

The blood protein Vitronectin forms a four-bladed propeller that is a hub for multiple functions and binds pathogenic bacteria.
Abstract
Vitronectin (Vn) is a major component of blood that controls many processes central to human biology. It is a drug target and a key factor in cell and tissue engineering applications, but despite long-standing efforts, little is known about the molecular basis for its functions. Here, we define the domain organization of Vn, report the crystal structure of its carboxyl-terminal domain, and show that it harbors the binding site for the Yersinia pestis outer membrane protein Ail, which recruits Vn to the bacterial cell surface to evade human host defenses. Vn forms a single four-bladed β/α-propeller that serves as a hub for multiple functions. The structure explains key features of native Vn and provides a blueprint for understanding and targeting this essential human protein.
INTRODUCTION
Vitronectin (Vn) interacts with a wide range of ligands to regulate hemostasis, cell adhesion, tissue remodeling, tumor metastasis, and immunity. Its ligands include integrins, complement factors, growth factors, cytokines, and anticoagulants (1), and pathogens bind Vn to acquire protection from complement-mediated lysis (2). Since its discovery in 1967 (3), Vn has been the focus of intensive studies aimed at understanding the molecular basis for its functions (1), targeting its interactions for therapeutic intervention (4), and harnessing its adhesive properties for cell and tissue engineering applications (5).
Vn circulates as an intact ~75-kDa molecule or as two disulfide-linked 65- and 10-kDa polypeptides produced by proteolysis after Arg398 (6, 7), with the bulk mass contributed by N-linked glycosylation at three sites. The sequence of mature Vn (8, 9) begins after cleavage of its secretion signal (Fig. 1A). The 44-residue somatomedin B (SMB) domain regulates plasminogen activation (10), and the contiguous ArgGlyAsp (RGD) motif (residues 64 to 66) mediates binding to integrin receptors (11). Notably, however, the interaction with integrin requires intact Vn for stabilization (12), underscoring the importance of obtaining molecular data for the entire protein. The SMB domain represents the only structure determined for Vn (13). This structure has provided important insights but accounts for only a small fraction of the 459-residue sequence and does not offer a complete view of its broad functional spectrum.
Fig. 1. Domain organization and NMR of Vn.
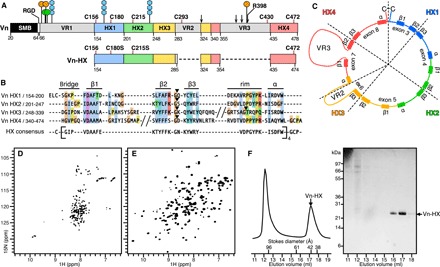
All domain representations are scaled to sequence. The HX domain is colored by repeat unit (HX1, blue; HX2, green; HX3, yellow; HX4, red). (A) Vn starts at Asp20 and comprises an SMB domain (black), RGD sequence (white), HX domain (colored by HX repeat), and three variable regions VR1 to VR3 (gray). The six Cys in the C-terminal domain (pink lines) and sites for phosphorylation (orange circles), sulfation (green circles), N-glycosylation (blue hexagons), and protease cleavage (arrows) are shown. Vn-HX starts at Glu154. (B) Alignment of Vn HX1-HX4 sequence repeats (Clustal color scheme). VR2 and VR3 are omitted for clarity (slashes). Structural elements (bridge-β1-β2-β3-rim-α) of the HX repeats are depicted above the alignment. Black arrowheads mark intron positions. The HX consensus was derived for Vn and other HX proteins of known structure (fig. S2). (C) HX structural repeat units viewed in the frame of the exon structure (dashed wedges). VR2 and VR3 form insertions in exons 6 and 7. (D and E) Solution NMR 1H/15N spectra of 15N-labeled Vn-HX in 6 M urea (D) or refolded in nondenaturing buffer (E). (F) Size exclusion chromatography of folded Vn-HX and SDS–polyacrylamide gel electrophoresis (PAGE) analysis of the eluted fractions. Vn-HX elutes with an apparent Stokes diameter of ~42 Å.
The rest of the protein contains binding sites for many biomedically important ligands but has eluded structural characterization due to its conformational complexity and the repetitive nature of its primary sequence, which has confounded both model prediction and the biosynthetic production of discrete protein domains for structural and functional studies. Here, we describe the structure of the C-terminal domain of Vn and show that it harbors the binding site for the Yersinia pestis outer membrane protein Ail, which recruits Vn to the bacterial cell surface to survive and multiply in infected human hosts.
RESULTS AND DISCUSSION
The sequence of Vn comprises four hemopexin-like repeats
Vn shares homology with proteins of the hemopexin (HX) family, defined by the presence of quadruplex sequence repeats (HX1 to HX4) that fold as a single four-bladed β/α-propeller, known as the HX domain (14–16). In the absence of experimental data, Vn structural models have been proposed on the basis of sequence alignment with known HX protein structures (17–20). The models, however, vary widely with respect to their assignments of HX repeats and have numbers of HX repeats ranging from 5 to 10, not compatible with either the fourfold symmetry or packing constraints of the HX domain. We took a broader bioinformatic approach, guided by evolutionary conservation of the Vn sequence, the molecular constraints of propeller structures, and correlation of the sequence repeats with the exon/intron organization of the Vn gene.
