

ABSTRACT
Children with Hutchinson–Gilford progeria syndrome (HGPS) succumb to myocardial infarction and stroke in their teen years. Endothelial dysfunction is an early event in more common forms of atherosclerosis. Endothelial pathobiology may contribute to HGPS, but a comprehensive characterization of endothelial function in HGPS has not been performed.
iPSCs derived from fibroblasts of HGPS patients or unaffected relatives were differentiated into endothelial cells (ECs). Immunofluorescent signal of the pluripotent stem cell markers SSEA4, Oct4, Sox2 and TRAI-60 was similar in HGPS or control iPSCs. Following the differentiation, FACS analysis and immunocytochemistry for CD31 and CD144 revealed a smaller percentage of ECs from HGPS iPSCs. Immunostaining for Lamin A revealed nuclear dysmorphology in HGPS iPSC-ECs. Furthermore, these cells were significantly larger and rounded, and they proliferated less, features which are typical of senescent endothelial cells. HGPS iPSC-ECs manifested less Dil-Ac-LDL uptake; less DAF-2DA staining for nitric oxide generation and formed fewer networks in matrigel in vitro. In immunodeficient mice injected with iPSC-ECs, HGPS iPSC-ECs generated a sparser vascular network compared to the control, with reduced capillary number. Telomere length (T/S ratio) of HGPS iPSC-EC was reduced as assessed by mmqPCR.
iPSC-ECs derived from HGPS patients have dysmorphic appearance, abnormal nuclear morphology, shortened telomeres, reduced replicative capacity and impaired functions in vitro and in vivo. Targeting the endothelial abnormality in patients with HGPS may provide a new therapeutic avenue for the treatment of this condition.
Abbreviations: HGPS: Hutchinson–Gilford progeria syndrome; ZMPSTE24: Zinc metallopeptidase STE24; FTI: Farnesyltransferase inhibitors; VSMCs: Vascular smooth muscle cells; iPSC: Induced pluripotent stem cells; EC: Endothelial cells; hTERT: Human telomerase reverse transcriptase; VEGF: vascular endothelial growth factor; DAF-FM DA: 3-Amino, 4-aminomethyl-2’,7’-difluorofluorescein diacetate; BMP4: Bone Morphogenetic Protein 4; mmqPCR: mono chrome multiplex PCR; SCG: single-copy gene; CSI: Cell shape index.
KEYWORDS: Progeria, endothelium, dysfunction, telomeres, iPSC, endothelial cells
Introduction
Hutchinson-Gilford progeria syndrome (HGPS) is a rare autosomal disorder of accelerated aging (OMIM: 176,660) [1]. Children are normal at birth but the first years of life are characterized by failure to thrive with low growth rates [2]. During childhood, more characteristic features of HGPS begin to appear including alopecia, reduced subcutaneous adipose tissue, osteoporosis, and sclerodermatous skin [3]. The disease is also characterized by rapid vascular aging, with death occurring in the teen years typically secondary to myocardial infarction or stroke [3].
A de novo mutation (C1824T) in the LMNA gene encoding lamin A causes the condition. Lamin A is a nuclear matrix protein that influences nuclear structure and function [4]. As prelamin A, it undergoes post-translational modifications, including farnesylation of the cysteine at the C-terminal CaaX motif, endoproteolytic cleavage of the three amino acids aaX, methylation of C-terminal cysteine, and endoproteolytic removal of the farnesyl group [5]. The hydrophobicity of the farnesyl group suggests that this modification favors the precise positioning of the final Lamin A to the nuclear envelope [6]. The mutation causes a splicing defect that deletes 150 bp in exon 11 (Δ150 LMNA). This deletion includes the proteolytic cleavage site at which zinc metalloprotease ZMPSTE24 removes the farnesyl group at the C-terminus [1]. Thus, progerin is persistently farnesylated, causing its accumulation in the nuclear envelope, progressive appearance of a number of cellular alterations including severe growth defects and disruption of nuclear architecture and cellular function [7]. Farnesyltransferase inhibitors (FTIs [8]) inhibit progerin farnesylation and have been shown to improve both structure and function of progerin-containing cells in vitro [9]; in murine models of HGPS, the FTIs reduce cardiovascular defects [10], and extend lifespan [11,12]. Subsequently, a clinical trial with lonafarnib indicated that treatment with the FTI improved vascular compliance and had benefits on the auditory and skeletal system [13]. A follow-up study suggested that the treatment improved lifespan by about 2 y[14].
Although HGPS mouse models, in which the lamin A gene is deleted, mutated, and/or overexpressed [15–18], have disclosed important information on the molecular mechanisms of the HGPS, none of them recapitulate all the features seen in the human disease. Human-induced pluripotent stem cell (iPSC) have greatly improved the modeling of human disease in vitro [19–22]. Studies on HGPS iPSC or iPSC-derived cells may complement information obtained in vivo. For example, in a previous report [23] HGPS iPSCs-derived vascular smooth muscle cells (VSMCs) showed increased sensitivity to various stimuli, including hypoxia, and perturbation of contractile properties due to calponin sequestration, whereas mesenchymal stem cells (MSCs) were not able to restore angiogenesis in hindlimb ischemia in mice [23]. A recent report in an atherosclerosis-prone mouse model of HGPS showed that VSMC-specific expression of progerin is sufficient to accelerate atherosclerosis, trigger plaque vulnerability, and reduce lifespan in mice [24] .
Endothelial dysfunction is a primary determinant of atherosclerosis and may also have an important role in HGPS. Previous observations from studies of lamin A defective ECs are quite discordant. For example, Zhang et al. [23] showed that HGPS-iPSC derived ECs are not functionally different compared to control, whereas Bonello-Palot et al. [25] demonstrated that prelamin A accumulation in progenitors and mature ECs derived from human umbilical vein induces premature senescence and functional impairment. Thus, the characterization of the endothelial defect in HGPS is incomplete and the results are mixed.
In this report, we have generated and extensively characterized HGPS iPSC-derived ECs. We have performed a comprehensive characterization of EC structure and function. We found a broad derangement of EC structure and function, including nuclear morphology, telomere length [26], generation of nitric oxide [27], Dil-Ac-LDL uptake, replicative capacity, as well as angiogenesis in vitro and in vivo. To conclude, there is a substantial derangement in HGPS EC, which could contribute to the premature vascular disease and death in these patients. Furthermore, iPSC-derived ECs may be an attractive model to study molecular mechanisms and test possible therapies for HGPS.
