

Stress-induced trafficking of ceramide synthase 1 to mitochondria is mediated by previously unidentified protein, p17/PERMIT.
Abstract
How lipid metabolism is regulated at the outer mitochondrial membrane (OMM) for transducing stress signaling remains largely unknown. We show here that this process is controlled by trafficking of ceramide synthase 1 (CerS1) from the endoplasmic reticulum (ER) to the OMM by a previously uncharacterized p17, which is now renamed protein that mediates ER-mitochondria trafficking (PERMIT). Data revealed that p17/PERMIT associates with newly translated CerS1 on the ER surface to mediate its trafficking to the OMM. Cellular stress induces Drp1 nitrosylation/activation, releasing p17/PERMIT to retrieve CerS1 for its OMM trafficking, resulting in mitochondrial ceramide generation, mitophagy and cell death. In vivo, CRISPR-Cas9–dependent genetic ablation of p17/PERMIT prevents acute stress-mediated CerS1 trafficking to OMM, attenuating mitophagy in p17/PERMIT−/− mice, compared to controls, in various metabolically active tissues, including brain, muscle, and pancreas. Thus, these data have implications in diseases associated with accumulation of damaged mitochondria such as cancer and/or neurodegeneration.
INTRODUCTION
The bioactive sphingolipid ceramide is both a structural component of biological membranes and a signaling molecule that induces cell death and tumor suppression (1). Ceramide-mediated cell death has been linked to mitochondrial dysfunction in various cell and tissue types. Although there is some evidence of ceramide transport to mitochondria to induce mitochondrial stress (2), mechanisms of ceramide generation in mitochondrial membranes are largely unknown.
Many stress stimuli, including oxidative stress inducers and anticancer drugs, mediate endogenous ceramide generation, resulting in ceramide stress and cell death (3, 4). De novo generation of ceramides is catalyzed by the ceramide synthases 1 to 6 (CerS1 to CerS6) (5–7), which exhibit fatty acyl chain length selectivity for ceramide generation. For example, while CerS6 produces mainly C16-ceramide, CerS1 selectively generates C18-ceramide (5–7). CerS1/C18-ceramide mediates cell death and tumor suppression by, at least in part, selective activation of autophagy and/or mitophagy in various cancer cell types, including head and neck squamous cell carcinoma (HNSCC) and acute myeloid leukemia (AML) (8, 9). In many cell types and tumors, mitochondrial dysfunction leads to tumor suppression by reducing adenosine 5′-triphosphate (ATP) generation, which depletes key macromolecules, including nucleotides or amino acids such as glutamine (10). Ceramide-dependent cell death via mitophagy or mitochondrial dysfunction has also been reported recently in various cell types, such as glioblastoma cells (11), cardiomyocytes (12), and lung epithelial cells (13). In HNSCC and AML cells, stress-induced mitophagy is dependent on the localization of de novo generated C18-ceramide at the outer mitochondrial membrane (OMM), which then directly binds light chain 3 (LC3) protein, involving I35 and F52 residues (9, 14). Ceramide-LC3 binding results in the recruitment of autophagosomes for the execution of mitophagy (9, 14). However, mechanisms that regulate generation and accumulation of CerS1-generated C18-ceramide at the OMM to mediate mitophagy in response to cellular stress remain unknown. In addition, although the molecular and structural details of ceramide transport from endoplasmic reticulum (ER) to Golgi by ceramide transporter (CERT) or four phosphate adaptor protein (FAPP2) for sphingomyelin (SM) or glucosylceramide (GlcCer) synthesis have been described (15, 16), the mechanisms underlying trafficking or translocation of ceramide and CerS enzymes from ER to mitochondria for signal transduction remain to be elucidated.
Mitochondrial morphology and function are regulated by dynamic cellular processes involving protein trafficking and import to mitochondria from various organelles, including ER. For example, recruitment of dynamin-related protein 1 (Drp1) to the OMM is involved in inducing mitochondrial fission (17–19). In a recent study, a genome-wide screen in yeast helped identify factors involved in intracellular sorting of the mitochondrial inner membrane protein Oxa1 via a novel pathway, termed ER-SURF (ER surface–mediated protein targeting), for targeting of mitochondrial membrane proteins (20). The ER-SURF was shown to retrieve mitochondrial proteins from the ER membranes for rerouting to mitochondria with the aid of the ER-localized chaperone Djp1. Thus, Djp-mediated ER-SURF allows cells to use the expanse of ER surfaces as a mechanism to mediate mitochondrial protein targeting (20). It is also known that alterations of mitochondrial quality control mechanisms such as mitophagy have important biological implications in regulating tumor growth and neurodegeneration. However, it remains to be determined whether ER-SURF is also operational in mammalian cells, and whether, for example, via regulation of mitochondrial metabolism or stress signaling, it executes any physiological functions in tumor suppression or neurodegeneration.
In the work described here, we determined how trafficking of a metabolic enzyme (CerS1), but not the lipid molecule (ceramide) itself, from the ER surface to the OMM is facilitated for the generation of ceramide to mediate cellular stress and cell death. We have identified a novel protein, p17, now renamed protein that mediates ER-mitochondria trafficking (PERMIT), that retrieves newly translated CerS1 from the ER surface for trafficking to the OMM through ER-mitochondria contact sites or mitochondria-associated membranes (MAMs), leading to mitochondrial ceramide generation and mitophagy-mediated cell death. Our data suggest that trafficking of the metabolic enzyme CerS1 is mediated by p17/PERMIT, inducing ceramide synthesis and stress signaling in mitochondrial membrane of various metabolically active tissues and tumors. We submit that alterations of p17/PERMIT-CerS1–dependent mitochondrial lipid metabolism constitute a novel link between tumor suppression and neurodegenerative disorders.
RESULTS
CerS1-dependent C18-ceramide generation mediates mitochondrial stress signaling
To determine the roles of CerS1/C18-ceramide in mediating mitochondrial stress signaling, we ectopically expressed V5-tagged wild-type (WT) CerS1WT-V5 or catalytically inactive CerS1H138A-V5 (which does not produce ceramide) in human HNSCC UM-SCC-22A cells and measured their effects on ceramide generation and mitophagy. Mass spectrometry–based lipidomics analyses were used to confirm increased C18-ceramide generation (16-fold) in response to CerS1WT-V5, but not CerS1H138A-V5, induction (+Tet, 72 hours) compared to controls (−Tet) (fig. S1A). Induction of CerS1WT-V5 but not CerS1H138A-V5 increased autophagosome formation and recruitment to mitochondria (Fig. 1, A and B). In addition, CerS1 induction decreased (87%) UM-SCC-22A cell survival/proliferation compared to controls, whereas CerS1H138A-V5 had no effect on autophagosome induction (fig. S1B). Figure S1 (C and D) shows the levels of CerS1WT-V5, CerS1H138A-V5, and CerS6WT-V5 proteins and mRNA. Tet-induced expression of CerS6WT-V5 (72 hours), which generates C16-ceramide in the ER, had no effect on the lipidation of autophagosomal protein LC3 (LC3-II formation) compared to controls in UM-SCC-22A cells (fig. S1, B and C). Measurement of ceramide distribution between cytosolic and mitochondrial fractions demonstrated that the majority of CerS1WT-generated C18-ceramide was localized in isolated mitochondria in response to CerS1 induction (Fig. 1C). Immunofluorescence and transmission electron microscopy (TEM) with immunogold staining using anti-ceramide antibody showed that, compared to uninduced (−Tet) controls, expression (+Tet, 72 hours) of CerS1WT-V5 increased the localization/accumulation of ceramide in mitochondrial membranes (Fig. 1D). TEM with immunogold labeling showed that the accumulation of ceramide in mitochondrial membranes was associated with autophagosomes (Fig. 1E). Expression of acid ceramidase–green fluorescent protein (GFP), which hydrolyzes ceramide, decreased ceramide signal in response to sodium selenite (SoSe) exposure compared to vector-only GFP-transfected cells, indicating the selective signal detection for ceramide using anti-ceramide antibody (fig. S1E). These data suggest that Tet-dependent induction of CerS1WT-V5/C18-ceramide, but not CerS1H138A-V5 or CerS6WT-V5, mediates mitochondrial accumulation of C18-ceramide and mitophagy.
Fig. 1. Induction of CerS1/C18-ceramide generation results in mitophagy.
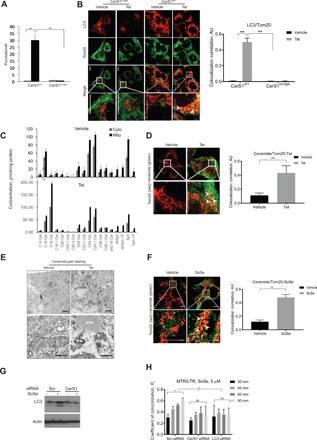
(A) Autophagic response evaluated by Cyto-ID in UM-SCC-22A-Tet On cells induced for expression of CerS1WT or CerS1H138A. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (B) Confocal images of UM-SCC-22A-Tet On cells induced for expression of CerS1WT (right) or CerS1H328A noncatalytic mutant (left) stained for LC3 (red) and mitochondria (MitoTracker, green). Images represent at least three independent experiments. Right panel shows quantification of colocalization estimated by calculating coefficient of colocalization (Rc) using Fiji Software. Scale bars, 100 μm (throughout the manuscript unless specifically noted). AU, arbitrary units. (C) Ceramide profiles of mitochondrial and soluble fractions of UM-SCC-22A cells upon treatment with vehicle or Tet were measured by lipidomics. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (D) Left: Confocal images of UM-SCC-22A-Tet On cells induced for expression of CerS1WT stained for ceramide (green) and mitochondria (Tom20, red). Images are representative of at least three independent experiments. Right: Quantification of the left panel. Colocolization correlation was estimated by calculating coefficient of colocalization using Fiji Software. Scale bars, 100 μm. (E) TEMs show fusion of mitochondria, gold-labeled with ceramide antibodies, in UM-SCC-22A-Tet On cells Tet-induced for expression of CerS1WT (+Tet) compared to untreated (−Tet) control. APH, autophagosome; M, mitochondria; sER, smooth ER. Top panel, 20,000× magnification; bottom panel, 80,000× magnification. Scale bars, 2 μm and 800 nm, respectively. Images represent at least three independent experiments. (F) Confocal images of UM-SCC-22A cells induced for expression of CerS1WT by SoSe and stained for ceramide (green) and mitochondria (Tom20, red). Images represent three independent experiments. Right: Quantification of left panel. Coefficient of colocalization was estimated using Fiji Software. Scale bars, 100 μm (throughout the manuscript unless specifically noted). (G) LC3 protein abundance in control (Scr) and CerS1 small interfering RNA (siRNA)–treated cells incubated with 5 μM SoSe for 3 hours. Images represent at least three independent experiments. (H) Quantification of confocal images of cells with silenced CerS1 or silenced LC3 UM-SCC-22A cells coloaded with 0.5 μM MTR for 60 min and LTG (0.5 μM for 20 min) upon treatment with 5 μM SoSe. Time points were selected to illustrate the onset and completion of mitochondrial digestion by autophagy. Data are means ± SD (n = 3 independent experiments, **P < 0.01). ns, not significant.
To determine whether induction of endogenous CerS1 plays a role in mediating mitochondrial stress signaling, we treated UM-SCC-22A cells with the known stress inducer, SoSe (5 μM, 3 hours), and measured its effects on CerS1 mRNA/protein abundance, mitophagy, and cell death. SoSe exposure increased CerS1 mRNA and protein (fig. S1F) and also induced ceramide accumulation in mitochondria (Fig. 1F). Short hairpin–mediated RNA (shRNA)–mediated knockdown (95%) of CerS1 (fig. S1G) almost completely prevented SoSe-mediated LC3-II formation and mitophagy (Fig. 1G, left and right panels). SoSe exposure resulted in mitophagy within 30 to 60 min, leading to degradation of mitochondria at 3 hours (Fig. 1H). Effects of SoSe on mitophagy were largely prevented by CerS1 or LC3 knockdown (Fig. 1H and fig. S1G). These data reveal that induction of endogenous CerS1/C18-ceramide in response to SoSe mediates mitochondrial stress signaling, resulting in mitophagy.
Mitochondrial localization of CerS1 mediates mitophagy
To determine whether increased mitochondrial ceramide accumulation (see Fig. 1, C to E) was linked to CerS1 trafficking to mitochondria from ER, we measured the colocalization of CerS1 with OMM protein Tom20 in response to CerS1 induction (+Tet) or SoSe exposure (Fig. 2, A and B). CerS1-V5 induction (+Tet) or SoSe exposure resulted in increased colocalization of CerS1 with Tom20 compared to controls (−Tet or vehicle treatment) (Fig. 2, A and B). Increased mitochondrial localization of CerS1-V5 in response to +Tet induction (0, 24, 48, and 72 hours) or endogenous CerS1 in response to SoSe exposure (0, 0.5, 1.5, and 3 hours) was confirmed after isolation of ER and mitochondria (Fig. 2, C and D). After 3 hours of SoSe exposure, CerS1 was largely localized in mitochondria (Fig. 2E).