We first examined the evolutionary conservation of Vn (fig. S1). Since its first appearance with jawed vertebrates, Vn has been highly conserved except for three variable regions (VR1 to VR3). VR1 is a highly variable sequence immediately after the RGD motif that contains sites for phosphorylation, sulfation, and N-glycosylation. VR2 is only somewhat variable and contains one of the thrombin cleavage sites. VR3 is highly variable. It spans ~80 residues in human Vn but is substantially expanded (~200 residues) in bony fishes while substantially shorter (~40 residues) in birds and cartilaginous fishes. VR3 contains the heparin binding site, as well as Arg398, and several other proteolytic cleavage sites. Notably, cleavage after Arg398 does not lead to dissociation of the 65/10-kDa chains of circulating Vn, indicating that VR3 does not contribute appreciably to conformational stability. VR1 to VR3 are predicted to harbor flexible/disordered segments (fig. S1).
Next, we compared Vn with other HX family proteins of known structure (fig. S2). The structures of β/α-propellers are highly conserved, constrained as they are by their number of blades and distinct circular symmetries (21). In the four-bladed version of the HX family (14–16), each blade coincides with a bridge-β1-β2-β3-rim-α structural unit: The bridge connects the propeller outer rim to β1 at the propeller center, a three-stranded β-sheet (β1 to β3) radiates from the center back to the rim, and the rim and α-helix connect to the next blade. A disulfide bond links the start of the first blade to the end of the last in all known animal HX domains. The β1 sequences have distinct signatures limited to residues with short side chains that can fit in the closely packed propeller center. We identified only four such sequence motifs for Vn (consensus VDAAF) that we assigned to β1 (Fig. 1B). Sequence conservation decreases toward the propeller rim where packing is less constrained, but other regions with distinct HX signature can be discerned. We assigned β2 and β3 to two richly aromatic penta/hexapeptide motifs connected by a tight turn (consensus KTYFFK-GN-KYWRY), and we assigned the α-helix to a polar tripeptide motif flanked by hydrophobic/aromatic residues (consensus ISDFW). The bridge and rim are more variable but distinguished by their abundance of Gly and Pro, including a signature YPK consensus in the rim. In all, we identified four HX repeats spanning residues 155 to 478. The full set of four is flanked by Cys156 and Cys472, both of which are 100% conserved through evolution (fig. S1).
Viewed in the frame of the Vn gene, each of our assigned HX repeats is split by an intron at the β2-β3 turn (Fig. 1B), revealing the genetic repeat unit to be β3-rim-α-bridge-β1-β2, a circular permutation of the structural repeat (Fig. 1C). Exons 4 and 5 each encode one complete genetic repeat, exons 6 and 7 combine to encode a third, and exons 8 and 3 complete each other to form the fourth. This arrangement reinforces the interdependence of the four repeats as a single genetically encoded domain. Segments containing VR2 and VR3 are flanked by introns of identical phase compatible with shuffling into an ancestral HX gene (fig. S3). They are unique to Vn and appear to form insertions reminiscent of the integrin α subunit cap subdomains that confer specialized functionality (22).
Vn forms a single HX domain
On the basis of our analysis, we engineered a Vn polypeptide (Vn-HX, Fig. 1A) spanning residues 154 to 474 but lacking VR2 and VR3. Vn-HX readily expressed in Escherichia coli. It could be purified under denaturing conditions and then renatured by removing the denaturant in the presence of a disulfide exchanger. The nuclear magnetic resonance (NMR) spectra obtained with 15N-labeled Vn-HX show that it undergoes a marked transformation from an unfolded state in the presence of denaturant, where the 1H/15N NMR signals (Fig. 1D) cluster around 8 parts per million (ppm) (1H) and 120 ppm (15N), to a homogeneously folded state, where the 1H/15N signals (Fig. 1E) are highly dispersed, as expected for ordered β structures. Folded Vn-HX is monomeric (Fig. 1F) and could be crystallized for x-ray diffraction, allowing us to solve and refine the three-dimensional structure to 1.9-Å resolution (Table 1).
Table 1. Crystallographic data and refinement statistics.
| Data collection* | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 40.56, 97.36, 49.16 |
| α, β, γ (°) | 90.00, 99.90, 90.00 |
| Resolution (Å) | 26.96–1.90 (1.94–1.90)* |
| Rsym or Rmerge | 0.071 (0.326) |
| I/σI | 7.2 (2.0) |
| Completeness (%) | 98.1 (96.1) |
| Redundancy | 2.3 (2.3) |
| Refinement | |
| Resolution (Å) | 24.21–1.90 (1.96–1.90) |
| No. of reflections | 28,986 (2453) |
| Rwork/Rfree | 0.1793/0.2102 (0.2481/0.2940) |
| No. of atoms (nonhydrogen) | |
| Protein | 3012 |
| Ligand/ion | 30 |
| Water | 168 |
| B factors | |
| Protein | 44.24 |
| Ligand/ion | 57.02 |
| Water | 41.98 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.832 |
*One crystal was used for the structure. Values in parentheses are for highest-resolution shell.
The crystallographic asymmetric unit contains two copies. Vn-HX forms a single HX domain: a four-bladed β/α-propeller circularized by a Cys156-Cys472 disulfide bond (Fig. 2, A to C). The bridges connecting α-β1 and the turns connecting β2-β3 form a topologically smooth surface at the propeller top (defined as the N-terminal end of β1), while longer loops protrude from the bottom forming a more complex structural landscape. The bottom is also more flexible, with larger differences between the two molecules in the asymmetric unit, higher B factors, and missing electron density for the β1-β2 loop and outer rim of HX3 (fig. S4). The rims have some β character and form distinct β-bulges in HX1, HX2, and HX4.
