

Abstract
Polymers that are refillable and sustain local release will have a great impact in both preventing and treating local cancer recurrence as well as addressing non-resectable disease. Polymerized cyclodextrin (pCD) disks, which reload drug into molecular “pockets” in vivo using affinity interactions, have previously shown to localize doxorubicin (Dox) to treat glioblastoma multiforme. However, one concern is whether drug refilling is influenced by competition from local biomolecules. In addition the impact of polymer form on drug refilling is unknown. Herein, different pCD formulations are synthesized from γ-cyclodextrin (γ-CD) and are compared in vitro using competitive drug filling/refilling assays. Data reveals that affinity-based drug refilling occurs as a function of both polymer form and sustained release polymeric liquid (SRPL) dilution factor, pointing to surface/volume ratio, as well as CD pocket density, and distance between pocket effects. In vitro refilling experiments with cholesterol demonstrated no interference with Dox filling of the CD polymer, while albumin presence only slightly reduced Dox filling of pCD-γ-MP (microparticle) and pCD-γ-SRPL forms, but not pCD-γ-disks. Moreover, whole serum competition did not inhibit filling or refilling of pCD-γ-MP with Dox at multiple concentrations and filling times, which indicates that this polymer (re)filling is primarily driven by affinity-based interactions that can overcome physiological conditions that may limit other drug delivery approaches. This was supplemented by isolating variables through docking simulations and affinity measurements. These results attest to the efficiency of in vivo or in situ polymer filling/refilling in the presence of competitive biological molecules achieved partially through high affinity drug to polymer interactions.
Graphical Abstract
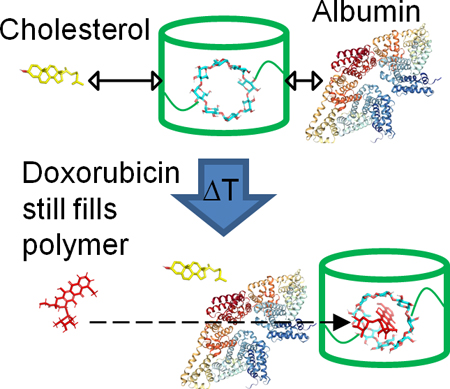
Competition of albumin and cholesterol which may bind cyclodextrin polymers is explored for effect on drug filling efficiency.
1. Introduction
Doxorubicin (Dox; trade name Adriamycin®) treatment has demonstrated strong anti-tumor effects in combination with chemotherapy or other anti-neoplastic agents for breast cancer, AIDS-related Kaposi’s sarcoma, ovarian cancer, multiple myeloma, bladder cancer, advanced gastric cancer, and small cell lung cancer.1–4 Dox is an highly effective anti-cancer drug which acts through intercalating DNA, ROS generation, and disruption of topoisomerase-II-mediated DNA repair.5 However, the conventional use of Dox as well as liposomal Dox and pegylated liposomal Dox still pose significant adverse effects including cardiac, hematological, hepatic, and dermal toxicities associated with treatment in clinical trials.6–8 In a retrospective multi-center analysis, Dox-related congestive heart failure was estimated to occur in 48% of patients at a cumulative dose of 700 mg/m2 following treatments for small cell lung carcinoma and breast carcinoma.9 Accordingly, the clinical use of Dox has been restricted due to dose-dependent cardiotoxicity and even greater restrictions are applied with the presentation of risk factors including congestive heart failure, hypertension, age, and co-administration with other cardiotoxic drugs.6, 10
As a solution to limit off-target toxicity, while maintaining delivery of an effective Dox concentration within the tumor, a local delivery strategy has been developed using polymerized cyclodextrin (pCD).11, 12 A high concentration of γ-cyclodextrin (γ-CD) in the synthesized polymer disk enables drug/cyclodextrin inclusion complexes to form repeatedly, which allows for higher drug filling and mediates a longer release time on the order of weeks to months over traditional diffusion or degradation based drug release mechanisms.13–15 The inclusion with pCD can even preserve drug bioactivity16 which effectively lengthens the therapeutic window to potentially eliminate slowly dividing cancer stem cells. The combination of extended drug release at effective concentrations with preserved bioactivity due to affinity interactions makes this technology an attractive anti-cancer drug delivery platform. Moreover, localizing the delivery to the tumor tissue directly results in less drug in circulation that may cause cardiotoxicity or hepatic toxicity.17, 18 Yet, the pCD-disk is limited to situations and locations amenable to disk implantation, such as post-resection therapy.
Recently, the pCD platform has been adapted to other formats for antibiotic and joint supplement applications, namely as pCD-microparticle (pCD-MP) and pCD-sustained release polymeric liquid (pCD-SRPL) forms.19–21 However, the question remains ‘How does a polymer’s form influence drug refilling?’ While each polymer form maintains its own unique advantages, such as injectability and viscosity, herein drug filling efficiency is directly compared across polymer forms with an anti-cancer therapy. We test the improvement in drug filling afforded by each polymer form by affinity interactions with Dox as well as the effect of MP and SRPL dilution factors on drug filling efficiency. This characterization will inform future approaches for which affinity-based release has the unique properties to better treat disseminated disease, dense tumors with heterogeneous vasculatures, and tissues which are conventionally difficult to access.