Fig. 2. Mitochondrial import of CerS1 from smooth ER mediates lethal mitophagy.
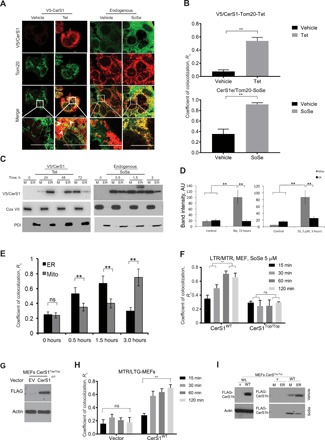
(A) Confocal images of UM-SCC-22A cells induced for expression of CerS1 by Tet or SoSe and stained for CerS1 (green) and mitochondrial marker Tom20 (red) compared with nontreated cells. Yellow shows colocalization. Images represent three independent experiments. (B) Quantification of images in (A). Data are means ± SD (n = 3 different optical fields, **P < 0.01). (C) Distribution of CerS1 between mitochondria (M) and ER in UM-SCC-22A cells induced for CerS1 expression with Tet (left) or SoSe (right). Cox IV mitochondrial marker shows equal protein loading. Images are representative of three independent experiments. (D) Quantification of (C) at 72 hours for Tet-treated cells (left) and at 3 hours for SoSe-treated cells (right). Data are means ± SD (n = 3 independent experiments, **P < 0.01). (E) Quantification of confocal images of UM-SCC-22A cells treated with 5 μM SoSe for indicated periods of time and labeled with ER (PDI) and mitochondrial (Tom20) markers. Quantification of colocalization was performed with Fiji ImageJ software using at least three random fields of view from three independent experiments. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (F) Quantification of the confocal images of live MEFs, from CerS1WT and CerS1Top/Top animals, treated with SoSe for indicated periods of time. MEFs were coloaded with 0.5 μM MTR for 60 min and LTR (0.5 μM for 20 min) upon treatment with 5 μM SoSe. Time points selected to illustrate onset and completion of mitochondrial digestion by autophagy. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (G) Protein abundance of transiently expressed CerS1WT in MEFsTop/Top was detected by Western blotting. Actin was used as loading control. (H) Quantification of confocal microphotographs of live MEFs from animals expressing nonactive CerS1 mutant (MEFs CerS1Top/Top) and transiently transfected with empty vector control (CerS1Top/Top) or CerS1WT and treated with SoSe and stained with LTG and MTR. Time points selected to illustrate onset and completion of mitochondrial digestion by autophagy. Quantification was done using at least three images from three independent experiments. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (I) Mitochondrial versus ER localization of transiently expressed FLAG-tagged CerS1WT in CerS1Top/Top MEFs after induction with SoSe. Images represent at least three independent experiments.
As an additional control, we measured the effects of SoSe on mitophagy and CerS1 localization in mouse embryonic fibroblasts (MEFs) isolated from WT and CerS1-toppler (CerS1Top/Top) mice, which express catalytically inactive CerS1 (21, 22). SoSe exposure (15, 30, 60, and 120 min) induced mitophagy in WT but not in CerS1Top/Top MEFs (Fig. 2F). Reconstitution of active human CerS1WT-FLAG expression in CerS1Top/Top MEFs restored mitophagy compared to vector-transfected controls (Fig. 2, G and H), further supporting the idea that CerS1/C18-ceramide plays a distinct and selective role in the induction of mitophagy. In addition, SoSe exposure also enhanced mitochondrial localization of CerS1WT-FLAG in WT-MEFs but not in CerS1Top/Top MEFs (Fig. 2I). Together, these data support the notion that ceramide stress exerted by CerS1-V5 induction or SoSe exposure mediates increased CerS1 localization to the mitochondria (both endogenous and ectopically expressed/V5-tagged enzymes).
Identification of residues involved in mitochondrial trafficking of CerS1
To determine the mechanisms involved in mitochondrial trafficking of CerS1 to mediate mitochondrial stress signaling, we first identified the amino acid residues of CerS1 that are involved in this process. Alignment of amino acid residues of CerS1 and CerS6 showed that, while CerS6 contains a Hox domain within residues 85 to 103 (23), which is also conserved in CerS2 to CerS5 enzymes, CerS1 does not have this domain (fig. S2A): It is replaced in CerS1, within residues 60 to 76, by a distinct stretch of amino acids not present in the CerS2 to CerS6 enzymes (fig. S2B). To determine whether these distinct residues (amino acids 60 to 76) are involved in increased mitochondrial localization of CerS1, we generated a deletion mutant of CerS1 lacking these residues (FLAG-CerS1Δ60–76) (fig. S2C) and examined its effects on ceramide generation and subcellular localization with or without SoSe exposure compared to FLAG-CerS1WT. Expression of the FLAG-CerS1Δ60–76 mutant had no effect on C18-ceramide but induced C24-ceramide generation in response to SoSe compared to vehicle-treated controls, suggesting that the deletion mutation did not cause any misfolding/degradation of CerS1 (fig. S2D). Compared to vehicle-treated controls, SoSe exposure induced the mitochondrial localization/transport of FLAG-CerS1WT but not of FLAG-CerS1Δ60–76 (fig. S2, E to G). Deletion of residues 60 to 76 forced the accumulation of FLAG-CerS1Δ60–76 mutant in the ER, whereas trafficking of FLAG-CerS1WT was increased from ER to mitochondria in response to SoSe, detected using anti-FLAG and anti–11beta-HSD1 (hydroxysteroid dehydrogenase 1, ER marker) antibodies (fig. S2, F and G). In addition, while reconstitution of FLAG-CerS1WT restored SoSe-mediated mitophagy and decrease in mitochondrial membrane potential, FLAG-CerS1Δ60–76 had no effect on these processes in the absence or presence of SoSe in UM-SCC-22A cells stably transfected with shRNA to target endogenous CerS1 (fig. S2, H and I). Moreover, we used mitochondrial membrane uncouplers FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) and DNP (2,4-dinitrophenol) as positive controls to measure their effects on the decrease in mitochondrial membrane potential in UM-SCC-22A cells (fig. S2J). Of note, the FCCP-mediated decrease in mitochondrial membrane potential had no effect on the mitochondrial transport of CerS1 without SoSe exposure (fig. S2K). These data suggest that mitochondrial transport of CerS1 is regulated upstream of decrease in mitochondrial membrane potential, which is possibly due to mitophagy. Overall, these data demonstrate that amino acids 60 to 76 of CerS1 are key for the trafficking of CerS1 to mitochondria to induce mitophagy, leading to a reduction in mitochondrial membrane potential in response to SoSe-mediated cellular stress.
To further validate the role of amino acids 60 to 76 (not present in CerS6, which contains a Hox domain instead) in the induction of mitochondrial translocation of CerS1, we performed domain swapping between CerS1 and CerS6 and examined their subcellular localization in response to SoSe exposure. We generated a mutant CerS6, in which the Hox domain was deleted and replaced with amino acids 60 to 76 from CerS1 (FLAG-CerS6ΔHox/+CS1-60–76) and measured the effects of its expression on ER or mitochondrial localization compared to FLAG-CerS6WT in UM-SCC-22A cells in the absence or presence of SoSe (Fig. 3A, left panel). The FLAG-CerS6WT remained in the ER with or without SoSe, whereas the localization of the FLAG-CerS6ΔHox/+CS1-60–76 in mitochondria was increased in response to SoSe compared to vehicle-treated controls (Fig. 3A, right panel). Immunofluorescence with anti-FLAG and anti-Tom20 antibodies confirmed these results, showing increased mitochondrial localization of the FLAG-CerS6ΔHox/+CS1-60–76 mutant in response to SoSe compared to vehicle-treated controls, while there was no visible mitochondrial presence of FLAG-CerS6WT (Fig. 3B). Ectopic expression of the FLAG-CerS6ΔHox/+CS1-60–76 mutant resulted in increased accumulation (twofold) of C16-ceramide in mitochondria-enriched cellular fractions in response to SoSe compared to FLAG-CerS6WT (Fig. 3C and fig. S3, A and B). Moreover, ectopic expression of the FLAG-CerS6ΔHox/+CS1-60–76 mutant, but not FLAG-CerS6WT, induced mitophagy (Fig. 3, D to F) in response to SoSe in UM-SCC-22A cells stably expressing shRNA against endogenous CerS1. Equal protein abundance of FLAG-CerS6WT and FLAG-CerS6ΔHox/+CS1-60–76 mutant in UM-SCC-22A/shCerS1 cells was confirmed by Western blotting (Fig. 3G). As expected, reconstitution of CerS1WT and FLAG-CerS6ΔHox/+CS1-60–76 mutant, but not CerS6WT, restored mitophagy induction in response to SoSe in MEFs isolated from CerS1Top/Top mice, compared to vector-transfected controls (fig. S3C). These data indicate that amino acids 60 to 76 play key roles in mitochondrial CerS1 import to mediate mitophagy in response to SoSe-induced cellular stress. These data also reveal that the mitochondrial generation of ceramide by CerS enzymes in mitochondrial membranes (such as CerS1 or mutant FLAG-CerS6ΔHox/+CS1-60–76), and not the specific fatty acid chain length of ceramide (C18-ceramides versus C16-ceramides), is key for the induction of mitophagy in response to cellular stress.
Fig. 3. Insertion of CerS1 60 to 76 residues to CerS6 mediates mitochondrial import of CerS6.
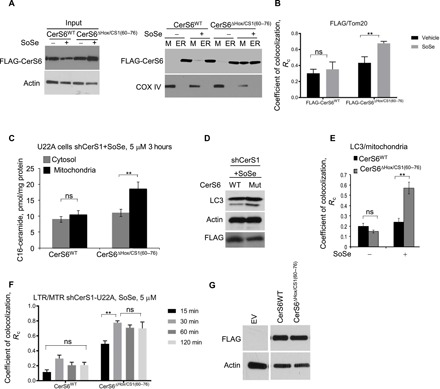
(A) Cellular lysates of UM-SCC-22A cells transfected with FLAG-tagged CerS6WT or CerS6 mutant with Hox domain replaced by the 60 to 76 amino acid sequence of CerS1 [CerSΔHox/CS1(60–76)] and treated with SoSe (5 μM, 3 hours) were differentially centrifuged, and resulting mitochondrial and ER fractions were analyzed using SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Mitochondrial marker Cox IV was used as loading control. Images represent three independent experiments. (B) Quantification of confocal images of UM-SCC-22A treated as in (A) and labeled with FLAG antibody and mitochondrial marker Tom20. Quantification of FLAG-CerS6WT or FLAG-CerS6ΔHox/CS1(60–76) colocalization with Tom20 was performed using ImageJ software. Images represent at least three independent experiments. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (C) Abundance of C16-ceramide in mitochondrial and cytosolic fractions of shCerS1 UM-SCC-22A cells ectopically expressing CerS6WT and CerS6ΔHox/CS1(60–76) measured using mass spectrometry lipidomics. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (D) Abundance of LC3 lipidation in cells treated as in (C). Levels of FLAG and actin indicate equal expression levels of FLAG-tagged CerS6WT and CerS6ΔHox/CS1(60–76) proteins and equal protein loading. Images represent at least three independent experiments. (E) Quantification by ImageJ of confocal images of cells treated as in (C) and stained with LC3 and Tom20 antibodies. At least three random fields of view from at least three independent experiments were analyzed. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (F) CerS6-Hox translocated to mitochondria induces lethal mitophagy. Quantification of confocal images of live shCerS1 UM-SCC-22A cells ectopically expressing WT or ΔHox/CS1 (60 to 76) mutant of CerS6 coloaded with MTR (0.5 μM for 60 min) and LTR (0.5 μM for 20 min) upon treatment with 5 μM SoSe. Time points selected to illustrate onset and completion of mitochondrial digestion by autophagy. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (G) Abundance of CerS6WT-FLAG and mutant proteins in UM-SCC-22A cells was confirmed by Western blotting using anti-FLAG antibody. Empty vector (EV)–transfected cells were used as negative controls.
p17/PERMIT mediates CerS1 trafficking to mitochondria to induce mitophagy
First, to determine whether any known transporters of ceramide are involved in increased localization of CerS1-generated C18-ceramide on the OMM in response to SoSe-mediated cellular stress, we measured the mitochondrial localization of ceramide after knocking down CERT or Fapp2, which transports ceramide from ER to Golgi for generation of SM or GlcCer, respectively (19, 20). shRNA-mediated knockdown of CERT or Fapp2 (fig. S3, D and E) had no effect on the increased mitochondrial accumulation of CerS1 or ceramide in response to SoSe (fig. S3F). Moreover, knockdown of CERT or Fapp2 had no effect on the induction of mitophagy in response to SoSe (fig. S3F). As expected, CERT or Fapp2 knockdown decreased the accumulation of ceramide in the Golgi compared to scrambled (Scr)–shRNA–transfected controls, detected by immunofluorescence and confocal microscopy using anti-ceramide and anti-GM130 (Golgi marker) antibodies (fig. S3G). Thus, these data suggest that mitochondrial ceramide stress induction and mitophagy might be independent of ceramide transport to mitochondria in response to SoSe.
