

Mitochondrial membrane lipids adapt to exercise or disuse, underscoring an essential role for PE in energy transduction.
Abstract
Exercise capacity is a strong predictor of all-cause mortality. Skeletal muscle mitochondrial respiratory capacity, its biggest contributor, adapts robustly to changes in energy demands induced by contractile activity. While transcriptional regulation of mitochondrial enzymes has been extensively studied, there is limited information on how mitochondrial membrane lipids are regulated. Here, we show that exercise training or muscle disuse alters mitochondrial membrane phospholipids including phosphatidylethanolamine (PE). Addition of PE promoted, whereas removal of PE diminished, mitochondrial respiratory capacity. Unexpectedly, skeletal muscle–specific inhibition of mitochondria-autonomous synthesis of PE caused respiratory failure because of metabolic insults in the diaphragm muscle. While mitochondrial PE deficiency coincided with increased oxidative stress, neutralization of the latter did not rescue lethality. These findings highlight the previously underappreciated role of mitochondrial membrane phospholipids in dynamically controlling skeletal muscle energetics and function.
INTRODUCTION
Low aerobic capacity is a stronger risk factor for all-cause mortality compared to other common risk factors such as hypertension, type 2 diabetes, and smoking (1). Skeletal muscle mitochondrial respiration is the largest contributor for whole-body aerobic capacity (2), which in turn is influenced by mitochondrial density and activities of the electron transport system (ETS). Changes in physical activity robustly alter skeletal muscle mitochondrial content and maximal aerobic capacity (3, 4). This proliferation or diminishment of mitochondrial biomass must coincide with synthesis or degradation of mitochondrial enzymes and structural lipids. While processes that regulate mitochondrial enzymes are well described (5, 6), it is unknown how composition of mitochondrial lipids changes in response to these adaptations.
Lipids of the inner mitochondrial membrane (IMM) are largely phospholipids with only trace amounts of sphingolipids and cholesterol (7). They consist of phosphatidylcholine (PC; 38 to 45%), phosphatidylethanolamine (PE; 32 to 39%), cardiolipin (CL; 14 to 23%), phosphatidylinositol (PI; 2 to 7%), phosphatidylserine (PS) (8), phosphatidylglycerol (PG), and lyso-PC (all less than 3%) (9). These phospholipids not only give rise to the shape of IMM but also are essential for activities of the enzymes of the ETS (9, 10). In particular, PE and CL are conical shaped phospholipids that promote the formation of cristae where ETS enzymes reside. These nonbilayer lipids lessen torsional strain of the IMM by localizing into the negatively curved inner leaflet (11–13). They also bind with high affinity to mitochondrial respiratory complexes and regulate their functions (9, 14). Human mutations that promote loss of mitochondrial PE or CL are detrimental to health (15–17).
In this study, we set out to describe changes in the skeletal muscle mitochondrial phospholipidome that occur with exercise or disuse. Mitochondrial PE emerged as a key lipid signature that was induced by alterations in physical activity. We then pursued the cellular consequences of changes in muscle mitochondrial PE per se in skeletal muscle–specific tamoxifen-inducible gain- or loss-of-function mouse models. Greater mitochondrial PE, in the absence of changes in the abundance of ETS enzymes, was sufficient to increase the capacity for oxidative phosphorylation. Loss of mitochondrial PE proved fatal because of metabolic and contractile failure in the diaphragm muscle.
RESULTS
Mitochondrial PE increases skeletal muscle respiratory capacity
Endurance exercise training induces a robust proliferation of skeletal muscle mitochondria to increase aerobic capacity (3), but it is unknown whether training coincides with qualitative changes in mitochondrial phospholipid composition (9). C57BL6/J mice were subjected to 5 weeks of graded treadmill training, which promoted skeletal muscle mitochondrial biogenesis (fig. S1A). Phospholipid analyses of these mitochondria revealed a disproportionately greater increase in PE compared to other phospholipids (Fig. 1, A and B). High-capacity running (HCR) rats, which have been selectively bred for their intrinsic exercise capacity, demonstrate protection from a wide range of metabolic and cardiovascular diseases compared to low-capacity running (LCR) rats (18). Skeletal muscle mitochondria from HCR rats contained more PE than did LCR (fig. S1B). These observations led us to examine the possibility that an increase in mitochondrial PE contributes to increased aerobic capacity in exercise-trained mice or HCR rats.
Fig. 1. Skeletal muscle mitochondrial PE promotes oxidative capacity.
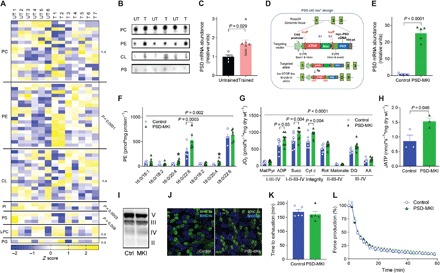
(A to C) Untrained (UT, n = 6) or trained (T, n = 7 or 8) C57BL6/J mice. (A) Skeletal muscle mitochondrial phospholipidome. LPC, lyso-PC; PG, phosphatidylglycerol; n.s., not significant. (B) Mitochondrial phospholipids quantified by thin-layer chromatography (TLC). (C) Skeletal muscle PSD mRNA. (D to L) Studies on PSD-MKI mice (n = 3 to 9). (D) Generation of mice with conditional knock-in of PSD. 5′UTR, 5′ untranslated region. (E) Skeletal muscle PSD mRNA. (F) Muscle mitochondrial PE. (G and H) Rates for oxygen consumption or ATP production in permeabilized muscle fibers with Krebs cycle substrates. DQ, duroquinol; AA, antimycin A. (I) Protein abundance of respiratory complexes II to V. (J) Myosin heavy chain (MHC) fiber-type distribution. (K) Endurance running test. (L) Ex vivo twitch endurance test. Means ± SEM.
Mitochondrial PE is synthesized primarily by the enzyme PS decarboxylase (PSD) that resides in the IMM (19, 20). Skeletal muscle PSD expression was greater in exercise-trained mice compared to sedentary mice (Fig. 1C) and in HCR rats compared to LCR rats (21). Overexpression of PSD in murine C2C12 myotubes increased the maximal O2 consumption rate (fig. S1C), suggesting that an increased amount of mitochondrial PE enhances respiratory capacity. To study the effects of increased mitochondrial PE in vivo, we generated mice with tamoxifen-inducible skeletal muscle–specific overexpression of PSD (PSD-MKI) (Fig. 1D). This strategy successfully yielded mice with skeletal muscle–specific PSD overexpression (Fig. 1E) and elevated mitochondrial PE (Fig. 1F). High-resolution respirometry/fluorometry experiments revealed that PSD overexpression increased the rates of O2 consumption and adenosine 5′-triphosphate (ATP) production (Fig. 1, G and H), effects that were not due to increased mitochondrial mass, abundance of ETS enzymes, supercomplex formation (Fig. 1I and fig. S1, D to F), or changes in fiber type (Fig. 1J). However, the increase in respiratory capacity did not increase treadmill endurance performance (Fig. 1K) or skeletal muscle force generating capacity ex vivo (Fig. 1L and fig. S1, G and H). PSD-MKI and control mice also did not differ in body weight or composition, food intake, or energy expenditure (fig. S1, I to M). Thus, an increase in muscle mitochondrial PE can increase oxidative capacity but not to an extent that influences endurance. An increase in muscle endurance likely requires concomitant improvements in contractile elements and substrate mobilization.