Fig. 2. Structure and topology of Vn-HX.
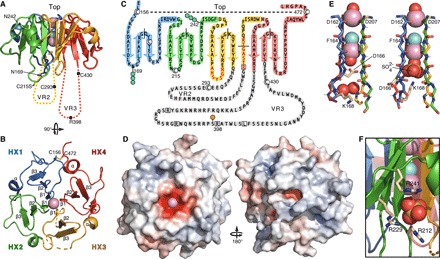
Colors denote structural repeat units HX1 (blue), HX2 (green), HX3 (yellow), and HX4 (red). Spheres denote metal (Na+, pink) or Cl− (cyan) ions and O (red) or S (yellow) atoms. Broken lines denote gaps in the protein chain due to missing electron density. Dotted lines denote VR2 (yellow) and VR3 (red). (A and B) Side and top views of the structure, showing the positions of the two N-glycosylation sites (Asn242 and Asn169) and cysteines. (C) Schematic topology of the Vn C terminus showing β-strands (arrows), α-helices (cylinders), VR2 and VR3 residues deleted in Vn-HX (gray highlight), residues with missing electron density (red letters), exon breaks (black solid lines), Cys (white circles), disulfide bond (dashed line), and sites for phosphorylation (orange circles), N-glycosylation (blue hexagons), and protease cleavage (white squares). (D) Top and bottom views with surface colored by electrostatic potential from −10 kT/e (red) to +10 kT/e (blue). (E) Backbone structure of the channel for molecule A (left) and molecule B (right) of the asymmetric unit, showing key side chains (wheat). (F) SO42− binding site on the surface of Vn-HX. The positively charged surface groove is lined by Arg212, Arg229, and Arg241 at the HX2-HX3 repeat interface.
The β1 aspartates produce a pole of negative electrostatic potential at the propeller top, while the bottom presents a heterogeneous mix of positively and negatively charged grooves and protrusions (Fig. 2D). The four β1 strands contribute backbone carbonyl oxygens and amide hydrogens to coordinate two metal cations, either sodium or calcium, which were both present as the only metals in the crystallization solution, and a chloride anion in the propeller central channel (Fig. 2E). Sodium and calcium cannot always be readily distinguished because their binding sites share similar characteristics, including oxygen ligands and octahedral geometry with metal-oxygen distances distributed around the optimal range of 2.4 to 2.5 Å. In the structure of Vn-HX, the metal-oxygen distances range between 2.2 and 2.3 Å for the site at the top of the channel where the ligand arrangement around the metal is trigonal bipyramidal and between 2.3 and 2.4 Å for the site at the bottom of the channel, where the ligand arrangement is octahedral (fig. S5). We assigned the metal ions to Na+ because this is most consistent with key parameters of metal-binding sites (23), including the geometry of the first coordination shells and ion valence considerations, ion occupancy and completeness of the coordination sphere, and drops in R-free. Assignment to Na+ is also most consistent with the relative concentrations of Na+ and Ca2+ in the crystallization solution. Calcium-binding sites typically have higher number of electrons and higher charge. Thus, it is possible that a calcium ion occupies the binding site deeper in the Vn-HX channel where the coordination sphere is octahedral and can include a negatively charged SO42− anion (see below). The central channels of other HX domains (14–16) also contain metal ions (assigned to Na+ or Ca2+) and Cl− ions, although the reported identity, arrangement, and number of ions vary from structure to structure.
The two molecules of the asymmetric unit have different structures around the bottom of the channel, reflecting the conformational plasticity accessible to Vn (Fig. 2E and fig. S5). A water molecule capped by the Asp166 side chain carboxylate coordinates the metal ion in molecule A, while a SO42− anion capped by the Lys168 side chain amine coordinates the metal in molecule B. This rearrangement alters the length of HX1 β1 by one residue and transmits to the surface-exposed β1-β2 loops of HX1 and HX2. A second SO42− binds in a positively charged surface groove at the HX2-HX3 repeat interface (Fig. 2F). These SO42− binding sites are likely candidates for the interactions of Vn with sulfated ligands, including heparin and cholesterol.
The structure displays defining features of the native protein. As predicted, introns coincide with the surface-exposed β2-β3 turns of each blade and the rim of the HX3 blade (Fig. 2C). The two N-glycosylation sites are surface-exposed (Fig. 2, A and C), as are the primary binding site for insulin-like growth factor II (4) and the epitopes for two Vn antibodies that react with both Vn-HX and full-length Vn (Fig. 3, A and B). Rather than participating in the core fold, VR2 and VR3 extend from the propeller bottom, in line with their accessibility to proteases, protein kinase A phosphorylation, and bacterial pathogens (1). VR2 and VR3 share some sequence similarity with the cap extensions that protrude from the β-propeller of the integrin αIIb subunit headpiece (22), and solving their structures will be important for understanding the functionalities that map to these regions.
Fig. 3. Binding activity of Vn-HX.