Next, to determine whether mitochondrial ceramide accumulation is due to mitochondrial trafficking of CerS1 and whether this process is mediated by a protein transporter, we examined CerS1-interacting proteins in UM-SCC-22A cells in the absence or presence of SoSe-induced (5 μM, 3 hours) mitochondrial localization of CerS1. To do so, we used coimmunoprecipitation (co-IP) with anti-CerS1 antibody [or immunoglobulin G (IgG) as a negative control], followed by Coomassie blue staining (Fig. 4A). We identified a protein with a molecular mass of 15 to 20 kDa associated with CerS1 in SoSe-exposed but not in control cells (Fig. 4A). Identification of this band using trypsin digestion and mass spectrometry/proteomics revealed that it was a previously uncharacterized 17-kDa protein (RPL29P31, gene ID: 284064) of high (86%) amino acid homology to ribosomal L29 protein.
Fig. 4. p17/PERMIT mediates mitochondrial import of CerS1.
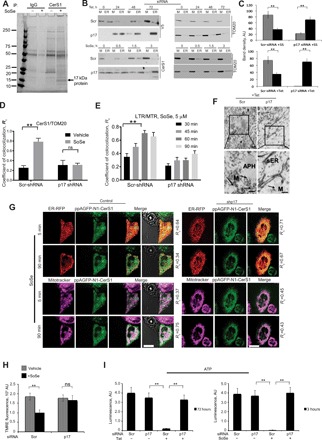
(A) SDS-PAGE analysis of proteins bound to CerS1 in UM-SCC-22A cells treated with SoSe. Binding to IgG was used to account for nonspecific binding. Protein-containing gel bands were excised and analyzed by mass spectrometry. (B) p17/PERMIT silencing prevents CerS1 mitochondrial translocation. Distribution of CerS1 between mitochondrial and ER fractions in UM-SCC-22A control cells and cells with knocked-down p17/PERMIT expressing CerS1 upon Tet (top) or SoSe (5 μM, 3 hours) (bottom) induction. (C) Quantification of (B) using ImageJ. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (D) Quantification of confocal images of scrambled (Scr) control UM-SCC-22A cells and cells with silenced p17 treated with vehicle or SoSe (5 μM, 3 hours) and stained with CerS1 and Tom20 antibodies. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (E) Quantification of confocal images of live control or silenced p17 UM-SCC-22A cells coloaded with MTR (0.5 μM for 60 min) and LTR (0.5 μM for 20 min) upon treatment with 5 μM SoSe. Time points selected to illustrate onset and completion of mitochondrial digestion by mitophagy. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (F) Electron micrographs of UM-SCC-22A-Tet On cells with silenced p17 or Scr control induced for V5-tagged CerS1 and detected using gold-labeled anti-V5 antibody. Bottom panels are higher-magnification images of top panels. Arrows indicate junction between autophagosome and V5-CerS1–labeled mitochondria (Scr-siRNA, +Tet) and absence of junction and labeled mitochondria in cells with silenced p17 (p17 siRNA, +Tet). Labeling (V5-CerS1) localizes in sER. Images are representative of at least three independent experiments. Scale bars, 2 μm. (G) Live cell imaging was performed to detect transport of CerS1-expressing photoactivatable GFP (405 nm) from ER to mitochondria in response to SoSe in cells expressing Scr-shRNA or p17-shRNA. Mitochondria and ER were labeled with MitoTracker (far red, 633 nm) and red fluorescent protein (RFP) (565 nm), respectively. Colocalization of CerS1-GFP with ER versus mitochondria was measured using Fiji software. Rc is colocalization coefficient. (H) Mitochondrial potential measured by TMRE fluorescence in Scr control and in silenced p17 UM-SCC-22A cells treated with SoSe and compared to vehicle-treated control. Data are means ± SD (n = 3 independent experiments, **P < 0.01). (I) ATP measurements in control and p17-silenced UM-SCC-22A cells induced for CerS1 expression either with Tet (left, at 72 hours) or SoSe (right, at 3 hours). Data are means ± SD (n = 3 independent experiments, **P < 0.01).
Then, to examine whether p17 is involved in CerS1 trafficking to mitochondria and mitophagy induction, we measured the effects of shRNA-mediated knockdown of p17 in these processes. We ectopically expressed FLAG-CerS1WT and knocked down p17 using shRNA (95%), confirmed by semiquantitative reverse transcription polymerase chain reaction (PCR) and Western blotting using anti-ribosomal protein L29 antibody (fig. S4, A and B). Decreased p17 abundance (fig. S4C) completely prevented mitochondrial localization of ectopically expressed (Tet-induced) V5-CerS1 (+Tet) and endogenous CerS1 in response to SoSe (5 μM, 3 hours) compared to controls (−Tet, Scr-shRNA–transfected and/or vehicle-treated controls) (Fig. 4, B to E). shRNA-mediated knockdown of p17 also abolished mitochondrial generation/accumulation of C18-ceramide compared to Scr-shRNA–transfected controls (fig. S4D) and decreased mitophagy (Fig. 4E). Inhibition of mitochondrial localization of Tet-induced V5-CerS1 in response to p17 knockdown was also detected using TEM with gold-labeled anti-V5 antibody, which resulted in the ER accumulation of CerS1 compared to controls (−Tet/Scr-shRNA–transfected cells) (Fig. 4F). The role of p17/PERMIT in mitochondrial trafficking of endogenous CerS1 in response to SoSe (5 or 90 min) was also shown using photoactivatable GFP-tagged CerS1 in cells transfected with Scr-shRNA or p17-shRNA (Fig. 4G). Mitochondrial localization of CerS1-GFP was detected after 90-min SoSe exposure and was attenuated in response to p17 knockdown (Fig. 4G and fig. S4E). Inhibition by p17 knockdown of mitochondrial localization of CerS1 in response to SoSe exposure is consistent with inhibition of the loss of mitochondrial membrane potential (Fig. 4H) and reduced ATP generation (Fig. 4I) compared to controls. Compared to controls, knockdown of p17 also inhibited LC3 activation, mitochondrial ceramide generation, and ceramide-LC3 association (fig. S4, F and G). These data demonstrate that p17 interacts with and mediates CerS1 trafficking to mitochondria for induction of mitophagy. Thus, we refer to p17 herein as the protein that mediates ER-mitochondria trafficking (p17/PERMIT).
p17/PERMIT interacts with CerS1 via its 28R-29Y-30E-R68-R107-R111 residues
To determine how p17/PERMIT interacts with CerS1 for its mitochondrial localization, we expressed FLAG-CerS1WT and FLAG-CerS1Δ60–76 or FLAG-CerS6WT and FLAG-CerS6ΔHox/+CS1-60–76, in which the CerS6 Hox domain is replaced with CerS1 residues 60 to 76 (Fig. 5, A and B), in UM-SCC-22A cells and examined their association with p17/PERMIT by co-IP with or without SoSe. The data showed that p17/PERMIT interacted with GFP-CerS1WT and FLAG-CerS6ΔHox/+CS1-60–76 but not with FLAG-CerS1Δ60–76 or FLAG-CerS6WT in response to SoSe compared to vehicle-treated controls (Fig. 5, A and B), establishing that p17/PERMIT interacts with CerS1 through residues 60 to 76.
Fig. 5. Amino acids 28R-29Y-30E of p17/PERMIT are key for CerS1 import to mitochondria.
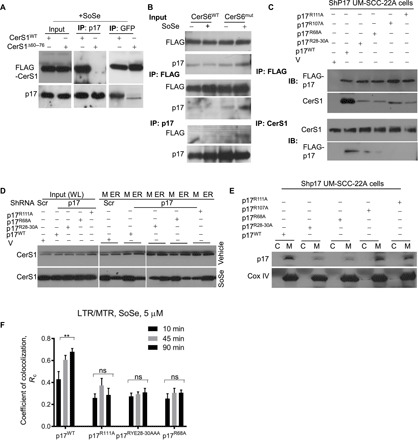
(A) shCerS1 UM-SCC-22A cells were transfected with FLAG-tagged CerS1WT or CerS1Δ60–76 and treated with SoSe. Precleared cell lysates (500 μg of protein) were immunoprecipitated with anti-p17 or anti-FLAG antibodies, and proteins were resolved and identified using SDS-PAGE and Western blotting. Images represent at least three independent experiments. (B) shCerS1 UM-SCC-22A cells were transfected with FLAG-tagged CerS6WT or CerS6ΔHox/CS1(60–76) and treated with SoSe. Precleared cell lysates (500 μg of protein) were immunoprecipitated with p17 or FLAG antibodies, and proteins were resolved and identified using SDS-PAGE and Western blotting. Images represent at least three independent experiments. (C) shp17 UM-SCC-22A cells were transfected with empty vector, p17WT, and p17 mutants, in which residues identified in fig. S6 were substituted with alanine (p17R28A;Y29A;E30A also labeled as p17RYE28-30AAA; p17R68A; p17R107A; p17R111A). Resulting lysates were subjected to IP with FLAG or CerS1 antibodies, and proteins were resolved and identified by SDS-PAGE and Western blotting. Images represent at least three independent experiments. (D) Stably knocked-down scrambled control or p17 shRNA UM-SCC-22A cells were transfected with either empty vector or p17 mutants as in (C). Resulting lysates were subjected to differential centrifugation, and distribution of CerS1 protein between mitochondrial and ER fractions was analyzed. Images represent at least three independent experiments. (E) Lysates of cells treated as in (D) were subjected to differential centrifugation to separate mitochondrial and ER fractions, and distribution of p17/PERMIT was analyzed by Western blotting. Images represent at least three independent experiments. (F) Quantification of live cell imaging of shp17 UM-SCC-22A cells transfected with either empty vector or p17WT or p17 mutants (p17R111A, p17RYE28-30AAA, and p17R68A) and coloaded with MTR (0.5 μM for 60 min) and LTR (0.5 μM for 20 min) upon treatment with 5 μM SoSe. Time points selected to illustrate onset and completion of mitochondrial digestion by autophagy. Data are means ± SD (n = 3 independent experiments, **P < 0.01).
To pinpoint the p17/PERMIT residues involved in its interaction with CerS1 we first used the ZDOCK and Phyre2 programs to predict a possible structural model of p17/PERMIT based on its amino acid sequence (fig. S5, A and B). These studies suggested that p17/PERMIT residues R28, Y29, E30, R68, R107, and/or R111 might be involved in CerS1 association (fig. S5, A and B). Next, we generated several mutants of p17/PERMIT by replacing these residues with alanine (fig. S5C) and then examined their effects on CerS1 interaction, mitochondrial localization of CerS1, and mitophagy in UM-SCC-22A cells in the presence/absence of SoSe. Reciprocal co-IP studies showed that mutations of p17/PERMIT with triple RYE28-30AAA conversions or R68A, R107A, or R111A point mutations almost completely abolished CerS1 interaction compared to p17/PERMITWT (Fig. 5C). Extracts obtained from vector-only–transfected cells (V) were used as negative controls (Fig. 5C).
