

Abstract
Approximately 75% of all disease relevant human proteins including those involved in intracellular protein-protein interactions (PPIs) are undruggable with the current drug modalities (i.e., small molecules and biologics). Macrocyclic peptides provide a potential solution to these undruggable targets, because their larger sizes (relative to conventional small molecules) endow them the capability of binding to flat PPI interfaces with antibody-like affinity and specificity. Powerful combinatorial library technologies have been developed to routinely identify cyclic peptides as potent, specific inhibitors against proteins including PPI targets. However, with the exception of a very small set of sequences, the vast majority of cyclic peptides are impermeable to the cell membrane, preventing their application against intracellular targets. This review examines the common structural features that render most cyclic peptides membrane impermeable as well as the unique features that allow the minority of sequences to enter the cell interior by passive diffusion, endocytosis/endosomal escape, or other mechanisms. We also present the current state of knowledge about the molecular mechanisms of cell penetration, the various strategies for designing cell-permeable, biologically active cyclic peptides against intracellular targets, and the assay methods available to quantify their cell-permeability.
Keywords: Cyclic peptide, Drug discovery, Macrocycle, Permeability, Protein-protein interaction, Undruggable targets
Graphical Abstract
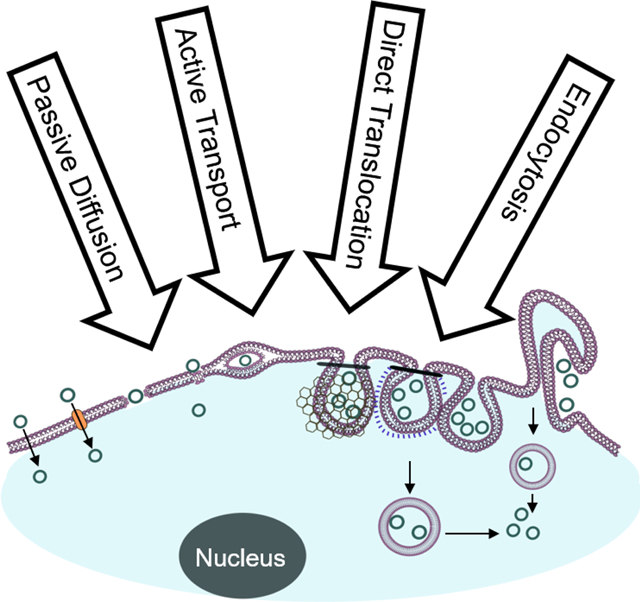
1. INTRODUCTION
There has been a major surge in cyclic peptide research since the beginning of this millennium. A SciFinder search using the term “cyclic peptide” (March 26th, 2019) produced 10,169 entries/publications, among which 7,222 entries were published since 2000 and 3,952 during the current decade alone (Figure 1). A major driving force behind this surge is undoubtedly the success as well as “failure” of the drug industry. On the one hand, the remarkable success of biologic drugs (e.g., monoclonal antibodies) has led to a paradigm shift from the “small molecule-centric” to a “modality-agnostic” approach to drug discovery in the pharmaceutical industry and, as a consequence, greater acceptance of peptides as a drug modality. On the other hand, there is growing realization that current drug modalities, including both small molecules and conventional biologics, are inadequate for the majority of yet untapped therapeutic opportunities, which represent ~75% of all therapeutically relevant proteins.1 Among the undruggable targets, the most prominent class is the proteins involved in intracellular protein-protein interactions (PPIs). This is because PPIs usually do not involve well-defined hydrophobic pockets at the interface for small-molecule drugs to bind, whereas biologics such as monoclonal antibodies cannot cross the cell membrane to reach the target. Meanwhile, two major breakthroughs in peptide science and technology occurred during the past two decades - the first enables the discovery of potent macrocyclic peptide ligands against essentially any protein target, while the second allows such ligands to be effectively delivered into the interior of mammalian cells. The combination of these factors makes macrocyclic peptides an exciting drug modality for the decades to come, especially for targeting intracellular PPIs.
Figure 1.

Publications in the cyclic peptide field by year (left Y-axis) and cumulative total (right Y-axis).