Loss of mitochondrial PE promotes rapid atrophy and ventilatory failure
Skeletal muscle disuse rapidly reduces mitochondrial mass and function. Reduced mitochondrial function precedes disuse-induced muscle atrophy (22) and might contribute to the mechanism for skeletal muscle loss (23). We subjected C57BL6/J mice to a 2-week hindlimb unloading (HU) (24), which promoted robust muscle loss (Fig. 2A). Phospholipid analyses of skeletal muscle mitochondria revealed that disuse promotes an accelerated loss of PE (Fig. 2B), concomitant with reduced PSD mRNA (Fig. 2C). To model the loss of mitochondrial PE in vitro, we performed lentivirus-mediated knockdown of PSD in C2C12 myotubes (fig. S2A). Reduction in mitochondrial PE (fig. S2B) robustly reduced mitochondrial respiratory capacity (fig. S2, C and D) in the absence of changes in ETS enzymes (fig. S2, E to G), suggesting that lack of PE suppresses activities of membrane-bound ETS enzymes (fig. S2, H and I). Because global knockout of PSD is embryonically lethal (25), we generated mice with tamoxifen-inducible skeletal muscle–specific knockout of PSD (PSD-MKO) (Fig. 2, D and E). Skeletal muscle mitochondria from PSD-MKO mice were selectively depleted in PE esterified with polyunsaturated fatty acids (Fig. 2, F and G, and fig. S3, A and B). Notably, tamoxifen-induced knockout promoted rapid weight loss (Fig. 2H), kyphosis (Fig. 2I), and ultimately death between 6 and 8 weeks after tamoxifen injection (Fig. 2J). The lethality of PSD knockout was likely induced by ventilatory failure in respiratory muscles, a common symptom in mitochondrial diseases (26), as evidenced by reduced breathing rate and SpO2 (Fig. 2, K and L). Cardiomyopathy, pulmonary edema, low bone density, hypophagia, or hypomobility did not explain the premature death in PSD-MKO mice (fig. S3, C to K). Reduction in body weight was manifested in both lean and fat mass loss (fig. S3L) and in weights of individual muscles including diaphragm (Fig. 2M and fig. S3M). The loss in muscle weights was explained by reduction in cross-sectional area of individual muscle fibers (Fig. 2N and fig. S3, N and O). The diaphragm displayed substantial fibrosis (Fig. 2O) and loss of force-generating capacity (Fig. 2P and fig. S3, P and Q). Thus, acute loss of mitochondrial PE promotes rapid loss of skeletal muscle mass and function that is reminiscent of atrophy found in disuse in limb muscles, as well as that in respiratory muscles during mechanical ventilation (27).
Fig. 2. Deficiency of mitochondrial PE promotes atrophy and respiratory failure.
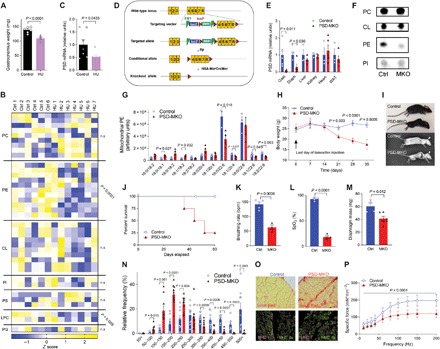
(A to C) Control (Ctrl; n = 6 or 7) and hindlimb-unloaded (HU; n = 7) C57BL6/J mice. (A) Gastrocnemius weight. (B) Skeletal muscle mitochondrial phospholipidome. (C) Skeletal muscle PSD mRNA. (D to P) Studies on PSD-MKO mice. (D) Generation of PSD-MKO mice. (E) PSD mRNA levels in multiple tissues (n = 5 to 6). (F) TLC analysis of mitochondrial phospholipids. (G) Muscle mitochondrial PE (n = 3 to 4). (H) Body weights after tamoxifen injection (n = 9 to 22). (I) Kyphosis in PSD-MKO mice. (J) Kaplan-Meier survival curve. (K and L) Breathing rate and peripheral capillary oxygen saturation (SpO2) 6 weeks after tamoxifen injection (n = 3). bpm, breaths per minute. (M to P) Diaphragm 4 weeks after tamoxifen injection. (M) Diaphragm weight (n = 4 to 6). (N) Distribution of fiber cross-sectional area (n = 9). (O) Fibrosis and fiber type. (P) Force-frequency curve (n = 4 to 6). Means ± SEM.
Loss of PE induces mitochondrial dysfunction and oxidative stress
As PSD generates PE for the IMM, the underlying cause of lethal myopathy in PSD-MKO mice is also likely due to changes to mitochondria. Mitochondria from PSD-MKO mice appeared to have disorganized and less dense cristae (Fig. 3A and fig. S4A), similar to findings in PSD-depleted Chinese hamster ovary cells (28) and global PSD knockout mice (25). These changes occurred in the absence of alterations in abundance of proteins involved in mitochondrial fusion and fission (fig. S4, B and C). High-resolution respirometry and fluorometry experiments revealed a robust reduction in the rates of O2 consumption and ATP production in PSD-MKO muscles (Fig. 3, B and C, and fig. S4, D and E), without changes in abundance of ETS enzymes (Fig. 3D and fig. S4F). PE molecules are bound to ETS complexes I to IV, likely facilitating conformational changes and acting as an allosteric activator (29–32). Enzyme activity assays revealed that activities of ETS complexes I to IV, but not V, were lower in muscles from PSD-MKO mice than in control muscles (Fig. 3E). Oxidative phosphorylation is also dependent on assembly of respiratory supercomplexes (33, 34), and PE appears to be essential for this process (28). Reduction in mitochondrial PE essentially eliminated the formation of respiratory supercomplexes (Fig. 3F). Together, these observations suggest that mitochondrial PE deficiency stagnates efficient electron transfer in the ETS. In turn, inefficiency in electron transfer is predicted to promote electron leakage that causes superoxide production (35, 36). H2O2 production was markedly higher in PSD-MKO muscles than in control muscles under various substrate conditions (Fig. 3G and fig. S4, G to K). Elevated oxidative stress also increased reactive lipid aldehydes such as 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) (Fig. 3, H and I, and fig. S4L) (37), oxidized glutathione (Fig. 3J), and counteroxidative response proteins (fig. S4, M and N) in PSD-MKO muscles to a greater extent than in control muscles. As oxidative stress has been implicated in skeletal muscle atrophy (38, 39), we further tested the mechanistic link among mitochondrial PE deficiency, oxidative stress, and respiratory failure in PSD-MKO mice.