Last, we determined the effects of the p17/PERMIT residues involved in CerS1 interaction on mitochondrial localization of CerS1 in response to SoSe. We ectopically expressed FLAG-tagged WT and mutant p17/PERMIT proteins in UM-SCC-22A cells, which were stably transfected with shRNAs to knock down endogenous p17/PERMIT, and examined ER-mitochondrial localization of CerS1 using subcellular fractionation and Western blotting. The RYE28-30AAA, R68A, R107A, or R111A conversions in p17/PERMIT largely inhibited mitochondrial translocation of CerS1 compared to p17/PERMITWT in response to SoSe (Fig. 5D). Further, FLAG-p17/PERMITWT, FLAG-p17/PERMITR107A, and FLAG-p17/PERMITR111A were translocated to mitochondria in response to SoSe, whereas mutations of p17/PERMIT with R68A or RYE28-30AAA conversions also inhibited the mitochondrial translocation of p17/PERMIT in response to SoSe (Fig. 5E). These data suggest that (i) the CerS1-interacting residues 28RYE30 and R68 of p17/PERMIT are involved in the mitochondrial translocation of both CerS1 and p17/PERMIT, and (ii) although its R107 and R111 residues are important for p17/PERMIT’s interaction with CerS1, they are not involved in the mitochondrial translocation of p17/PERMIT itself in response to SoSe.
p17/PERMIT-CerS1 interaction mediates mitochondrial stress signaling
Next, we measured the effects of the p17/PERMIT-CerS1 complex on mitophagy induction. In experiments on UM-SCC-22A/sh-p17/PERMIT cells’ response to SoSe, inhibition of the CerS1-p17/PERMIT interaction and attenuation of mitochondrial localization of CerS1 by RYE28-30AAA or R111A conversions of p17/PERMIT prevented mitophagy (Fig. 5F and fig. S5D) compared to FLAG-p17/PERMITWT. These findings are consistent with the inhibition of mitochondrial accumulation of ceramide by the ectopic expression of FLAG-p17/PERMITRYE28-30AAA or FLAG-p17/PERMITR111A mutants compared to FLAG-p17/PERMITWT in response to SoSe in UM-SCC-22A/sh-p17/PERMIT cells (fig. S5, E and F).
To establish the direct interaction between p17/PERMIT and CerS1 in vitro, we used recombinant purified p17WT-His and p17RYE28-AAA-His (isolated from Escherichia coli using column chromatography/fast protein liquid chromatography) and partially purified CerS1WT-FLAG and CerS1Δ60–76-FLAG proteins (isolated from UM-SCC-22A cells using anti-FLAG antibody conjugated agarose beads and immunoprecipitation) (fig. S5, G and H, right panels). The binding between WT p17/PERMIT and CerS1, and not their mutants, in vitro was confirmed by co-IP and Western blotting using anti-His or anti-FLAG antibodies (fig. S5, G and H, right panels). Mutant p17/PERMIT or CerS1 proteins were used as negative controls (fig. S5, G and H, right panels). Purification of recombinant p17/PERMIT (WT and mutant) proteins was confirmed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and Coommasie blue staining (fig. S5, I and J). These data demonstrate that CerS1-p17/PERMIT directly interacts with each other in vitro and in cells and that inhibition of the CerS1-p17/PERMIT interaction prevents SoSe-mediated mitochondrial translocation of CerS1 and mitophagy.
The p17/PERMIT-CerS1 complex is recruited to OMM through MAMs
Because MAMs are known to be involved in protein and lipid trafficking between ER and mitochondria, we sought to establish whether mitochondrial translocation of CerS1 by p17/PERMIT is mediated through MAMs during cellular stress. First, we measured the localization of CerS1 and p17/PERMIT in mitochondria-enriched versus MAM-enriched fractions in the absence or presence of SoSe in UM-SCC-22A cells. SoSe exposure induced the localization of CerS1 in mitochondria through MAMs, as SoSe exposure almost completely eliminated the CerS1-p17/PERMIT localization in MAMs compared to controls (fig. S6A), indicating that p17/PERMIT enables the trafficking of CerS1 from the ER surface to mitochondria through MAMs in response to cellular stress.
Because the translocation of multispanning membrane proteins, such as CerS1, from ER to mitochondria might be difficult, we hypothesized that cellular stress–induced p17/PERMIT-mediated translocation of CerS1 through the ER surface, involving MAMs, to mitochondria might require newly translated protein. A similar process was previously demonstrated for SURF-mediated protein import to mitochondria from ER (20). To test this hypothesis, we measured the mitochondrial localization of CerS1 in the presence/absence of cycloheximide. Inhibition of protein synthesis by cycloheximide largely abrogated the mitochondrial localization of CerS1 in response to SoSe compared to vehicle-treated controls (fig. S6B). This suggests that, in response to SoSe, p17/PERMIT mediates the mitochondrial import of newly translated CerS1 but not the transport through the ER surface and MAMs of already-existing CerS1 protein in the ER.
Next, we sought to determine how the p17/PERMIT-CerS1 complex is recruited to mitochondria. We found that the N terminus of p17/PERMIT contains a putative mitochondria-targeting signal (MitoProt 1.0) (24) within residues 1 to 25. We generated mutants of p17/PERMIT containing deletion of residues 1 to 20 (Δ20), and R14A/W16A or L21A/R25A conversions, and measured their effects on p17/PERMIT-CerS1 association and trafficking to mitochondria, with or without SoSe. While expression of p17/PERMITWT-FLAG enhanced CerS1 association and its mitochondrial import, p17/PERMITΔ20 or p17/PERMITL21A/R25A mutations completely prevented these processes in response to SoSe compared to vector-transfected UM-SCC-22A cells (fig. S6, C and D, and fig. S6, E and F, respectively). Notably, p17/PERMITR14A/W16A mutations had no preventive effect on CerS1 association or its mitochondrial import compared to p17/PERMITWT (fig. S6, E and F), revealing that residues L21-R25 provide the mitochondrial targeting signal for p17/PERMIT, leading to recruitment of the p17/PERMIT-CerS1 complex to the OMM. To confirm the accumulation of the p17/PERMIT-CerS1 complex in the mitochondrial membrane, we measured the interaction between CerS1 or p17/PERMIT and OMM protein Tom20 (fig. S6, G and H). We found that, upon treatment with SoSe, the p17/PERMIT-CerS1 complex associates with Tom20, but this interaction is not observed in vehicle-treated cells (fig. S6, G and H). These data were further supported by immunofluorescence, in which the colocalization of p17/PERMIT-FLAG and Tom20 in response to SoSe exposure was detected in UM-SCC-22A cells compared to vector-transfected and vehicle-treated controls (fig. S6, I and J). Overall, these data suggest that p17/PERMIT, via its residues L21-R25, plays a key role for trafficking of CerS1 to the OMM in response to SoSe-mediated cellular stress.
p17/PERMIT-mediated mitochondrial import of CerS1 is regulated by Drp1 nitrosylation
Because CerS1/C18-ceramide–dependent mitophagy is regulated by Drp1 activation (9, 14), we then determined whether p17/PERMIT-mediated mitochondrial import of CerS1 is regulated by Drp1. We first assessed the effects of shRNA knockdown of Drp1 on mitochondrial localization of CerS1 and mitophagy in response to V5-CerS1 induction (+Tet). Knockdown of Drp1 (fig. S7A) prevented the mitochondrial import of Tet-induced V5-CerS1 compared to −Tet controls (Fig. 6A). To determine how Drp1 is induced, we measured phosphorylation of Drp1 at Ser637 and Ser616, the residues known to be involved in activation of Drp1 (fig. S7B). Expression of a dominant-negative mutant of Drp1 (K38A) or nitrosylation-lacking mutant (C644A) also inhibited V5-CerS1 translocation to mitochondria and mitophagy (+Tet, 72 hours) compared to controls (−Tet) (Fig. 6, B and C). This is consistent with inhibition of the p17/PERMIT-CerS1 association in response to ectopic expression of Drp1C644A compared to Drp1WT in UM-SCC-22A cells (Fig. 6D), suggesting that the p17/PERMIT-dependent mitochondrial localization of CerS1 is regulated by Drp1, involving its C644. Because C644 of Drp1 is known to be nitrosylated for activation and mitochondrial fission induction (25), we next investigated the involvement of Drp1 nitrosylation in regulation of the p17/PERMIT-Drp1 and p17/PERMIT-CerS1 interactions. To achieve this, we generated a nitrosylation mimic mutant of Drp1 (C644W) and measured its effects on the p17/PERMIT-CerS1 interaction. GFP-Drp1C644W did not associate with p17/PERMIT, whereas GFP-Drp1C644A interacted strongly with p17/PERMIT with or without SoSe exposure compared to vector-transfected controls (Fig. 6E and fig. S7C). Knocking down CerS1 decreased nitrosylation of Drp1 (fig. S7D), suggesting a ceramide-mediated mechanism. Ectopic expression of GFP-Drp1C644W, but not GFP-Drp1C644A (equal expression confirmed by Western blotting; Fig. 6E and fig. S7, E and F), in UM-SCC-22A cells stably transfected with shRNA against endogenous Drp1 resulted in constitutive mitochondrial localization of CerS1 even in the absence of SoSe. Together, these results provide the following mechanistic explanation: Nitrosylation of Drp1 at C644 releases p17/PERMIT to associate with CerS1, resulting in import of newly translated CerS1 to the OMM for the induction of ceramide-dependent mitophagy.
Fig. 6. p17/PERMIT-mediated mitochondrial import of CerS1 is induced by Drp1 nitrosylation at C644.
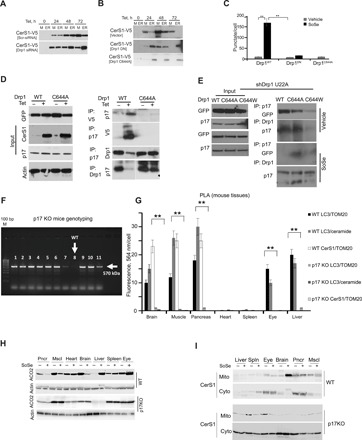
(A) Scr-siRNA–treated control and Drp1 siRNA UM-SCC-22A-Tet On cells were induced for CerS1WT expression for the indicated period of time. Resulting lysates were subjected to differential centrifugation, and V5-tagged CerS1 expression in mitochondrial and ER fractions was evaluated. Images represent three independent experiments. (B) UM-SCC-22A CerS1-Tet On cells were transiently transfected with empty vector or Drp1DN or Drp1C644A mutants and simultaneously induced for CerS1WT expression. Mitochondrial and ER fractions were analyzed for CerS1-V5 expression. Images represent three independent experiments. (C) Autophagic response evaluated by Cyto-ID in UM-SCC-22A-Tet On cells treated as in (B). Quantification was done using three random fields of view from three independent experiments. (D) UM-SCC-22A-Tet On cells were transiently transfected with empty vector or GFP-Drp1WT or GFP-Drp1C644A mutants and simultaneously induced for CerS1 expression with Tet. Precleared lysates were immunoprecipitated with V5, p17, or Drp1 antibodies. Interactions between p17 and CerS1 or between p17 and Drp1 were analyzed using SDS-PAGE and Western blotting. Images represent at least three independent experiments. (E) Effects of the ectopic expression of GFP-Drp1WT or GFP-Drp1C644A or GFP-GFP-Drp1C644W mutants on p17 interaction in the absence or presence of SoSe was detected by co-IP in UM-SCC-22A cells that express shRNA against endogenous Drp1 (right). Equal expression and immunoprecipitation of proteins are shown as input (left). Data shown represent three independent studies. (F) Genotyping of the p17/PERMIT−/− mice was performed by PCR using genomic DNA. (G) Mitophagy induction in the absence or presence of SoSe measured in brain, muscle, pancreas, heart, spleen, eye, and liver tissues obtained from WT and p17/PERMIT−/− mice by PLA to detect the association of LC3 with ceramide and CerS1 with Tom20 using anti-LC3, anti-Tom20, anti-ceramide, or anti-CerS1 antibodies. PLA images shown are for brain, muscle, and pancreas. Quantification of PLA signals by PLA analysis software as described by the manufacturer (n = 3 mice per group). (H) Induction of mitophagy by SoSe in WT and p17/PERMIT−/− mice detected in brain, muscle, and pancreatic tissues measured by ACO2 expression with Western blot (n = 3 mice per group). Actin was used as loading control. (I) Levels of CerS1 in liver, spleen, eye, brain, pancreas, and muscle mitochondrial and soluble fractions of p17 KO versus WT mice treated with vehicle or SoSe for 3 hours.
p17/PERMIT-CerS1–mediated mitophagy requires ceramide-LC3 association
Because Drp1-mediated fission and LC3-autophagosome formation are known to be important for ceramide-dependent mitophagy (9, 14), we performed experiments to uncover the roles of Pink1, upstream mediator of Drp1-dependent fission, and ATG5/LC3, regulators of autophagosome formation, in this process. To achieve this, we quantified mitophagy and mitochondrial transport of CerS1 in SoSe-exposed HeLa cells expressing CRISPR-Cas9 to silence DRP1, PINK1, or ATG5 and compared them to control CRISPR-Cas9–expressing cells. Silencing of DRP1, PINK1, and ATG5 almost completely abrogated SoSe-mediated mitophagy and mitochondrial localization of CerS1 (fig. S8, A to E). However, reconstitution of PINK1 or ATG5 in their respective cells, expressing shRNAs against endogenous proteins, restored mitophagy and CerS1 mitochondrial import in response to SoSe-mediated stress (fig. S8, D and E).
