine statistical significance. For experiments with >2 groups, the Kruskal-Wallis test was used, followed by Mann-Whitney U tests to compare individual genetically modified groups with the WT or TRIF mutant control group when appropriate. The Fisher exact test was used to determine if the proportion of positive test results was different. These analyses were done using GraphPad Prism (San Diego, Calif., USA). p < 0.05 was considered statistically significant.
Results
IFN-γ Production Is Impaired in TRIF Mutant Mice during Klebsiella Pneumonia
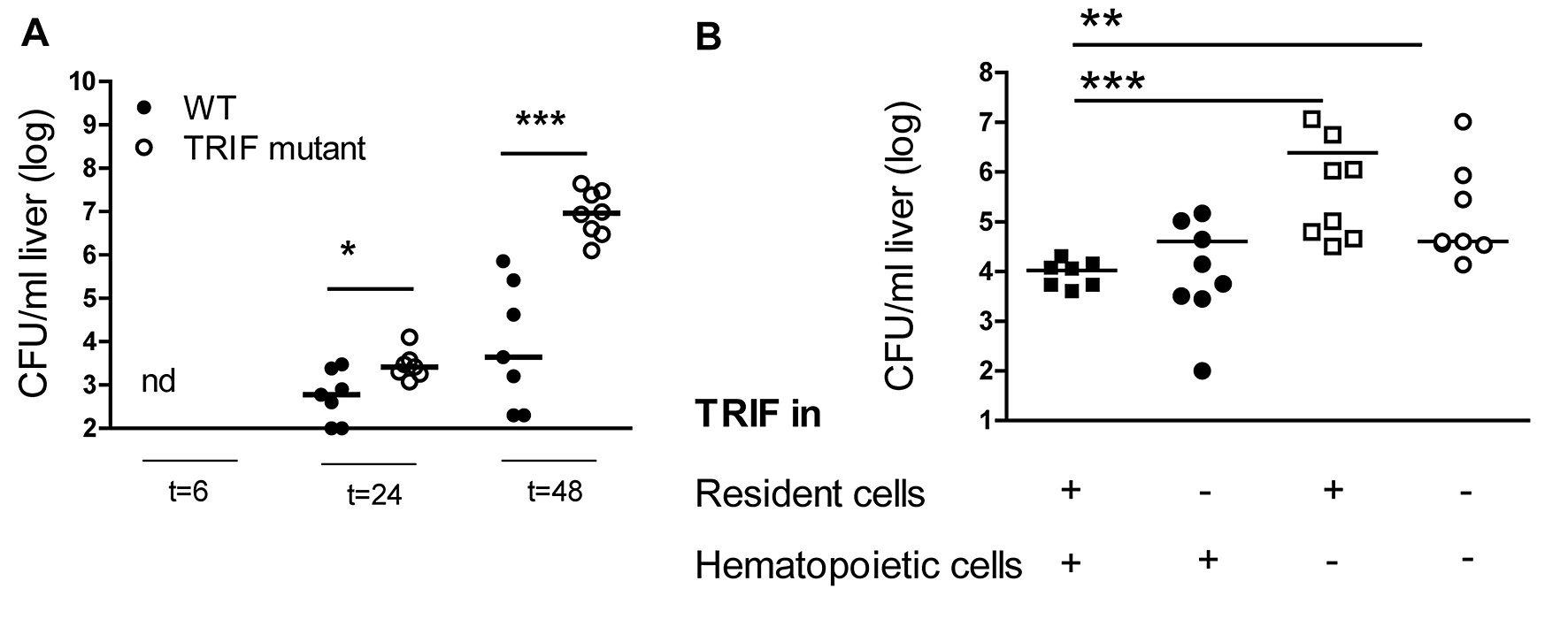
In our previous studies on the role of TRIF during K. pneumoniae airway infection, we demonstrated that TRIF mutant mice have an impaired antibacterial defense, illustrated by a significantly higher bacterial load in the lungs, blood and spleen [8]. In these investigations, we also observed higher bacterial loads in the livers of TRIF mutant mice and TRIF bone marrow chimeras lacking TRIF in hematopoietic cells (online suppl. fig. 1A, B; for all online suppl. material, see www.karger.com/doi/10.1159/000430913). In a multiplex cytokine assay performed on whole-lung homogenates, we noticed that IFN-γ levels remained undetectable in TRIF mutantmice throughout the infection (<5 pg/ml) whereas in WT mice, lung IFN-γ concentrations increased after Klebsiella inoculation, peaking after 24 h (p < 0.05–0.001 for the difference between groups; fig. 1a). TRIF mutantmice also showed strongly reduced IFN-γ mRNA expression in the lungs during Klebsiella pneumonia (p < 0.01 vs. WT mice; fig. 1b).