Moreover, affinity-based CD polymers maintain a distinct advantage over traditional drug delivery platforms as they are capable of drug refilling post-implantation in vivo.11, 20, 22 The ability to refill CD polymers with drug multiple times in vivo has potential to provide multiple delivery windows, which could be utilized for chemotherapy treatments to especially address cancer recurrence. Glioblastoma multiforme is an invasive brain cancer with a 3-year survival rate of <10% and a recurrence rate of 80–90%.23, 24 Implanted pCD-disks have demonstrated proof-of-concept for refilling with Dox to treat glioblastoma multiforme in animal models at later time points via local injection of bolus drug resulting in another extended drug release window.11 However, the effect of local biomolecules competing for polymer refilling over time has not been tested. It is hypothesized that the if the CD pockets are obstructed by circulating biological molecules over time, then a secondary injection of free drug will be prevented from efficiently refilling into the polymer and giving another window of extended local release. Albumin and cholesterol are of significant concern due to their ubiquitous presence in the body, cholesterol’s pretence for sequestering CD, and albumin’s nature as a hydrophobic transporter as seen in previous studies with retinoids, Warfarin, and Naproxen.25–29 Docking simulations for binding of cholesterol and albumin to γ-CD as well as experimental affinity measurements between cholesterol and albumin relative to Dox using surface plasmon resonance (SPR) are performed to explore how drug-CD affinity interactions may impact refilling in the presence of local biomolecules.
2. Materials and Methods
2.1. Materials
γ-cyclodextrin (γ-CD) prepolymer, lightly crosslinked with epichlorohydrin, and octakis (6-deoxy-6-amino) γ-cyclodextrin octahydrochloride were purchased from CycloLab (Budapest, Hungary). Ethylene glycol diglycidyl ether was purchased from Polysciences, Inc. (Warrington, PA). Dextran (15–25k molecular weight), hexamethylene diisocyanate, and cholesterol were purchased from Sigma-Aldrich (St. Louis, MO). Doxorubicin hydrochloride, agarose, bovine serum albumin and all other reagents, solvents, and chemicals were purchased from Thermo Fisher Scientific (Waltham, MA).
2.1. Disk Synthesis
To make polymer disks, vacuum dried epichlorohydrin-crosslinked γ-CD prepolymer (or dextran for low-affinity control) was dissolved in dimethylsulfoxide at a 25% w/v solution and heated. 1,6-hexamethylene diisocyanate was added and the solution vortexed for 2 minutes. The solution was poured into a Teflon dish and allowed to crosslink until solidified. The polymer film was then cut into disks using an 8mm circular biopsy punch and the resulting disks were washed in sequence with excess dimethylsulfoxide for one day, 50/50 dimethylsulfoxide and deionized water the next day, then deionized water alone for 3 days before drying.
2.2. Microparticle Synthesis
Microparticles formulation started with epichlorohydrin-crosslinked γ-CD prepolymer (or dextran for low-affinity control) solubilized in 0.2 M potassium hydroxide (25% w/v) and heated to 60°C for 10 minutes. Light mineral oil in a beaker was warmed with a Tween85/Span85 solution (24%/76%) and mixed on a stir plate. Next, ethylene glycol diglycidyl ether was added drop-wise and the solution was vortexed before pouring into the oil/Span/Tween85 mixture, increasing the temperature to 70°C. The polymerized cyclodextrin microparticles were formed after 3 hours. The microparticles were then centrifuged from the oil mixture, washed with excess hexanes twice and deionized water twice. The microparticles were resuspended, frozen, and lyophilized to complete dryness before further use.
2.3. Sustained Release Polymeric Liquid Synthesis
A fluidic cyclodextrin polymer formulation was made using the method designed by Rivera-Delgado, et al., 2018 21. Epichlorohydrin-crosslinked γ-CD prepolymer (or dextran for low-affinity control) that was first dried for at least 4 hours in a vacuum oven at 70°C in a 20 ml scintillation vial. After drying, dimethylsulfoxide was added to the vial, the solution was capped with nitrogen, and allowed to dissolve while mixing with a stir bar. The vial was then immersed in a preheated light mineral oil bath at 55°C until equilibrated while continuously stirring. 1,6-hexamethylene diisocyanate was added as a crosslinker, the vial was recapped with excess nitrogen gas, and tightly sealed before returning to the oil bath with continuous stirring. Upon formation of a viscous gel solution, the reaction was stopped by the addition of excess deionized water, vortexed, and placed on a rotator overnight. Then the polymer solution was placed in a 6,500 Dalton molecular weight cutoff dialysis bag and dialyzed against excess deionized water for 3 days. The polymer was then frozen to −80°C and lyophilized to dryness before use in further experiments.
2.4. Polymer Imaging and FTIR Analyses
Polymers were imaged with a 12 megapixel camera (Samsung, Seoul, South Korea) in a saline solution to show bulk form and characteristics. An AmScope stereomicroscope with a 0.5x lens and LED light source (United Scope LLC, Irvine, CA) was used to show dried polymers at the micro scale. Infrared (IR) spectroscopy was performed using an Excalibur FTS 3000 Fourier-Transform Infrared (FTIR) Spectrophotometer (BioRad, Hercules, CA, USA). The samples were prepared by mortar and pestle grinding dried samples with potassium bromide (KBr) powder and then compressing the mixtures to form scanning samples in transmission mode. Scans were run from 4,000 to 600cm-1.