To delineate the roles of Drp1 and LC3 in this process, we used HeLa cells expressing CRISPR-Cas9 against Drp1 and shRNA against LC3 (single knockdown versus double knockdown). Then, we measured the effects of reconstitution of WT versus various mutant forms of Drp1, including C644A and C644W, or LC3, including F52A, which is deficient in ceramide binding, on mitophagy induction and CerS1 translocation to mitochondria. Data showed that reconstitution of constitutively active Drp1C644W induced mitophagy with or without SoSe, while Drp1WT, but not inactive mutant Drp1C644A, required SoSe-mediated stress to induce mitophagy in cells expressing shRNA-targeting Drp1 (fig. S9A). However, knockdown of both Drp1 and LC3 required the reconstitution of both Drp1WT and LC3WT for induction of SoSe-mediated mitophagy (fig. S9, A and B). Reconstitution of a mutant of LC3 with F52A conversion was unable to restore mitophagy even in the presence of WT-Drp1 (fig. S9, D and E). LC3 and Drp1 protein reconstitutions are shown in fig. S9C. Overall, these data show that Drp1 activation via nitrosylation at C644 and ceramide-LC3 association via F52 are both required for mitophagy induction in response to SoSe-mediated mitochondrial localization of CerS1.
Loss of p17/PERMIT prevents acute stress-induced mitophagy in metabolically active tissues
We then sought to determine the physiological roles of p17/PERMIT in the induction of CerS1-dependent mitophagy in vivo. To achieve this, we generated p17/PERMIT−/− mice using CRISPR-Cas9 (Fig. 6F). Genetic ablation of p17/PERMIT did not result in any obvious phenotype in p17/PERMIT−/− mice at 6 months. Then, to assess whether p17/PERMIT plays any roles in the regulation of stress-induced signaling and mitophagy, we exposed age-matched WT (p17/PERMIT+/+) and p17/PERMIT−/− mice to SoSe (0.02 mg/kg) for 3 hours, and then we measured mitophagy in various major metabolically active organs (26) by proximity ligation assay (PLA) for detection of the association between ceramide-LC3, LC3-Tom20, and Tom20-CerS1 (Fig. 6, G and H). Degradation of ACO2 in these tissues was also measured as a marker for mitophagy (Fig. 6I). Notably, while SoSe exposure induced mitophagy in brain, muscle, pancreas, eye, and liver, but not in the heart or spleen of WT mice (Fig. 6, G to I), loss of p17/PERMIT protected against SoSe-induced mitophagy in brain, muscle, and pancreas but had no protective effect in eye or liver tissues (Fig. 6, G to I). These data are consistent with the abrogation of mitochondrial localization of CerS1 in response to SoSe that we observed in brain tissues obtained from p17/PERMIT−/− compared to WT mice (Fig. 6J). Thus, p17/PERMIT plays a role in regulating CerS1/ceramide-mediated mitophagy in vivo in a tissue-specific manner, including selectively in the brain, as well as in muscle and pancreas.
DISCUSSION
Our data suggest that mitochondrial ceramide stress is mediated by trafficking of newly translated CerS1 to the OMM, from the ER through MAMs, by a novel protein, p17/PERMIT. Mitochondrial localization involves a unique sequence within amino acids 60 to 76 of CerS1 and R28, Y29, E30, R68, R107, and R111 of p17/PERMIT. Mechanistically, our data suggest that, under stress-free conditions, Drp1 is associated with p17/PERMIT, forming an inactive complex in the cytosol. Cellular stress, induced by either SoSe or C18-ceramide, mediates Drp1 activation, which is dependent on the S-nitrosylation (25) of the C644, which then releases p17/PERMIT to retrieve newly translated CerS1 from the ER. Drp1, activated by nitrosylation of C644, then induces mitochondrial fission, resulting in mitochondrial membrane damage. This results in the translocation of the p17/PERMIT-CerS1 complex to the damaged OMM, involving residues 1 to 25 of p17/PERMIT, which appears to be important for Tom20 association. Mitochondrial import of CerS1 results in C18-ceramide generation, inducing recruitment of LC3-II–dependent autophagosomes, involving the F52 of LC3-II (18), and mediating mitophagy (summarized in fig. S10). Moreover, CerS1/p17/PERMIT-dependent mitophagy was induced by acute stress in metabolically active tissues (brain, muscle, and pancreas) of WT mice but was prevented in these tissues of p17/PERMIT−/− mice.
Sphingolipid ceramide transport from ER to Golgi is regulated by CERT or Fapp2 for the generation of SM or GlcCer, respectively (15, 16). An explanation of how ceramide accumulation and generation in mitochondria are regulated, however, has been lacking. In this study, we identified a novel amino acid sequence of CerS1 that is key for p17/PERMIT-dependent trafficking of CerS1 for the generation of C18-ceramide at the OMM, leading to autophagosome recruitment and mitophagy. All of the CerS enzymes except CerS1 contain a Hox domain of uncertain function (23). Our data suggest that the amino acid residues of CerS1 involved in its p17/PERMIT-dependent mitochondrial localization are replaced in CerS2 to CerS6 enzymes by a Hox domain, preventing their interaction with p17/PERMIT. This replacement abrogates translocation of CerS2 to CerS6 from ER to mitochondria by p17/PERMIT and the consequent mitophagy in response to Drp1-dependent fission and cellular stress. Of note is the fact that insertion of the CerS1 MTS to replace the Hox domain of CerS6 (FLAG-CerS6ΔHox/+CS1-60–76) induced p17/PERMIT-dependent translocation of the mutant CerS6 from ER to mitochondria. The mitochondrial translocation of the FLAG-CerS6ΔHox/+CS1-60–76 resulted in generation of C16-ceramide, and not C18-ceramide, at the OMM, leading to mitophagy in response to cellular stress. These data suggest that the mitochondrial translocation of CerS enzymes to generate ceramide stress in mitochondria, but not a particular fatty acid chain length of the ceramide, is required for induction of lethal mitophagy. However, it remains unknown whether various other enzymes, which contain amino acid sequences with high homology to the residues involved in mitochondrial localization of CerS1, are also imported to mitochondria by p17/PERMIT in response to Drp1-mediated fission and mitochondrial stress signaling.
Mitochondrial localization of CerS1 in response to cisplatin was previously reported to induce cell death (27). A number of studies showed that CerS1, CerS2, and CerS6 are found in isolated cerebral mitochondria from neuronal cells, playing an important role in Sirt3 regulation (28). There are also reports that CerS6-CerS2 or other heterodimeric CerS complexes with mitochondrial functions exist (29). It is possible that these CerS complexes might be localized to mitochondria, without involvement of p17/PERMIT, via ER-mitochondrial communication through MAMs, where ER and mitochondrial membranes might be in close proximity (30), but this needs to be further evaluated. Notably, Drp1 is known not only to induce fission but also to stabilize mitochondria-ER contact sites, which might play a role in the transport of the p17/PERMIT-CerS1 complex to mitochondria (31). In this connection, the mitochondrial outer membrane protein FUNDC1 has been shown to interact with LC3 to recruit autophagosomes to damaged mitochondria in response to hypoxia. FUNDC1-mediated mitophagy is regulated by its interaction with Drp1 through MAMs, leading to mitochondrial fission (2). This process is similar to ceramide stress-dependent mitophagy, which is also dependent on Drp1. Further, in this regard, we have identified a novel protein, p17/PERMIT, which binds and mediates the translocation of CerS1 to mitochondria in response to cellular stress. There are proteins such as StAR (steroidogenic acute regulatory protein) known to be involved in protein-protein association and mitochondrial import (32), but trafficking by p17/PERMIT of any enzymes involved in (sphingo)lipid metabolism in mitochondria has not been described previously. However, in a recent study, transport from ER to inner mitochondrial membranes of a protein having multiple transmembrane domains was shown to be mediated by a mechanism called ER-SURF, which is completed in yeast through the expanse of the ER surface through the function of a protein chaperone Djp1 (20). Whether ER-SURF–mediated protein trafficking to mitochondria occurs in mammalian cells and whether this process has any physiological or signaling roles remain unknown. Nevertheless, the p17/PERMIT-mediated trafficking of CerS1 to the mitochondria appears to be regulated via an ER-SURF–like mechanism, but this needs further investigation.
Our results with regard to the p17/PERMIT-mediated trafficking of CerS1 to the OMM to induce mitophagy were also consistent in vivo: Short-term exposure of WT mice to SoSe selectively induced mitophagy in brain, muscle, pancreas, eye, and liver tissues but not in the heart or spleen. Genetic loss of p17/PERMIT prevented SoSe-induced mitophagy in tissues obtained from brain, muscle, and pancreas but not in those from the eye or liver. These data suggest that mitophagy in the eye and liver is regulated independently of p17/PERMIT. The mice lacking p17/PERMIT did not exhibit any obvious phenotype at 6 months. However, as alterations of Drp1-mediated fission are associated with neurological disorders such as Parkinson’s and Alzheimer’s diseases, dysfunctional mitochondrial trafficking of CerS1 in response to p17/PERMIT loss might have important physiological implications in the induction of these neurological disorders in response to aging-associated stress signaling. This needs to be further examined.
In summary, data presented here uncover a novel mechanism for the trafficking by p17/PERMIT of a metabolic enzyme CerS1 from the ER surface to mitochondria to mediate ceramide-dependent mitophagy and stress signaling. Our findings have broad implications in the regulation of mitochondrial localization of a metabolic enzyme to induce lipid metabolism and stress signaling in the context of cancer growth regulation and neurological disorders, in which dysfunctional mitochondria accumulation and function are known to play key roles.
MATERIALS AND METHODS
Cell lines and culture conditions
A549 cells were obtained from the American Type Culture Collection (RRID:CVCL_0023); HNSCC cell lines UM-SCC-1, UM-SCC-47, and UM-SCC-22A were obtained from T. Carey (Department of Otolaryngology/Head and Neck Surgery, University of Michigan, Flint, MI, USA; RRID:CVCL_7707, RRID:CVCL_7759, and RRID:CVCL_7731, respectively); and UM-SCC-22A-Tet On V5-CerS1WT, UM-SCC-22A-Tet On V5-CerS1H138A, and UM-SCC-22A-Tet On V5-CerS6WT were generated as described earlier (14). shp17 and shDrp1 UM-SCC-22A cells were generated by stable transduction with pLKO-shDrp1 and pLKO-shp17 of UM-SCC-22A cells. HeLa Scr knockout (KO), HeLa ATG5 KO, HeLa Drp1 KO, and HeLa Parkin KO cells (33) were a gift from R. Youle (NINDS, Bethesda, MD; RRID: CVCL_0058). ShSCR-HeLa-SCRKO, shLC3-HeLa-Scr KO, shSCR-HeLa-Drp1KO, and shLC3-HeLa-Drp1KO were generated by stable transduction of HeLa ScrKO and HeLa Drp1KO with pLKO-shScr and pLKO-shLC3. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) with 10% fetal bovine serum (FBS) (Atlanta Biologicals) and 1% penicillin and streptomycin (Gibco). WT, CerS1, and CerS1-deficient (Top/Top mutant) MEFs were obtained from K. Argraves [Medical University of South Carolina (MUSC)]. MEFs were cultured in DMEM and incubated at 37°C with 5% CO2. Immortal MEFs from WT and Top/Top mutant mice were generated from the fifth passage of primary MEFs by transfection with the SV40 1 pBSSVD2005 plasmid using Fugene and splitting five times by 1/10 split. Stable clones of UM-SCC-22A-TetOn cells expressing V5-CerS1WT, V5-CerS1H138A, and V5-CerS6WT were induced for expression of the proteins for indicated periods of time with doxycycline (2 μg/ml) (Sigma-Aldrich). UM-SCC-22A and A549 cells were treated with either vehicle or 5 μM SoSe (Sigma-Aldrich) for 3 hours. For nitrosylation studies, UM-SCC-22A cells were treated for the indicated periods of time with vehicle or 1 mM NO- and superoxide donor SIN-1 chloride (Cayman Chemical Company).
Cell viability assays
Cell viability was assessed by trypan blue exclusion assay, by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay using cell Titer 96 kit (Promega), or by measuring ATP using CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s directions.
Quantitative PCR
Total RNA isolation was performed using RNeasy (Qiagen), and 1 μg of total RNA was used for complementary DNA (cDNA) synthesis using the iScript cDNA Synthesis Kit (Bio-Rad). TaqMan probes (CerS1, Hs00162669; CerS2, Hs00184211; CerS4, Hs00219999; CerS5, Hs00908758_g1; and CerS6, Hs00831972) were used for quantitative PCR (qPCR) (Thermo Life Technologies).