Fig. 3. PE deficiency in skeletal muscle mitochondria.
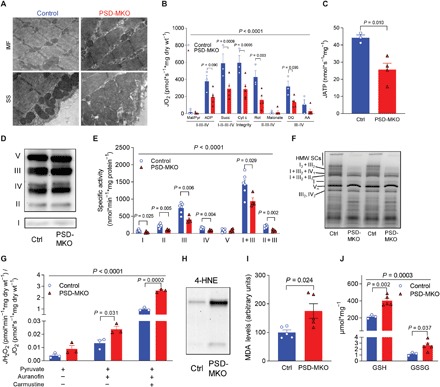
(A) Electron micrograph of subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria. (B and C) Rates of oxygen consumption and ATP production in permeabilized fibers with Krebs cycle substrates (n = 3 to 5). (D) Protein abundance of respiratory complexes I to V. (E) Activities of respiratory enzymes (n = 4 to 6). (F) Blue native gel of isolated mitochondria revealing supercomplexes (n = 4). High–molecular weight supercomplexes (HMW SCs). (G) Mitochondrial H2O2 production and emission with pyruvate normalized to O2 consumption (n = 3). (H) 4-HNE. (I) MDA (n = 5). (J) Reduced glutathione (GSH) and oxidized glutathione (GSSG) (n = 3 to 5). Means ± SEM.
Neutralization of mitochondrial reactive oxygen species does not rescue metabolic defects induced by mitochondrial PE deficiency
In skeletal muscle, excess superoxide dismutase converts superoxide to H2O2 (40, 41). To neutralize H2O2 produced in mitochondrial PE-deficient mice, we crossed the PSD-MKO mice with mice that overexpressed mitochondria-targeted catalase (mCAT) (Fig. 4A and fig. S5A). This strategy yielded mice (mCAT × PSD-MKO) with suppressed skeletal muscle H2O2 production (Fig. 4B and fig. S5B). However, mCAT overexpression did not rescue the lethality of PSD-MKO mice (Fig. 4C), nor did it ameliorate muscle atrophy (Fig. 4D and fig. S5C), force-generating capacity (Fig. 4E and fig. S5D), or oxidative capacity (Fig. 4F and fig. S5E). Thus, while mitochondrial PE deficiency increases oxidative stress, it is not directly responsible for the contractile and metabolic defects in PSD-MKO mice. This finding was somewhat unexpected because oxidative stress is predicted to activate an array of downstream pathways, many of which overlap with defects observed in PSD-MKO mice (38, 39). To understand the biological processes in PSD-MKO mice that might explain their lethality, we performed deep-sequencing analyses on diaphragms from control, PSD-MKO, and mCAT × PSD-MKO mice. Transcripts for 6026 genes were differentially expressed between control and PSD-MKO diaphragms, and some of which were rescued with mCAT overexpression (fig. S5F). However, a large majority of differentially expressed genes were not rescued with mCAT (fig. S5G), consistent with our observations that deficiency in mitochondrial PE triggers events independent of oxidative stress. Among the 68 pathways that were statistically significantly affected between control and PSD-MKO diaphragms, 45 of them remained altered in mCAT × PSD-MKO diaphragms (Fig. 4G). PSD deletion activated pathways for proteasome and ubiquitin-mediated proteolysis but not lysosome or apoptosis, suggesting that mitochondrial PE deficiency likely promotes muscle atrophy via proteasomal degradation (Fig. 4H). PSD deletion also induced activation of transcriptional and translational pathways that were not reversed in mCAT × PSD-MKO diaphragms. As expected, mCAT overexpression suppressed activation of antioxidant pathways including glutathione metabolism and peroxisomal genes.
Fig. 4. Overexpression of mitochondrial catalase does not rescue PSD deficiency.
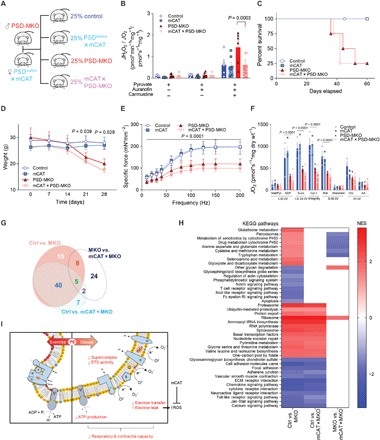
(A) PSD-MKO mice were crossed with mCAT transgenic mice to generate mCAT × PSD-MKO mice. (B) Mitochondrial H2O2 production and emission with pyruvate normalized to O2 consumption (n = 5 to 7). (C) Kaplan-Meier survival curve. (D) Body weights after tamoxifen injection (n = 6 to 9). (E) Force-frequency curve of diaphragm muscles (n = 4 to 11). (F) Rates of oxygen consumption in permeabilized fibers with Krebs cycle substrates (n = 4 to 10). (G and H) Pathway analyses for differentially expressed genes between control, PSD-MKO, and mCAT × PSD-MKO diaphragms (n = 3 to 4). (G) Area-proportional Venn diagram of differentially activated pathways. (H) Normalized enrichment scores (NES) of differentially activated pathways. ECM, extracellular matrix. (I) Schematic illustration of the consequences of mitochondrial PE deficiency. ROS, reactive oxygen species. Means ± SEM.
DISCUSSION
Phospholipid molecules are largely insoluble in the aqueous cytosol, and their intracellular movements are relatively limited. Thus, the membrane phospholipid composition of different organelles is highly distinct, thereby creating a biophysical environment unique to each subcellular location. In mitochondria, the high concentration of PE not only promotes membrane curvature in the cristae but also is essential for efficient electron transfer and oxidative phosphorylation (12, 28, 42). Recent reports identified loss-of-function mutations in the human PISD gene, which encodes the PSD enzyme, that promote severe mitochondrial dysfunction and are characterized by congenital cataracts, short stature, facial dysmorphism, platyspondyly, ataxia, and/or intellectual disability (15, 17). Combined with data presented in the current manuscript, these findings indicate a critical role that mitochondrial PE plays in health and disease.
In skeletal muscle, inhibition of mitochondria-autonomous synthesis of PE via PSD causes robust skeletal muscle atrophy and ventilatory failure because of fibrosis and loss of contractility in the diaphragm muscle. The underlying cause appears to be due to a system failure of mitochondrial circuitry that is evident in reduced activities of complexes I to IV, low supercomplex formation, and elevated electron leak (Fig. 4I). We predict that these defects trigger proteosomal degradation pathways that promote muscle atrophy and weakness. In contrast, such a phenotype does not occur when PE synthesis via the cytidine 5′-diphosphate (CDP)–ethanolamine pathway on the endoplasmic reticulum (ER) is inhibited in skeletal muscle (43, 44), suggesting that pools of PE made by PSD and CDP-ethanolamine pathway are functionally distinct. How selected phospholipids (such as PC, PI, and PA) are transported from ER to mitochondria but PE is unable to do so is not understood (12, 20). Furthermore, it is known that PE generated by PSD is readily transported from mitochondria to ER (45). Thus, it remains possible that some of the defects in the PSD-MKO mice are due to disturbances in ER function caused by a lack of PE or an accumulation of PS at the ER. Pathways involved in ribosome and protein translation were significantly altered with PSD deletion (Fig. 4H), suggesting that ER stress may be involved in the atrophy phenotype. Nevertheless, note that mitochondrial PE deficiency did not promote changes in protein abundance for ETS enzymes or mitochondrial dynamics (Fig. 3D and fig. S4, B and C).