Fig. 1.

TRIF mediates IFN-γ production during K. pneumoniae airway infection. WT and TRIF mutant mice (n = 7–8 per group) were infected with about 104 CFU K. pneumoniae and sacrificed at designated time points. IFN-γ levels in the lungs of the mice were determined by cytometric bead assay (a) and qRT-PCR (b). Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. * p < 0.05, ** p < 0.01, Mann-Whitney U test. ### p < 0.001, Fisher exact test.
IFN-γ Production in Response to Klebsiella Is TLR4-Dependent via Both Myd88 and TRIF
Next, we stimulated splenocytes, as a source of IFN-γ producing cells, with growth-arrested K. pneumoniae in vitro. In a pilot study, we observed significantly impaired IFN-γ secretion by TRIF mutantcells stimulated with either 2 × 105 or 2 × 106 bacteria (data not shown). We repeated this experiment, this time including splenocytes of Tlr4-/- and Myd88-/- mice in addition to those from TRIF mutant and WT mice. IFN-γ production in response to growth-arrested K. pneumoniae was most severely impaired in Myd88-/- cells, followed by Tlr4-/- and then TRIF mutant cells (p < 0.05 to 0.01 compared to WT cells; fig. 2a). In addition, we stimulated cells with LPS derived from K. pneumoniae or ultrapurified LPS derived from E. coli, and found that IFN-γ release by Tlr4-/-, Myd88-/- and TRIF mutant cells was virtually absent (p < 0.01 vs. WT cells; fig. 2b).
Fig. 2.

IFN-γ secretion by splenocytes is dependent on TLR4, MyD88 and TRIF. Splenocytes derived from WT, Tlr4-/-, Myd88-/- and TRIF mutant mice were stimulated with different concentrations of growth-arrested K. pneumoniae and LPS derived from E. coli or K. pneumoniae (n = 4–6 for each condition). IFN-γ levels were determined after 48 h. Data are expressed as mean (SE). ** p < 0.01, Mann-Whitney U test (performed post hoc after Kruskal-Wallis test).
Antibacterial Defense of TRIF MutantMice Can Be Restored by Local Treatment with IFN-γ
To test if the strongly reduced pulmonary IFN-γ levels contributed functionally to the impaired antibacterial defense of TRIF mutant mice, we treated WT and TRIF mutantmice with IFN-γ intranasally 30 min before and 24 h after infection with Klebsiella; we used 48 h of infection as a predefined end point, since this was the point at which the enhanced growth of Klebsiella in TRIF mutantrelative to WT mice was clearest [8]. TRIF mutantmice treated with vehicle displayed undetectable pulmonary IFN-γ concentrations, confirming the results presented in figure 1a. TRIF mutantmice administered with rIFN-γ had lung IFN-γ levels that were similar to those measured in WT mice (fig. 3a); WT mice that received rIFN-γ had significantly higher levels than WT mice treated with vehicle (p < 0.05). We reproduced the previously described phenotype in TRIF mutantmice [8], showing a 100- to 1,000-fold higher bacterial load in the lungs relative to WT mice, together with increased bacterial dissemination to the blood and spleen (p < 0.001; fig. 3b-d). Importantly, we observed a spectacular improvement of antibacterial defense in rIFN-γ-treated TRIF mutantmice compared to vehicle-treated TRIF mutantmice (p < 0.01-0.001), as reflected by bacterial loads similar to in WT mice in all organs. Of note, we observed no effect on bacterial burdens in WT mice treated with rIFN-γ compared to vehicle-treated WT mice (fig. 3b-d).
Fig. 3.

Administration of rIFN-γ via the airways restores antibacterial defense in TRIF mutant mice. WT and TRIF mutant mice were infected with about 104 CFU K. pneumonia; 50 ng recombinant IFN-γ or vehicle was administered intranasally 30 min before infection and 24 h afterwards (n = 8 mice each group). Mice were sacrificed after 48 h of infection. IFN-γ levels in lung homogenates 48 h after infection (a) are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. Bacterial loads are shown in the lung (b), blood (c) and spleen (d) 48 h after infection. Each symbol represents an individual mouse, horizontal lines represent medians. ** p < 0.01, *** p < 0.001 vs. WT mice treated with vehicle, ## p < 0.01, ### p < 0.001 vs. TRIF mutant mice treated with vehicle, Mann-Whitney U test, and Fisher exact test was used for comparison between TRIF mutant groups (performed post hoc after Kruskal-Wallis test).