1.1. Cyclic Peptides as an Emerging Drug Modality
Cyclic peptides (including cyclic depsipeptides and bicyclic peptides) possess a number of favorable properties as therapeutic agents and research tools. First, cyclization of peptides reduces the conformational freedom, greatly enhancing their metabolic stability and binding affinity/specificity to target molecules. In general, cyclic peptides of smaller ring sizes (≤10 aa) are relatively resistant to proteolytic degradation.2 Second, medium-sized cyclic peptides (6–15 aa; MW = 500–2000 Da), which are most commonly used, are typically 3- to 5-times larger than conventional small-molecule drugs (MW≤500) and engage larger surface areas on target proteins. Consequently, cyclic peptides have the capacity to recapitulate the exceptional affinity and specificity of proteins, even against targets that do not have any binding pocket. Indeed, many of the naturally occurring as well as synthetic macrocyclic peptides have demonstrated antibody-like affinity and specificity to their intended targets.3–5 Third, cyclic peptides have enormous structural diversity. By using just the 20 proteinogenic amino acids, one can generate ~208 or ~2.5 × 1010 different cyclooctapeptides. The structural diversity (and protease resistance) can be further increased by incorporation of non-proteinogenic amino acids (e.g., d- and Nα-methylated amino acids) and/or varying the ring size. In fact, cyclic peptide libraries of up to 1014 different compounds have been experimentally generated.6 This level of structural diversity greatly surpasses that of the human antibody repertoire and therefore, in principle, it should be possible to generate potent and specific cyclic peptide ligands against any protein target of interest. Fourth, compared to proteins, cyclic peptides retain some of the attributes of small molecules such as stability, lower risk of immune response, synthetic accessibility, and lower cost of production. Finally, as will be described in detail below, cyclic peptides can be rendered cell-permeable and therefore capable of targeting intracellular proteins including those involved in PPIs.
It should come as no surprise that cyclic peptides, cyclic depsipeptides, and bicyclic peptides are broadly distributed in nature and exhibit a wide range of biological activities, including anticancer, antibacterial, antiviral, antifungal, anti-clotting, and anti-inflammatory properties. To date, >1000 cyclic peptides have been discovered in nature,7–9 some of the cyclic peptides have become highly successful drugs, including vancomycin (antibacterial), daptomycin (antibacterial), cyclosporine A (an immunosuppressant for transplantation), and caspofungin (antifungal). Inspired by the natural products, chemists have developed numerous methodologies to prepare cyclic peptides through N-to-C, sidechain-to-sidechain, or main chain-to-sidechain cyclization.10 Some of the synthetic cyclic peptides, such as integrilin (eptifibatide, for treating heart diseases), octreotide (a somatostatin mimetic used to treat acromegaly and diarrhea), cyclic RGD peptides, disulfide cyclized linaclotide, and hydrocarbon-stapled peptides against the MDM2-p53 interaction have also been approved by FDA for clinical use or in late-stage clinical trials.11
1.2. Enabling Technologies for Discovery of Cyclic Peptide Ligands
Despite their enormous potential, until recently cyclic peptides had been underexploited as a drug modality, primarily because no technology was available to tap into this massive “treasure trove”.12 Except for stapled peptides13 and some special cases, rational design of cyclic peptidyl ligands against protein targets is generally a challenging task, especially when structural information is not available or if binding (e.g., PPI) is mediated by multiple, noncontiguous epitopes. Methods that proved effective for small-molecule drug discovery, e.g., fragment-based drug design, generally do not work well with macrocyclic peptides. Very recently, there has been some success in designing cyclic peptide ligands by using computational approaches,14 but its generality remains to be determined. In the absence of rational design approaches, many investigators have been pursuing combinatorial library approaches, i.e., a large number of cyclic peptides of different sequences are simultaneously synthesized and then rapidly screened for binding to (or other biological activities against) a target of interest. These efforts have finally led to the development of several powerful combinatorial library technologies over the past two decades. This represents a major breakthrough/milestone in peptide science and technology, as these technologies have now made it possible to discover potent and selective cyclic peptidyl ligands against essentially any protein target including those involved in PPIs. These methods are briefly introduced below; for more details, the reader is referred to several recent reviews15–17 or the original publications cited in this review.