In conclusion, our findings reveal that alteration in PE composition represents a key adaptive response to exercise or disuse in skeletal muscle mitochondria. Gain- or loss-of-function studies show that changes in mitochondrial PE modulate oxidative capacity independently of changes in abundance of ETS proteins. A deficiency of mitochondrial PE in the diaphragm muscle is detrimental to its metabolic and contractile function, leading to ventilatory failure and lethality. While these defects were associated with increased oxidative stress, its neutralization with mCAT did not prevent any of the dysfunction induced by PSD deficiency. These findings also raise a possibility that reduced mitochondrial PE represents a mechanism by which disuse promotes the loss of muscle mass and function associated with muscle atrophy. Changes in mitochondrial phospholipid composition appear to be an important regulatory mechanism by which physical activity modulates mitochondrial energetics in skeletal muscle.
MATERIALS AND METHODS
Rodent models
PSD conditional knock-in (PSDcKI+/+) mice were generated by inserting myc-tagged mouse Pisd complementary DNA (cDNA) into a Rosa26 locus. Pisd cDNA was preceded by a CAG promoter and loxP-flanked stop codon for tissue-specific ectopic expression of PSD. These mice were then crossed with HSA-MerCreMer mice (tamoxifen-inducible α-human skeletal actin Cre, courtesy of K. Esser, University of Florida) to generate PSD-MKI (PSDcKI+/−, HSA-MerCreMer+/−) and control (PSDcKI+/−, no Cre) mice. Mouse embryonic stem (ES) cells that carried loxP sites flanking exons 4 to 8 of the mouse PSD gene were purchased from the European Conditional Mouse Mutagenesis Program. The cells were microinjected into C57BL/6J blastocysts and transplanted in pseudopregnant females to produce PSD conditional knockout (PSDcKO+/+) mice. These mice were crossed with HSA-MerCreMer mice to generate PSD-MKO (PSDcKO+/+, HSA-MerCreMer+/−) and control (PSDcKO+/+, no Cre) mice. mCAT transgenic mice were purchased from the Jackson laboratory (stock no. 0161971) and crossed with PSD-MKO mice to generate the mCAT × PSD-MKO (mCAT+/−, PSDcKO+/+, HSA−MerCreMer+/−), PSD-MKO, mCAT (mCAT+/−, PSDcKO+/+, no Cre), and control (PSDcKO+/+, no Cre) mice. Cre control mice (PSDcKO−/− or PSDcKI−/−, HSA-MerCreMer+/−) and tamoxifen-untreated control mice displayed no difference in phenotype to loxP control mice. All mice were bred onto C57BL/6J background and were born at normal Mendelian ratios. Both male and female mice were studied with no difference in phenotypes. HCR and LCR rats were maintained and studied at the University of Michigan. All animals were fasted 4 hours before tissue collection. All protocols were approved by Institutional Animal Care and Use Committees at University of Utah, East Carolina University, and University of Michigan.
Exercise training
Male C57BL/6J mice were kept untrained (n = 5) or underwent treadmill training (5 days per week, 12 m/min, 2 to 6% incline, n = 8) for 5 weeks. The mice were then euthanized, and tissues were dissected ~40 hours after the last exercise session.
Hindlimb unloading
Male C57BL/6J mice underwent 2 weeks of HU or were ambulatory controls. The HU (two mice per cage) were subjected to a modified unloading method based on the traditional Morey-Holton design for studying disuse atrophy in rodents (24). Body weight and food intake were monitored every other day to ensure that mice did not experience excessive weight loss due to malnutrition or dehydration. At the end of day 14 of HU, mice were fasted for 4 hours and anesthetized for tissue collection.
RNA quantification
For quantitative polymerase chain reaction (qPCR) experiments, mouse tissues or cells were lysed in 1 ml of TRIzol (Thermo Fisher Scientific), and RNA was isolated using standard techniques. The iScript cDNA Synthesis Kit was used to reverse transcribe total RNA, and qPCR was performed with SYBR Green reagents (Thermo Fisher Scientific). Prevalidated primer sequences were obtained from mouse primer depot (https://mouseprimerdepot.nci.nih.gov/). All mRNA levels were normalized to RPL32. For RNA sequencing, diaphragm RNA was isolated with the RNeasy Kit (74104, Qiagen). RNA library construction and sequencing were performed by the High-Throughput Genomics Core at the Huntsman Cancer Institute, University of Utah. RNA libraries were constructed using the Illumina TruSeq Stranded Total RNA Sample Prep Kit, and contaminating ribosomal RNAs were removed using Ribo-Zero Gold. Sequencing was performed using a NovSeq2 with 25 million reads per sample. Pathway analyses were performed by the Bioinformatics Core at the Huntsman Cancer Institute, University of Utah using the KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway Database. For differentially expressed genes, only transcripts with Padj < 0.05 and baseMean > 100 are included. For pathway analyses, the area-proportional Venn diagram was drawn with eulerAPE (46). KEGG pathways that were differentially expressed between control and PSD-MKO diaphragms were stratified to those that were or were not rescued by mCAT overexpression.
Mitochondrial isolation
Tissues were minced in ice-cold mitochondrial isolation medium (MIM) buffer [300 mM sucrose, 10 mM Hepes, 1 mM EGTA, and bovine serum albumin (BSA; 1 mg/ml) (pH 7.4)] and gently homogenized with a Teflon pestle. The homogenate was centrifuged at 800g for 10 min at 4°C. The supernatant was transferred to another tube and centrifuged again at 12,000g for 10 min at 4°C. The crude mitochondrial pellet was suspended in 15% Percoll (diluted with MIM buffer), and a discontinuous Percoll gradient was prepared consisting of 50, 22, and 15% Percoll layers. Mitochondria were carefully layered on top of the gradient and spun at 22,700 rpm for 10 min at 4°C in an ultracentrifuge (SureSpin 630 rotor, Thermo Fisher Scientific). The purified mitochondrial fraction was collected at the 50 to 22% Percoll interface. To remove excess Percoll, the collected mitochondrial fraction was diluted with MIM and spun for 3 min at 10,000g. This step was repeated twice, and the final mitochondrial pellet was suspended in MIM buffer for experiments.