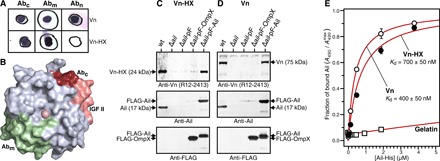
(A) Antibody (Ab) binding activity detected by Western dot blots of Vn and Vn-HX with anti-Vn antibodies directed to residues 141 to 154 (Abn), 209 to 258 (Abm), or 446 to 472 (Abc). The sequence of Vn-HX starts at Glu154 and does not react with Abn. (B) Top view of Vn-HX. Surface exposed epitopes for Abm (green) and Abc (ruby and pink) map to HX2 and HX4, respectively. The binding site for insulin-like growth factor II (4) maps to the surface-exposed outer rim of HX4 (pink). (C and D) Ail-mediated microbial binding activity assayed by co-sedimentation of Vn-HX (C) or full-length Vn (D) with Ail-expressing Y. pestis bacterial cells. Immunoblots were probed with anti-Vn, anti-Ail, or anti-FLAG antibodies. Y. pestis cell types were wild-type expressing native ail (wt); ail deletion mutant (Δail); and Δail complemented with plasmid expressing FLAG tag (Δail-pF), FLAG-tagged OmpX (Δail-pF-OmpX), or FLAG-tagged Ail (Δail-pF-Ail). Arrows mark positions of Vn, Vn-HX, Ail, FLAG-Ail, and FLAG-OmpX. (E) Vn-HX binds purified Ail. Plates preadsorbed with Vn-HX (5 μg/ml) (black circles), full-length Vn (5 μg/ml) (white circles), or gelatin (20 μg/ml) (white squares) were incubated overnight with increasing concentrations of purified refolded His-tagged Ail. Binding was detected with ELISA using a mouse anti-His antibody, by measuring light absorbance at 490 nm (A490). Each data point represents the mean ± SEM of three independent experiments. Binding curves with dissociation constants of Kd = 700 ± 50 nM (Vn-HX) and Kd = 400 ± 50 nM (Vn) were estimated by fitting (red lines; R2 = 0.99) the ELISA data relative to the maximum fraction of bound Ail (A490max) in each titration. No Ail binding is observed to plates preadsorbed with gelatin.
The Vn HX domain is circularized by two conserved disulfide-linked cysteines
The disulfide bond pattern of Vn has been examined extensively (1, 19, 24, 25), with conflicting results that reflect the potential for dynamic rearrangement by regulatory extracellular enzymes and redox-dependent functional states of the protein (25). The structure shows that Cys156 and Cys472 form the signature circularizing disulfide of the HX domain, in line with their 100% conservation in all Vn orthologs. Cys180Ser (mutated in Vn-HX) is buried with a cluster of aromatic side chains and not accessible to other cysteines. Consistent with its position in the structure, it is only 50% conserved, often substituted with Phe, and thus a likely candidate for one of the two free thiols estimated for Vn. Cys215Ser, by contrast, is in a solvent-exposed loop on the propeller bottom (Fig. 2, A and C) and topologically close to Cys293 (deleted with VR2) for disulfide bond formation, while Cys430 (deleted with VR3) is topologically distant from other cysteines. Both Cys215 and Cys293 are 100% conserved, while the consensus is 80% for Cys430.
The HX domain is held together by aromatic and proline side chains, packed to favor ring stacking interactions. Evolutionarily conserved prolines stabilize outward facing aromatic side chains and exposed backbone sites at the propeller surface. Of these, Pro205, Pro240, Pro459, and Pro462 adopt cis-amide conformations that change the direction of the backbone, enabling the partially positively charged faces of their rings to interact favorably with the negatively charged π faces and CH groups of aromatic residues and with backbone carbonyl groups (fig. S6). We propose that the integrity of the 65/10-kDa two-chain form of Vn is maintained by the core structure of its HX domain, with the chains additionally tethered by the Cys156-Cys472 disulfide bond. Since VR3 is not a structural part of the HX domain, the two-chain split generated by proteolysis at Arg398 is not expected to disrupt the core fold of the propeller. The extent of Arg398-Ala399 bond cleavage is genetically determined by the identity of the amino acid at position 400 (26). A Met400Thr polymorphism decreases susceptibility to cleavage and increases the risk of hemangioblastoma in patients with the Von Hippel–Lindau gene defect (27), pointing to a potential regulatory function of VR3.
The Vn HX domain is recruited to the surface of Y. pestis cells through its interaction with the outer membrane protein Ail
Our interest in Vn was motivated by the discovery that it is recruited from human serum to the surface of Y. pestis through its interaction with the bacterial outer membrane protein Ail (28), a process linked to human infection by this deadly pathogen. Bacterial-bound Vn can function as a bridge linking bacteria to select integrin-expressing cells, and Vn is a principal regulator of both the complement and coagulation pathways, both targeted by pathogens to enhance their survival and dissemination in infected hosts.
To determine whether the Vn HX domain contains the binding site for Ail, we incubated purified Vn-HX with Y. pestis cells and analyzed binding by co-sedimentation. Vn-HX co-sediments with wild-type Y. pestis expressing native Ail but not with the ail deletion mutant Y. pestis(Δail) (Fig. 3C). Binding is restored when Y. pestis(Δail) is engineered to express FLAG-tagged Ail, but not FLAG tag alone or FLAG-tagged OmpX, another Y. pestis outer membrane protein that has no known virulence activity. Notably, the binding profile of Vn-HX is identical to that of full-length Vn (Fig. 3D), indicating that the binding site for Ail is contained in the HX domain. Heparin can bind Vn (1) and has been proposed to bind Ail (29), raising the question that it might serve as a bridge for the intermolecular interactions of either protein. In a previous study (28), however, we showed that the addition of heparin has no effect on the co-sedimentation of Vn with Ail-expressing bacteria and that purified Ail embedded in lipid bilayer membranes binds purified Vn in a dose-dependent manner.