2.5. Agarose Gel Drug Filling Assays
Polymer filling was determined by adapting a previously validated model for determining antibiotic refilling of polymer disks in tissue-like conditions [11]. Each well of a 6 well polystyrene plate was filled with 3 ml of a warm 0.3% w/v agarose solution and allowed to solidify. Next, 3 equally sized pockets were resected from the agarose and filled with either pCD-γ-disk, pCD-γ-MP, or pCD-γ-SRPL in phosphate-buffered saline (PBS), controlled by equal weight of polymer per pocket (20 mg each). More warm agarose solution was poured over the wells to maintain the pCD forms in their individual locations. A 6 mm biopsy punch was used to create a consistent hole in the middle of each well and Dox solution (2 mM in PBS with 0.1% DMSO) was pipetted into the middle at time zero. The absorbance of Dox into each polymer was determined by area scan with a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek Instruments, Inc., Winooski, VT) at each indicated time point. Absorbance change due to drug diffusion alone was measured in an area of the well without a pocket. Results were calculated by subtraction of initial baseline absorbance of polymers and then normalized to an area averaged diffusion-only measurement giving fold improvement in drug filling over standard diffusion. For experiments comparing γ-CD and dextran or different polymer concentrations, the gels were constructed using the same method as above, but with only two polymer pockets.
2.6. Molecular Docking Simulations
Molecular structure data files for Dox (CID: 31703), cholesterol (CID: 5997), bovine serum albumin (CID: 4F5S) and γ-CD (CID: 5287407) were downloaded from the PubChem database. Structures were converted to PDBQT format. γ-CD was used as a host for Dox and cholesterol docking in PyRx (Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA), while albumin was simulated as the host when paired with γ-CD to account for its larger structure,. The Autodock Vina algorithm was used to predict the strength of the interaction 30, 31.
2.7. Surface Plasmon Resonance
The binding strength between γ-CD monomer with Dox, cholesterol, and albumin was measured experimentally through surface plasmon resonance (SPR) with a Biacore X100 system (GE Healthcare Bio-Sciences, Pittsburgh, PA). Conditions used were based upon previous optimization for small molecule drugs binding to cyclodextrins 32, 33. The surface of a sensor chip CM-3 was conjugated with EDC (0.4 M) and NHS (0.1 M) followed by 10 mM 6-amino-6-deoxy γ-cyclodextrin (CycloLab) suspended in HBS-N buffer (a HEPES balanced salt solution with pH 7.4). The remaining functional groups were capped with ethanolamine. A multi-cycle kinetic experiment was performed separately with the following solutions and running buffers: Dox in diH2O, albumin in diH2O, and cholesterol in 10% ethanol and diH2O. The surface was regenerated with 50 mM sodium hydroxide between samples to fully dissociate any remaining bound analyte. The differential responses between the channels were fit to both steady state affinity and a 1:1 kinetics binding model using Biacore evaluation software. Goodness of fit was verified by U-value <25 as specified in the manufacturer’s instructions.
2.8. Albumin and Cholesterol Blocking pCD from Dox Refilling
CD polymer (20 mg) was mixed with physiological concentrations of either bovine serum albumin (29.2 mg/ml 27 in PBS and has a similar tertiary structure to human serum albumin 34), cholesterol (0.4215 mg/ml 35 in PBS with 10% ethanol), or solvent control (PBS with 10% ethanol) and incubated for 72 hours at 37°C. This solution was removed and Dox (1 mg/ml in PBS) was added to simulate a delayed local drug injection for filling of polymers in the presence of potentially inhibitory endogenous molecules (albumin and cholesterol). After 24 hours, the Dox solution was removed, polymers were washed to remove unbound Dox, and the remaining loaded Dox was leeched into DMSO over 3 weeks with 6 solvent exchanges. After determining any individual effects from these highly competitive and abundant molecules, full concentration human serum (MP Biomedicals, Santa Ana, CA) was tested in separate experiments. Drug filling and refilling at lower Dox concentrations (18.4 μM, 9.2 μM) and with shorter Dox incubation times (1 hour, 30 minutes) to demonstrate clinical versatility. The total amount of Dox leeched from each polymer was determined with a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek Instruments, Inc.) and normalized to the total amount recovered from solvent controls.
3. Results
3.1. Synthesizing Multiple Forms of Polymerized Cyclodextrin
Three unique polymer forms were synthesized from the same base γ-CD pre-polymer by different sets of optimized procedures and are herein compared. The pCD-γ-disks were synthesized by cross-linking Y-CD into a film, evaporating the solvent away, and using a biopsy punch to create custom sized disks for drug delivery applications (Figure 1A). The CD disk platform enables affinity-based drug delivery,22, 36, 37 but is not injectable via syringe and therefore limited in application.
Figure 1.
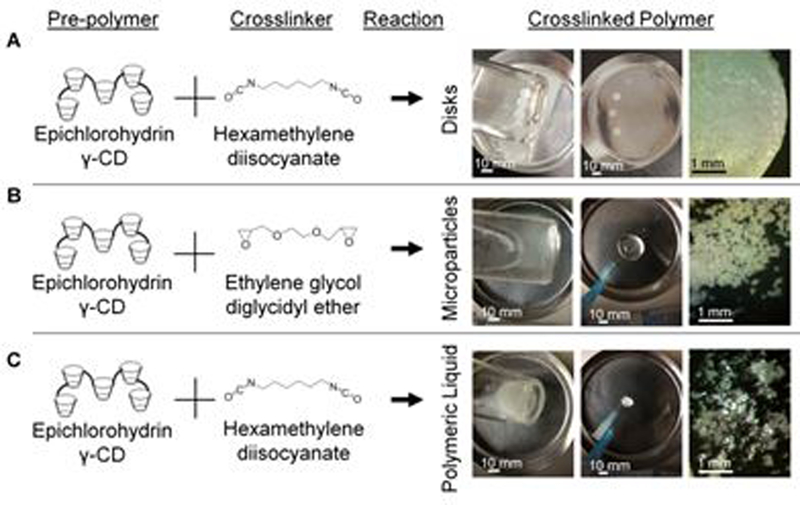
CD prepolymer is capable of crosslinking into various forms including (A) disks, (B) microparticles, and (C) polymeric liquid. Scale bars are applicable to all images per column.