Lipid extraction, TLC, and lipid mass spectrometry
Mitochondrial lipids were extracted using a modified Bligh-Dyer extraction. Resuspended lipids were then used for phospholipid quantification by thin-layer chromatography (TLC) or mass spectrometry. The TLC plates were developed using chloroform:glacial acetic acid:methanol:water (65:35:5:2) as mobile phase for PC and PS, and chloroform:glacial acetic acid:methanol:water (85:25:5:2) as mobile phase for PE, CL, and PI. The plates were dried, sprayed with charring solution, and heated at 190°C for ~15 min. Intensity of the charred lipid spots was measured using the Odyssey Infrared Imager. Mass spectrometry analyses of phospholipids for exercise training, disuse, and PSD-MKI samples were performed at the University of Utah Metabolomics Core, with untargeted (Agilent 6530 UHPLC-QTOF mass spectrometer) and targeted (UHPLC-QQQ mass spectrometer) platforms. Mass spectrometry analyses of HCR or LCR and PSD-MKO samples were performed at the University of Michigan Nutrition and Obesity Research Center Metabolomics Core using an ABSCIEX 5600 TripleTOF mass spectrometer. For untargeted comprehensive lipidomics (exercise training, HCR or LCR, and disuse), quantities are expressed as z scores.
Cell culture
C2C12 myoblasts were grown and maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS), and penicillin/streptomycin (100 μg/ml). Once 90 to 100% confluent, C2C12 myoblasts were differentiated into myotubes using low-glucose DMEM [glucose (1 g/liter), l-glutamine, and sodium pyruvate (110 mg/liter)], 2% horse serum, and penicillin/streptomycin (100 μg/ml). Human embryonic kidney 293T cells were maintained in high-glucose DMEM, 10% FBS, and penicillin/streptomycin (100 μg/ml). The overexpression or lentivirus-mediated knockdown of PSD was performed as previously described (47). Vectors were sourced from OriGene (Rockville, MD) for PISD-expressing plasmid (MR206380), Sigma-Aldrich (St. Louis, MO) for short hairpin RNA (shRNA) for mouse PISD (no. TRCN0000115415), and Addgene (Cambridge, MA) for psPAX2 (ID no. 12260), pMD2.G (ID no. 12259), and scrambled shRNA plasmid (ID no. 1864).
Mitochondrial respiration measurements
Respiration in permeabilized muscle fiber bundles and isolated mitochondria was performed as previously described (48, 49). Briefly, a small portion of freshly dissected red gastrocnemius muscle tissue was placed in buffer X [7.23 mM K2EGTA, 2.77 mM Ca K2EGTA, 20 mM imidazole, 20 mM taurine, 5.7 mM ATP, 14.3 mM phosphocreatine, 6.56 mM MgCl2.6H2O, and 50 mM K-MES (pH 7.1)]. Fiber bundles were separated and permeabilized for 30 min at 4°C with saponin (30 μg/ml) and immediately washed in buffer Z [105 mM K-MES, 30 mM KCl, 10 mM K2HPO4, 5 mM MgCl2.6H2O, BSA (0.5 mg/ml), and 1 mM EGTA (pH 7.4)] for 15 min. After washing, high-resolution respiration rates were measured using an OROBOROS Oxygraph-2k. The muscle fibers were suspended in buffer Z with 20 mM creatine and 10 μM blebbistatin to inhibit myosin adenosine triphosphatases during respiration measurements. A variety of respiration protocols were used in permeabilized fibers and isolated mitochondria. For the mixed substrate protocol, the chamber was hyperoxygenated to ~300 μmol and started with the addition of malate (0.5 mM), followed by sequential additions of pyruvate (5 mM), adenosine 5′-diphosphate (ADP) (2 mM), succinate (10 mM), cytochrome c (10 μM), rotenone (5 μM), malonate (5 mM), duroquinol (0.25 mM), and antimycin A (2 μM). For the fatty acid oxidation protocol, the chamber was not hyperoxygenated and the protocol started with the addition of malate (0.5 mM) followed by sequential additions of palmitoyl-l-carnitine (50 μM) and ADP (2 mM). For complex IV–mediated respiration, rotenone (5 μM), malonate (5 mM), and antimycin A (2 μM) were added to inhibit complexes I to III, after which ascorbate [2 mM; prevents autooxidation of N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD)], TMPD (0.5 mM), and KCN (20 mM) were sequentially added. When respiration experiments were complete, fiber bundles were washed in distilled H2O to remove salts and then freeze-dried in a lyophilizer (Labconco). Dry weight was measured, and respiration rate was expressed relative to fiber weight. When isolated mitochondria respiration rates were measured, the respiration rate was normalized to the total protein content in the chamber.
Oxygen consumption rates in C2C12 cells were measured with the Seahorse Flux Analyzer XF24 or XFe96 (Seahorse Bioscience, Billerica, MA). The cells were plated at ~20,000 to 40,000 cells per well and then differentiated into myotubes. On the day of the experiment, medium was switched to the XF Assay Medium Modified DMEM (pH 7.4) containing added glucose (10 mM), pyruvate (200 mM), and glutamine (200 mM) for 1 hour. Subsequently, basal and maximal respiration rates were measured as previously described (48).
H2O2 emission and production
The Amplex UltraRed (10 μM)/horseradish peroxidase (3 U/ml) detection system was used to measure mitochondrial H2O2 emission and production fluorometrically (excitation/emission, 565:600, HORIBA Jobin Yvon Fluorolog) at 37°C (49). Permeabilized muscle fibers were placed into a glass cuvette with Amplex UltraRed reagents and buffer Z (with 1 mM EGTA and 23 U superoxide dismutase). Initially, an 8-min background rate was obtained followed by the addition of palmitoyl-l-carnitine/malate (50 μM/1 mM) into the cuvette for measurement of H2O2 emission rate. For maximal H2O2 production rate, auranofin (1 μM) and carmustine (100 μM) were titrated into the cuvette to inhibit thioredoxin reductase and glutathione reductase, respectively. The fiber bundles were washed in distilled H2O and freeze-dried, and dry weight was measured. H2O2 rates were expressed relative to fiber weight. Rates were then corrected for O2 consumption, which was measured with an Oxygraph-2k machine in the presence of identical substrates.
Western blotting
Tissues or cells were homogenized in lysis buffer, nutated at 4°C for 1 hour, and centrifuged at 4°C for 15 min at 12,000g, and the supernatant was transferred to a new tube. Western blotting was performed as previously described (10), and samples were analyzed for protein abundance of FoxO1 (no. 2880, Cell Signaling Technology), 4-HNE (ab48506, Abcam), DRP-1 (ab56788, Abcam), pDRP-1 (no. 3455, Cell Signaling Technology), Mfn-2 (ab56889, Abcam), Nrf2 [Developmental Studies Hybridoma Bank (DSHB)], FoxO3 (DSHB), PRDX4 (DSHB), citrate synthase (ab96600, Abcam), and catalase (ab1877, Abcam).