To confirm that the binding interaction is directly mediated by Vn and Ail and involves the HX domain, we performed an enzyme-linked immunosorbent assay (ELISA) with purified Ail and surface-adsorbed Vn or Vn-HX. The data (Fig. 3E) show that Ail binds Vn-HX and full-length Vn, while no binding is observed for surface-adsorbed gelatin. Estimates of the apparent dissociation constant (Kd) reflect similar, albeit weaker, affinity of Ail for Vn-HX (Kd ~700 nM) compared with Vn (Kd ~400 nM), suggesting that other regions of Vn may contribute to strengthen the binding interaction in vivo. The data, therefore, demonstrate that Ail and Vn interact directly without the need for an intermediary bridge ligand and that the Vn HX domain contributes substantially to the binding affinity.
Conclusions
The Vn HX domain thus appears to provide a scaffold for a wide range of Vn interactions. In summary, the structure described here provides a blueprint for understanding the multiple functions of Vn, as well as new opportunities for advancing the applications of this important human protein in medicine and biotechnology. The clear definition of Vn sequence and domain organization helps guide mutagenesis studies and the biosynthetic preparation of Vn polypeptides for the many bioengineering applications that rely on immobilized Vn. Moreover, identification of the Vn domain responsible for the interaction with Ail provides an important molecular platform for understanding the roles of Vn in Y. pestis pathogenesis and for the development of medical countermeasures.
MATERIALS AND METHODS
Sequence analysis
Protein sequences were obtained from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov) and analyzed in Jalview (30). Initial sequence alignments were generated with Clustal (31) and then refined by several rounds of manual editing, guided by the known constraints of four-bladed β/α-propeller structures (14–16, 21) and the gene structure. Sequence identity, conservation, and consensus were calculated with Jalview. Disordered protein regions were identified with DISEMBL (32), using the “Remark 465” disorder predictor, which is based on comparison to Protein Data Bank (PDB) x-ray structures with nonassigned electron densities.
Protein expression, purification, and refolding
The DNA sequence encoding Vn-HX was cloned into the Nde I and Xho I sites of a pET23a plasmid vector. The sequence encompasses Vn residues 154 to 285, 324 to 354, and 435 to 474 and includes two mutations: C180S and C215S. The Vn-HX plasmid was transformed into E. coli BL21(DE3) cells (New England Biolabs), and cells were grown at 37°C, in M9 minimal medium, to a cell density corresponding to OD600 (optical density at 600 nm) of 0.8. Gene expression was induced by adding isopropyl-β-d-thiogalactopyranoside to 1 mM. Cells were grown for an additional 4 hours at 37°C and then harvested by centrifugation (6000g for 10 min at 4°C). Cells from 1000 ml of culture were suspended in 30 ml of lysis buffer [50 mM tris-HCl (pH 8), 100 mM NaCl, 20 mM EDTA, 25% (w/v) sucrose, and 1 mM dithiothreitol (DTT)], flash-frozen in liquid N2, stored at −80°C overnight, and then lysed by three passes through a French Press (Glen Mills). The soluble fraction was removed by centrifugation (24,000g for 30 min at 4°C). The resulting pellet was washed twice with 30 ml of lysis buffer supplemented with 2% (v/v) Triton X-100 and then once with 30 ml of lysis buffer and finally incubated with 30 ml of urea buffer [20 mM tris-HCl (pH 8) and 6 M urea] overnight at room temperature. The insoluble fraction enriched in Vn-HX was collected by centrifugation and then dissolved in guanidine buffer [20 mM tris-HCl (pH 8), 6 M guanidine, and 10 mM DTT] for refolding and purification.
Vn-HX was folded at 4°C by dropwise dilution from an initial concentration of 2 to 3 mg/ml in guanidine buffer to a final concentration of 0.1 mg/ml in folding buffer [20 mM tris-HCl (pH 8), 500 mM ArgCl, 300 mM NaCl, 50 mM CaCl2, 5 mM β-mercaptoethanol, 1 mM hydroxyethyldisulfide, and 0.05% Brij-35]. The folding reaction was allowed to proceed for 72 hours with gentle stirring, then precipitants were removed by centrifugation, and the supernatant was dialyzed overnight against final buffer [20 mM MES buffer (pH 6.5), 300 mM NaCl, and 2 mM CaCl2]. The protein solution was concentrated 60-fold using a Vivaspin 20 device with 10-kDa cutoff (GE Healthcare) and then purified by size exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare). The protein identity was confirmed by electrospray ionization mass spectrometry (Agilent 6230 TOFMS). The concentration was determined by measuring ultraviolet absorbance at 280 nm.
NMR spectroscopy
The bacterial growth medium was supplemented with (15NH4)2SO4 (Cambridge Isotopes) to produce 15N-labeled protein for NMR. The samples contained ~100 μM 15N-labeled Vn-HX in final buffer supplemented with 5% 2H2O. Two-dimensional NMR heteronuclear single quantum coherence spectra were acquired at 25°C on a Bruker AVANCE 600 MHz spectrometer equipped with a 1H/15N/13C triple-resonance cryoprobe. The data were processed and analyzed using TopSpin (Bruker).