Results
We utilized iPSCs derived from fibroblasts originally collected from HGPS patients (167–1Q and 003–1D) and father or mother (168–1P and 090–1B, were used as control cell line, hereafter referred to as Non-HGPS). HGPS and Non-HGPS iPSC colonies appeared morphologically similar by light microscopy (Movie 1, Figure 1(a)) in cells from both patients. Western blotting analysis for pluripotent stem cell-specific markers showed a similar expression of Sex determining region Y-box 2 (Sox2) and octamer-binding transcription factor 4 (Oct4, also known as POU5f1) in the two groups (Figure 1(b)). Furthermore, immunostaining for the pluripotent stem cell-specific markers: stage-specific embryonic antigen (SSEA4), Oct4, Sox2, and TRAI-60 (podocalyxin) showed no difference between the HGPS and non-HGPS iPSCs (Figure 1(c)). Subsequently, HGPS and non-HGPS iPSCs at the same passage, that was between 22 and 24, were differentiated to endothelial lineage, a process that requires about 2 wk using our standardized protocol (Figure 2(a)). Experiments with cell lines of both patients showed no difference. Real-time PCR on differentiated cells confirmed the presence of cells of mesodermal lineage, although the cell population was heterogeneous with markers of endoderm and ectoderm as well (Figure 2(b)). Accordingly, at this stage, we sorted double-positive cells for the endothelial markers CD31 and CD144. We observed a smaller percentage of double-positive cells generated from HGPS iPSCs compared to control (61.7% vs 89.3%; p ≤ 0.01; Figure 2(c)). Furthermore, real-time PCR of FACS sorted CD31+-CD144+ double-positive cells revealed a lower expression of endothelial markers in HGPS compared to Non-HGPS (Figure 3(a)). HGPS iPSC-ECs also exhibited morphological features that were different from non-HGPS iPSC-ECs (Figure 3(b)), discussed below in more detail. Immunofluorescence for CD31 and CD144 confirmed the different morphology of HGPS iPSC-ECs compared to Non-HGPS iPSC-ECs (Figure 3(c)).
Figure 1.
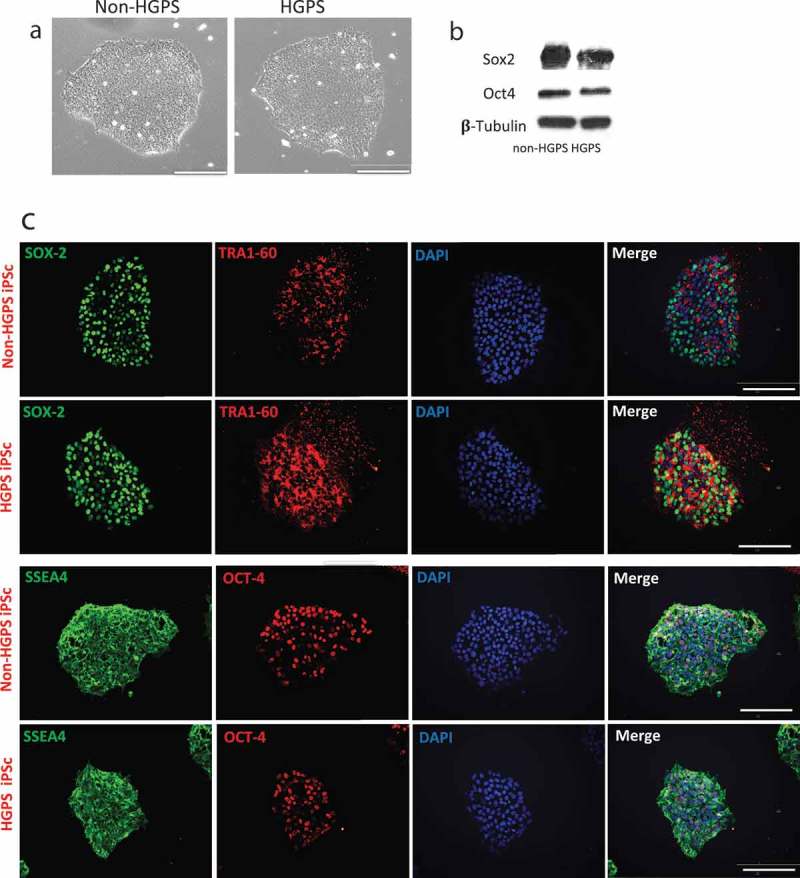
Characterization of iPSCs. (a) Brightfield images of iPSC (HGPS 167–1Q and non-HGPS 168–1P) colonies cultured on matrigel coated dishes. (b) Western blotting analysis for pluripotency markers Sox2 and Oct4 in HGPS and non-HGPS iPSC. (c) Immunofluorescence staining for Oct4, SSEA–4, SOX2 and TRA1–60 in HGPS (167–1Q) and non-HGPS (168–1P) iPSCs show no difference in the expression of pluripotent stem cell-specific markers. White bar length 100 um. N = 3 experiments per donor cells.
Figure 2.
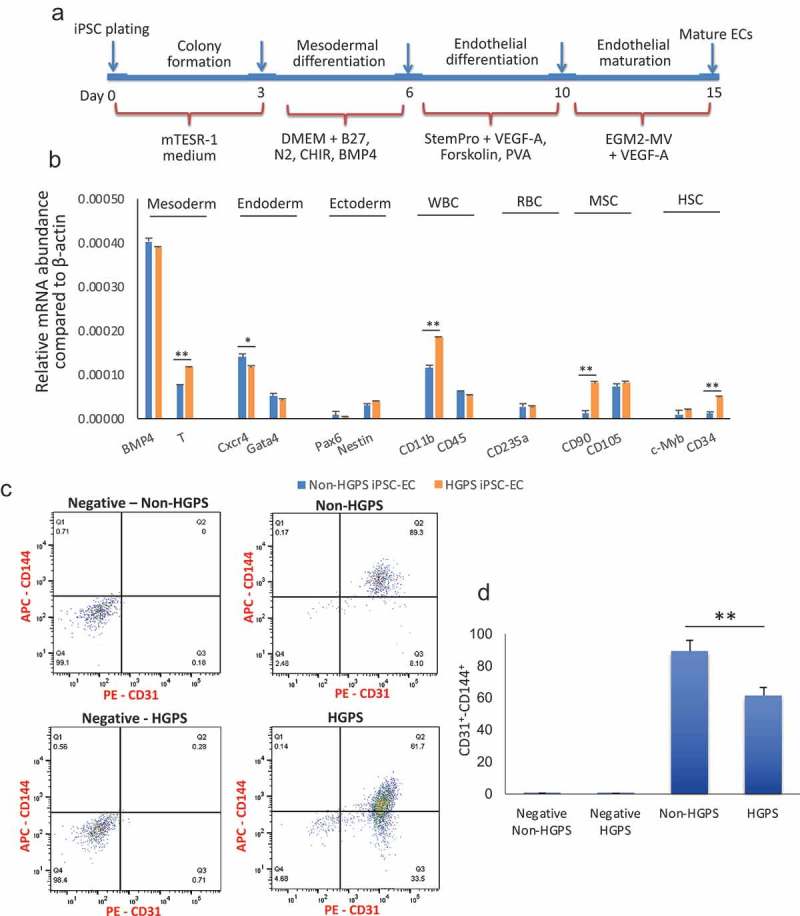
Differentiation and purification of iPSCs-derived ECs. (a) Protocol for differentiation of iPSC in endothelial cells. (b) Real-time PCR for markers of mesoderm, endoderm, ectoderm, white blood cells (WBC), red blood cells (RBC), mesenchymal stem cells (MSC) and hematopoietic stem cells (HSC) after differentiation and just prior to sorting for CD31+ CD144+ cells. (c–d) FACS analysis for the endothelial markers CD31 and CD144 reveals that both HGPS and non-HGPS iPSCs differentiate to endothelial cells, although the percentage of mature endothelial cells is lower in HGPS. In negative samples, no 1st antibodies were added. N = 3 experiments per donor cells, Student t-test, *p ≤ 0.05, **p ≤ 0.01.
Figure 3.