Impact of IFN-γ Treatment on the Inflammatory Response to Pneumonia
To obtain insight into the extent of local inflammation at the primary site of infection in TRIF mutantand WT mice, and the effect on it of rIFN-γ treatment, we semiquantitatively scored lung histopathology of tissue samples harvested 48 h after infection, focusing on key histological features characteristic for severe pneumonia. While total lung histopathology scores were not different between groups, rIFN-γ-treated TRIF mutantand WT mice had more signs of bronchitis and less signs of pleuritis than their respective vehicle-treated controls (p < 0.05 vs. controls; table 1; fig. 4a-f). Neutrophil recruitment to the lungs, measured as the percentage of Ly-6-positive lung cell surface, was significantly higher in vehicle-treated TRIF mutant mice than in vehicle-treated WT mice at this late stage of infection (p < 0.01). Administration of rIFN-γ reduced total neutrophil numbers in lung tissue of TRIF mutant mice, similar to those measured in WT mice (p < 0.05 compared to vehicle-treated TRIF mutant mice); rIFN-γ treatment did not influence lung neutrophil counts in WT mice). Moreover, when the Ly-6 staining was studied in detail, the number of intrabronchial neutrophils appeared to be greater after rIFN-γ treatment (fig. 4e-h). We next determined the effect of rIFN-γ treatment on the induction of the proinflammatory cytokines TNF-α, IL-1β and IL-6, the anti-inflammatory cytokine IL-10 and the chemokines CXCL1, CXCL2 and CCL2 in whole-lung homogenates. TRIF mutantmice demonstrated reduced levels of TNF-α, IL-1β, CXCL1, CXCL2 and CCL2 relative to WT mice (p < 0.05-0.001; table 2). Treatment of TRIF mutant mice with rIFN-γ partially restored the inflammatory profile with the exception of IL-1β. TNF-α and CXCL2 were not significantly different from vehicle-treated WT mice and the levels of CXCL1 and CCL2 were still significantly lower, although differences were smaller (p < 0.05-0.01 vs. vehicle-treated WT mice). The change in levels of inflammatory cytokines and chemokines after treatment with rIFN-γ of TRIF mutantmice was significant for TNF-α, IL-6, CXCL2 and CCL2 compared to vehicle-treated TRIF mutantmice (p < 0.05-0.001 between groups; table 2).
Table 1.
Histological scores: WT and TRIF mutant mice were infected with 1 × 104 CFU K. pneumoniae and 50 ng recombinant IFN-γ was administered intranasally upon infection and after 48 h
| WT vehicle | WT rIFN-γ | TRIF mutant vehicle | TRIF mutant rIFN-γ | |
|---|---|---|---|---|
| Total pathology score: lung | 14.5 (0.6) | 13.8 (0.8) | 14.5 (1.2) | 13.1 (0.6) |
| Pneumonia % of lung surface | 15 (3) | 6 (4) | 22 (6) | 7 (3) |
| Interstitial inflammation | 3.1 (0.1) | 3.0 (0.5) | 2.8 (0.7) | 2.4 (0.3) |
| Edema | 2.8 (0.2) | 2.5 (0.3) | 3.4 (0.5) | 3.0 (0.2) |
| Endothelialitis | 2.5 (0.2) | 2.9 (0.1) | 3 (0.2) | 2.6 (0.2) |
| Bronchitis | 2.9 (0.1) | 3.5 (0.2)* | 2.6 (0.3) | 3.8 (0.2)# |
| Pleuritis | 1.8 (0.3) | 1.3 (0.2)* | 1.5 (0.3) | 0.8 (0.2)# |
| Ly-6-positive % of total lung surface | 2.3 (0.4) | 2.1 (0.5) | 8.6 (1.2)** | 3.9 (0.8)# |
Total pathology score is the sum of the histological subscores (determined as described in Methods). Data are mean (SE) of 7–8 mice per group.
p <0.05,
p <0.01, vs. vehicle-treated WT mice.
p <0.05, rIFN-γ-treated TRIF mutant mice vs. vehicle-treated TRIF mutant mice.
Fig. 4.