Crystallization
A solution of Vn-HX (6 mg/ml) was dialyzed overnight into 20 mM MES (pH 6.5), 100 mM NaCl, and 1 mM CaCl2 and then cleared by centrifugation. The final protein concentration was 4 mg/ml. Initial crystallization trials were conducted in 96-well sitting drop plates (MRC crystallization plates, Molecular Dimensions), and initial crystals were obtained at room temperature, in conditions C4 and C8 of the Morpheus HT-96 screen, each containing 90 mM NPS salts (30 mM NaNO3, 30 mM Na2HPO4, and 30 mM (NH4)2SO4], 12.5% (v/v) 2-methyl-2,4-pentanediol, 12.5% (w/v) PEG-1,000, and 12.5% (w/v) PEG-3350, plus 100 mM imidazole/MES buffer (pH 6.5) (condition C4) or 100 mM Hepes/Mops buffer (pH 7.5) (condition C8). Crystals were optimized using the Additive Screen HT (Hampton Research).
The final crystal used for data collection grew at room temperature, after mixing 1 μl of Vn-HX [4 mg/ml in 20 mM MES (pH 6.5), 100 mM NaCl, and 1 mM CaCl2] with 1 μl of precipitant solution [90 mM imidazole/MES buffer (pH 6.5), 81 mM NPS salts, 11.25% (v/v) 2-methyl-2,4-pentanediol, 11.25% (w/v) PEG-1000, 11.25% (w/v) PEG-3350, and 3% (w/v) d-(+)-trehalose] and equilibration in a sitting drop plate with 50 μl of precipitant solution. The crystal was harvested and directly transferred into a 100 K nitrogen cryo-stream for data collection.
Data acquisition and structure determination
Data were collected on an FR-E superbright (Rigaku) x-ray generator and processed to 1.9-Å resolution in CCP4 (33). Phases were obtained using Phaser (34) in Phenix (35). The most homologous known structures share only ~30% sequence identity; thus, a trimmed ensemble model of five structures was generated for molecular replacement. The model was generated using Sculptor (36) and Ensembler, from the structures of human matrix metalloproteases (PDB 1RTG chain, PDB 3C7X chain A and PDB 2MQS chain A), and rabbit hemopexin (PDB 4RT6 chain B and PDB 1QHU chain A). Phaser found two copies of Vn in the asymmetric unit with a top Translation Function Z-score of 16.5, indicative of a definitive correct solution. Phenix.AutoBuild (35) was used for initial model building, followed by several rounds of manual model inspection and correction in Coot (37) and refinement by phenix.refine (35). The CheckMyMetal server (23) was used to assess consistency of the modeled Ca2+/Na+ and Cl− ions. We were not able to fully discriminate between Ca2+ and Na+ ions, which were both present in the crystallization solution. In the structure, we built the metal ions as Na+, most consistent with their relative concentrations of Ca2+ and Na+, the drops in R-free, and the ligand-binding geometry, as analyzed by the CheckMyMetal server. MolProbity (38) and the PDB validation server were used for structure validation throughout refinement. The final MolProbity score is 1.13 (100th percentile), 98.6% of all residues are in the favored region of the Ramachandran plot, and 1.4% are in the allowed region, with no outliers. Poisson-Boltzmann electrostatics were calculated in PyMOL using APBS (39). Illustrations were prepared using PyMOL. The structure coordinates and data have been deposited in the PDB (code: 6O5E).
Western dot blots
Vn (Sigma) or Vn-HX were adsorbed onto a nitrocellulose membrane (LC2001, Life Technologies) by direct addition from solution. The proteins were probed with three primary antibodies specific for residues 141 to 154 (TA321171, OriGene Technologies), 209 to 258 (ABIN1454094, Antibodies online), or 446 to 472 (LS-C407672, LifeSpan Biosciences).
Bacterial binding of Vn and Vn-HX
Whole-cell binding assays were performed as described (28). Briefly, Y. pestis strains were grown in heart infusion broth containing 2.5 mM CaCl2 for 3 hours at 37°C, then harvested by centrifugation (10,000g for 5 min at 4°C), washed in ice-cold phosphate-buffered saline (PBS), and suspended in PBS to an OD620 of 5. Washed bacteria (250 μl) were added to an equal volume of 1 μM human Vn (Innovative Research) or 1 μM Vn-HX in PBS. Binding reactions were incubated for 30 min at room temperature. Bacteria and co-sedimenting proteins were collected by centrifugation (10,000g for 5 min at 4°C), washed three times with 1 ml of ice-cold PBS containing 0.05% Tween 20, and lysed by boiling in 100 μl of SDS-PAGE sample buffer [50 mM tris-HCl, 2% SDS, 5% glycerol, and 1% β-mercaptoethanol (pH 6.8)]. Bacterial lysates and co-sedimenting proteins were subjected to SDS-PAGE and immunoblot analysis with rabbit polyclonal anti-Vn (R12-2413, Assay Biotech), anti-Ail (40), or anti-FLAG M2 (Sigma) antibodies.