To expand the potential clinical applications for CD affinity-based drug delivery, pCD-γ-MPs were synthesized (Figure 1B) using a water-in-oil suspension in which γ-CD pre-polymer was dissolved in potassium hydroxide, mixed with a bi-functional crosslinker, and then added to a spinning mixture of Tween85, Span85, and light mineral oil. The average particle diameter achieved can be tailored from 15 to 800 microns,16, 19 by varying the stirring speed among other parameters. The pCD-γ-MPs are insoluble and are non-degradable for over 30 days.16
Maintaining the same base γ-CD pre-polymer in non-aqueous solvent and stopping the reaction at its solution-gel transition point forms a CD polymer that is a fluid at room temperature, maintains a fluid form after injection, yet sustains drug release similar to solid polymers (Figure 1C).21 This shear thinning polymer can be useful for injectable drug delivery but also provides tissue bulking as well as other physical properties.
The chemical structures of each polymer form was characterized by FTIR (Supplemental Figure 1). The representative bonds were similar among each polymer with γ-CD incorporation (3,600–3,000 cm−1 (v(O-H), 2,928 cm−1 (v(C-H)); and polymer crosslinking (1,701 cm−1 (v(C=O), 1,541 cm−1 (δ(N-H)) evident.
3.2. Comparing Doxorubicin Filling Efficiency Among Cyclodextrin Polymer Forms
A competitive assay developed previously for determining antibiotic refilling by local injection near affinity-based polymers in an agarose gel simulating tissue-like conditions was used to compare Dox filling into the three polymer forms.22 The polymers were placed into individual pockets to maintain spatial separation, yet compete for binding of the free Dox diffusing through the agarose gel (Figure 2A). Total filling into polymers was compared to the free drug in the agarose gel as measured by absorbance change over time at 510 nm. The pCD-γ-MP loaded Dox 5.7-fold above diffusion levels, while the pCD-γ-SRPL and pCD-γ-disks loaded an average of 2.6- and 2.1-fold above background drug diffusion (Figure 2B). Overall, drug sequestering into all polymer forms was significantly greater than Dox diffusion into the gel alone.
Figure 2.
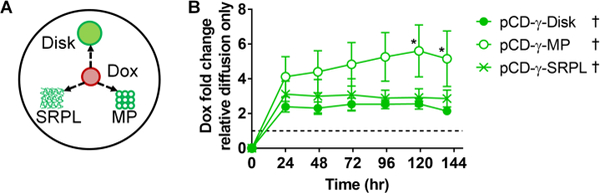
(A) Dox was injected near different polymer forms in an agarose gel and (B) the change in absorbance relative to drug diffusion into the gel was measured longitudinally. n=4 per group and bars represent standard error of the mean. * indicates significant difference by two-way ANOVA with Tukey’s test post-hoc versus disk group at respective time points. † indicates significance (p<0.05) for all time points compared to Dox diffusion only (dotted line at x=1) by Wilcoxon signed-rank test.
3.3. CD Inclusion Increases Dox Filling into Polymers
The competitive gel assay (Figure 3A) was further utilized to determine the effect on Dox filling efficiency due to CD incorporation in polymers versus chemically similar, low-affinity dextran polymers. By comparing changes in absorbance, Dox loaded more rapidly into pCD-γ-disks than dextran disks with ~2-fold increased filling in the first 1.5 hours after drug application, but over time equilibrated to a similar level signal as dextran disks and diffusion background (Figure 3B). However, peak filling ratio in pCD-γ-MP and pCD-γ-SRPL occurred at 24 hours indicating a difference in filling rate due to the combined effects of polymer form in our in vitro model (Figure 3B).
Figure 3.
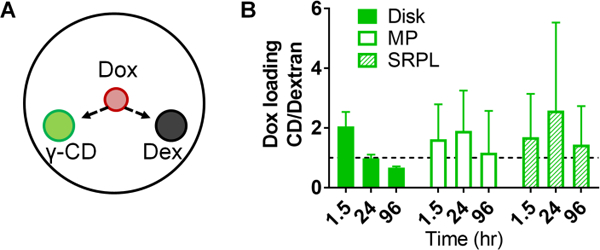
(A) Effects of affinity on Dox filling was determined by comparing (B) the relative change in absorbance of γ-CD polymers greater than one indicates greater filling over low-affinity dextran control polymers of the same form. n=4 per group, bars represent standard error of the mean, and no significant difference was found between groups by two-way ANOVA with Tukey’s test post-hoc.