Effect of IFN-γ treatment on lung pathology. WT and TRIF mutant mice were infected with about 104 CFU K. pneumonia; 50 ng recombinant IFN-γ or vehicle was administered intranasally 30 min before infection and 24 h afterwards. Mice were sacrificed after 48 h of infection. Representative lung histology of WT mice treated with vehicle (a), WT mice treated with rIFN-γ (b), TRIF mutantmice treated with vehicle (c) and TRIF mutantmice treated with rIFN-γ (d). HE. a-d Upper panel: arrows indicate signs of bronchitis. ×10. Lower panels: stars indicate pleuritis. ×20. Representative lung histology (Ly-6 staining, indicating neutrophils) of lungs of WT mice treated with vehicle (e), WT mice treated with rIFN-γ (f), TRIF mutantmice treated with vehicle (g) and TRIF mutantmice treated with rIFN-γ (h). ×10.
Table 2.
Inflammatory response
| WT Vehicle | WT rIFN-γ | TRIF mutant vehicle | TRIF mutant rIFN-γ | |
|---|---|---|---|---|
| TNF-α | 892 (245) | 760 (57) | 191 (48)** | 501 (115)# |
| IL-1β | 7,434 (642) | 4,950 (753)* | 4,168 (731)** | 4,525 (557)** |
| IL-6 | 1,914 (451) | 2,030 (624) | 2,446 (398) | 1,507 (216)# |
| IL-10 | 14 (2) | 11 (1) | 14 (2) | b.d. |
| CXCL1 | 12,586 (1,899) | 9,453 (1,645) | 3,625 (871)** | 4,255 (828)* |
| CXCL2 | 20,553 (6,546) | 38,048 (7,157) | 6,432 (1,532)* | 28,943 (5,785)### |
| CCL2 | 4,619 (541) | 3,718 (366) | 1,841 (210)*** | 2,126 (240)**' # |
WT and TRIF mutant mice were infected with 1 × 104 CFU K. pneumoniae and 50 ng recombinant IFN-γ was administered intranasally upon infection and after 48 h. Homogenates were prepared from right lungs. Cytokine and chemokine levels are presented in pg/ml of lung homogenate. Data are mean (SE) of 7–8 mice per group. b.d. = Below detection.
p <0.05,
p <0.01,
p <0.001, vs. vehicle-treated WT mice.
p <0.05,
## p <0.01,
p <0.001, rIFN-γ-treated TRIF mutant mice vs. vehicle-treated TRIF mutant mice.
IFN-γ Deficiency Protects TRIF MutantMice from Liver Injury
Klebsiella-induced pneumonia-derived sepsis is associated with hepatocellular injury, as reflected by increased plasma concentrations of AST and ALT [8, 28]. TRIF mutantmice had lower AST and ALT plasma levels 48 h after infection than WT mice (p < 0.01; fig. 5a, b) as well as fewer signs of liver inflammation as determined by liver histopathology scores (p < 0.01; fig. 5c; online suppl. fig. 2). Remarkably, rIFN-γ treatment significantly increased the levels of AST and ALT in TRIF mutant mice compared to vehicle-treated TRIF mutant mice (p < 0.01-0.001), and these were similar to those measured in WT mice. In WT mice, rIFN-γ treatment reduced transaminase levels, significantly so for AST (p < 0.05; fig. 5a).
Fig. 5.

TRIF mutant mice have attenuated liver injury that increases after rIFN-γ treatment. WT and TRIF mutant mice were infected with about 104 CFU K. pneumonia; 50 ng rIFN-γ or vehicle was administered intranasally 30 min before infection and 24 h afterwards. Mice were sacrificed after 48 h of infection. AST (a) and ALT (b) plasma levels and liver histopathology were scored (see Methods) (c) expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. * p < 0.05, ** p < 0.01, *** p < 0.001, Mann-Whitney U test (performed post hoc after Kruskal-Wallis test).
Discussion
K. pneumoniae is a clinically important Gram-negative bacterium in pneumonia and one of the pathogens that causes major concern because of increasing antimicrobial resistance rates, which limit therapeutic options [2, 3, 4, 29]. Previous research has documented the importance of TLR signaling for host defense during K. pneumoniae pneumonia, notably that of TLR4, TLR2 and TLR9 [25, 30, 31]. We and other study groups have previously described the pivotal role of the TLR-adaptors MyD88 and TRIF in this [8, 32]. Given our discovery that in the absence of TRIF lung levels of IFN-γ were undetectable during the course of K. pneumoniae airway infection, we explored the functional importance here; our main findings were that TRIF is indeed crucial for IFN-γ production in response to K. pneumonia, together with TLR4 and MyD88, and that reconstitution of TRIF mutant mice with rIFN-γ improves antibacterial defense to the level of WT mice, but at the expense of enhanced liver injury.