Specific activity of mitochondrial enzymes
The specific activities of electron transport chain enzymes were determined using spectrophotometric methods. Briefly, myoblasts or isolated mitochondria were freeze-thawed two to three times to disrupt the outer mitochondrial membrane and allow access of substrates to enzymes. The specific activity of each complex was measured at 37°C using reagents and substrates specified previously (50). For experiments involving C2C12 cells, activities of complexes I and V were measured in isolated mitochondria, whereas activities of other complexes were measured in whole cells. Citrate synthase activity was measured on a 96-well plate using a commercially available kit (CS0720, Sigma-Aldrich).
Blue native PAGE
Isolated mitochondria suspended in MIM were solubilized (0.5 to 2 mg) in 2% digitonin for 15 min on ice and then centrifuged at 20,000g for 30 min at 4°C. The supernatant was collected and placed into a new tube, and protein content was measured. Approximately 25 to 35 μg of mitochondrial protein was suspended in a mix of native polyacrylamide gel electrophoresis (PAGE) 5% G-250 sample buffer and 1× native-PAGE sample buffer (total volume of 10 to 20 μl). The samples and standards were then loaded onto a native PAGE 3 to 12% Bis-Tris Gel (BN1001BOX, Thermo Fisher Scientific), and electrophoresis was performed at 150 V for 3 hours on ice. The gel was then placed in fixative solution (40% methanol and 10% acetic acid), microwaved on high for 45 s, and then shaken on an orbital shaker for 15 min at room temperature. After incubation, the gel was placed in destaining solution (8% acetic acid), microwaved on high for 45 s, and then incubated overnight at 4°C on an orbital shaker. The gel was scanned for densitometry.
Glutathione quantification
Levels of reduced glutathione (GSH) and oxidized glutathione disulfide (GSSG) were measured using high-performance liquid chromatography (HPLC) (Shimadzu Prominence HPLC system). Freshly dissected gastrocnemius muscle was hand homogenized with a glass pestle in buffer containing Trizma base (50 mmol/liter), boric acid (20 mmol/liter), l-serine (20 mmol/liter), and N-ethylmaleimide (NEM) (10 mmol/liter). The homogenate was then frozen until processing, in which the sample was split into two parts for detection of GSH and GSSG. For GSH measurement, the homogenate was deproteinated with 1:10 (v/v) 15% trichloroacetic acid, centrifuged for 5 min at 20,000g, and the supernatant was transferred to an autosampler vial for measurement of GSH by HPLC using 92.5% of a 0.25% (v/v) glacial acetic acid mixed with 7.5% pure HPLC-grade acetonitrile. Ultraviolet chromatography was used to measure the GSH-NEM conjugate at a wavelength of 265 nmol/liter (SPD-20A, Shimadzu).
MDA quantification
MDA content was quantified in fresh gastrocnemius muscles using a lipid peroxidation assay kit (ab118970, Abcam) according to the manufacturer’s instruction. Rates of appearance of MDA-thiobarbituric acid adduct were quantified colorimetrically at 532 nm using a spectrophotometer.
Bone density, body composition, and indirect calorimetry
Bone density of the femur was determined using microcomputed tomography (SCANCO Medical AG). Fat and lean body mass were measured using the EchoMRI-500 (EchoMRI, Houston, TX). Whole-body oxygen consumption, respiratory exchange ratio, and physical activity levels were determined using the TSE LabMaster System (TSE Systems, Chesterfield, MO). Measurements were taken over a 4- to 5-day period, with the first 1 and 2 days of data excluded for acclimatization to the new environment. Data represent averages of two or three 12-hour light or dark cycles.
Echocardiography
Echocardiography measurements were made as previously described (49). Briefly, mice were anesthetized with a 0.5 to 2% isoflurane in an oxygen mixture and kept on a heated monitoring plate to maintain body temperature. Heart rate was kept between 400 to 500 beats per minute for all measurements to ensure physiological relevance. The Vevo 2100 High-Resolution In Vivo Imaging System (VisualSonics) was used with a 30-MHz transducer for echocardiography recordings. B-mode recordings from transthoracic long-axis view were used to measure left ventricular volume during diastole and systole. These measurements were used to calculate ejection fraction, stroke volume, and cardiac output.
Electron microscopy
Freshly dissected skeletal muscle was immediately placed in a 2% glutaraldehyde and 0.1 M cacodylate fixation mix and cut into longitudinal sections ~2 mm in diameter and ~3 to 4 mm in length. Tissues were stored in a fixation mixture at 4°C until all tissues were collected. The tissues were washed three times for 10 min in 0.1 M phosphate buffer, fixed in 1% osmium tetroxide for 1 hour, and then washed three times for 10 min in 0.1 M phosphate buffer. The fixed tissues were dehydrated in sequential steps of 25, 50, 75, and 100% alcohol (twice each step) for 15 min each. The tissue was then embedded in increasing concentrations of Spurr’s media including 30% (for 30 min), 70% (overnight), 100% (2 hours), and 100% (30 min). Each tissue was placed in a flat-bottom embedding mold filled 50% with Spurr’s medium and then polymerized at ~60°C overnight. The tissues were then cut through the transverse plane and imaged with a JEOL 1200EX transmission electron microscope equipped with a Soft Imaging System MegaView III charge-coupled device camera.
Muscle strength experiments
The extensor digitorum longus and diaphragm muscles were dissected as previously described (51) and tied into the Horizontal Tissue Bath System from Aurora Scientific Inc. (model 801C). Muscles were stimulated with a 20-V twitch train and stretched until optimal length for force production was reached. Muscles were then allowed to equilibrate for 5 min. For force-frequency curves, muscles were stimulated with frequencies ranging from 10 to 200 Hz (0.1-ms pulse, 330-ms train, and 2 min between trains). To estimate fatigability, muscles were given a 40-Hz stimulus for 330 ms, with 1 s between contractions for 200 contractions. Fatigability was further assessed using a twitch protocol over 60 min (0.2-ms twitch and 120 twitches/min). Forces produced by electrically stimulated muscle contractions were recorded in real time via a force transducer (model 400A, Aurora Scientific Inc.). Specific force was calculated using cross-sectional area of the muscle tissue (millinewton per square millimeter), as estimated from the weight and length of the muscle.
Histology
Frozen muscle or lung tissues were embedded in optimal cutting temperature compound and were sectioned (10 μm) with a cryostat (Microtome Plus). Muscle sections were used for myosin heavy chain (MHC) isoform immunofluorescence (IF) and Sirius Red staining to examine fibrosis. For MHC IF, sections were incubated with MHCI (BA.D5), MHCIIa (SC.71), MHCIIb (BF.F3; all three from DSHB, University of Iowa), or laminin (L9393, Sigma-Aldrich) and imaged at the University of Utah Cell Imaging Core. Negative-stained fibers were considered to be MHCIIx. Master Tech Picro Sirius Red was used for Sirius Red staining. Myofiber cross-sectional area was quantified using semi-automatic muscle analysis using segmentation of histology: a MATLAB application (SMASH) alongside ImageJ software. Lung tissues were used for hematoxylin and eosin staining.