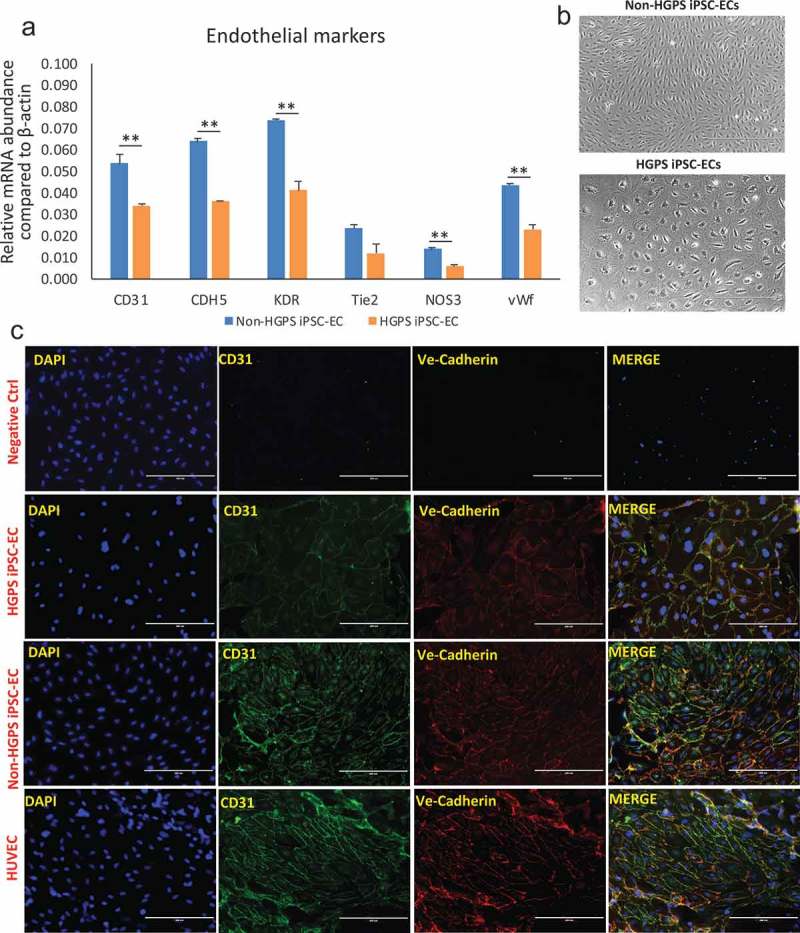
Characterization of iPSCs-derived ECs. (a) Real-time PCR for endothelial markers CD31, CDH5, KDR, Tie2, NOS3 and vWF in non-HGPS and HGPS FACS purified iPSC-derived EC. (b) Brightfield images of non-HGPS and HGPS purified iPSC-derived EC, showing different shape and size (see analysis in Figure 5(b–c)). (c) Immunofluorescence staining for endothelial markers CD31 and VE-Cadherin (CD144). Non-HGPS iPSC-ECs with no 1st antibodies added were used as negative control (ctrl); HUVECs were used as positive control.
Images show HGPS 167–1Q and non-HGPS 168–1P cells. N = 3 experiments per donor cells, Student t-test, **p ≤ 0.01.
Next, we asked whether HGPS iPSC-EC exhibited features of premature senescence, including reduced telomere length and cell proliferation. Nuclear dysmorphology is a feature of HGPS somatic cells. We observed, using fluorescence microscopy and Lamin A immunostaining, the prevalence of nuclear dysmorphology in HGPS iPSC-ECs by comparison to non-HGPS iPSC-ECs or HUVECs (Figure 4(a)). Protein analysis confirmed that HGPS iPSC-ECs express Progerin and less Lamin A compared to Non-HGPS cells (Figure 4(b)). Senescent endothelial cells are larger and rounder by comparison to healthy endothelial cells. Immunostaining revealed that HGPS iPSCs-EC have a size and shape consistent with senescence (Figure 4(c–d)). We quantified this observation by assessing average cell “roundness” using the Cell Shape Index (CSI) as described [28]. The HGPS iPSC-ECs manifested a higher CSI (0.55 ± 0.15 vs. 0.41 ± 0.17 for control; p ≤ 0.01), confirming that HGPS iPSCs-EC had a rounder shape. In addition, HGPS iPSC-EC had a greater area (p ≤ 0.01) (Figure 4(c–d)). Monochrome Multiplex Quantitative PCR (mmqPCR) was used to assess telomere length. We observed that the HGPS iPSC-ECs have reduced T/S (Telomere/Single copy gene) ratios, meaning shortened telomeres compared to non-HGPS iPSC-ECs (p ≤ 0.01) at the same cell passage (Figure 4(e)). Senescence cells have reduced replicative capacity. Accordingly, to analyze real-time changes in cell number we used the xCELLigence instrument to monitor cell replication. The xCELLigence instrument monitors the impedance to a current passing through the membrane on which the cells are growing. As the monolayer increases in confluence, impedance rises. The impedance value is plotted as a parameter called Cell Index (CI). The HGPS iPSC-ECs (red curve) exhibited a reduced CI compared to control (green curve) (Figure 4(f)). These observations were consistent with a reduced proliferation of the HGPS iPSC-EC.
Figure 4.
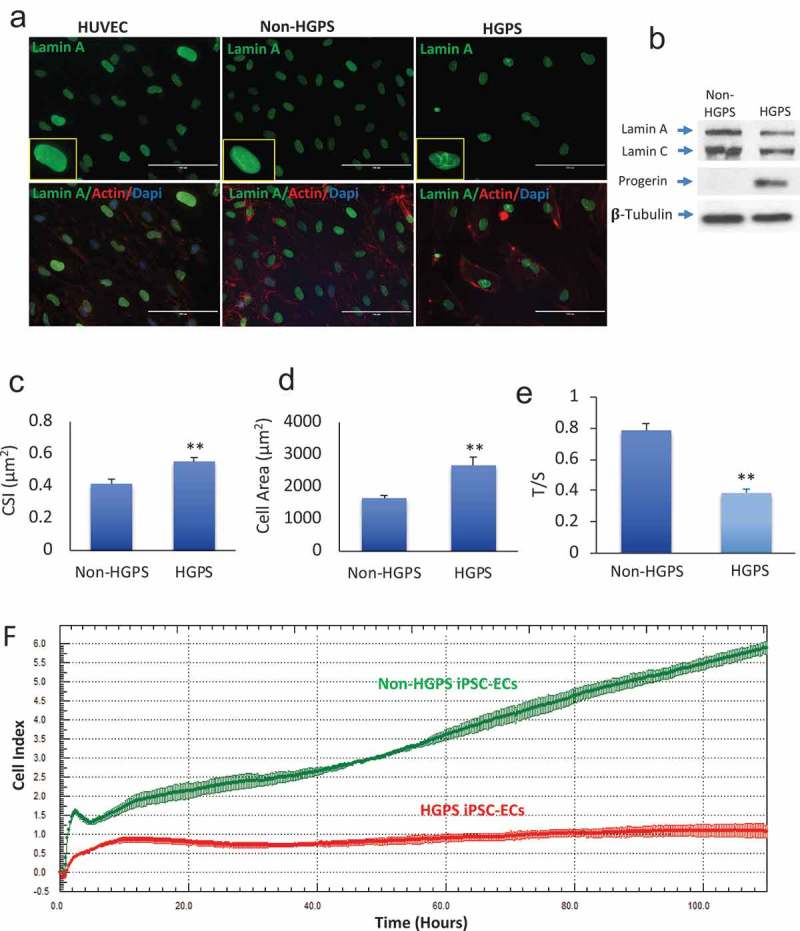
Senescence features in HGPS iPSC-derived ECs. (a) Immunofluorescence images showing the expression of lamin A in control HUVEC, non-HGPS and HGPS iPSC-EC. Nuclear dysmorphologies were observed in HGPS iPSC-ECs (right) compared to non-HGPS iPSC-ECs (left). (b) Western blotting for Lamin A/C and Progerin in Non-HGPS and HGPS iPSC-ECs. (c–d). HGPS iPSC-ECs were larger (cell area) and retain a rounded-shaped morphology [cell shape index (CSI)] compared to controls. (e) Bar graph of telomere length assessed by Monochrome Multiplex Quantitative PCR. HGPS iPSC-ECs have shortened telomeres compared to non-HGPS iPSC-ECs at the same passage, as shown by the reduced T/S ratios in HGPS iPSC-ECs. (f) Real-time cell analyzer profiles showed that HGPS iPSC-ECs (red curve) have a reduced cell index compared to non-HGPS iPSC-ECs (green curve).