3.4. Polymer Concentration Critical for Sustained Release Polymeric Liquid But Not Microparticle Drug Filling
In another application of the agarose gel drug filling assay, different dilutions of either pCD-γ-MP or pCD-γ-SRPL were injected to observe competition for drug filling based upon local CD polymer concentration. The pCD-γ-MP at 100 mg/ml demonstrated a trend for slightly greater Dox filling than the pocket with 1 mg/ml pCD-γ-MP, yet the lower concentration of polymer was sufficient to result in filling at least 2-fold Dox greater than diffusion alone (Figure 4A). Conversely, the pCD-γ-SRPL was only able to achieve enhanced affinity-based sequestering of Dox at the 100 mg/ml concentration, but was not able to localize drug at 1 mg/ml (Figure 4B). The CD is not excluding Dox, but rather the negative result is indicative of polymer dissociation into the surrounding gel due in part to the plate shaking during the experiment and the low concentration SRPL being less cohesive. The 1 mg/ml SRPL dissociation results in a decreasing signal as polymer leaves the 1 mg/ml area (resulting in absorbance decreasing below the baseline value), and results in a negative fold change (Supplemental Figure 2). When comparing the max filling for each concentration and polymer form, the 1 mg/ml concentration of pCD-γ-SRPL is unable to localize drug while the 1 mg/ml concentration of pCD-γ-MP loads almost 5 times more Dox relative to the background diffusion signal (Figure 4C). At 100 mg/ml the pCD-γ-SRPL loads almost 2.5 times more Dox and pCD-γ-MP loads 5.5 times more Dox as compared to background (Figure 4C). The compact nature of the pCD-γ-MP and its close, rigid network of CD molecules is hypothesized to lend to the enhanced filling and concentration-independence over the pCD-γ-SRPL form.
Figure 4.
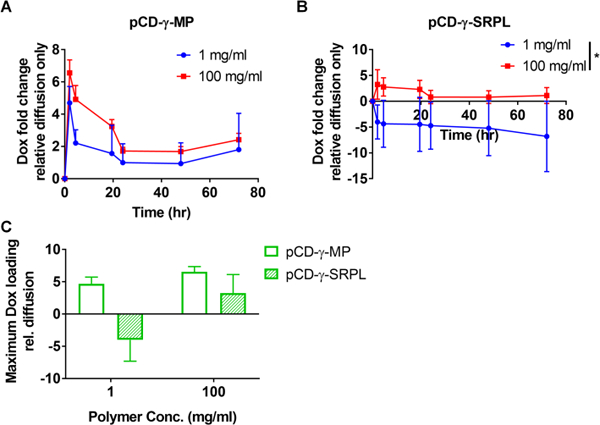
Effect of local concentration of (A) pCD-γ-MP and (B) pCD-γ-SRPL on Dox filling efficiency was compared in agarose gels and measured via absorbance. (C) Comparison between particle and SRPL maximum drug filling capacities indicate SRPL localizing of drug is concentration dependent. n=2 per group; bars represent standard error of the mean. * indicates significance by Student’s t-test between group averages.
3.5. Effects of Cholesterol and Albumin on Dox Refilling of CD Polymers In Vitro
One other concern is whether drug refilling is influenced by competition from local biomolecules. Previous studies indicated the potential for albumin and cholesterol to interfere with Dox refilling into the CD polymers.25, 26, 28 A pre-incubation assay was setup to quantify ability of pCD forms to reload drug in presence of local biomolecules in vitro. Briefly, after pre-incubation with either cholesterol or albumin at physiological levels,27, 35 Dox was then introduced to the polymers for 24 hours before washing and subsequent drug leeching measurements from the polymers. Results indicated that pCD-γ-disks maintained the same Dox filling efficiency despite pre-incubation with albumin or cholesterol (Figure 5A). For the pCD-γ-MP we found that cholesterol did not adversely affect subsequent Dox filling and even with albumin pre-incubation Dox filling was 91% of controls (Figure 5B). The only substantial effect was observed for the pCD-γ-SRPL polymer form in which albumin reduced Dox filling to 77% of controls but cholesterol did not interfere (Figure 5C). While albumin and cholesterol were of primary concern due to their abundance in interstitial tissue where CD polymers would potentially be implanted, we also tested the ability of CD polymers to refill in the presence of whole human serum as well. Results indicated that pCD-γ-MP co-incubated with serum achieved 97% of the Dox filling relative to control polymers using Dox concentrations of 10 ug/ml (Figure 5D). Following drug leeching, the same polymer was refilled for 30 minutes using 5 ug/ml Dox co-incubated with whole human serum and demonstrated no inhibition of refilling efficiency compared to controls (Figure 5D).
Figure 5.
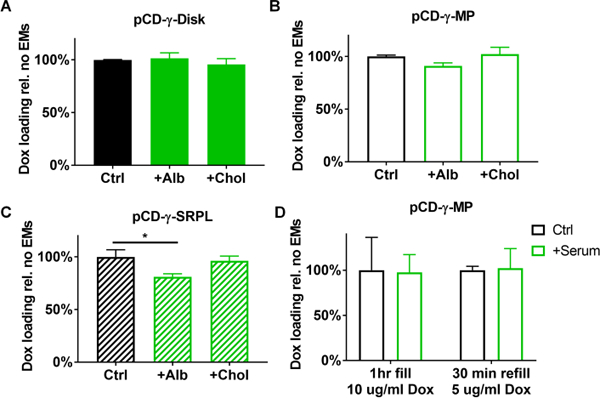
Impact of pre-incubation with cholesterol and albumin on Dox filling in (A) pCD-γ-disks, (B) pCD-γ-MP, and (C) pCD-γ-SRPL was determined from total drug leeched relative to solvent controls. (D) Full concentration human serum was co-incubated with pCD-γ-MP and Dox for polymer filling and later refilling at lower concentrations and shorter times versus solvent only controls. * indicates significance by one-way ANOVA with Tukey’s test post-hoc; n=3–6 per group and bars represent standard error of the mean.