Based on their methods of generation, cyclic peptide libraries can be classified into biologically vs chemically synthesized libraries. Among ribosomally synthesized libraries, phage display was the first method developed.18 In the early designs, peptide cyclization was usually mediated by disulfide formation between a pair of cysteine residues N- and C-terminal to the peptide sequence of interest. A major limitation of this method was that the cyclic peptide ligands obtained cannot be used against intracellular targets. Winter and co-workers later extended the phage display technology to generate bicyclic peptide libraries.19 In this case, each library member is designed to contain three invariant cysteine residues; after being displayed on the phage surface, the peptide is treated with a trifunctional alkylating agent [e.g., tris(bromomethyl)benzene], which converts the peptide into a bicyclic structure through the formation of three thioether bonds. Bicyclic peptides of this type have proven extremely potent inhibitors against enzymes [e.g., a 1.5 nM inhibitor against protease kallikrein19] as well as specific ligands for cell-surface receptors.20,21 Benkovic and co-workers generated N-to-C cyclized peptide libraries inside living cells by utilizing the protein splicing properties of split inteins.22,23 The cyclic peptides produced in situ can directly modulate a cellular event(s) inside the cell. Tavassoli and co-workers have used this technology to discover small cyclic peptide inhibitors against several protein dimerization events.24–26 Finally, mRNA display has been used to generate linear peptide libraries by in vitro translation and the peptides are then chemically cyclized by using a bifunctional reagent.27,28 A drawback of the biological libraries is that they are generally limited to the 20 proteinogenic amino acids. To alleviate this drawback, Suga, Szostak, and co-workers integrated the mRNA display technology with a codon reassignment strategy to incorporate a variety of unnatural amino acids into displayed peptides, including an N-chloroacetyl-d-tryptophan at the N-terminus, which spontaneously cyclizes with an internal cysteine residue to form cyclic peptides covalently attached to their coding mRNAs.4,29,30 The latter technology, called “RaPID (for Random nonstandard Peptide Integrated Discovery)”,6 is capable of producing and screening 1014 different cyclic peptides in solution, against a protein target of interest. Because of the exceptionally high library diversity, cyclic peptide ligands of very high affinity and specificity (KD in the pM to low nM range) can often be directly isolated from the libraries.
Cyclic peptide libraries can be readily synthesized (chemically) in the one bead-one compound (OBOC) format by using Lam and Houghten’s split-and-pool synthesis.31,32 The challenge lies in post-screening hit identification, because main-chain cyclized peptides cannot be sequenced by Edman degradation or tandem mass spectrometry (or at least not reliably).33,34 This problem was solved by employing two different encoding strategies, much like the way biologically synthesized libraries are encoded by DNA or RNA. Pei and co-workers designed one bead-two compound (OBTC) libraries, in which each library bead displays a unique cyclic peptide on its surface and contains a linear peptide of the same sequence in the interior as an encoding tag.35 Hit identification was readily achieved by sequencing the linear encoding peptide inside the bead by Edman degradation and/or mass spectrometric methods.36 They later extended the methodology to synthesize bicyclic peptide libraries, in which each peptide contained three fixed, orthogonally protected amino groups, one at the N-terminus, one on the side chain of a C-terminal 2,3-diaminopropionic acid (Dap) residue, and the third on the side chain of an internal lysine, ornithine, or Dap residue.37 Following selective deprotection of the three amines, the peptides were converted into bicyclic structures by the formation of three amide bonds between the amines and a small-molecule scaffold (e.g., trimesic acid). In an alternative encoding strategy, Liu and colleagues carried out DNA-templated synthesis (DTS), where each building block is covalently attached to a “transfer” DNA oligonucleotide, which brings the building block to the vicinity of another building block attached to a template DNA through DNA hybridization.38,39 Reaction between the two building blocks is greatly accelerated due to the proximity effect. After coupling of all building blocks and cyclization, each macrocycle is covalently linked to a unique DNA template that serves as the “blueprint” for its construction. The macrocycle-DNA library is typically screened for binding to a target of interest, and the most potent binders were identified by deep sequencing of the enriched DNA library, assuming that tighter binding results in greater enrichment of the corresponding macrocycle-DNA adduct. A distinct advantage of chemically synthesized libraries is that almost any unnatural amino acid (or non-peptidic building block) can be incorporated into the library, greatly enhancing the structural diversity and metabolic stability of the library compounds. There is also a broader selection of cyclization methods available for chemically synthesized libraries.