Earlier, we and others described the susceptible phenotype of TRIF-deficient mice in Klebsiella pneumonia, marked by a clearly impaired antibacterial defense with a 100- to 1,000-fold increase in bacterial load 48 h after infection, a finding that we have reproduced here [8, 25, 32]. The early inflammatory response of mice that are partially or fully deficient for TRIF is characterized by impaired neutrophil influx, probably as a result of impaired CXCL1 secretion and lower levels of TNF-α and IL-6. However, during the course of the infection and in response to a higher bacterial load, all of these cytokines gradually increased in spite of (partial) TRIF deficiency [8]. Notably, in this study, CXCL1, CXCL2 and TNF-α levels were still reduced in TRIF mutant mice 48 h after infection, while lung neutrophil numbers as determined by immunohistochemistry were significantly higher. This is probably due to the very high bacterial numbers present in TRIF mutant mice at this time point, which leads to tissue injury and neutrophil attraction via mechanisms apart from what is provided by the chemoattractant gradient by the aforementioned mediators. Remarkably, however, IFN-γ levels remained virtually undetectable in TRIF mutant mice throughout, which formed the rationale for our study. We hypothesized that deficient IFN-γ production could, at least in part, be responsible for the impaired antibacterial defense of TRIF mutant mice, considering that IFN-γ is a powerful pleiotropic cytokine that, during bacterial infection, can enhance leukocyte attraction, pathogen recognition, processing and presentation of antigens and microbicidal effector cell functions [10]. We extended our in vivo observation of decreased IFN-γ levels in TRIF mutant mice by demonstrating that also under controlled conditions, with equal amounts of growth-arrested bacteria, the capacity of TRIF mutantsplenocytes to secrete IFN-γ is impaired. Moreover, IFN-γ production was critically dependent on MyD88 and TLR4. This is not surprising, since it is well known that these innate immune sensors are highly important for the induction of the inflammatory response to K. pneumoniae, and that the phenotype of Myd88-/- and Tlr4-/- mice is more severe than that of TRIF mutantmice during in vivo infection [8, 25]. However, the role of these receptors, specifically in the induction IFN-γ, in response to pathogens is less well known. In agreement with this report, TRIF-deficient mice have been shown to produce lower IFN-γ levels during Aspergillus airway infection in vivo [33]. Our results suggest that TLR2-dependent signals play a role in response to K. pneumoniae in addition to TRIF, MyD88 and TLR4, since IFN-γ levels secreted by TRIF mutantand Tlr4-/- cells gradually increased along with increasing bacterial concentrations, which is in line with the role of TLR2 during infection with Klebsiella in vivo [25].
We observed a spectacular effect on bacterial load after the reconstitution of TRIF mutant mice with rIFN-γ, which coincided with a partial recovery of the inflammatory cytokine profile. The importance of IFN-γ during K. pneumoniae infection has been demonstrated previously, since Ifn-γ-/- mice displayed an impaired antibacterial defense and increased mortality [11, 12, 25]. The other way around, in a rat model of ethanol intoxication followed by Klebsiella airway infection, adenoviral expression of IFN-γ improved antibacterial defense [34]. Likewise, conditional adenoviral expression of IFN-γ improved clearance of Klebsiella from the lungs in mice [35]. Strikingly, in our study, there was no effect of rIFN-γ on bacterial loads in WT mice, suggesting that local rIFN-γ administration is only beneficial when it compensates for a clearly deficient production. In addition, in WT mice, rIFN-γ treatment did not affect lung cytokine concentrations (with the exception of IL-1β) whereas in TRIF mutant mice, it increased the levels of TNF-α, IL-6, CXCL1, CXCL2 and CCL2. The mechanism by which rIFN-γ improves bacterial defense in TRIF mutant mice might be by enhancing the bacterial killing capacity of alveolar macrophages [36]. Unfortunately, the Klebsiella strain used here cannot be killed by macrophages or neutrophils in vitro (our own observations), illustrating its high virulence and precluding further in vitro analyses. Improved monocyte and macrophage function was also presumed to play a role in human clinical trials where treatment with rIFN-γ demonstrated beneficial effects in Mycobacterium tuberculosis and M. avium infections as well as with leishmaniasis and fungal sepsis, although the exact mechanisms are currently unknown [17, 18, 19, 20, 21]. Recently, however, in fungal sepsis patients, it was demonstrated that the ex vivo cytokine response was enhanced in patients treated with rIFN-γ [17].