Images show HGPS 167–1Q and non-HGPS 168–1P cells. N = 3 experiments per donor cells, Student t-test, **p ≤ 0.01.
Senescent cells often exhibit an impairment of lineage-specific cell processes. The uptake of acetylated LDL (AcLDL) is a function of EC that can be assessed using fluorescently (Dil)-labeled Ac-LDL. In the HGPS iPSC-EC, Ac-LDL uptake was impaired, as reflected by a reduced fluorescence intensity in this assay (p ≤ 0.01) (Figure 5(a–b)). Endothelium-derived nitric oxide (NO) is a major regulator of EC homeostasis [29], and its generation is reduced in senescent cells [30]. Accordingly, we used DAF-FM DA staining to assess NO generation, which studies revealed that HGPS iPSC–ECs generated less nitric oxide (NO; p ≤ 0.01) (Figure 5(c–d)). In addition, measurement of Nitrate/Nitrite, two end-products of NO, confirmed a reduced synthesis of NO in HGPS iPSC–ECs compared to control (Figure 5(e)).
Figure 5.
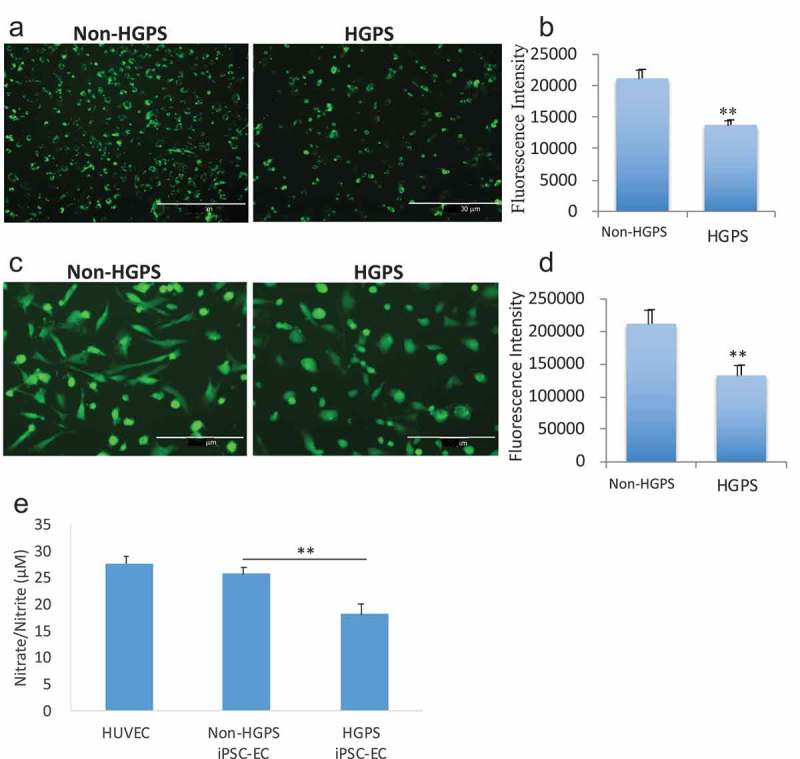
Analysis of iPSCs-derived ECs functionality. (a) Immunofluorescence images showing the uptake of acetylated low-density lipoprotein in HGPS iPSC-EC and control. Fluorescence intensity is reported graphically in (b). (c) Immunofluorescence images of nitric oxide (NO), measured by DAF-FM staining, generated in HGPS and non-HGPS iPSC-ECs. Fluorescence intensity is reported graphically in (d). (e) Nitrate/Nitrite assay as measurement of nitric oxide. Images show HGPS 167–1Q and non-HGPS 168–1P cells. N = 3 experiments per donor cells, Student t-test was used in (b) and (d), ANOVA test was used in (e). **p ≤ 0.01.
Angiogenesis is a characteristic function of ECs. Accordingly, we assess the angiogenic capacity of HGPS iPSC-ECs in vitro and in vivo. Using an in vitro matrigel assay we observed that HGPS iPSC-ECs formed fewer network structures (Figure 6(a–b)). In addition, analysis of neovascularization in vivo using the Matrigel plug assay revealed a reduced neovascularization in matrigel plugs containing HGPS iPSC-ECs (Figure 6(c)); reduced CD31 immunostaining (p ≤ 0.01) (Figure 6(d–e)) and reduced lumina containing red blood cells (HE staining) (p ≤ 0.01) (Figure 6(f–g)). These data indicate that HGPS iPSC-ECs are functionally defective cells, resembling senescent cells.
Figure 6.
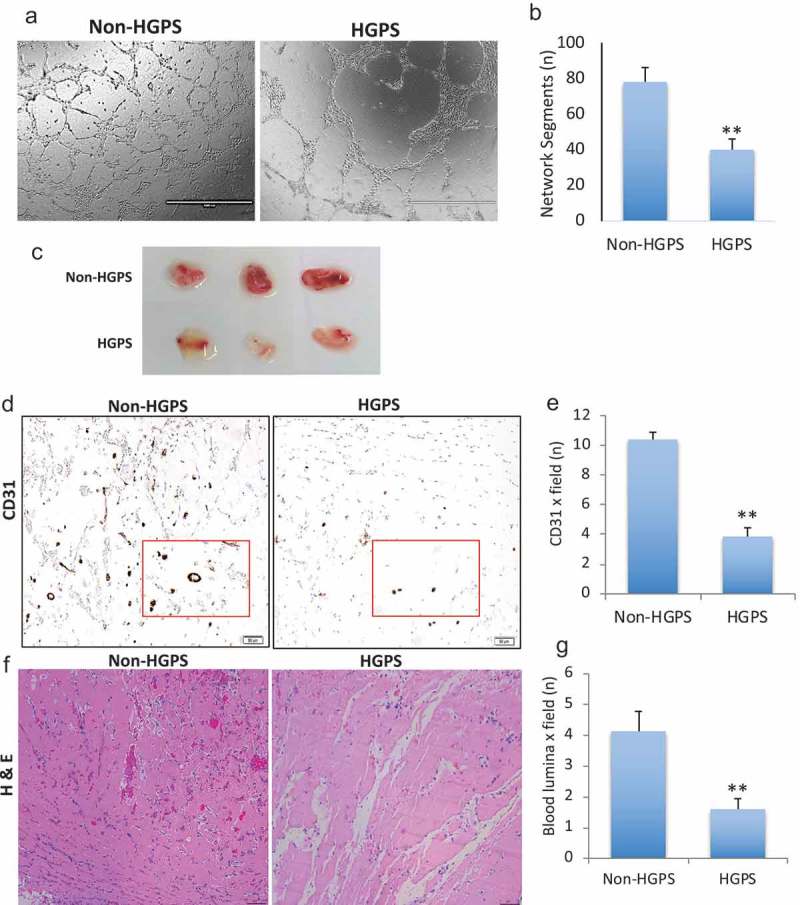
Measure of angiogenesis by iPSCs-derived ECs. (a) Representative images of vascular network formation (in-vitro matrigel assay) showing that HGPS iPSC-derived ECs formed fewer network segments compared to control. Results are reported graphically in (b). (c–g). Reduced neovascularization, shown by in-vivo matrigel plug assay, was observed in HGPS iPSC-ECs compared to non-HGPS iPSC-ECs. Images of CD31-immunostained sections (D) and HE stained section (F) with results graphically reported in (e) and (g), respectively. Images are derived from experiments with HGPS 167–1Q and non-HGPS 168–1P cells. N = 3 experiments per donor cells, Student t-test, **p ≤ 0.01.