In sum, there is clearly a demand for alternative drug modalities for targeting challenging proteins (e.g., PPIs). Cyclic peptides provide excellent candidates for meeting this demand, because of their favorable physiochemical properties and the advent of powerful technologies that enable the rapid discovery of potent, specific cyclic peptide ligands against essentially any protein target. The remaining challenge is how to deliver cyclic peptides, the vast majority of which are impermeable to the plasma membrane of the cell, into the cell interior, where most PPIs take place. It is the latter, the cell-permeability of cyclic peptides, that will be the focus of this review. Other worthy topics, e.g., the oral bioavailability of cyclic peptides, will not be covered here; interested readers are directed to several excellent recent reviews on the topic.40–42
2. WHAT MAKES MOST CYCLIC PEPTIDES MEMBRANE-IMPERMEABLE?
The capacity to cross the cell membrane is essential for any compound if the intended target is located inside the cytosol (or nucleus) of the cell. Owing to the wealth of intracellular therapeutic targets, small molecule drugs are continuously designed and optimized with cellular permeability in mind. Over the years, certain structural features and physicochemical properties, e.g., low molecular weight (MW <500) and lipophilicity, have been empirically observed to facilitate membrane penetration and oral bioavailability. These features/properties, often called the “drug-likeness” of a compound, have become a guiding principle in small-molecule drug discovery. Based on these empirical rules established for small molecules, any cyclic peptide of 6 or more residues would not be expected to have significant membrane permeability or oral bioavailability. While this is indeed true for the vast majority of cyclic peptides of medium to large ring sizes (i.e., ≥6 aa), a very small fraction of them, of both natural and synthetic origins, have demonstrated good cell-permeability and even oral bioavailability. Therefore, understanding the mechanisms by which cyclic peptides penetrate the cell membrane and the physiochemical determinants of cell-permeability are of paramount importance. In this section, we will discuss the current understanding of the physical mechanisms behind the membrane permeability of small molecules, the physiochemical properties that influence permeability, and how most peptides violate the traditional rules of drug-likeness. In Section 3, we will discuss how the small fraction of cyclic peptides achieve membrane permeability and the structural features conducive to high permeability.
2.1. How Do Small Molecules Cross the Cell Membrane?
Small molecules cross the cell membrane primarily by two different mechanisms: energy-independent passive diffusion and energy-dependent active transport. The two mechanisms are not mutually exclusive for a given compound.
2.1.1. Passive Diffusion.
Many small molecules are capable of crossing the plasma membrane by passive diffusion, an energy-independent process. Passive diffusion can be conceptualized as the partitioning of a molecule among the aqueous extracellular environment, the hydrophobic cell membrane, and finally the aqueous cytosol. The process of passive diffusion is driven by the net concentration gradient of a given compound, flowing from regions of higher concentration (extracellular) to lower concentration (intracellular). Derivation of Fick’s first law of diffusion for passive diffusion across a plasma membrane gives:43
where dM/dt is the amount of compound transferred across a membrane over a time t, kf is the membrane-water partition coefficient for a given compound, S is the total membrane surface area, ΔC is the difference in extra/intracellular concentration, d is the thickness of the membrane, and MW is the compound’s molecular weight. According to this equation, the permeability of a compound is inversely related to its size (molecular weight), with smaller molecules having greater membrane diffusion rates. Permeability is also directly related to the compound’s membrane-water partition coefficient (kf), which is a measure of lipophilicity. Dickson et al. further characterized the kinetic steps in membrane permeation, identifying desolvation as a key step for initial membrane partitioning, whereas lipophilicity dictates how readily the compound transitions out of the membrane into the cytosol.44 Optimization of lipophilicity is thus a routine starting point for ensuring drug-likeness and will be covered in detail in section 2.2 of this review. Mechanistically, the process of passive diffusion is the same for all plasma membranes, but lipid composition has been demonstrated to affect the absolute rate of passive diffusion, although compounds displaying high diffusion rates are still able to effectively enter different cell types.45,46 Diffusion across the blood-brain barrier (BBB) is also an outstanding question in the field and is beyond the scope of this discussion, interested readers are directed to comprehensive treatments of the subject in the literature.47,48