Plasmids and transfections
PAGFP-N1-CerS1 was generated by subcloning human CerS1 sequence from pcDNA3.1-FLAG-HA-CerS1 using Infusion HD Cloning Kit (Clontech) according to the manufacturer’s instructions. pMD2.G and psPAX2 were a gift from D. Trono (Addgene plasmids #12259 and #12260). pCMV6-AC-CalR-mRFP-ER was obtained from OriGene (RC100108). SV40 1: pBSSVD2005 was a gift from D. Ron (Addgene plasmid #21826). pSelect-LC3-GFP was a gift from A. Cowart (Virginia Commonwealth University, Richmond, VA). pEGFP-Drp1WT, pEGFP-Drp1K38A(DN), and pEGFP-Drp1C644A were a gift from S. A. Lipton (Burnham Institute for Medical Research, La Jolla). pEGFP-Drp1C644W was generated from pEGFP-Drp1WT using Q5 site-directed mutagenesis kit (New England Biolabs) according to the manufacturer’s instructions. Primer sequences used for generation of C644W Drp1 mutants were as follows: forward, 5′-CGGGAACAGCGATGGGAGGTTATTGAACGA-3′; reverse, 5′-TCGTTCAATAACATCCCAATCTCGCTGTTCCCG-3′. The pIRES2CerS1Δ60–76 was generated from pIRES2CerS1WT using site-directed mutagenesis and the following primers: forward, 5′-CTC GGGCGGCGCCAGGTGCGC-3′; reverse, 5′-GCGGCCACTGCGGCGCCTCTTTC-3′. pcDNA3.1-FLAG-CerS6ΔHox/CS1(60–76) was generated from pcDNA3.1-FLAG-CerS6WT using site-directed mutagenesis in two steps: (i) To delete the Hox domain, the following primers were used: forward, 5′-CCGCCCAATGCCATTTCCAAGCAACTGGAC-3′; reverse, 5′-GTCCAGTTACTTGGCAATGGCATTGGGCGG-3′. (ii) To insert CS1 (60 to 76), the following megaprimers were used: forward, 5′-CAGGCCAATGGACCACAAATTGCTCCGCCCAATGCCATTCTGCTGCTGCTGGCGCTCGGCGCGCTGGGCTGGACCGCCCTGCGCTCCTCCAAGCAACTGGACTGGGATGTTCGAAGCATTCAGCGC-3′; reverse, 5′-GCGCTGAATGCTTCGAACATCCCAGTCCAGTTGCTTGGAGGAGCGCAGGGCGGTCCAGCCCAGCGCGCCGAGCGCCAGCAGCAGCAGAATGGCATTGGGCGGAGCAATTTGTGGTCCATTGGCCTG-3′; p17 cDNA was amplified from total cDNA by PCR using the following primers: forward, 5′-ATG GCCAAGTCCAAGAACCAGAGCACAA-3′; reverse, 5′-CTACTCTGAAGCCTTTGTAGGGGCCTGG-3′. The resulting cDNA was inserted in vector PCR 2.1 using the TA-cloning kit (Thermo Life Technologies) and subcloned into FLAG-HA-pcDNA3.1 (a gift from A. Antebi, Addgene plasmid #52535) using the following primers: forward, 5′-CTGGCGGCTCGAATGGCCAAGTCCA-3′; reverse, 5′-TAGATGCATGCTCGACTACTCTGACTCTGAAGCCT-3′ and the Infusion HD Cloning Kit (Clontech), according to the manufacturer’s instructions. p17R68A, p17R107A, p17R111A, p17RYE28-30AAA, p17RKW14-16AAA, p17LKKPR21-25AAA, and p17Δ20 constructs were generated from FLAG-HA-pcDNA3.1-p17WT using the following primers: p17R68A: forward, 5′-CAGTCCCGAAAAGCCCAAAGAAATGG-3′; reverse, 5′-CCATTTCTTTGGGCTTTTCGGGACTG-3′; p17R107A: forward, 5′-CGTGAGTGCAGCTGCTGAGGCTATC-3′; reverse, 5′-GATAGCCTCACGTGCACTCACG-3′; p17R111A: forward, 5′-CAAGCTTGGGAAGGCTGCTCATGCC-3′; reverse, 5′-GGCATGAGCAGCCTTCCCAAGCTTG-3′; p17RYE28-30AAA: forward, 5′-CGTGCTCATGCCGCCATTGTCAAGG-3′; reverse, 5′-CCTTGACAATGGCGGCATGAGCAGG-3′; p17RKW14-16AAA: forward, 5′-CAGCACAAACAACCAGTCCGCAGCAGCGCAAAGAAATGGTCTC-3′; reverse, 5′-GAGACCATTTCTTTGCGCTGCTGCGGACTGGTTGTTTGTGCTG-3′; p17LKKPR21-25AAA: forward, 5′-GAAAATGGCAAAGAAATGGTGCGGCGGCGGCCTCACAAAGATACG-3′; reverse, 5′-CGTATCTTTGTGAGGCCGCCGCCGCACCATTTCTTTGCCATTTTC-3′; p17Δ20: forward, 5′-CACACTGGCGGCCGCTCGAATGCAATCCCTTAAGGGGGTGG-3′; reverse, 5′-CCACCCCCTTAAGGGATTGCATTCGAGCGGCCGCCAGTGTG-3′.
Antibodies
The antibodies used for Western blotting in this study were as follows: ICT1 (cat# ab55259, RRID:AB_2122854; Abcam), NUBPL (cat# ab69150, RRID:AB_2153651; Abcam), COXIV (cat# ab131177, RRID:AB_11154973; Abcam), Tom20 (cat# sc-17764, RRID:AB_628381; Santa Cruz Biotechnology), LASS1 (sc-65096; Santa Cruz Biotechnology), LASS4 (sc-65116; Santa Cruz Biotechnology), LASS5 (sc-135038; Santa Cruz Biotechnology), LASS6 (sc-65127; Santa Cruz Biotechnology), COX17 (cat# sc-100521, RRID:AB_2085114; Santa Cruz Biotechnology), Parkin (cat# sc-32282, RRID:AB_628104; Santa Cruz Biotechnology), CERT (cat# XW-7119, RRID:AB_735674; ProSci), LC3B (cat# 2775, RRID:AB_915950; Cell Signaling Technology), ACO2 (cat# 6922S, RRID:AB_10828218; Cell Signaling Technology), GFP (cat# 14-6774-63, RRID:AB_468332; Thermo Fisher Scientific), protein disulfide isomerase (PDI) (cat# NB100-1921, RRID:AB_10001061; Novus), 11beta-HSD1 (cat# NBP1-32027; Novus), cystein S-nitrosylated (SNO-Cys) (cat# C9002-75; U.S. Biological Life Sciences), DLP1 (cat# 611112; BD Transduction Lab), V5 (cat# R96025; Invitrogen), actin (cat# A2066, RRID:AB_476693; Sigma-Aldrich), ceramide (MD15B4; Enzo), GM130 (cat# sc-30100, RRID:AB_2232778; Santa Cruz Biotechnology), ATG5 (cat# 12994, RRID:AB_2630393; Cell Signaling Technology), ribosomal protein L29 (first antibody, cat# sc-103166; Santa Cruz Biotechnology; second antibody, cat# ab88514, RRID:AB_2042834; Abcam).
Stable shRNA and siRNA silencing
ShRNA against p17 (SHRPL29P31202925, TRCN0000202925), Drp1 (SHCLNG-NM_012062, TRCN0000010593), CerS1 (SHCLNG-NM_021267, TRCN0000168362), CerS2 (SHCLNG-NM_029789, TRCN0000193773), CerS5 (SHCLNG-NM_147190, TRCN0000433716), CerS6 (SHCLNG-NM_172856, TRCN0000086348), LC3 (SHCLNG-NM_032514, TRCN0000243387), and nontargeting shRNA-containing plasmids were provided by the MUSC shRNA Shared Technology Resource from Sigma shRNA Mission Library. 293T cells were cotransfected with pCMV-psPAX2 and pMD2 plasmids using the viral transduction protocol as described by the RNA Interference Consortium. The viral supernatants were added to UM-SCC-22A cells, and selection was performed using puromycin (1 μg/ml) for 14 days. Small interfering RNAs (siRNAs) against Cers1, Drp1, CERT, and LC3 were purchased from Dharmacon and used for transfections with Oligofectamine for 48 hours. Nontargeting Scr-siRNAs (Dharmacon) (34) were used as controls. Efficiency of siRNAs was evaluated by quantitative real-time PCR and/or Western blotting. β-Actin or ribosomal RNA was used as internal controls.
Immunoprecipitation and Western blotting
Cellular lysates in radioimmunoprecipitation assay (RIPA) buffer containing a protease inhibitor cocktail (Sigma-Aldrich) were normalized by the total protein level and analyzed by SDS-PAGE and immunoblotting with corresponding antibodies. For immunoprecipitation, precleared cytosolic fractions were incubated overnight with 2 μg of corresponding antibodies at 4°C, followed by 1-hour incubation with Protein A/G Agarose (Santa Cruz Biotechnology) (50 μl of a 50% slurry). Resin was washed three to five times, and pulled-down proteins were analyzed by SDS-PAGE and Western blotting with corresponding antibodies.
Immunofluorescence
UM-SCC-22A cells (50,000 per well) were plated on glass coverslips in a six-well plate for 18 hours. Cells were fixed and permeabilized using 4% paraformaldehyde (20 min) and 0.1% Triton X-100 in 1× PBS (phosphate-buffered saline) (pH 7.4) for 10 min. The cells were then blocked with 1% BSA (bovine serum albumin)/PBS (pH 7.4) for 1 hour. Cells were incubated for 18 hours at 4°C with antibodies specific for ceramide, LC3, Tom20, V5, GFP, FLAG, CerS1, GM130, or 11beta-HSD1 (1:50) in blocking solution, followed by Alexa Fluor 488– or Alexa Fluor 594–conjugated secondary antibodies (1:1000) for 1 hour. Immunofluorescence was performed using a Leica TCS SP2 AOBS confocal microscope, an Olympus FV10i microscope with 543- and 488-nm channels for visualizing green and red fluorescence, or a Zeiss LSM 880 NLO Quasar confocal/multiphoton microscope with a Fast Airyscan super-resolution detector. Images were taken at 63× magnification. At least three random fields were selected for image quantification.
Monitoring CerS1 dynamics by photoactivation of GFP
Photoactivation experiments were conducted as previously described (35). PA-GFP imaging was performed in multitracking mode on a Zeiss LSM 880 NLO laser scanning confocal microscope (Carl Zeiss, Thornwood, NY) with a 63× Plan Apochromat 1.4 NA objective and a 413/488 dichroic mirror. In brief, pPAGFP-N1 or pPAGFP-N1-CerS1 plasmids were cotransfected with pCMV6-AC-CalR-mRFP-ER in HeLa cells. After 48 hours, post-transfection cells were loaded with MitoTracker Far Red (Invitrogen) for 45 min. Immediately before imaging, 5 μM SoSe was added to the cells. After three initial frames, cells were irradiated by high energy (1 μW) of 405 laser light. Images were shot at 5 and 90 min after photoactivation.
Ultrastructural analysis using TEM
After removal of culture medium, UM-SCC-22A cells were fixed in 2% (w/v) glutaraldehyde in 0.1 M cacodylate buffer. After post-fixation in 2% (v/v) osmium tetroxide, specimens were embedded in Epon 812, and sections were cut orthogonally to the cell monolayer with a diamond knife. Thin sections were visualized in a JEOL 1010 TEM. In immunoelectron microscopy studies, after fixing with 4% paraformaldehyde (PFA), cells were permeabilized with 0.1% Triton X-100 for 10 min at room temperature (RT), washed, and blocked with 1% BSA in PBS for 20 min. Corresponding primary antibodies (1:50 dilution) were added to cells and incubated overnight at 4°C. After washing with PBS, Nanogold (1.4 nm) (Nanoprobes)–conjugated anti-mouse or anti-rat Fab fragments (1:200) were incubated with cells for 1 hour at RT. After post-fixation with 1% glutaraldehyde in PBS for 10 min at RT, LI Silver enhancement was performed for 5 min. After rinsing with H2O, specimens were embedded in Epon 812, and sections were cut orthogonally to the cell monolayer with a diamond knife. Thin sections were visualized in a JEOL 1010 TEM.
Subcellular fractionation
For mitochondria preparation, the Mitochondria Isolation Kit (ab110171; Abcam) was used, according to the manufacturer’s instructions. Briefly, trypsinized cells were collected and pelleted by centrifugation at 1000g. After freezing and thawing several times, cells were resuspended in Buffer A and incubated for 10 min on ice, followed by dounce homogenization. After centrifugation, the supernatant was saved and the pellet was resuspended in Buffer B to the same volume as Buffer A, following the same rupturing steps. Combined supernatants were centrifuged at 12,000g for 15 min at 4°C to obtain the mitochondrial fraction. For the membrane fraction, after obtaining the mitochondrial pellet, the supernatant was collected and centrifuged at 100,000g for 1 hour in an ultracentrifuge. The pellet was resuspended in wash buffer and recentrifuged for 45 min. The membrane pellet was then resuspended in lysis buffer [250 mM sucrose, 20 mM Hepes (pH 7.4), 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 0.1% SDS, 1 mM dithiothreitol, and protease inhibitor (PI) cocktail]. Western blotting was used to detect the purity of the fractions with Tom20 as a mitochondrial marker, actin as a cytosolic marker, and PDI as a marker for ER.