In our study, TRIF mutant mice treated with rIFN-γ also had higher plasma IFN-γ levels than TRIF mutant mice treated with vehicle, even though rIFN-γ was instilled locally in the airways. Hence, although it is likely that the reduced bacterial loads at distant body sites in rIFN-γ-treated TRIF mutant mice, at least in part, are the consequence of lower bacterial burdens at the primary site of infection, we cannot exclude an additional systemic effect of local rIFN-γ treatment. Another aspect of the inflammatory response that we observed in our study is that while total lung histopathology scores did not differ between groups, rIFN-γ-treated Trif-/- and WT mice had more signs of bronchitis and lower pleuritis scores when compared to their respective vehicle-treated controls, possibly indicating a redistribution in the pattern of inflammatory cell migration. This might be secondary to a higher intrabronchial rIFN-γ concentration after intranasal administration, resulting in an increased attraction of inflammatory cells to the intrabronchial and intra-alveolar compartment (fig. 4). Possibly, this contributed to better containment of the infection.
In this and in our previous study, we demonstrated significantly lower levels of AST and ALT in mice (partially) deficient for TRIF, despite higher bacterial loads in the blood and liver [8]. Although liver bacterial loads were not determined in rIFN-γ-treated mice, it is unlikely that the increased hepatocellular injury in these animals was caused by higher bacterial burdens in the liver, considering the reduced Klebsiella numbers in the blood and spleen. This illustrates the double-edged sword character of the innate immune response that is, on the one hand, essential for early antibacterial defense, but on the other, contributes to collateral tissue damage in sepsis as illustrated in previous studies [37, 38, 39]. Strikingly, the reconstitution of TRIF mutant mice with rIFN-γ caused deteriorated liver injury. This suggests that IFN-γ is involved in inflammation-driven liver injury, as was proposed before in an intravenous model of K. pneumoniae sepsis in Ifn-γ-/- mice [12]. However, the lower AST levels in WT mice treated with rIFN-γ are more difficult to explain and require further investigation. Possibly, the increased plasma levels of the anti-inflammatory cytokine IL-10 in rIFN-γ-treated WT mice (albeit not significant) played a role here.
In conclusion, we demonstrate a crucial role for TRIF in IFN-γ production during K. pneumoniae pneumonia. TRIF-mediated IFN-γ release is essential for an adequate innate immune response as reflected by the fact that the strongly impaired antibacterial defense of TRIF mutant mice can be restored by the reconstitution of IFN-γ levels in the lungs by local treatment. These data provide new insight into how TRIF mediates protective immunity during Gram-negative infection.
Supplementary Material
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
Acknowledgments
We thank Regina de Beer, Joost Daalhuisen and Marieke ten Brink for expert technical assistance. This work was supported by the AMC Graduate School of Medical Science (M.H.P.v.L).
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Kollef MH, Shorr A, Tabak YP, Gupta V, Liu LZ, Johannes RS. Epidemiology and outcomes of health-care-associated pneumonia: results from a large US database of culture-positive pneumonia. Chest. 2005;128:3854–3862. doi: 10.1378/chest.128.6.3854. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organisation World Health Observatory top ten causes of death, 2012
- 4.Schwaber MJ, Carmeli Y. Mortality and delay in effective therapy associated with extended-spectrum beta-lactamase production in Enterobacteriaceae bacteraemia: a systematic review and meta-analysis. J Antimicrob Chemother. 2007;60:913–920. doi: 10.1093/jac/dkm318. [DOI] [PubMed] [Google Scholar]
- 5.Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. 2008;358:716–727. doi: 10.1056/NEJMra074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 8.van Lieshout MH, Blok DC, Wieland CW, de Vos AF, van ‘t Veer C, van der Poll T. Differential roles of MyD88 and TRIF in hematopoietic and resident cells during murine Gram-negative pneumonia. J Infect Dis. 2012;206:1415–1423. doi: 10.1093/infdis/jis505. [DOI] [PubMed] [Google Scholar]