2.1.2. Receptor-Mediated Uptake.
Small molecule uptake via active transport is an area of intense research and is supported by the range of biologically necessary transporters that are intended to shuttle key nutrients and metabolites into the cell. It is estimated that the average mammalian cell membrane contains hundreds of these transporters, many of which are poorly characterized or lack an identified endogenous ligand.49 Extensive surveys of the literature have been carried out to evaluate small molecules which have been proposed to have significant cellular uptake as a result of interaction with carrier proteins.50 Carrier-mediated active transport has been suspected to play a significant role for the cellular uptake of many compounds due to large discrepancies between permeability data derived from artificial permeability assays (such as PAMPA) and values observed in vivo.51 Exemplar carrier proteins that have been implicated in facilitating small molecule cellular uptake include the solute carrier (SLC) family, which encompasses a range of specialized carrier protein subfamilies. For example, Hsiang and co-workers showed that statins such as lovastatin and atorvastatin are transported by OATP2/SLCO2B1, a hepatic organic anion transporting polypeptide, which is part of the solute carrier organic anion (SLCO) subfamily naturally responsible for the transport of thyroid hormone and taurocholic acid.52 Other SLC family members have been implicated in the transport of therapeutics such as memantine, propranolol, and cimetidine.53–55 The SLC family function as facilitative transporters, enabling transport of charged cargo with the electrochemical gradient, or in some situations act as secondary active transporters with simultaneous transport with and against the gradient for net favorable change in free energy. In these situations, small molecules possessing net charges are able to “hijack” the receptors due to their physiochemical similarity to natural substrates and be transported. Carrier proteins are ubiquitously expressed across all tissue families, implicating them in uptake across virtually all cell types.56 For further reading of carrier protein-mediated cellular uptake, readers are referred to several excellent recent reviews.57–59
2.2. Physiochemical Properties that Impact Permeability
Understanding the mechanistic basis for passive diffusion facilitates the evaluation of properties whose optimization is likely to predispose a given compound to membrane permeability. Lipinski et al. laid forth a set of empirical guidelines for predicting oral bioavailability.60 Termed the “Rule of Five” (Ro5), they predicted that compounds with molecular weights below 500 g/mol, CLogP below 5, fewer than 5 hydrogen bond donors (HBD), and fewer than 10 hydrogen bond acceptors (HBA) are more likely to be orally bioavailable. Given that transcellular transport is likely the primary mechanism by which most small-molecule drugs traverse the gut epithelium, the oral bioavailability of a compound should be positively correlated with its cell membrane permeability (i.e., its capacity to cross the plasma membrane of a cell). We shall also point out that, while the expectation is that an orally bioavailable drug should be cell-permeable, this is not always the case. Paracellular transport through tight junctions in gut epithelium or transcytosis can introduce compounds into circulation without intracellular localization. Conversely, cell-permeable compounds may not be able to survive the gut’s harsh environment and therefore may lack oral bioavailability. The necessity of these parameters can be rationalized through the impact of molecular weight on the rate of diffusion, as evidenced in the previous equation, as well as the direct relationship between the membrane-water partition coefficient and CLogP. In addition to molecular weight, early work by Xiang and Anderson demonstrated clear size dependence on partitioning into ordered bilayers, with larger solutes having reduced membrane permeability.61 These findings were rationalized in the context of solute and bilayer entropy loss incurred upon membrane insertion. Diffusion through the hydrophobic plasma membrane interior necessitates desolvation of water molecules coordinated to the solute through hydrogen bonds. Compounds with significant numbers of HBD and HBAs also tend to be more polar, affecting their membrane-water partition coefficient. Further extrapolation of the Ro5 to other physiochemical parameters has provided investigators with additional predictive tools for membrane permeability and oral bioavailability. Palm et al. suggested that polar surface area (PSA) is a highly effective predictor for oral bioavailability.62 PSA is determined by evaluating the 3D conformation and molecular surface of a given molecule, followed by calculating how much of the molecular surface is composed of polar atoms. PSAs greater than 130 Å are strongly correlated with poor oral bioavailability, even for compounds that adhere to other Ro5 properties. Further refinement of PSA include exposed polar surface area (EPSA), based on exposed polarity and intended for the design of passively permeable cyclic peptides by considering conformational changes occurring in solution.63 Kihlberg and colleagues evaluated the predictive power of multiple molecular descriptors for passive diffusion and identified that a novel descriptor, solvent accessible 3D PSA, had the highest correlation with experimentally observed permeability for macrocycles.64 Large-scale analysis of physiochemical properties related to passive permeability carried out by Jacobson and co-workers found that desolvation energy had the highest correlation across a diverse compound data set.65 Interestingly, they also found that entropic loss on membrane insertion was correlated with permeability, supporting the value of cyclization and conformational preorganization on passive uptake. Veber and co-workers also noted that molecular flexibility, as extrapolated from the total number of rotatable bonds, contributes to oral bioavailability, with more rigid compounds (rotatable bonds <10) being more permeable.66 Further predictors have been proposed, such as number of aromatic rings, molar refractivity, and others.67,68