3.6. Predicting Affinity of Doxorubicin, Albumin, and Cholesterol for γ-CD
To further test if affinity interactions could result in drug refilling in the presence of local biomolecules, molecular docking was performed in silico to predict the relative affinity of each molecule for γ-CD. Smaller affinity (KD) values indicate a slower dissociation and, consequently, a longer binding time between the indicated molecule and γ-CD. Dox, albumin, and cholesterol binding to γ-CD was simulated with Autodock Vina software with binding energy minimization (Figure 6A). The strongest simulated conformations are shown and results indicate the strongest average affinity (lower KD) for Dox (17.8 μM) versus albumin (67.1 μM) or cholesterol (173 μM) for γ-CD (Figure 6B). However, from the range of affinities predicted for albumin and cholesterol it is hypothesized that free Dox would eventually replace these endogenous molecules binding to γ-CD.
Figure 6.
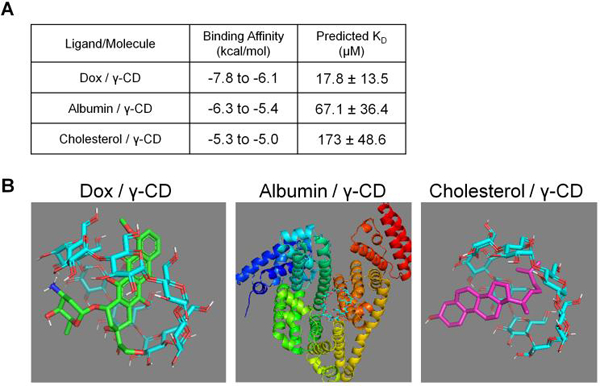
(A) PyRx binding simulation results of Dox, albumin, and cholesterol each individually binding with γ-CD. (B) Molecular models representing the highest affinity conformations for each simulated ligand/molecule pair in PyRx with Vina Autodock software. Note the scale is different and biological assemblies were simulated for Albumin / γ-CD.
3.7. Experimental Determination of Doxorubicin, Albumin, and Cholesterol Affinity for γ-CD
For final verification, the binding of Dox, albumin, and cholesterol to γ-CD was measured directly using SPR. The interactions between each individual flowing molecule at a range of concentrations and surface bound 6-amino-6-deoxy-γ-CD was determined with a Langmuir 1:1 kinetics model (Figure 7A).38 Reported KD values were within the model confidence interval.33 Results indicated that Dox maintained the strongest affinity (0.409 μM), while albumin (24.0 μM) and cholesterol (372 μM) demonstrated much weaker binding to the surface modified γ-CD (Figure 7B). While there are some differences between the estimated simulations and experimental data, the trends indicate that albumin has a higher affinity for γ-CD than cholesterol, but both molecules have a lower affinity than the Dox-γ-CD complex.
Figure 7.
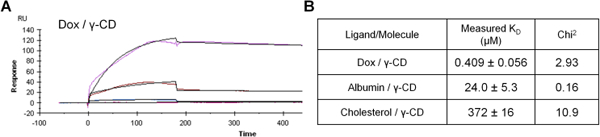
(A) Example binding response of flowing Dox binding to 6-amino-6-deoxy γ-cyclodextrin measured by SPR. (B) Results of Dox, albumin, and cholesterol binding with γ-CD.
Discussion
In this work, synthesis methods have been adapted for forming polymer disks, polymeric microparticles, and a sustained release polymeric liquid with γ-CD pre-polymer conserved as the fundamental structural unit (Figure 1). For minimally invasive procedures, the injectability of both pCD-γ-MP and pCD-γ-SRPL was demonstrated (Figure 1). While the underlying chemical structures of each polymer is the same, the degree of crosslinking is inherently different, creating highly cross-linked CD disks, densely packed CD microparticles and loosely associated SRPL, with different surface/volume ratios, as well as CD pocket densities, and distance between pockets, which manifest as differences in drug filling of these affinity-based polymers (Figures 2, 4, and 5). In quantifying functionality, in vitro competitive drug filling assays in agarose gels revealed that pCD-γ-MP achieved the highest Dox filling versus the pCD-γ-disk and pCD-γ-SRPL, but all CD polymer forms loaded at least twice as much drug as in the media surrounding the polymers (Figure 2). These results demonstrate that CD inclusion is capable of improving the filling capacity of multiple different polymer forms (disks, particles, polymeric liquid) to various degrees.
Affinity-based polymers demonstrate unique trends different than those which generally govern drug delivery for other polymers. For example, swelling rate has little effect on affinity based drug delivery as dissociation rates are the primary factor of drug release from CD polymers 12, 39. Additionally, affinity-based filling effects for drugs of interest may be improved through addition of functional groups that could provide higher CD affinity 40, 41 and molecular imprinting during polymer synthesis,42 although these concepts have yet to be demonstrated for MP and SRPL polymer forms. Dextran was used as a low-affinity control polymer and acted as a baseline to show the improvement in filling in a time-dependent manner under the same assay conditions versus γ-CD inclusion (Figure 3).
Our results also address the importance of considering polymer dilutions for clinical injection efficacy. An improvement in Dox filling capacity and localization was observed to be concentration-dependent for pCD-γ-SRPL (Figure 4), which was similar to a previous finding regarding affinity-based corticosteroid filling and release.21 However, a 100 fold difference in pCD-γ-MP concentration exhibited only a slight effect on Dox filling, possibly afforded by the compact spherical structure which increases the internal CD density as opposed to the loose chains of the SRPL (Figure 4). Because affinity-based drug filling and release is proposed to occur as a function of local CD density, caution to polymer dilution, surface area/volume ratio, and distance between pockets needs to be considered when developing and testing new affinity-based polymer platforms in the future.