Discussion
The seminal finding in this paper is that there is a global impairment of endothelial functions in HGPS iPSC-ECs. We find that HGPS iPSC-ECs have a senescent phenotype, as characterized by their cellular and nuclear dysmorphology, reduced telomere length, and impaired proliferative capacity; together with an impairment of angiogenic processes in vitro and in vivo. These endothelial aberrations are likely to be involved in the development of the premature vascular disease observed in HGPS, and thus ECs derived from HGPS iPSCs may represent an excellent in vitro cell model for testing new therapies.
The premature aging observed in HGPS is associated with a de novo mutation in the LMNA gene. The mutation results in the generation of an abnormal lamin protein that is permanently farnesylated. The aberrant farnesylation leads to accumulation of progerin in the nuclear envelope and a dysmorphic nucleus, with disorganization of nuclear lamina and heterochromatin [31–33]. HGPS patients exhibit premature aging associated with accelerated coronary and carotid artery disease, which cause death typically in the early teens [1,34].
Clinical trials have tested the efficacy of the farnesyltransferase inhibitors (FTI) lonafarnib, alone or in combination with statins (pravastatin) that blocks protein prenylation, including farnesylation, and reduces the formation of progerin, and bisphosphonates (zoledronate), but benefits have been modest [3,13,14]. Although the recent development of CRISPR/Cas9-based therapy for HGPS seems promising [35], the therapeutic options are limited.
Accordingly, a greater understanding of the pathobiology that leads to the premature death of these children might inform improved therapies. Previous observations of the vascular disease in HGPS described intimal lesions that may be obstructive, deposition of extracellular matrix, thinning of the media, calcification, and adventitial thickening [36,37], which are features that are consistent with atherosclerosis. A few studies have focused on the pathobiology affecting vascular smooth muscle cells in Progeria [23,24]. By contrast, there has been an insufficient characterization of the endothelial cell dysfunction in Progeria. This is a critical gap in knowledge, as in adults, the process of atherosclerosis begins with impairment of endothelial processes [38]. The earliest stages of an atherosclerotic lesion, i.e. focal permeation and trapping of circulating lipoprotein particles in the sub-endothelial space [39], results from endothelial activation in lesion-prone areas of the arterial vasculature [40]. This endothelial dysfunction initiates a complex pathogenic sequence that later also involves immune and vascular smooth muscle cells. Because endothelial dysfunction is a characteristic feature of vascular disease, and is a major contributor to the initiation and progression of vascular disease, we chose to characterize HGPS iPSC-EC.
In this report, we have differentiated and characterized HGPS iPSC-derived endothelial cells, contrasting their function compared to iPSC-derived endothelial cells from a related non-HGPS individual (the patients’ father). First, we confirmed that HGPS and control iPSC have similar expression levels of the iPSCs pluripotency markers SSEA4, OCT-4, SOX-2, and TRA-1–60, as previously shown [23,41].
Then, we differentiated iPSCs, at the same passage number, to endothelial cells (EC). We used cells at passage number between 22 and 24 because cells at higher passages, in particular above 28, showed a more frequent loss of pluripotency and reduced endothelial cells yield after differentiation. At the end of the differentiation protocol, the yield of EC was lower and the expression of EC markers impaired in HGPS iPSC, suggesting that the effect of progerin may begin during development at the stage of differentiation and maturation of EC. It is known that aging reduces the expression and activity of CD144 (VE-cadherin), a main endothelial marker, in the adherens junctions of aged rat arteries, that contributes to endothelial dysfunction [42]. Senescence can be induced by a plethora of stimuli that cause persistent DNA damage signaling and drives the phenotype of senescent cells, that may include the reduced expression of endothelial markers in HGPS endothelial cells. Indeed, progerin seems to affect the activation of the DNA damage response pathway and dysregulation of this pathway may be responsible for the development of cardiovascular pathology in HGPS patients [43]. Consistent with a senescent state, the HGPS iPSC-EC had a “fried egg” appearance, i.e. they were larger and rounder as quantified by cell index and cell area.
HGPS iPSC-ECs displayed the nuclear lobulation which is a common feature of HGPS cells. The nuclear lamina is a meshwork of intermediate filaments composed of A- and B-type lamins [44] and has been shown to be critical for nuclear stability, particularly in tissues that are exposed to mechanical forces such as muscle fibers [45] and endothelial cells subject to the pulsatile nature of blood flow [46]. The alterations in the nuclear shape in HGPS cells appear to affect chromatin stability and gene expression [47], and may also affect the organization of nuclear pore complexes that normally regulate the trafficking of protein and RNA across the nuclear envelope [48].
We observed that HGPS iPSC-ECs have a reduced production of nitric oxide, a phenomenon also observed in aged endothelial cells [30]. Telomeres play a prominent role in aging and cell senescence [49]. Human telomeres are sequences of TTAGGG repeats that undergo progressive shortening with cell divisions as a consequence of the inability of DNA polymerases to fully replicate the DNA lagging strand. Eventually, telomere erosion will elicit a DNA damage response, resulting in growth arrest and senescence. Telomere reverse transcriptase (TERT), an enzyme present in stem cells, can add telomeric repeats to telomere ends by copying a template sequence of its RNA component (TERC) [50]. A previous study has shown that stable expression of the human telomerase reverse transcriptase (hTERT), associated with an increase in telomere length, restored endothelial cells eNOS and NO activity in senescent cells [30]. Thus, the telomere erosion in HGPS may be linked to eNOS expression and activity. Also linked to telomere erosion is an impairment in cell proliferation. We measured the proliferative capacity of HGPS iPSC-EC in real-time and noted a significant reduction in cell growth within 48 h after seeding. HGPS iPSC-ECs also displayed impaired network formation on matrigel and reduced neovascularization in vivo in the matrigel plug assay, demonstrating the inability for the cells to promote angiogenesis.