Isolation of mitochondria and MAMs
MAMs isolation was performed according to a published protocol (36). In brief, U22A cells were grown to 95% confluency in four culture flasks. After washing cells with PBS and homogenizing in isolation buffer [225 mM mannitol, 75 mM sucrose, 0.1 mM EGTA, and 30 mM tris-HCl (pH 7.4)], homogenate was centrifuged at 600g for 5 min. After discarding unbroken cells and nuclei, the supernatant was centrifuged twice at 7000g for 10 min and once at 10,000g for 10 min. The mitochondrial pellet was resuspended in ice-cold mitochondria resuspending buffer (MRB) [250 mM mannitol, 5 mM Hepes (pH 7.4), and 0.5 mM EGTA] and subjected to Percoll gradient [225 mM mannitol, 25 mM Hepes (pH 7.4), 1 mM EGTA, and 30% (v/v) Percoll]. After a 1-hour centrifugation at 95,000g, a dense band containing purified mitochondria located at the bottom and a MAM band located above the mitochondrial band were collected. After additional washing steps, followed by several rounds of centrifugation at 100,000g, pure mitochondria and MAM were isolated, resuspended in small volumes of MRB, and analyzed by SDS-PAGE, followed by Western blot with corresponding antibodies.
Fractionation of mitochondrial membrane
Mitochondria were isolated, and submitochondrial fractionation was performed as previously described (37). Briefly, the pellet of crude mitochondria was prepared as described above. Following washing with SHE buffer [250 mM sucrose, 10 mM Hepes, and 1 mM EGTA (pH 7.4)] and centrifugation at 16,000g for 10 min, the mitochondrial pellet was resuspended in swelling buffer [10 mM KH2PO4 (pH 7.4)] at a concentration of 200 μg of mitochondrial protein per milliliter and incubated on ice for 20 min. Then, an equal volume of shrinking buffer [10 mM KH2PO4 (pH 7.4), 32% (wt/vol) sucrose, 30% (wt/vol) glycerol, and 10 mM MgCl2] was added and centrifuged at 10,000g for 10 min. The supernatant containing the OMM and the mitochondrial intermembrane space fractions were transferred to a fresh tube. The mitoplast pellet was washed with 1:1 mixture of swelling and shrinking buffer and centrifuged at 10,000g for 10 min at 4°C. After washing, the mitoplasts were resuspended in swelling buffer and incubated on ice for 20 min. OMM/IMS (intermembrane space)- and IMM (inner mitochondrial membrane)–containing fractions were centrifuged at 14,000g for 1 hour. The resulting pellet contains OMMs and IMMs. The supernatant contains the IMS proteins. The supernatant was concentrated with centrifugal concentrators (Millipore). Fractions were resuspended in RIPA buffer and analyzed by SDS-PAGE and Western blot with corresponding antibodies, including mitochondrial membrane and intermembrane markers.
Lipidomics analysis of sphingolipids
Lipid extraction and analysis were performed by the Analytical Unit in the Lipidomics Shared Resource at MUSC. Briefly, cells were lysed with RIPA buffer, and further preparation of samples and advanced analyses of endogenous bioactive sphingolipids were performed on a Thermo Fisher TSQ Quantum liquid chromatography/triple-stage quadrupole mass spectrometer system operating in a multiple reaction monitoring (MRM)–positive ionization mode, as previously described (9, 14). Lipid levels were normalized by the level of protein present in samples (pmol/mg protein).
Detection of LC3 activation by Cyto-ID
LC3 activation was assessed with the Cyto-ID Autophagy Detection kit (Enzo) in live cells using fluorescence microscopy, according to the manufacturer’s instructions. Briefly, cells were seeded into six-well plates at a density of 1.25 million per well and treated immediately with vehicle/Dox (2 μg/ml) for 48 hours for Tet On induction, or the next day with vehicle/5 μM SoSe for 3 hours. After this 48- or 3-hour treatment, cells were washed, stained with Green autophagosome detection reagent and Hoechst 33342 nuclear stain, and analyzed by confocal microscopy using fluorescein isothiocyanate and Hoechst filters.
Detection of mitophagy by live cell imaging
Cells were seeded in 35-mm glass-bottomed dishes (MatTek Corporation) the day before the experiment in complete growth media (38). On the day of the experiment, cells were loaded with MitoTracker Red (MTR) (0.5 μM) for 60 min in a humidified atmosphere at 37°C in the growth media. Afterward, the cells were washed and incubated with LysoTracker Green (LTG) (0.5 μM) for 20 min. After MTR and LTG had been loaded, one-third of the initial concentration of LTG was kept in the media for the duration of the experiment. On the microscope stage, culture media (CM) (38) was replaced with CM plus 5 μM SoSe. Time series of live cell images were collected every 1 to 2 min for 150 min after addition of SoSe on the Olympus FluoView FV10i LIV laser scanning confocal microscope in the MUSC Cell and Molecular Imaging Core Facility.
Measurement of mitochondrial potential
Mitochondrial potential was measured using the tetramethylrhodamine ethyl ester (TMRE) mitochondrial membrane potential assay kit (ab113852; Abcam), following the manufacturer’s protocol. Briefly, UM-SCC-22A cells were grown overnight in 96-well, black microplates with clear bottom and treated for 3 hours either with vehicle or with 5 μM SoSe, 20 μM FCCP, or DNP (positive controls) for 10 min. Following washing with PBS, 500 nM TMRE in complete media was added to the cells, and the plate was incubated for an additional 20 min at 37°C. After washing with PBS/0.2%BSA, TMRE fluorescence was measured using a microplate reader with Ex/Em = 549/575.
Molecular modeling and docking simulations
Modeling, simulations, and visualizations to model the CerS1-p17/PERMIT complex and predict which p17 amino acid residues are involved in its interaction with CerS1 were performed using ZDOCK (37), Phyre2 (39), and Predict server (40), which identifies protein residues responsible for the interaction with other proteins. Predictions of p17 mitochondrial targeting sequence were done using MitoProt 1.0 (24).
Proximity ligation assay
Tissues were fixed, embedded in paraffin, and mounted on glass slides. After deparaffinization and rehydration, slides were incubated with 0.2% glycine for 5 min to quench autofluorescence, followed by incubation with various antibodies (10 to 20 μg/ml) at 4°C for 18 hours. PLA was performed using the Duolink in situ hybridization kit as described by the manufacturer (Sigma-Aldrich). Images were quantified by Duolink Image Tool Software (Olink Bioscience).
Recombinant p17/PERMIT cloning, expression, and purification
The cDNA sequence encoding full-length WT p17/PERMIT was cloned into a pET28b(+) vector using Nde I and Sac I restriction sites. The resulting construct (pET28b-p17) produces a 175-residue (19.1 kDa) fusion protein containing a thrombin-cleavable N-terminal 6×His tag, preceding the 155–amino acid p17/PERMIT coding sequence. This plasmid was used as a template for QuikChange site-directed mutagenesis (Agilent) to generate a triple-alanine mutant (pET28b-p17/PERMITR28–30A) of the RYE residues at positions 28 to 30 (R28A, Y29A, and E30A). Both WT and mutant p17/PERMIT constructs were used to transform E. coli BL21(DE3) cells. Cells were grown to ODA595 = 0.8 in 1 liter of LB media, and protein expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 hours at 37°C. Since >90% of His6-p17/PERMIT and His6-p17/PERMITR28–30A were expressed in the insoluble fraction, a urea-denaturating/on-column refolding purification was used for both constructs. Briefly, cells were pelleted by centrifugation for 20 min at 4°C (10,000g) and resuspended in 40 ml of Lysis Buffer A, containing 20 mM tris (pH 7.4), 500 mM NaCl, 3 mM beta-mercaptoethanol (βME), deoxyribonuclease I (DNase I) (2.5 μg/ml) (#D5025; Sigma-Aldrich), 1× phenylmethylsulfonyl fluoride (PMSF), and one PI tablet (#A32965; Pierce). Cells were lysed by sonication and centrifuged at 4°C for 30 min (15,000g) to pellet the His6-p17–containing insoluble fraction. The supernatant was discarded, and the pellet was resuspended in 40 ml of Lysis/Wash Buffer B [6 M urea, 20 mM tris (pH 7.4), 500 mM NaCl, 20 mM imidazole 3 mM βME, 1× PMSF, and 1× PI tablet]. Following a second round of sonication and centrifugation, the urea lysate was applied to an equilibrated 1-ml HisTrap FF affinity column (GE Healthcare) and the column was washed with 20 volumes (c.v.) of Buffer B. His tag–immobilized p17 was refolded on-column by applying a linear gradient of 6 to 0 M urea over 30 c.v. at a flow rate of 0.5 ml/min. Renatured protein was liberated from the column with a 0 to 100% linear gradient of Elution Buffer C [20 mM tris (pH 7.4), 500 mM NaCl, 500 mM imidazole, 3 mM βME, and 1× PI tablet] over 10 c.v. Eluted fractions were analyzed by SDS-PAGE, and fractions containing His6-p17 at purity >80% were combined, EDTA was added to 500 μM, and fractions were concentrated to ~3 ml using an Amicon Ultra-15 (10,000 molecular weight cut off) centrifugal concentrator (Millipore). Contaminating proteins were removed by size exclusion chromatography (SEC) by applying the concentrated sample to a preparative-scale HiLoad 26/600 Superdex 75 pg column pre-equilibrated with SEC Buffer D [1× PBS (pH 7.4), 500 μM EDTA, 3 mM βME, and 1× PI tablet]. Isocratic elution at a flow rate of 2.5 mg/ml resulted in a monodisperse peak containing His6-p17/PERMIT (WT and mutant) at >95% purity. Protein concentration was determined by ultraviolet absorbance at 280 nm using the molecular weight (MW) and extinction coefficient (ε) for each construct (His6-p17/PERMIT MW = 19,356 g/mol, ε = 8480 M−1 cm−1; His6-p17/PERMITR28–30A MW = 19,120 g/mol, ε = 8480 M−1 cm−1). A typical purification yielded approximately 2.5 g of purified protein per liter of rich media. Purified protein was stored as 200-μl aliquots at −80°C.
Detection of direct interaction between partially purified FLAG-tagged CerS1 and recombinant His-p17/PERMIT proteins in vitro
A549 cells transfected with the hCerS1WT-FLAG, hCerS1Δ60–76-FLAG, or empty vector were lysed by freeze-thawing in a lysis buffer containing 50 mM tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 0.5% NP-40 with a protease inhibitor cocktail (1:500). Cell lysates (400 μg of protein) were incubated with 150 μl of magnetic beads conjugated with antibody against FLAG (Sigma-Aldrich) for 18 hours at 4°C with agitation. After extensive washes, CerS1-FLAG containing beads were incubated with 30 μg of His-tagged p17/PERMITWT or His-tagged p17/PERMITR28–30A recombinant purified proteins in 150 μl of binding buffer containing 50 mM tris (pH 7.5), 137 mM NaCl, 1 mM MgCl2, 2.7 mM KCl, 15 mM NaF, and 0.5 mM NaV3O4 overnight at 4°C with agitation. This was followed by several washes after proteins bound to the beads were eluted using 1× SDS loading buffer without βME at 95°C for 3 min. The resulting eluate was loaded on the Criterion TGX Stain-Free Precast Gel (Bio-Rad), followed by Western blot analysis with anti-FLAG (Sigma-Aldrich) or anti-His (Invitrogen) antibodies.
Vertebrate animals and generation of p17/PERMIT−/− KO mice
Vertebrate animal studies were performed using protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the MUSC. Mouse Rpl29-ps2 (p17) KO model (C57BL/6) was created using CRISPR-Cas–mediated genome engineering by Cyagen Biosciences Inc. (Santa Clara, CA). The mouse Rpl29-ps2 gene (Gene ID: 432721; Ensembl: ENSMUSG00000094772) is located on mouse chromosome 13. The Rpl29-ps2 gene was selected as target site. Cas9 mRNA and guide RNA generated by in vitro transcription were then injected into fertilized eggs for KO mouse production. The founders were genotyped by PCR, followed by DNA sequencing analysis. The positive founders were bred to the next generation (F1) and subsequently genotyped by PCR and DNA sequencing analysis. Mouse Rpl29-ps2-F, 5′-TGACTCCATCACAATGGACAACATTG-3′; mouse Rpl29-ps2-R, 5′AAGTTTCAAATGCCGAAGTCCTTCC-3′.