2.3. What Makes Peptides Generally Impermeable to the Cell?
Applying the conventional membrane permeability/oral bioavailability predictors to peptides reveals how peptides are intrinsically incompatible with the concept of “drug-likeness”. The physiochemical obstacles to membrane permeability are intrinsic to the peptide structure - with each peptide bond contributing a HBD and two HBAs, a cyclic pentapeptide consisting of only nonpolar side chains already reaches the boundary of the Ro5. Indeed, Burton et al. evaluated a series of linear peptides and found that the primary obstacle to membrane permeability is the energy required for amide desolvation.69 Given that the average length of therapeutic peptides in clinical use is about 20 residues,70 they violate essentially all Ro5 guidelines for molecular weight, HBA, HBD, PSA, rotatable bonds, and CLogP. Additionally, amino acid side-chains tend to be flexible and only three possess desirable aromatic rings to enhance permeability. Combined, these factors present clear obstacles to peptide membrane permeability. Accordingly, unmodified peptides (including cyclic peptides) of ≥5 aa are generally impermeable to the cell membrane (at least with respect to passive diffusion). To overcome these obstacles, nature and investigators have employed a number of strategies to enhance the membrane permeability of peptides, as discussed in the following section.
3. HOW DO SOME CYCLIC PEPTIDES ACHIEVE CELL-PERMEABILITY?
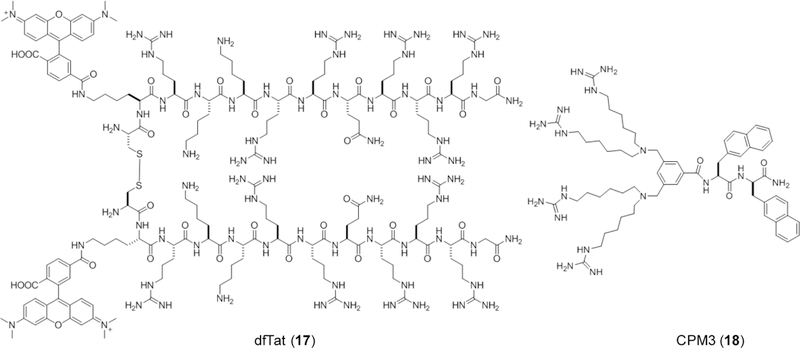
Despite violating some or all of the Ro5 parameters, some naturally occurring as well as synthetic peptides (especially cyclic peptides) have demonstrated good cell-permeability. The mechanisms by which cyclic peptides enter the cell have been the subject of intense research over the past two decades. The results of these studies suggest that cyclic peptides can enter cells through a number of different mechanisms, with some perhaps utilizing multiple pathways to achieve cell penetration. Some cyclic peptides, particularly those consisting of predominantly hydrophobic residues, are capable of directly crossing the plasma membrane by passive diffusion, while others (usually positively charged) enter mammalian cells by endocytic uptake followed by endosomal escape. Some cyclic peptides have been shown to bind to cell surface protein transporters and are actively transported into cells. Still other cyclic peptides have demonstrated cell-permeability, apparently by directly traversing the plasma membrane (direct translocation), but their precise mechanism of cellular entry remains poorly defined. The following sections provide examples of cyclic peptides that attain cell-permeability through distinct, unique pathways and the mechanisms underlying them.
3.1. Cell-Permeability by Passive Diffusion
Nature is an excellent source of biologically active cyclic peptides, some of which are capable of crossing the cell membrane by passive diffusion. Romidepsin (1, Figure 2) is a bicyclic depsipeptide of five residues and is largely Ro5 compliant. It is thus not surprising that romidepsin is membrane permeable and acts against an intracellular target (histone deacetylase).71 Cyclosporine A (CsA, 2), on the other hand, is far beyond the conventional Ro5 in molecular weight, HBD and HBA, and yet is membrane permeable by passive diffusion and has respectable oral bioavailability.72 The unusual capacity of CsA to cross the cell membrane by passive diffusion has inspired extensive mechanistic studies in recent decades, with the hope of gaining mechanistic insights to guide the design of cell-permeable cyclic peptides with novel biological activities. Researchers also systematically modified the structures of several simpler model cyclic peptides of 5–7 residues (and other macrocycles) to determine the relationship between structure and cell-permeability/oral bioavailability. Several key structural features have emerged from these studies and are discussed below, although a complete understanding of cyclic peptide permeability (by passive diffusion) will require additional studies.