We observed that the relative telomere length was significantly decreased in HGPS iPSC-EC. Telomere erosion is a key feature of aging. Advanced age is a major risk factor for cardiovascular disease (CVD) [51] and the health of the vasculature [52] is directly associated with the increase of CVD risk. A loss of telomeric DNA repeats below a critical threshold contributes to genome instability [53,54]. This also causes deficiencies in the repair of DNA double-strand breaks, in particular through the non-homologous end-joining recombination, an error-prone mechanism for repairing lost telomeres [55].
Notably, progerin accumulates in tissues of normal-aged individuals [31], including the artery wall [36]. Therefore, progeria may be a model to elucidate cell and molecular mechanisms driving normal aging and associated CVD. In addition, cardiovascular risk factors such as hypercholesterolemia, diabetes, obesity, hypertension, and smoking are normally absent in HGPS patients. Thus, cells derived from these patients provide a unique model to isolate age-related mechanisms that impair cardiovascular health.
In conclusion, we have characterized the endothelial structure and function in HGPS. We find a broad derangement in endothelial processes that are characteristic of senescence. The iPSC-derived endothelial cells of HGPS patients are a useful model to characterize the endothelial pathobiology in HGPS and to understand its role in the premature vascular disease in this condition.
Material and methods
Maintenance of human HGPS iPSC
The human HGPS iPSC lines were obtained from the Progeria Research Foundation (PRF) Cell and Tissue Bank and were maintained on Matrigel (BD Biosciences)-coated plates (Corning) in mTeSR1 medium (STEMCELL Technologies cat. 85850) according to their protocol. The iPSCs were passaged every 4 d using RELSR dissociation reagent (STEMCELL Technologies cat. 05873). All cells were cultured in humidified incubators at 37°C and 5% CO2. Pluripotency of HGPS iPSC was characterized by morphology and immunostaining of pluripotency markers.
Differentiation of endothelial cells (EC) from iPSC
EC differentiation was carried out using an in vitro monolayer endothelial differentiation of iPSC based on a modification of a previously established protocol [56,57]. In brief, the iPSCs at passage between 22 and 24 were grown to 80% confluence, and placed in differentiation medium Advanced DMEM/F12 (ThermoFisher Scientific cat. 11320033), supplemented with Wnt agonist CHIR 99021 5 µM (Selleck, cat. S2924), bone morphogenetic protein-4 (BMP4, 25 ng/ml) (Peprotech cat120–05), B27 supplement (ThermoFisher Scientific cat. 17504044), and N2 supplement (ThermoFisher Scientific cat. 17502048) for 3 d. Then, cells were dissociated with HyQtase (GE Healthcare cat. SV30030.01) and plated in StemPro media (ThermoFisher Scientific cat. 10639011), supplemented with forskolin 5 µM (LC Laboratories cat. F-9929), vascular endothelial growth factor (VEGF) 50ng/mL (Peprotech cat. 100–20), and polyvinyl alcohol 2 mg/mL (Sigma-Aldrich cat. 360627) for 4 d. Finally, cells were washed with PBS, and cultured in endothelial growth media (EGM-2MV, Lonza cat. CC-3202) supplemented with additional VEGF (100 ng/ml) for 5 more days and maintained at 37°C, and 5% CO2 in a humidified incubator. Cells were passaged once they reached 80–90% confluence.
Flow cytometry characterization of iPSC-EC
Cells were dissociated with HyQtase, washed with PBS and blocked with 5% bovine serum albumin (BSA). Cells were then incubated with either Alexa Fluor 488-conjugated CD31 mAb (PE mouse anti-human, BD Pharmingen, cat. 555446) or PE-conjugated CD144 mAb (FITC mouse anti-human, BD Pharmingen, cat. 560411) for 30 min. Isotype-matched antibody served as negative control. The cells were washed with 5% BSA and then analyzed using a FACSAria (BD) flow cytometer. Data were analyzed by Flowjo software.
Fluorescence-activated cell sorting (FACS)
After differentiation, the iPSC-ECs were purified using FACS. Cells were dissociated into single cells with HyQtase for 5 min at 37°C, washed with PBS containing 5% BSA and passed through a 40-μm cell strainer. Cells were then incubated with either Alexa Fluor 488-conjugated CD31 antibody or PE-conjugated CD144 antibody for 30 min. Isotype-matched antibody served as negative control. The purified iPSC-ECs were expanded in EGM-MV media.
Immunofluorescence staining
For iPSC and staining, the Pluripotent Stem Cell 4-Marker Immunocytochemistry Kit (ThermoFisher Scientific, A24881) was used, according to manufacturer instructions. iPSC-ECs were seeded at a density of 10000 x cm2 2. At 80% of confluence, they were fixed with 4% paraformaldehyde (ThermoFisher Scientific cat. 28908), and blocked with 1% normal goat serum (Abcam cat. ab7481) and stained for anti-human CD31 (1:250, Abcam cat. ab28364), anti-human CD144 (1:250, Abcam cat. ab166715), overnight at 4°C. After washes with PBS, the cells were treated with Alexa Fluor-488 or −594 secondary antibodies (1:500) and co-stained with DAPI (Vector Laboratories, 1:10000). Images were captured using the EVOS M5000 Imaging System (ThermoFisher Scientific).
Cell size and morphology were analyzed using ImageJ to determine size and circularity. Cell shape index (CSI) was calculated using the equation: (4π X area)/(perimeter2 2), where a perfect circle would have a value of 1, whereas a straight line would have a value of 0.
RNA extraction and RT-PCR
mRNA was extracted from cells using column purification (RNeasy Mini Kit, Qiagen, cat. 74104). The mRNA was reverse transcribed into cDNA using qScript cDNA Synthesis Kit (Quanta Bio, cat. 95047.) Primers (IDT Technologies), see table S1 for the full primers list, targeting specific genes and SYBR Green PCR kit (Invitrogen, Carlsbad, CA) were used for real-time qPCR with the QuantStudio 12 k Flex system (Applied Biosystems, Foster City, CA) following the manufacturer’s instructions. Genes expression was expressed as relative fold changes using the ΔCt method of analysis and normalized to β-actin.
Protein extraction and western blot analysis
Cultured cells were collected and solubilized in RIPA buffer (25 mmol/L Tris-HCl pH 7.6, 150 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitor cocktail. Protein concentration was measured using BCA assay. Samples were loaded on polyacrylamide gel electrophoresis (4–15% gradient) for 2 h and transferred on PVDF membranes for 2 h. Membranes were blocked with nonfat milk 5% in PBST (PBS+0.1% Tween) for 1 h at room temperature and probed with primary antibody overnight at 4°C. Antibody used were: Lamin A mAb (mice, ThermoFisher Scientific, cat. MA1–06101), Oct4 mAb (rabbit, ThermoFisher Scientific, cat. 701756), Sox2 mAb (mice, ThermoFisher Scientific, cat. MA1–014), β-tubulin polyclonal Ab (rabbit, Abcam, cat. Ab6046). Membranes were washed three times (5 min per wash) with PBST. HRP-conjugated goat anti-mouse or rabbit antibodies were incubated for 1 h at room temperature. PVDF membranes were washed three times with PBST for 5 min. Antigen-antibody complexes were then detected by exposure for 4 min to the enhanced chemiluminescence solution (ECL; Amersham). Then, the membrane was placed down on a film layer, which had been arranged inside a film cassette, covered with another film layer and exposed to photographic film (BioMax XAR Film Kodak, Sigma-Aldrich) for an adequate exposure time. The film was developed and immunoreactivity (band density) was quantified by using densitometry (source: http://rsbweb.nih.gov/ij/docs/user-guide.pdf) using ImageJ. β-tubulin was used as loading control.