Statistical analysis
All data are presented as means ± SD, and group comparisons were performed with a two-tailed Student’s t test. P < 0.05 was considered statistically significant. Data were reported as means ± SD or SE as indicated. The mean values were compared using two-sided Student’s t test or analysis of variance (ANOVA), and P > 0.05 was considered nonsignificant (ns); *P ≤ 0.05 and **P ≤ 0.01 were considered significant.
Supplementary Material
Acknowledgments
We thank the members of the Ogretmen Laboratory for technical support and helpful discussions. We also thank A. Bai and J. Pierce for help in ceramide measurements at the Lipidomics Shared Resource facility, MUSC. We thank R. Youle [National Institutes of Health (NIH)] for providing the HeLa cells. Funding: This work was supported by research funding from the NIH (CA088932, CA214461, CA173687, DE016572, and P01 CA203628 to B.O.). The core facilities used were supported by NIH (C06 RR015455), Hollings Cancer Center Support Grant (P30 CA138313), or Center of Biomedical Research Excellence (Cobre) in Lipidomics and Pathobiology (P30 GM103339). The Zeiss 880 microscope was funded by a shared instrumentation grant (S10 OD018113). Ethics statement: This study was performed in strict accordance with the recommendations in the Guide for the Care and the Use of Laboratory Animals of the NIH. All of the animals were handled according to approved IACUC protocol (2018-00412) of the MUSC. Author contributions: Conception and design: N.O. and B.O. Methodology development: N.O., J.K., J.J.L., B.M.R., and B.O. Data acquisition: N.O., J.K., B.M.R., S.P.S., and M.G. Data analysis and interpretation: N.O., J.K., S.P.S., and M.G. Manuscript writing, review, and revision: N.O., R.H.J., and B.O. Administrative, technical, or material support: N.O. and B.O. Study supervision: B.O. Competing interests: N.O. and B.O. are inventors on a provisional patent application covering ceramide-selenium analogs as therapeutic agents owned by the MUSC Foundation for Research Development (no. 62/692,241, filed 29 June 2018). B.O. is the founder and Chief Scientific Officer of Lipo-Immuno Tech LLC, which is responsible for commercialization of ceramide-selenium analog drugs to induce mitophagy. The authors declare that they have no other competing interests. Data and materials availability: Plasmids and small molecules can be provided by B.O. pending scientific review and a completed material transfer agreement (ogretmen@musc.edu). All data needed to evaluate the conclusions in the paper are present in the manuscript and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax1978/DC1
Fig. S1. Roles of CerS1 versus CerS6 in ceramide generation and mitophagy induction.
Fig. S2. CerS1 amino acids 60 to 76 are critical for Cers1 import to mitochondria.
Fig. S3. Ceramide transport by CERT or FAPP2 is not involved in mitophagy.
Fig. S4. CerS1 mitochondrial import is mediated by p17/PERMIT.
Fig. S5. Mitochondrial import of CerS1 requires p17/PERMIT and involves amino acids RYE28-30.
Fig. S6. p17/PERMIT-mediated CerS1 import to mitochondria induces mitophagy.
Fig. S7. Roles of Drp1 nitrosylation at C644 in mitochondrial localization of CerS1.
Fig. S8. Mitochondrial localization of CerS1 induces mitophagy via ATG, LC3, and Drp1 in response to SoSe.
Fig. S9. Mitochondrial CerS1-dependent mitophagy is induced via activation of LC3 and Drp1.
Fig. S10. Summary of the hypothesis and proposed mechanism for the mitochondrial trafficking of CerS1 by p17/PERMIT.
REFERENCES AND NOTES
- 1.Hannun Y. A., Obeid L. M., Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Edvardson S., Yi J. K., Jalas C., Xu R., Webb B. D., Snider J., Fedick A., Kleinman E., Treff N. R., Mao C., Elpeleg O., Deficiency of the alkaline ceramidase ACER3 manifests in early childhood by progressive leukodystrophy. J. Med. Genet. 53, 389–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chalfant C. E., Rathman K., Pinkerman R. L., Wood R. E., Obeid L. M., Ogretmen B., Hannun Y. A., De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J. Biol. Chem. 277, 12587–12595 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Morad S. A. F., Levin J. C., Shanmugavelandy S. S., Kester M., Fabrias G., Bedia C., Cabot M. C., Ceramide–antiestrogen nanoliposomal combinations—Novel impact of hormonal therapy in hormone-insensitive breast cancer. Mol. Cancer Ther. 11, 2352–2361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pewzner-Jung Y., Ben-Dor S., Futerman A. H., When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J. Biol. Chem. 281, 25001–25005 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Laviad E. L., Albee L., Pankova-Kholmyansky I., Epstein S., Park H., Merrill A. H. Jr., Futerman A. H., Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 283, 5677–5684 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Deng X., Yin X., Allan R., Lu D. D., Maurer C. W., Haimovitz-Friedman A., Fuks Z., Shaham S., Kolesnick R., Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science 322, 110–115 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simanshu D. K., Kamlekar R. K., Wijesinghe D. S., Zou X., Zhai X., Mishra S. K., Molotkovsky J. G., Malinina L., Hinchcliffe E. H., Chalfant C. E., Brown R. E., Patel D. J., Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 500, 463–467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dany M., Gencer S., Nganga R., Thomas R. J., Oleinik N., Baron K. D., Szulc Z. M., Ruvolo P., Kornblau S., Andreeff M., Ogretmen B., Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128, 1944–1958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinberg S. E., Chandel N. S., Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 11, 9–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panda P. K., Naik P. P., Meher B. R., Das D. N., Mukhopadhyay S., Praharaj P. P., Maiti T. K., Bhutia S. K., PUMA dependent mitophagy by Abrus agglutinin contributes to apoptosis through ceramide generation. Biochim. Biophys. Acta. Mol. Cell Res. 1865, 480–495 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Law B. A., Liao X., Moore K. S., Southard A., Roddy P., Ji R., Szulc Z., Bielawska A., Schulze P. C., Cowart L. A., Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. 32, 1403–1416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizumura K., Justice M. J., Schweitzer K. S., Krishnan S., Bronova I., Berdyshev E. V., Hubbard W. C., Pewzner-Jung Y., Futerman A. H., Choi A. M. K., Petrache I., Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. FASEB J. 32, 1880–1890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sentelle R. D., Senkal C. E., Jiang W., Ponnusamy S., Gencer S., Panneer Selvam S., Ramshesh V. K., Peterson Y. K., Lemasters J. J., Szulc Z. M., Bielawski J., Ogretmen B., Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8, 831–838 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., Nishijima M., Molecular machinery for non-vesicular trafficking of ceramide. Nature 426, 803–809 (2003). [DOI] [PubMed] [Google Scholar]
- 16.D’Angelo G., Polishchuk E., Di Tullio G., Santoro M., Di Campli A., Godi A., West G., Bielawski J., Chuang C.-C., van der Spoel A. C., Platt F. M., Hannun Y. A., Polishchuk R., Mattjus P., de Matteis M. A., Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 449, 62–67 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S. O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y.-i., Taguchi N., Morinaga H., Maeda M., Takayanagi R., Yokota S., Mihara K., Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Murakawa T., Yamaguchi O., Hashimoto A., Hikoso S., Takeda T., Oka T., Yasui H., Ueda H., Akazawa Y., Nakayama H., Taneike M., Misaka T., Omiya S., Shah A. M., Yamamoto A., Nishida K., Ohsumi Y., Okamoto K., Sakata Y., Otsu K., Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6, 7527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yonashiro R., Ishido S., Kyo S., Fukuda T., Goto E., Matsuki Y., Ohmura-Hoshino M., Sada K., Hotta H., Yamamura H., Inatome R., Yanagi S., A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 25, 3618–3626 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen K. G., Aviram N., Laborenz J., Bibi C., Meyer M., Spang A., Schuldiner M., Herrmann J. M., An ER surface retrieval pathway safeguards the import of mitochondrial membrane proteins in yeast. Science 361, 1118–1122 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Senkal C. E., Ponnusamy S., Manevich Y., Meyers-Needham M., Saddoughi S. A., Mukhopadyay A., Dent P., Bielawski J., Ogretmen B., Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J. Biol. Chem. 286, 42446–42458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao L., Spassieva S. D., Jucius T. J., Shultz L. D., Shick H. E., Macklin W. B., Hannun Y. A., Obeid L. M., Ackerman S. L., A deficiency of ceramide biosynthesis causes cerebellar purkinje cell neurodegeneration and lipofuscin accumulation. PLOS Genet. 7, e1002063 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mesika A., Ben-Dor S., Laviad E. L., Futerman A. H., A new functional motif in Hox domain-containing ceramide synthases: Identification of a novel region flanking the Hox and TLC domains essential for activity. J. Biol. Chem. 282, 27366–27373 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Claros M. G., MitoProt, a Macintosh application for studying mitochondrial proteins. Comput. Appl. Biosci. 11, 441–447 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Cho D.-H., Nakamura T., Fang J., Cieplak P., Godzik A., Gu Z., Lipton S. A., S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McWilliams T. G., Prescott A. R., Montava-Garriga L., Ball G., Singh F., Barini E., Muqit M. M. K., Brooks S. P., Ganley I. G., Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439–449.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min J., Mesika A., Sivaguru M., Van Veldhoven P. P., Alexander H., Futerman A. H., Alexander S., (Dihydro)ceramide synthase 1–regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol. Cancer Res. 5, 801–812 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Novgorodov S. A., Riley C. L., Keffler J. A., Yu J., Kindy M. S., Macklin W. B., Lombard D. B., Gudz T. I., SIRT3 deacetylates ceramide synthases: Implications for mitochondrial dysfunction and brain injury. J. Biol. Chem. 291, 1957–1973 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen S. A., Calvert A. E., Volpert G., Kouri F. M., Hurley L. A., Luciano J. P., Wu Y., Chalastanis A., Futerman A. H., Stegh A. H., Bcl2L13 is a ceramide synthase inhibitor in glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 111, 5682–5687 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y., Amano A., Yoshimori T., Autophagosomes form at ER–mitochondria contact sites. Nature 495, 389–393 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Prudent J., Zunino R., Sugiura A., Mattie S., Shore G. C., McBride H. M., MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell 59, 941–955 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Bose H. S., Lingappa V. R., Miller W. L., Rapid regulation of steroidogenesis by mitochondrial protein import. Nature 417, 87–91 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Nezich C. L., Wang C., Fogel A. I., Youle R. J., MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 210, 435–450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barceló-Coblijn G., Martin M. L., de Almeida R. F. M., Noguera-Salvà M. A., Marcilla-Etxenike A., Guardiola-Serrano F., Lüth A., Kleuser B., Halver J. E., Escribá P. V., Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc. Natl. Acad. Sci. U.S.A. 108, 19569–19574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patterson G. H., Lippincott-Schwartz J., A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873–1877 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Wieckowski M. R., Giorgi C., Lebiedzinska M., Duszynski J., Pinton P., Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Pierce B. G., Wiehe K., Hwang H., Kim B.-H., Vreven T., Weng Z., ZDOCK server: Interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Picard M., McManus M. J., Csordás G., Várnai P., Dorn G. W. II, Williams D., Hajnóczky G., Wallace D. C., Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat. Commun. 6, 6259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T., Hönigschmid P., Schafferhans A., Roos M., Bernhofer M., Richter L., Ashkenazy H., Punta M., Schlessinger A., Bromberg Y., Schneider R., Vriend G., Sander C., Ben-Tal N., Rost B., PredictProtein—An open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 42, W337–W343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax1978/DC1
Fig. S1. Roles of CerS1 versus CerS6 in ceramide generation and mitophagy induction.
Fig. S2. CerS1 amino acids 60 to 76 are critical for Cers1 import to mitochondria.
Fig. S3. Ceramide transport by CERT or FAPP2 is not involved in mitophagy.
Fig. S4. CerS1 mitochondrial import is mediated by p17/PERMIT.
Fig. S5. Mitochondrial import of CerS1 requires p17/PERMIT and involves amino acids RYE28-30.
Fig. S6. p17/PERMIT-mediated CerS1 import to mitochondria induces mitophagy.
Fig. S7. Roles of Drp1 nitrosylation at C644 in mitochondrial localization of CerS1.
Fig. S8. Mitochondrial localization of CerS1 induces mitophagy via ATG, LC3, and Drp1 in response to SoSe.
Fig. S9. Mitochondrial CerS1-dependent mitophagy is induced via activation of LC3 and Drp1.
Fig. S10. Summary of the hypothesis and proposed mechanism for the mitochondrial trafficking of CerS1 by p17/PERMIT.