Ligand-binding ELISA
Binding assays were performed as described (28, 40). Briefly, 96-well plates (Nunc) were preadsorbed with Vn-HX (5 μg/ml), Vn (5 μg/ml; Sigma), or gelatin (20 μg/ml; Sigma), then blocked with tris-buffered saline (TBS) containing 3% milk, and washed with TBS supplemented with 0.05% Tween 20. Recombinant C-terminal His-tagged Ail (Ail-His) was purified, refolded, and concentrated to 80 μg/ml in buffer with 4 mM decylphosphocholine and then added in decreasing concentrations to the preadsorbed wells. After incubating overnight at 4°C, bound Ail-His was detected with mouse anti-His monoclonal antibody (Qiagen), secondary goat anti-mouse antibody conjugated to horseradish peroxidase (Sigma), and the horseradish peroxidase substrate o-phenylenediamine (Pierce).
Supplementary Material
Acknowledgments
We thank J. Burns, P. Czabotar, S. K. Dutta, J. Gulbis, P. Jennings, S. Opella, E. Ruoslahti, C. Shimizu, C. Singh, J. Smith, and Y. Tian for discussion and assistance. Funding: This study was supported by grants from the NIH (GM118186 and AI130009) and a postdoctoral fellowship from Canadian Institutes of Health Research (to K.S.). It used the Cancer Center Structural Biology Resource supported by grant CA030199. Author contributions: K.S. developed protein preparation, crystallized proteins, and performed NMR experiments. B.C.L. crystallized proteins, acquired and analyzed the x-ray diffraction data, and built the models. L.M.F. developed protein preparation and performed binding assays. Y.Y. assisted with protein characterization and performed NMR experiments. S.S.B. and G.V.P. designed and performed bacterial co-sedimentation experiments. F.M.M. designed experiments and protein sequences, performed sequence and data analysis, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The structure coordinates have been deposited in the PDB (code: 6O5E). All data needed to evaluate the conclusions are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax5068/DC1
Fig. S1. Alignment of orthologous Vn sequences.
Fig. S2. Alignment of the HX domains of human Vn, rabbit hemopexin, and human matrix metalloproteases.
Fig. S3. HX repeat and intron/exon structures of human Vn, hemopexin N- and C-terminal domains, and matrix metalloproteases.
Fig. S4. Structure, B factors, and surface electrostatics of Vn-HX.
Fig. S5. Geometry of the ion-binding sites in the central channel of Vn-HX.
Fig. S6. Conserved prolines stabilize outward facing aromatic side chains.
REFERENCES AND NOTES
- 1.Preissner K. T., Reuning U., Vitronectin in vascular context: Facets of a multitalented matricellular protein. Semin. Thromb. Hemost. 37, 408–424 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Singh B., Su Y.-C., Riesbeck K., Vitronectin in bacterial pathogenesis: A host protein used in complement escape and cellular invasion. Mol. Microbiol. 78, 545–560 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Holmes R., Preparation from human serum of an alpha-one protein which induces the immediate growth of unadapted cells in vitro. J. Cell Biol. 32, 297–308 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kashyap A. S., Shooter G. K., Shokoohmand A., Govern J. M., Sivaramakrishnan M., Croll T. I., Cane G., Leavesley D. I., Söderberg O., Upton Z., Hollier B. G., Antagonists of IGF:Vitronectin interactions inhibit IGF-I-induced breast cancer cell functions. Mol. Cancer Ther. 15, 1602–1613 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Braam S. R., Zeinstra L., Litjens S., Ward-van Oostwaard D., van den Brink S., van Laake L., Lebrin F., Kats P., Hochstenbach R., Passier R., Sonnenberg A., Mummery C. L., Recombinant vitronectin is a functionally defined substrate that supports human embryonic stem cell self-renewal via alphavbeta5 integrin. Stem Cells 26, 2257–2265 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Conlan M. G., Tomasini B. R., Schultz R. L., Mosher D. F., Plasma vitronectin polymorphism in normal subjects and patients with disseminated intravascular coagulation. Blood 72, 185–190 (1988). [PubMed] [Google Scholar]
- 7.Kubota K., Katayama S., Matsuda M., Hayashi M., Three types of vitronectin in human blood. Cell Struct. Funct. 13, 123–128 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Suzuki S., Oldberg A., Hayman E. G., Pierschbacher M. D., Ruoslahti E., Complete amino acid sequence of human vitronectin deduced from cDNA. Similarity of cell attachment sites in vitronectin and fibronectin. EMBO J. 4, 2519–2524 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenne D., Stanley K. K., Molecular cloning of S-protein, a link between complement, coagulation and cell-substrate adhesion. EMBO J. 4, 3153–3157 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seiffert D., Ciambrone G., Wagner N. V., Binder B. R., Loskutoff D. J., The somatomedin B domain of vitronectin. Structural requirements for the binding and stabilization of active type 1 plasminogen activator inhibitor. J. Biol. Chem. 269, 2659–2666 (1994). [PubMed] [Google Scholar]
- 11.Pytela R., Pierschbacher M. D., Ruoslahti E., A 125/115-kDa cell surface receptor specific for vitronectin interacts with the arginine-glycine-aspartic acid adhesion sequence derived from fibronectin. Proc. Natl. Acad. Sci. U.S.A. 82, 5766–5770 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orlando R. A., Cheresh D. A., Arginine-glycine-aspartic acid binding leading to molecular stabilization between integrin alpha v beta 3 and its ligand. J. Biol. Chem. 266, 19543–19550 (1991). [PubMed] [Google Scholar]