Nitric oxide production in cultured iPSC-ECs
iPSC-ECs were stained with DAF-FM DA (Thermo Scientific) to assess intracellular NO levels. Cultured iPSC-ECs were incubated with DAF-FM DA 10 μM at 37°C/5% CO2 for 30 min as recommended by the supplier. Excess probe was removed by washing with PBS and the cells were switched to fresh media prior to imaging. The intensity of the fluorescent signal (reflecting NO level) was measured using image J and plotted as mean of the fluorescent signal (integrated density) in each cell. As a second assay to measure nitric oxide, NO metabolites nitrite and nitrate (Cayman Chemical, Nitrate/Nitrite Assay Kit, cat. n. 780001) were detected. In brief, cell culture medium was first added with nitrate reductase that converts nitrate to nitrite. Then, Griess reagents added to the sample convert nitrite into a deep purple azo compound. Photometric measurement of the absorbance (540 nm wavelength) derived from this azo chromophore accurately determines nitrite concentration. Cellular nitrate/nitrite production is quantitated by subtracting the level of nitrate/nitrite present in the media alone from the total nitrate/nitrite level present during cell growth.
Dil-Ac-LDL uptake assay
iPSC-ECs were plated in 6-well plates. Uptake of Ac-LDL was assessed by incubating with ac-LDL-594 (Thermo Scientific) at 1:200 dilutions for 3 h. Then cells were washed with PBS and the mean fluorescence measured in n = 5 high power fields using image J and plotted as mean of the fluorescent signal (integrated density) in each cell.
Angiogenesis assay (vascular network formation)
Vascular network formation assays were carried out as previously described in our studies [58,59]. iPSC-ECs were seeded at 40000 cells per 24-well plate in growth factor reduced Matrigel (Corning) in EGM2-MV medium. The number of network segments was measured after 24 h in five random microscopic fields. ImageJ was used to process and analyze the images.
Real-time iPSC-ECs proliferation assay
Proliferation assays were carried out using the xCELLigence instrument (Roche) which was placed in a humidified incubator at 37°C and 5% CO2. iPSC-EC proliferation was monitored using 16-well plates (E-plate, Roche). iPSC-ECs were harvested and equal number of cells (3000–5000) was seeded into the wells with medium, in triplicate. The impedance value of each well was monitored by xCELLigence and expressed as a Cell Index (CI) value. CI was monitored every minute in the first 4 h, and every hour for the rest of the days.
Telomere length measurement by monochrome multiplex PCR (MMqPCR)
Cellular DNA was isolated using Qiagen® AllPrep® DNA/RNA/Protein Mini Kit following the manufacturer’s protocol. MMqPCR was performed in accordance with a previously published protocol by Cawthon [60]. Briefly, samples were run in triplicate for both control and HGPS iPSC-ECs. Samples and standards were pipetted into PCR tubes; standard was created via two-fold serial dilution. A PCR mix was added to each PCR tube and then vortexed. Samples and standards were transferred to a white LightCycler® 480 Multiwell plate (384 wells), pipetting 10 µL of the total PCR preparation mix into each well, creating experimental triplicates in addition to the biological triplicates detailed earlier. PCR runs were performed on a Roche LightCycler® 480 (Software: 1.5.1.62) using a preinstalled MMqPCR protocol. Raw data were pre-processed via Python script to differentiate different primers for the telg/telc primers and single-copy gene (SCG) primer (human β-globin). After preprocessing, data were re-uploaded to the Roche LightCycler® system for T/S ratio calculations.
In vivo matrigel angiogenesis assay
To assess the capacity of the iPSC-ECs to form capillary structures in vivo, matrigel was mixed with 5 × 105 5 iPSC-ECs in a final volume of 200 μl and injected subcutaneously into the lower abdominal region of SCID mice. Two Matrigel plugs were implanted per mouse. Five days later, the mice were sacrificed and the Matrigel plugs were retrieved, processed where appropriate, and analyzed accordingly. For CD31 immunohistochemistry of Matrigel plugs harvested from the mice, 5 μm-thick paraffin sections were deparaffinized and hydrated. The slides were incubated with the CD3 antibody overnight followed by conjugation to the secondary antibody and DAB staining. The Matrigel plug was fixed in paraformaldehyde 4% and embedded in paraffin. Sections of 5 μm were prepared and deparaffinized. After rehydration, the slides were stained with hematoxylin and eosin following a standard protocol.
Data analysis
Results were expressed as the mean±SEM. Each experiment was performed 3 times. The Shapiro–Wilk test was used to confirm the null hypothesis that the data follow a normal distribution. Statistical comparisons between two groups or multiple groups were then performed, respectively, via Student t-test or ANOVA test using PRISM 7 software. A P value <0.05 was considered significant.
Funding Statement
This work was supported by the National Institutes of Health, the Progeria Research Foundation, the Cullen Trust for Health Care and the American Heart Association.