Statistical analyses
Data are presented as means ± SEM. Statistical analyses were performed using GraphPad Prism 7.03 software. Independent samples t tests were used to compare two groups. For two-by-two comparisons, two-way analyses of variance (ANOVAs) were performed (main effect of genotype shown over a horizontal line), followed by appropriate post hoc tests corrected for multiple comparisons. All tests were two-sided, and P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by NIH grants DK107397, DK109888, DK095774, and AG063077 to K.F.; DK110656 to P.D.N.; AG050781 to M.J.D.; HL123647 and AT008375 to S.R.S.; AR071967 to C.M.K.; HL129632 to T.E.R.; DK099034 and DK097153 to C.F.B.; and DK109556 and DK110338 to T.D.H. This work was also supported by P&F Funding from P30 DK020579 at Washington University in St. Louis to K.F.; Larry H. & Gail Miller Foundation grant to P.J.F.; Uehara Memorial Foundation to H.E.; and American Heart Association grants 19PRE34380991 to J.M.J., 18PRE33960491 to A.R.P.V., and 16POST30980047 and 19POST343701052 to T.D.H. University of Utah Metabolomics Core Facility was supported by S10 OD016232, S10 OD021505, and U54 DK110858. Author contributions: T.D.H., J.M.J., and K.F. designed the study and wrote the manuscript. T.D.H. and J.M.J. performed or supervised all experiments. P.J.F., H.E., A.R.P.V., P.S., P.T.R., G.K., and M.J.D. assisted muscle functional and histological analyses. C.-T.L., T.E.R., J.R., and P.D.N. assisted with mitochondrial phenotyping. T.M.N., C.B.C., P.T.R., L.E.d.C.B., C.M.K., and S.B. assisted with mouse whole-body phenotyping. C.F.B., J.A.M., and J.E.C. performed mass spectrometry analyses. T.N.Z., S.R.S., and J.E.V. assisted with biochemical assays. T.D.H., J.M.J., E.J.W., and K.F. designed and generated the mouse models. A.P.R.V., T.E.R., D.G.M., S.B., J.R., S.R.S., J.E.V., M.J.D., and P.D.N. edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Mouse lines are available from K.F. upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax8352/DC1
Fig. S1. Skeletal muscle mitochondrial PE and oxidative capacity.
Fig. S2. Deficiency of muscle mitochondrial PE in vitro.
Fig. S3. Deficiency of skeletal muscle mitochondrial PE in vivo.
Fig. S4. PE deficiency in skeletal muscle mitochondria.
Fig. S5. Overexpression of mitochondrial catalase does not rescue muscle-specific PSD deficiency.
REFERENCES AND NOTES
- 1.Myers J., Prakash M., Froelicher V., Do D., Partington S., Atwood J. E., Exercise capacity and mortality among men referred for exercise testing. N. Engl. J. Med. 346, 793–801 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Bassett D. R. Jr., Howley E. T., Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med. Sci. Sports Exerc. 32, 70–84 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Holloszy J. O., Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 242, 2278–2282 (1967). [PubMed] [Google Scholar]
- 4.Henriksson J., Reitman J. S., Time course of changes in human skeletal muscle succinate dehydrogenase and cytochrome oxidase activities and maximal oxygen uptake with physical activity and inactivity. Acta Physiol. Scand. 99, 91–97 (1977). [DOI] [PubMed] [Google Scholar]
- 5.Lin J., Wu H., Tarr P. T., Zhang C.-Y., Wu Z., Boss O., Michael L. F., Puigserver P., Isotani E., Olson E. N., Lowell B. B., Bassel-Duby R., Spieglelman B. M., Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 418, 797–801 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Larsson N.-G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G. S., Clayton D. A., Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236 (1998). [DOI] [PubMed] [Google Scholar]
- 7.van Meer G., Voelker D. R., Feigenson G. W., Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spencer C. T., Bryant R. M., Day J., Gonzalez I. L., Colan S. D., Thompson W. R., Berthy J., Redfearn S. P., Byrne B. J., Cardiac and clinical phenotype in Barth syndrome. Pediatrics 118, e337–e346 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Heden T. D., Neufer P. D., Funai K., Looking beyond structure: Membrane phospholipids of skeletal muscle mitochondria. Trends Endocrinol. Metab. 27, 553–562 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mejia E. M., Hatch G. M., Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 48, 99–112 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Ikon N., Ryan R. O., Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta Biomembr. 1859, 1156–1163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calzada E., Avery E., Sam P. N., Modak A., Wang C., McCaffery J. M., Han X., Alder N. N., Claypool S. M., Phosphatidylethanolamine made in the inner mitochondrial membrane is essential for yeast cytochrome bc1 complex function. Nat. Commun. 10, 1432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teague W. E. Jr., Soubias O., Petrache H., Fuller N., Hines K. G., Rand R. P., Gawrisch K., Elastic properties of polyunsaturated phosphatidylethanolamines influence rhodopsin function. Faraday Discuss. 161, 383–395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pennington E. R., Funai K., Brown D. A., Shaikh S. R., The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 1039–1052 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao T., Goedhart C. M., Sam P. N., Sabouny R., Lingrell S., Cornish A. J., Lamont R. E., Bernier F. P., Sinasac D., Parboosingh J. S.; Care4Rare Canada Consortium, Vance J. E., Claypool S. M., Innes A. M., Shutt T. E., PISD is a mitochondrial disease gene causing skeletal dysplasia, cataracts, and white matter changes. Life Sci. Alliance 2, e201900353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barth P. G., Scholte H. R., Berden J. A., Van der Klei-Van Moorsel J. M., Luyt-Houwen I. E., Van’t Veer-Korthof E. T., Van der Harten J. J., Sobotka-Plojhar M. A., An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355 (1983). [DOI] [PubMed] [Google Scholar]
- 17.Girisha K. M., von Elsner L., Neethukrishna K., Muranjan M., Shukla S., Bhavani G. S., Nishimura G., Kutsche K., Mortier G., The homozygous variant c.797G>A/p.(Cys266Tyr) in PISD is associated with a Spondyloepimetaphyseal dysplasia with large epiphyses and disturbed mitochondrial function. Hum. Mutat. 40, 299–309 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Wisløff U., Najjar S. M., Ellingsen O., Haram P. M., Swoap S., Al-Share Q., Fernström M., Rezaei K., Lee S. J., Koch L. G., Britton S. L., Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 307, 418–420 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Zborowski J., Dygas A., Wojtczak L., Phosphatidylserine decarboxylase is located on the external side of the inner mitochondrial membrane. FEBS Lett. 157, 179–182 (1983). [DOI] [PubMed] [Google Scholar]
- 20.Shiao Y. J., Lupo G., Vance J. E., Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J. Biol. Chem. 270, 11190–11198 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Ren Y. Y., Koch L. G., Britton S. L., Qi N. R., Treutelaar M. K., Burant C. F., Li J. Z., Selection-, age-, and exercise-dependence of skeletal muscle gene expression patterns in a rat model of metabolic fitness. Physiol. Genomics 48, 816–825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kauffman F. C., Albuquerque E. X., Effect of ischemia and denervation on metabolism of fast and slow mammalian skeletal muscle. Exp. Neurol. 28, 46–63 (1970). [DOI] [PubMed] [Google Scholar]