Figure 2.

Examples of cyclic peptides that enter cells by passive diffusion.
3.1.1. Role of Nα-Methylation.
As discussed earlier, a common feature of cyclic peptides (and peptides in general) that impedes membrane permeability is the large number of HBDs and HBAs associated with the peptide backbone. Reduction of the number of HBDs and HBAs is therefore expected to increase the passive permeability of cyclic peptides. Indeed, membrane-permeable macrocyclic natural products often contain extensive Nα-methylation and/or substitution of ester functionality for peptide bonds (as in depsipeptides). For example, seven out of the eleven backbone amides of CsA are Nα-methylated (Figure 2). These observations led investigators to explore Nα-methylation as a general strategy to enhance membrane permeability of cyclic peptides. Biron et al. optimized the membrane permeability and oral bioavailability of cyclohexapeptidyl somatostatin analogs through extensive Nα-methylation.73 Starting from the previously identified Veber-Hirschmann peptide, cyclo(Pro-Phe-d-Trp-Lys-Thr-Phe),74 they synthesized and evaluated 30 of the 31 possible Nα-methylated derivatives, whereas the penta-Nα-methylated derivative could not be obtained. Of the 30 derivatives, only 8 retained substantial binding affinity toward the cognate SRIF receptor subtypes 2 and 5, revealing one of the drawbacks of Nα-methylation. Caco-2 permeability assay showed that Nα-methylation of d-Trp, Lys, and Phe resulted in a relatively permeable compound with a Papp value of 1.8 × 10−6 cm/s (compound 3, Figure 3), underscoring the validity of Nα-methylation in optimizing passive permeability. Nα-Methylation was also integral to optimizing the permeability of cyclohexapeptide Cilengitide derivatives.75 A combinatorial approach was employed to access all possible Nα-methylated derivatives of a cyclic poly-Ala peptide. The study identified key methylation motifs that conferred enhanced Caco-2 permeability (Papp > 1 × 10−5 cm/s), but no obvious correlation between the number of Nα-methylated positions and permeability was observed.76 Grafting an RGD motif onto this template while maintaining the stereochemistry and Nα-methylation pattern resulted in a biologically active cyclic peptide (compound 4, Figure 3) possessing reasonable passive permeability.77
Figure 3.

Examples of Nα-methylated cyclic peptides, with Nα-methylated positions highlighted in red.
It should be noted that Nα-methylation can result in dramatic conformational consequences. Nα-Methylation decreases the potential energy difference between cis- and trans- peptide bonds and greatly increases the probability of cis- peptide bonds in cyclic peptides. In fact, the conformational constraints exerted by an Nα-methylated residue are similar to those induced by proline.78 On the positive side, Nα-methylation may allow conformations inaccessible to an unmodified cyclic peptide. However, multiple Nα-methylation of a cyclic peptide may also result in a mixture of many conformers under physiological conditions, only one of which possesses the desired cell-permeability and/or target binding. Practically, these inseparable mixtures of unique conformers can render a compound unsuitable for translation into a clinical setting as the desired species cannot be isolated and could introduce unforeseen issues with off-target binding. Chatterjee et al. prepared a library of Nα-methylated cyclopentaalanine peptides as innately permeable scaffolds and observed multiple distinct backbone conformations.79 The scaffolds followed the general formula cyclo(d-Ala-Ala4) containing 1–4 Nα-methylated residues. Only 7 out of the 30 compounds tested had a clear conformational preference by NMR and often had Nα-methylation at the d-Ala residue. Interestingly, 4 of the compounds with preferred conformations contained cis peptide bonds, with the tetra-Nα-methylated compound containing two. This was caused by unfavorable steric interactions between the Nα-methyl group with adjacent residue β protons. In another example, Yudin and co-workers employed combinations of Nα-methylation, d-amino acids, and polar sidechains to develop cyclic peptides where sidechain-to-backbone hydrogen bonds effectively reduced the total number of HBAs and HBDs, thus improving permeability with the most permeable compound (5, Figure 3) demonstrating a -logPe of 5.4.80 Doedens et al. employed Nα-methylation to optimize the melanocortin receptor subtype selectivity of cyclic peptide MT-II, Ac-Nle-cyclo(5β→10ε)(Asp-His-d-Phe-Arg-Trp-Lys) (compound 6, Figure 3).81 The resulting tetra-Nα-methylated cyclic peptide retained tight