Acknowledgments
We thank Progeria Research Foundation and Coriell Cell Repositories for providing cell lines, as well as grants to J.P.C. from the Progeria Research Foundation (PRF 2015-58) and from the National Institutes of Health R01 HL133254, R01HL149303 and R01GM125632. G.M. was also supported by a grant from The Cullen Trust for Health Care, R.A.T. was also supported by a grant from the American Heart Association.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003. DOI: 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Walther B, Li Y, Thandavarayan R, et al. Progeria and accelerated cardiovascular aging. Cardiol Plus. 2018;3:81. [Google Scholar]
- [3].Gordon LB, Kleinman ME, Massaro J, et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with hutchinson-gilford progeria syndrome. Circulation. 2016. DOI: 10.1161/CIRCULATIONAHA.116.022188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Broers JLV. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006. DOI: 10.1152/physrev.00047.2005 [DOI] [PubMed] [Google Scholar]
- [5].Barrowman J, Hamblet C, George CM, et al. Analysis of prelamin a biogenesis reveals the nucleus to be a CaaX processing compartment. Mol Biol Cell. 2008;19:5398–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reddy S, Comai L, Lamin A.. farnesylation and aging. Exp Cell Res. 2012;318:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci. 2004. DOI: 10.1073/pnas.0402943101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006. DOI: 10.1194/jlr.R500012-JLR200 [DOI] [PubMed] [Google Scholar]
- [9].Rusinol AE. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J Cell Sci. 2006. DOI: 10.1242/jcs.03156 [DOI] [PubMed] [Google Scholar]
- [10].Capell BC, Olive M, Erdos MR, et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc Natl Acad Sci. 2008. DOI: 10.1073/pnas.0807840105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yang SH, Meta M, Qiao X, et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006. DOI: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fong LG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006. DOI: 10.1126/science.1124875 [DOI] [PubMed] [Google Scholar]
- [13].Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci. 2012. DOI: 10.1073/pnas.1202529109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gordon LB, Massaro J, D’Agostino RB, et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014. DOI: 10.1161/CIRCULATIONAHA.113.008285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sagelius H, Rosengardten Y, Hanif M, et al. Targeted transgenic expression of the mutation causing Hutchinson-Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J Cell Sci. 2008. DOI: 10.1242/jcs.022913 [DOI] [PubMed] [Google Scholar]
- [16].Varga R, Eriksson M, Erdos MR, et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci. 2006. DOI: 10.1073/pnas.0600012103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang SH, Bergo MO, Toth JI, et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci. 2005. DOI: 10.1073/pnas.0504641102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mounkes LC, Kozlov S, Hernandez L, et al. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003. DOI: 10.1038/nature01631 [DOI] [PubMed] [Google Scholar]
- [19].Lee G, Papapetrou EP, Kim H, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009. DOI: 10.1038/nature08320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Park IH, Arora N, Huo H, et al. Disease-specific induced pluripotent stem cells. Cell. 2008. DOI: 10.1016/j.cell.2008.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Svendsen CN. Back to the future: How human induced pluripotent stem cells will transform regenerative medicine. Hum Mol Genet. 2013. DOI: 10.1093/hmg/ddt379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Agarwal S, Loh Y-H, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010. DOI: 10.1038/nature08792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang J, Lian Q, Zhu G, et al. A human iPSC model of hutchinson gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011. DOI: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- [24].Hamczyk MR, Villa-Bellosta R, Gonzalo P, et al. Vascular smooth muscle–specific progerin expression accelerates atherosclerosis and death in a mouse model of Hutchinson-Gilford progeria syndrome. Circulation. 2018. DOI: 10.1161/CIRCULATIONAHA.117.030856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bonello-Palot N, Simoncini S, Robert S, et al. Prelamin A accumulation in endothelial cells induces premature senescence and functional impairment. Atherosclerosis. 2014. DOI: 10.1016/j.atherosclerosis.2014.08.036 [DOI] [PubMed] [Google Scholar]
- [26].Bernadotte A, Mikhelson VM, Spivak IM. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY). 2016. DOI: 10.18632/aging.100871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sukhovershin RA, Yepuri G, Ghebremariam YT. Endothelium-derived nitric oxide as an antiatherogenic mechanism: implications for therapy. Methodist Debakey Cardiovasc J. 2015. DOI: 10.14797/mdcj-11-3-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lai ES, Huang NF, Cooke JP, et al. Aligned nanofibrillar collagen regulates endothelial organization and migration. Regener Med. 2012. DOI: 10.2217/rme.12.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cooke JP. The pivotal role of nitric oxide for vascular health. Can J Cardiol. 2004;20 Suppl B:7B–15B. [PubMed] [Google Scholar]
- [30].Matsushita H, Chang E, Glassford AJ, et al. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001. DOI: 10.1161/hh2101.098443 [DOI] [PubMed] [Google Scholar]
- [31].Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006. DOI: 10.1126/science.1127168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005. DOI: 10.1038/nm1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pegoraro G, Kubben N, Wickert U, et al. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009. DOI: 10.1038/ncb1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003. DOI: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- [35].Santiago-Fernández O, Osorio FG, Quesada V, et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat Med. 2019;25:423–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Olive M, Harten I, Mitchell R, et al. Cardiovascular pathology in hutchinson-gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010. DOI: 10.1161/ATVBAHA.110.209460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stehbens WE, Wakefield SJ, Gilbert-Barness E, et al. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol. 1999. DOI: 10.1016/S1054-8807(98)00023-4 [DOI] [PubMed] [Google Scholar]
- [38].Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016. DOI: 10.1161/CIRCRESAHA.115.306301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Virmani R, Kolodgie FD, Burke AP, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000. DOI: 10.1161/01.ATV.20.5.1262 [DOI] [PubMed] [Google Scholar]
- [40].Stary HC. Natural history and histological classification of atherosclerotic lesions an update. Arterioscler Thromb Vasc Biol. 2000. DOI: 10.1161/01.ATV.20.5.1177 [DOI] [PubMed] [Google Scholar]
- [41].Liu GH, Barkho BZ, Ruiz S, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011. DOI: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chang F, Flavahan S, Flavahan NA. Impaired activity of adherens junctions contributes to endothelial dilator dysfunction in ageing rat arteries. J Physiol. 2017;595:5143–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kinoshita D, Nagasawa A, Shimizu I, et al. Progerin impairs vascular smooth muscle cell growth via the DNA damage response pathway. Oncotarget. 2017;8:34045–34056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gruenbaum Y, Wilson KL, Harel A, et al. Review: Nuclear lamins – structural proteins with fundamental functions. J Struct Biol. 2000. DOI: 10.1006/jsbi.2000.4216 [DOI] [PubMed] [Google Scholar]
- [45].Cohen TV, Hernandez L, Stewart CL. Functions of the nuclear envelope and lamina in development and disease: Figure 1. Biochem Soc Trans. 2008;36:1329–1334. [DOI] [PubMed] [Google Scholar]
- [46].Jufri NF, Mohamedali A, Avolio A, et al. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc Cell. 2015. doi: 10.1186/s13221-015-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dechat T, Pfleghaar K, Sengupta K, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008. DOI: 10.1101/gad.1652708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hetzer MW. The nuclear envelope. Cold Spring Harb Perspect Biol. 2010;2:a000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Aubert G, Lansdorp PM. Telomeres and Aging. Physiol Rev. 2008;88:557–579. [DOI] [PubMed] [Google Scholar]
- [50].De Lange T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005. DOI: 10.1101/gad.1346005 [DOI] [PubMed] [Google Scholar]
- [51].Hamczyk MR, Del Campo L, Andrés V. Aging in the cardiovascular system: lessons from hutchinson-gilford progeria syndrome. Annu Rev Physiol. 2018;80:27–48. [DOI] [PubMed] [Google Scholar]
- [52].Seals DR, Kaplon RE, Gioscia-Ryan RA, et al. You’re only as old as your arteries: translational strategies for preserving vascular endothelial function with aging. Physiology. 2014. DOI: 10.1152/physiol.00059.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Blasco MA. Telomeres and human disease: Ageing, cancer and beyond. Nat Rev Genet. 2005. DOI: 10.1038/nrg1656 [DOI] [PubMed] [Google Scholar]
- [54].Hoeijmakers JHJ. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. [DOI] [PubMed] [Google Scholar]
- [55].Khanna KK, Jackson SP. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat Genet. 2001. DOI: 10.1038/85798 [DOI] [PubMed] [Google Scholar]
- [56].Lian X, Bao X, Al-Ahmad A, et al. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of WNT signaling. Stem Cell Reports. 2014. DOI: 10.1016/j.stemcr.2014.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wanjare M, Hou L, Nakayama KH, et al. Anisotropic microfibrous scaffolds enhance the organization and function of cardiomyocytes derived from induced pluripotent stem cells. Biomater Sci. 2017. DOI: 10.1039/c7bm00323d [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sayed N, Wong WT, Ospino F, et al. Transdifferentiation of human fibroblasts to endothelial cells role of innate immunity. Circulation. 2015. DOI: 10.1161/CIRCULATIONAHA.113.007394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Meng S, Matrone G, Lv J, et al. LIM domain only 2 regulates endothelial proliferation, angiogenesis, and tissue regeneration. J Am Heart Assoc. 2016. DOI: 10.1161/JAHA.116.004117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009. DOI: 10.1093/nar/gkn1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.