- 13.Zhou A., Huntington J. A., Pannu N. S., Carrell R. W., Read R. J., How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat. Struct. Biol. 10, 541–544 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Faber H. R., Groom C. R., Baker H. M., Morgan W. T., Smith A., Baker E. N., 1.8 å crystal structure of the C-terminal domain of rabbit serum haemopexin. Structure 3, 551–559 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Libson A. M., Gittis A. G., Collier I. E., Marmer B. L., Goldberg G. I., Lattman E. E., Crystal structure of the haemopexin-like C-terminal domain of gelatinase A. Nat. Struct. Biol. 2, 938–942 (1995). [DOI] [PubMed] [Google Scholar]
- 16.Li J., Brick P., O’Hare M. C., Skarzynski T., Lloyd L. F., Curry V. A., Clark I. M., Bigg H. F., Hazleman B. L., Cawston T. E., Blow D. M., Structure of full-length porcine synovial collagenase reveals a C-terminal domain containing a calcium-linked, four-bladed beta-propeller. Structure 3, 541–549 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Hunt L. T., Barker W. C., Chen H. R., A domain structure common to hemopexin, vitronectin, interstitial collagenase, and a collagenase homolog. Protein Seq. Data Anal. 1, 21–26 (1987). [PubMed] [Google Scholar]
- 18.Jenne D., Stanley K. K., Nucleotide sequence and organization of the human S-protein gene: Repeating peptide motifs in the “pexin” family and a model for their evolution. Biochemistry 26, 6735–6742 (1987). [DOI] [PubMed] [Google Scholar]
- 19.Xu D., Baburaj K., Peterson C. B., Xu Y., Model for the three-dimensional structure of vitronectin: Predictions for the multi-domain protein from threading and docking. Proteins 44, 312–320 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Lynn G. W., Heller W. T., Mayasundari A., Minor K. H., Peterson C. B., A model for the three-dimensional structure of human plasma vitronectin from small-angle scattering measurements. Biochemistry 44, 565–574 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Springer T. A., An extracellular beta-propeller module predicted in lipoprotein and scavenger receptors, tyrosine kinases, epidermal growth factor precursor, and extracellular matrix components. J. Mol. Biol. 283, 837–862 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Xiao T., Takagi J., Coller B. S., Wang J.-H., Springer T. A., Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59–67 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng H., Cooper D. R., Porebski P. J., Shabalin I. G., Handing K. B., Minor W., CheckMyMetal: A macromolecular metal-binding validation tool. Acta Crystallogr. D Struct. Biol. 73, 223–233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skorstengaard K., Halkier T., Hojrup P., Mosher D., Sequence location of a putative transglutaminase cross-linking site in human vitronectin. FEBS Lett. 262, 269–274 (1990). [DOI] [PubMed] [Google Scholar]
- 25.Bowley S. R., Fang C., Merrill-Skoloff G., Furie B. C., Furie B., Protein disulfide isomerase secretion following vascular injury initiates a regulatory pathway for thrombus formation. Nat. Commun. 8, 14151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tollefsen D. M., Weigel C. J., Kabeer M. H., The presence of methionine or threonine at position 381 in vitronectin is correlated with proteolytic cleavage at arginine 379. J. Biol. Chem. 265, 9778–9781 (1990). [PubMed] [Google Scholar]
- 27.Huang J.-S., Lin C.-M., Cheng Y.-C., Hung K.-L., Chien C.-C., Chen S.-K., Chang C.-J., Chen C.-W., Huang C.-J., A vitronectin M381T polymorphism increases risk of hemangioblastoma in patients with VHL gene defect. J. Mol. Med. 87, 613–622 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Bartra S. S., Ding Y., Fujimoto L. M., Ring J. G., Jain V., Ram S., Marassi F. M., Plano G. V., Yersinia pestis uses the Ail outer membrane protein to recruit vitronectin. Microbiology 161, 2174–2183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamashita S., Lukacik P., Barnard T. J., Noinaj N., Felek S., Tsang T. M., Krukonis E. S., Hinnebusch B. J., Buchanan S. K., Structural insights into Ail-mediated adhesion in Yersinia pestis. Structure 19, 1672–1682 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clamp M., Cuff J., Searle S. M., Barton G. J., The Jalview Java alignment editor. Bioinformatics 20, 426–427 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Thompson J. D., Higgins D. G., Gibson T. J., CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linding R., Jensen L. J., Diella F., Bork P., Gibson T. J., Russell R. B., Protein disorder prediction: Implications for structural proteomics. Structure 11, 1453–1459 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., Coy A. M., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., Coy A. J. M., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bunkoczi G., Read R. J., Improvement of molecular-replacement models with Sculptor. Acta Crystallogr. D Biol. Crystallogr. 67, 303–312 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A., Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding Y., Fujimoto L. M., Yao Y., Plano G. V., Marassi F. M., Influence of the lipid membrane environment on structure and activity of the outer membrane protein Ail from Yersinia pestis. Biochim. Biophys. Acta 1848, 712–720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax5068/DC1
Fig. S1. Alignment of orthologous Vn sequences.
Fig. S2. Alignment of the HX domains of human Vn, rabbit hemopexin, and human matrix metalloproteases.
Fig. S3. HX repeat and intron/exon structures of human Vn, hemopexin N- and C-terminal domains, and matrix metalloproteases.
Fig. S4. Structure, B factors, and surface electrostatics of Vn-HX.
Fig. S5. Geometry of the ion-binding sites in the central channel of Vn-HX.
Fig. S6. Conserved prolines stabilize outward facing aromatic side chains.