Whole serum incubation studies were performed to further demonstrate the robustness of affinity-based CD polymers to load drug in physiological scenarios. Co-incubation of serum biomolecules while filling Dox for only 1 hour or refilling for 30 minutes resulted in no difference in affinity-based polymer drug loading (Figure 5D). Dox was filled into polymers with equal efficiency to controls regardless of drug concentration (Figure 5D). Filling versus refilling exhibited equivalent results as well (Figure 5D). Cellular toxicity of Dox released from our CD polymers and anti-tumor therapeutic efficacy including reduction of tumor growth and increasing overall survival has been demonstrated in a previous publication using cell culture and in vivo tumor xenograft models 11. Results indicate that even the lowest amount of Dox filled herein will have a therapeutic effect (Figure 5D).
Cholesterol was found to not block Dox filling of γ-CD polymers (Figure 5), likely due to the lower affinity of cholesterol for γ-CD relative to Dox for γ-CD (Figure 6 and 7). There was a slight decrease in Dox filling due to albumin for pCD-γ-MP and an even greater reduction for pCD-γ-SRPL (Figure 5), which may be due not only to albumin’s affinity for CD but also contributions from steric hindrance as the SRPL form has more surface area exposed than the MP form. Alternatively, other studies demonstrate a moderate affinity of Dox for albumin (although only in the 7–11 mM range), which may compete for the free drug versus the CD polymer and partially explain our results.43 Future studies should elucidate if the increased surface area of CD device coatings 44 results in similar blocking of refilling in solutions with common biomolecules, but recent surprising results indicate that drug-polymer refilling can still occur through bacterial biofilms.22 Also, how changes in local protein and molecular concentrations due to aging or pathological conditions, such as cancer or multiple sclerosis, may influence affinity-based drug refilling of polymers is yet to be determined.45–48
Molecular simulations are a useful tool for estimating affinity interactions to determine mechanisms. Herein, we leveraged docking simulations to show that albumin has a higher affinity for γ-CD than cholesterol, but that Dox still bound more strongly to γ-CD than either endogenous molecule (Figure 6). Results were confirmed experimentally with SPR (Figure 7). These data imply the potential effectiveness for in situ or in vivo polymer filling in the presence of biological molecules and sets precedence as a method that can be applied to other affinity-based systems.38
Conclusions
Herein, we developed three different polymeric forms from the same base cyclodextrin pre-polymer and found differences in polymer filling/refilling of the anti-cancer drug, Dox, over time. Results indicated that affinity-based filling was higher for the microparticles, yet all polymer forms maintained increased filling versus low-affinity control polymers and drug diffusion alone. Dilution-dependent Dox filling was observed for SRPL, but not microparticles. Affinity simulations and in vitro experiments isolated the effect of albumin and cholesterol on affinity-based filling of CD polymers, and indicated that drug refilling of pCD is not significantly affected by albumin and cholesterol, thus making in vivo CD implants more attractive for future clinical investigation.
Supplementary Material
Acknowledgements
The authors would like to acknowledge support from NIH T32DK083251–09 (NAR), R01GM12147–03 (HvR), and from an NIH Research Facilities Construction Grant (C06 RR12463–01). The authors would also like to acknowledge Gilmour Academy and Dr. Neena Goel for enabling Olivia Robida’s contributions through the Catalyst externship program.
Footnotes
Conflicts of interest
H.A. von Recum is a co-founder of Affinity Therapeutics but does not receive salary. The other authors have nothing to disclose.
References
- 1.Duggan ST and Keating GM, Drugs, 2011, 71, 2531–2558. [DOI] [PubMed] [Google Scholar]
- 2.Dieras V, Oncology (Williston Park, N.Y.), 1997, 11, 31–33. [PubMed] [Google Scholar]
- 3.Udhrain A, Skubitz KM and Northfelt DW, International journal of nanomedicine, 2007, 2, 345–352. [PMC free article] [PubMed] [Google Scholar]
- 4.Pisano C, Cecere SC, Di Napoli M, Cavaliere C, Tambaro R, Facchini G, Scaffa C, Losito S, Pizzolorusso A and Pignata S, Journal of drug delivery, 2013, 2013, 898146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE and Altman RB, Pharmacogenetics and genomics, 2011, 21, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shafei A, El-Bakly W, Sobhy A, Wagdy O, Reda A, Aboelenin O, Marzouk A, El Habak K, Mostafa R, Ali MA and Ellithy M, Biomedicine & Pharmacotherapy, 2017, 95, 1209–1218. [DOI] [PubMed] [Google Scholar]
- 7.Volkova M and Russell R 3rd, Current cardiology reviews, 2011, 7, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.August DA, Verma N, Vaertan MA, Shah R and Brenner DE, British journal of cancer, 1995, 72, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swain SM, Whaley FS and Ewer MS, Cancer, 2003, 97, 2869–2879. [DOI] [PubMed] [Google Scholar]
- 10.Von Hoff DD, Layard MW, Basa P, Davis HL Jr., Von Hoff AL, Rozencweig M and Muggia FM, Annals of Internal Medicine, 1979, 91, 710–717. [DOI] [PubMed] [Google Scholar]