- 9.van Lieshout MH, Anas AA, Florquin S, Hou B, Van't Veer C, de Vos AF, van der Poll T. Hematopoietic but not endothelial cell MyD88 contributes to host defense during Gram-negative pneumonia-derived sepsis. PLoS Pathog. 2014;10:e1004368. doi: 10.1371/journal.ppat.1004368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida K, Matsumoto T, Tateda K, Uchida K, Tsujimoto S, Iwakurai Y, Yamaguchi K. Protection against pulmonary infection with Klebsiellapneumoniae in mice by interferon-gamma through activation of phagocytic cells and stimulation of production of other cytokines. J Med Microbiol. 2001;50:959–964. doi: 10.1099/0022-1317-50-11-959. [DOI] [PubMed] [Google Scholar]
- 12.Moore TA, Perry ML, Getsoian AG, Newstead MW, Standiford TJ. Divergent role of gamma interferon in a murine model of pulmonary versus systemic Klebsiella pneumoniae infection. Infect Immun. 2002;70:6310–6318. doi: 10.1128/IAI.70.11.6310-6318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shinozawa Y, Matsumoto T, Uchida K, Tsujimoto S, Iwakura Y, Yamaguchi K. Role of interferon-gamma in inflammatory responses in murine respiratory infection with Legionella pneumophila. J Med Microbiol. 2002;51:225–230. doi: 10.1099/0022-1317-51-3-225. [DOI] [PubMed] [Google Scholar]
- 14.Easton A, Haque A, Chu K, Lukaszewski R, Bancroft GJ. A critical role for neutrophils in resistance to experimental infection with Burkholderia pseudomallei. J Infect Dis. 2007;195:99–107. doi: 10.1086/509810. [DOI] [PubMed] [Google Scholar]
- 15.Beck JM, Liggitt HD, Brunette EN, Fuchs HJ, Shellito JE, Debs RJ. Reduction in intensity of Pneumocystis carinii pneumonia in mice by aerosol administration of gamma interferon. Infect Immun. 1991;59:3859–3862. doi: 10.1128/iai.59.11.3859-3862.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buccheri S, Reljic R, Caccamo N, Meraviglia S, Ivanyi J, Salerno A, Dieli F. Prevention of the post-chemotherapy relapse of tuberculous infection by combined immunotherapy. Tuberculosis (Edinb) 2009;89:91–94. doi: 10.1016/j.tube.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, Monneret G, Venet F, Bleeker-Rovers CP, van de Veerdonk FL, Pickkers P, Pachot A, Kullberg BJ, Netea MG. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect Dis. 2014;14:166. doi: 10.1186/1471-2334-14-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao XF, Yang ZW, Li J. Adjunctive therapy with interferon-gamma for the treatment of pulmonary tuberculosis: a systematic review. Int J Infect Dis. 2011;15:e594–e600. doi: 10.1016/j.ijid.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Milanes-Virelles MT, Garcia-Garcia I, Santos-Herrera Y, Valdes-Quintana M, Valenzuela-Silva CM, Jimenez-Madrigal G, Ramos-Gomez TI, Bello-Rivero I, Fernandez-Olivera N, Sanchez-de la Osa RB, Rodriguez-Acosta C, Gonzalez-Mendez L, Martinez-Sanchez G, Lopez-Saura PA. Adjuvant interferon gamma in patients with pulmonary atypical mycobacteriosis: a randomized, double-blind, placebo-controlled study. BMC Infect Dis. 2008;8:17. doi: 10.1186/1471-2334-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray HW. Interferon-gamma in infection and immunoparalysis. Intensive Care Med. 1996;22((suppl 4)):S455. doi: 10.1007/BF01743723. [DOI] [PubMed] [Google Scholar]
- 21.Wunderink RG. Adjunctive therapy in community-acquired pneumonia. Semin Respir Crit Care Med. 2009;30:146–153. doi: 10.1055/s-0029-1202933. [DOI] [PubMed] [Google Scholar]
- 22.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 23.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 24.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 25.Wieland CW, van Lieshout MH, Hoogendijk AJ, van der Poll T. Host defence during Klebsiella pneumonia relies on haematopoietic-expressed Toll-like receptors 4 and 2. Eur Respir J. 2011;37:848–857. doi: 10.1183/09031936.00076510. [DOI] [PubMed] [Google Scholar]