- 23.Sandri M., Lin J., Handschin C., Yang W., Arany Z. P., Lecker S. H., Goldberg A. L., Spiegelman B. M., PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U.S.A. 103, 16260–16265 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon O. S., Nelson D. S., Barrows K. M., O’Connell R. M., Drummond M. J., Intramyocellular ceramides and skeletal muscle mitochondrial respiration are partially regulated by Toll-like receptor 4 during hindlimb unloading. Am. J. Physiol. Regul. Integr. Comp. Physiol. 311, R879–R887 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steenbergen R., Nanowski T. S., Beigneux A., Kulinski A., Young S. G., Vance J. E., Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 280, 40032–40040 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barends M., Verschuren L., Morava E., Nesbitt V., Turnbull D., McFarland R., Causes of Death in Adults with Mitochondrial Disease. JIMD Rep. 26, 103–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smuder A. J., Sollanek K. J., Nelson W. B., Min K., Talbert E. E., Kavazis A. N., Hudson M. B., Sandri M., Szeto H. H., Powers S. K., Crosstalk between autophagy and oxidative stress regulates proteolysis in the diaphragm during mechanical ventilation. Free Radic. Biol. Med. 115, 179–190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tasseva G., Bai H. D., Davidescu M., Haromy A., Michelakis E., Vance J. E., Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 288, 4158–4173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharpley M. S., Shannon R. J., Draghi F., Hirst J., Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Shinzawa-Itoh K., Aoyama H., Muramoto K., Terada H., Kurauchi T., Taehara Y., Yamasaki A., Sugimura T., Kurono S., Tsujimoto K., Mizushima T., Yamashita E., Tsukihara T., Yoshikawa S., Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 26, 1713–1725 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun F., Huo X., Zhai Y., Wang A., Xu J., Su D., Bartlam M., Rao Z., Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121, 1043–1057 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Nelson B. D., Fleischer S., Phospholipid requirements for the reconstitution of complex-III vesicles exhibiting controlled electron transport. Biochem. J. 194, 783–787 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Acin-Perez R., Fernandez-Silva P., Peleato M. L., Perez-Martos A., Enriquez J. A., Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Letts J. A., Sazanov L. A., Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 24, 800–808 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Wong H.-S., Dighe P. A., Mezera V., Monternier P.-A., Brand M. D., Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brand M. D., Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 100, 14–31 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Anderson E. J., Vistoli G., Katunga L. A., Funai K., Regazzoni L., Monroe T. B., Gilardoni E., Cannizzaro L., Colzani M., De Maddis D., Rossoni G., Canevotti R., Gagliardi S., Carini M., Aldini G., A carnosine analog mitigates metabolic disorders of obesity by reducing carbonyl stress. J. Clin. Invest. 128, 5280–5293 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powers S. K., Smuder A. J., Judge A. R., Oxidative stress and disuse muscle atrophy: Cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 15, 240–245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvani R., Joseph A. M., Adhihetty P. J., Miccheli A., Bossola M., Leeuwenburgh C., Berabei R., Marzetti E., Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol. Chem. 394, 393–414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsu J.-L., Hsieh Y., Tu C., O’Connor D., Nick H. S., Silverman D. N., Catalytic properties of human manganese superoxide dismutase. J. Biol. Chem. 271, 17687–17691 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Pereira B., Costa Rosa L. F. B., Safi D. A., Medeiros M. H. G., Curi R., Bechara E. J. H., Superoxide dismutase, catalase, and glutathione peroxidase activities in muscle and lymphoid organs of sedentary and exercise-trained rats. Physiol. Behav. 56, 1095–1099 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Osman C., Voelker D. R., Langer T., Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Funai K., Lodhi I. J., Spears L. D., Yin L., Song H., Klein S., Semenkovich C. F., Skeletal muscle phospholipid metabolism regulates insulin sensitivity and contractile function. Diabetes 65, 358–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selathurai A., Kowalski G. M., Burch M. L., Sepulveda P., Risis S., Lee-Young R. S., Lamon S., Meikle P. J., Genders A. J., McGee S. L., Watt M. J., Russell A. P., Frank M., Jackowski S., Febbraio M. A., Bruce C. R., The CDP-ethanolamine pathway regulates skeletal muscle diacylglycerol content and mitochondrial biogenesis without altering insulin sensitivity. Cell Metab. 21, 718–730 (2015). [DOI] [PubMed] [Google Scholar]
- 45.van der Veen J. N., Lingrell S., da Silva R. P., Jacobs R. L., Vance D. E., The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes 63, 2620–2630 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Micallef L., Rodgers P., eulerAPE: Drawing area-proportional 3-Venn diagrams using ellipses. PLOS ONE 9, e101717 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paran C. W., Verkerke A. R. P., Heden T. D., Park S., Zhou K., Lawson H. A., Song H., Turk J., Houmard J. A., Funai K., Reduced efficiency of sarcolipin-dependent respiration in myocytes from humans with severe obesity. Obesity 23, 1440–1449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heden T. D., Ryan T. E., Ferrara P. J., Hickner R. C., Brophy P. M., Neufer P. D., McClung J. M., Funai K., Greater oxidative capacity in primary myotubes from endurance-trained women. Med. Sci. Sports Exerc. 49, 2151–2157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson J. M., Ferrara P. J., Verkerke A. R. P., Coleman C. B., Wentzler E. J., Neufer P. D., Kew K. A., de Castro Brás L. E., Targeted overexpression of catalase to mitochondria does not prevent cardioskeletal myopathy in Barth syndrome. J. Mol. Cell. Cardiol. 121, 94–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sullivan E. M., Pennington E. R., Sparagna G. C., Torres M. J., Neufer P. D., Harris M., Washington J., Anderson E. J., Zeczycki T. N., Brown D. A., Shaikh S. R., Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome. J. Biol. Chem. 293, 466–483 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferrara P. J., Verkerke A. R. P., Brault J. J., Funai K., Hypothermia decreases O2 cost for ex vivo contraction in mouse skeletal muscle. Med. Sci. Sports Exerc. 50, 2015–2023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax8352/DC1
Fig. S1. Skeletal muscle mitochondrial PE and oxidative capacity.
Fig. S2. Deficiency of muscle mitochondrial PE in vitro.
Fig. S3. Deficiency of skeletal muscle mitochondrial PE in vivo.
Fig. S4. PE deficiency in skeletal muscle mitochondria.
Fig. S5. Overexpression of mitochondrial catalase does not rescue muscle-specific PSD deficiency.