- 11.Fu AS, von Recum H, Thesis Dissertation CASE WESTERN RESERVE UNIVERSITY, 2013. [Google Scholar]
- 12.Halpern JM, Gormley CA, Keech M and von Recum HA, Journal of materials chemistry. B, Materials for biology and medicine, 2014, 2, 2764–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang NX and von Recum HA, Macromolecular Bioscience, 2010, 11, 321–332. [DOI] [PubMed] [Google Scholar]
- 14.Halpern JM and von Recum HA, in Biomaterials and Regenerative Medicine, ed. Ma PX, Cambridge University Press, Cambridge, 2014. [Google Scholar]
- 15.Fu A. S. v. R., Horst A, in Engineering Polymer Systems for Improved Drug Delivery, 2014. [Google Scholar]
- 16.Rohner NA, Schomisch SJ, Marks JM and von Recum HA, Molecular Pharmaceutics, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolinsky JB, Colson YL and Grinstaff MW, Journal of controlled release : official journal of the Controlled Release Society, 2012, 159, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solorio L, Wu H, Hernandez C, Gangolli M and Exner AA, Therapeutic delivery, 2016, 7, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grafmiller KT, Zuckerman ST, Petro C, Liu L, von Recum HA, Rosen MJ and Korley JN, Journal of Surgical Research, 2016, 206, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cyphert EL, Learn GD, Hurley SK, Lu C.-y. and von Recum HA, Advanced Healthcare Materials, 2018, 7, 1870080. [DOI] [PubMed] [Google Scholar]
- 21.Rivera-Delgado E, Djuhadi A, Danda C, Kenyon J, Maia J, Caplan AI and von Recum HA, Journal of Controlled Release, 2018, 284, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cyphert EL, Zuckerman ST, Korley JN and von Recum HA, Acta Biomaterialia, 2017, 57, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loeffler JS, Alexander E 3rd, Hochberg FH, Wen PY, Morris JH, Schoene WC, Siddon RL, Morse RH and Black PM, International journal of radiation oncology, biology, physics, 1990, 19, 1455–1462. [DOI] [PubMed] [Google Scholar]
- 24.Hochberg FH and Pruitt A, Neurology, 1980, 30, 907–911. [DOI] [PubMed] [Google Scholar]
- 25.Monnaert V, Tilloy S, Bricout H, Fenart L, Cecchelli R and Monflier E, The Journal of pharmacology and experimental therapeutics, 2004, 310, 745–751. [DOI] [PubMed] [Google Scholar]
- 26.Ohtani Y, Irie T, Uekama K, Fukunaga K and Pitha J, European journal of biochemistry, 1989, 186, 17–22. [DOI] [PubMed] [Google Scholar]
- 27.Poulsen HL, Scandinavian Journal of Clinical and Laboratory Investigation, 1974, 34, 119–122. [PubMed] [Google Scholar]
- 28.Vesole SM, Case Western Reserve University, 2011. [Google Scholar]
- 29.Oss-Ronen L and Seliktar D, Advanced Engineering Materials, 2010, 12, B45–B52. [Google Scholar]
- 30.Trott O and Olson AJ, Journal of computational chemistry, 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dallakyan S and Olson AJ, Methods in molecular biology (Clifton, N.J.), 2015, 1263, 243–250. [DOI] [PubMed] [Google Scholar]
- 32.Edgardo R-D, Zhina S, Nick XW, Jonathan K, Sapna S, Michael K, Chris F, Adonis ZH and Horst A. v. R., Biomedical Materials, 2016, 11, 025022. [Google Scholar]
- 33.Rivera-Delgado E and von Recum HA, Molecular Pharmaceutics, 2017, 14, 899–907. [DOI] [PubMed] [Google Scholar]
- 34.Steinhardt J, Krijn J and Leidy JG, Differences between bovine and human serum albumins. Binding isotherms, optical rotatory dispersion, viscosity, hydrogen ion titration, and fluorescence effects, 1971. [DOI] [PubMed] [Google Scholar]
- 35.Parini P, Johansson L, Broijersen A, Angelin B and Rudling M, European journal of clinical investigation, 2006, 36, 98–104. [DOI] [PubMed] [Google Scholar]
- 36.Thatiparti TR and von Recum HA, Macromolecular Bioscience, 2009, 10, 82–90. [DOI] [PubMed] [Google Scholar]
- 37.A. S. Fu and H. A. von Recum, 2012.
- 38.Rivera-Delgado E, Sadeghi Z, Wang NX, Kenyon J, Satyanarayan S, Kavran M, Flask C, Hijaz AZ and von Recum HA, Biomedical Materials, 2016, 11, 025022. [DOI] [PubMed] [Google Scholar]
- 39.Merritt SR, Velasquez G and von Recum HA, Experimental eye research, 2013, 116, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cyphert EL, Fu AS and von Recum HA, Experimental Biology and Medicine, 2017, 242, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cyphert LE, Wallat DJ, Pokorski KJ and Von Recum AH, Antibiotics, 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juric D, Rohner NA and von Recum HA, Macromolecular Bioscience, 2019, 19, 1800246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agudelo D, Bourassa P, Bruneau J, Berube G, Asselin E and Tajmir-Riahi HA, PLoS One, 2012, 7, e43814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cyphert EL and von Recum HA, Experimental Biology and Medicine, 2017, 242, 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Félix-Redondo FJ, Grau M and Fernández-Bergés D, Aging and disease, 2013, 4, 154–169. [PMC free article] [PubMed] [Google Scholar]
- 46.Rohner NA, McClain J, Tuell SL, Warner A, Smith B, Yun Y, Mohan A, Sushnitha M and Thomas SN, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2015, 29, 4512–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rohner NA and Thomas SN, Journal of Controlled Release, 2016, 223, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LeVine SM, BMC neurology, 2016, 16, 47–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.