- 26.van ‘t Veer C, van den Pangaart PS, Kruijswijk D, Florquin S, de Vos AF, van der Poll T. Delineation of the role of Toll-like receptor signaling during peritonitis by a gradually growing pathogenic Escherichia coli. J Biol Chem. 2011;286:36603–36618. doi: 10.1074/jbc.M110.189126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Achouiti A, Vogl T, Urban CF, Rohm M, Hommes TJ, van Zoelen MA, Florquin S, Roth J, Van't Veer C., de Vos AF, van der Poll T. Myeloid-related protein-14 contributes to protective immunity in Gram-negative pneumonia derived sepsis. PLoS Pathog. 2012;8:e1002987. doi: 10.1371/journal.ppat.1002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Renckens R, Roelofs JJ, Bonta PI, Florquin S, de Vries CJ, Levi M, Carmeliet P, Van't Veer C., van der Poll T. Plasminogen activator inhibitor type 1 is protective during severe Gram-negative pneumonia. Blood. 2007;109:1593–1601. doi: 10.1182/blood-2006-05-025197. [DOI] [PubMed] [Google Scholar]
- 29.Vardakas KZ, Tansarli GS, Rafailidis PI, Falagas ME. Carbapenems versus alternative antibiotics for the treatment of bacteraemia due to Enterobacteriaceae producing extended-spectrum beta-lactamases: a systematic review and meta-analysis. J Antimicrob Chemother. 2012;67:2793–2803. doi: 10.1093/jac/dks301. [DOI] [PubMed] [Google Scholar]
- 30.Bhan U, Lukacs NW, Osterholzer JJ, Newstead MW, Zeng X, Moore TA, McMillan TR, Krieg AM, Akira S, Standiford TJ. TLR9 is required for protective innate immunity in Gram-negative bacterial pneumonia: role of dendritic cells. J Immunol. 2007;179:3937–3946. doi: 10.4049/jimmunol.179.6.3937. [DOI] [PubMed] [Google Scholar]
- 31.Branger J, Knapp S, Weijer S, Leemans JC, Pater JM, Speelman P, Florquin S, van der Poll T. Role of Toll-like receptor 4 in Gram-positive and Gram-negative pneumonia in mice. Infect Immun. 2004;72:788–794. doi: 10.1128/IAI.72.2.788-794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai S, Batra S, Shen L, Wakamatsu N, Jeyaseelan S. Both TRIF- and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J Immunol. 2009;183:6629–6638. doi: 10.4049/jimmunol.0901033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Luca A, Bozza S, Zelante T, Zagarella S, D'Angelo C, Perruccio K, Vacca C, Carvalho A, Cunha C, Aversa F, Romani L. Non-hematopoietic cells contribute to protective tolerance to Aspergillus fumigatus via a TRIF pathway converging on IDO. Cell Mol Immunol. 2010;7:459–470. doi: 10.1038/cmi.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolls JK, Lei D, Stoltz D, Zhang P, Schwarzenberger PO, Ye P, Bagby G, Summer WR, Shellito JE, Nelson S. Adenoviral-mediated interferon-gamma gene therapy augments pulmonary host defense of ethanol-treated rats. Alcohol Clin Exp Res. 1998;22:157–162. [PubMed] [Google Scholar]
- 35.Ruan S, Young E, Luce MJ, Reiser J, Kolls JK, Shellito JE. Conditional expression of interferon-gamma to enhance host responses to pulmonary bacterial infection. Pulm Pharmacol Ther. 2006;19:251–257. doi: 10.1016/j.pupt.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Serezani CH, Chung J, Ballinger MN, Moore BB, Aronoff DM, Peters-Golden M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am J Respir Cell Mol Biol. 2007;37:562–570. doi: 10.1165/rcmb.2007-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hochhauser E, Avlas O, Fallach R, Bachmetov L, Zemel R, Pappo O, Shainberg A, Ben AZ. Bone marrow and nonbone marrow Toll-like receptor 4 regulate acute hepatic injury induced by endotoxemia. PLoS One. 2013;8:e73041. doi: 10.1371/journal.pone.0073041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitazawa T, Tsujimoto T, Kawaratani H, Fukui H. Salvage effect of E5564, Toll-like receptor 4 antagonist on D-galactosamine and lipopolysaccharide-induced acute liver failure in rats. J Gastroenterol Hepatol. 2010;25:1009–1012. doi: 10.1111/j.1440-1746.2009.06145.x. [DOI] [PubMed] [Google Scholar]
- 39.Moore TA, Lau HY, Cogen AL, Monteleon CL, Standiford TJ. Anti-tumor necrosis factor-alpha therapy during murine Klebsiella pneumoniae bacteremia: increased mortality in the absence of liver injury. Shock. 2003;20:309–315. doi: 10.1097/01.shk.0000087203.34916.45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data
Supplementary data