binding to hMC1R (EC50 = 13 nM) while displaying virtually no activity towards other receptors. Hewitt et al. employed a combinatorial approach to rapidly access a diverse library of stereochemically diverse cyclic hexapeptides to explore the relationship between the extent of Nα-methylation on conformation and passive permeability.82 Screening for permeability using PAMPA followed by structural deconvolution revealed that a macrocycle (compound 6.7, Caco-2 Papp. = 23.3 × 10−6 cm/s) containing three contiguous Nα-methylated residues resulted in a conformation that minimized SASA and correspondingly reduced ΔGdesolv. These results support the notion that, in general, Nα-methylation is beneficial to passive permeability over their non-methylated counterparts as long as it does not prevent access to conformations containing transannular hydrogen bonds or sterically occluded amide groups (from solvent).83
Replacement of peptide bonds with peptoids (Nα-substituted glycine) can also dramatically enhance cellular uptake.84,85 This can be rationalized within the framework of current mechanistic understanding for passive membrane permeability, as this accomplishes the same effect as removal of an amide HBD through Nα-methylation with the added benefit of a functional handle for derivatization or target binding affinity. Nα-Methylation and substitution of peptoid moieties have been used in combination to improve the membrane permeability and/or target-binding potency of cyclic peptides, as observed with macrocycles targeting β-catenin and CXCR7.86,87
The above studies indicate that Nα-methylation of the peptide backbone (and substitution of esters or peptoids) can be an effective approach to reducing the number of HBDs and HBAs and increasing the passive diffusion of cyclic peptides across membranes. However, random Nα-methylation of one or more positions or simply per-methylating all peptide amides is unlikely to be effective. Rather, judicious placement of Nα-methylation at certain “strategic” position(s) to promote intramolecular hydrogen bonds (while permitting conformational switches) will likely be more productive (vide infra). Nα-Methylation should improve the proteolytic stability of cyclic peptides, but may cause solubility problems and mixtures of multiple conformations.
3.1.2. Intramolecular Hydrogen Bonding and Conformational Transition.
The incongruence between CsA’s primary structure and the conventional wisdom for ensuring passive membrane permeability led to significant investments in understanding and replicating CsA’s mechanisms for overcoming physicochemical constraints for cell-permeability. In addition to Nα-methylation, CsA exploits solvent dielectric-dependent conformational heterogeneity to eliminate additional HBDs and HBAs that would incur substantial desolvation energy penalties during passive diffusion across a lipid bilayer.88 Specifically, CsA exists in an “open” conformation in aqueous media, in which the backbone amides are free to interact with the bulk solvent to maintain solubility or make specific contacts with a target protein (cyclophilin) via hydrogen bonds (Figure 4). Upon entry into a lower dielectric media (such as a lipid bilayer), CsA transitions into a “closed” conformation that involves extensive intramolecular hydrogen bonding among the backbone amides, lowering its effective polar surface area.89 In essence, these conformational changes enable CsA to effectively diffuse into and out of the plasma membrane, achieving reasonable cell-permeability without overly sacrificing aqueous solubility. Facile interconversion between these two conformations is essential for effective membrane permeability. Witek et al. evaluated the rates of conformational conversion in CsA and a close analogue, cyclosporine E (CsE), which differs from CsA by missing a single Nα-methylation at Val11, but has markedly reduced membrane permeability.90 Relative to CsA, the rate of interconversion between the open and closed states in CsE is slowed by almost an order of magnitude, and it has been proposed as the most significant contributor to CsE’s reduced membrane permeability. Intramolecular hydrogen bonding facilitated by conformational transitions is also thought to contribute to the membrane permeability of PF1171, a family of naturally occurring cyclohexapeptides.91 Multiple intramolecular backbone hydrogen bonds have been observed by spectroscopic methods while in low dielectric solvents, and a strong correlation between the 3D topology and biological activity underscores the importance of membrane permeability for efficacy. Examination of macrocyclic natural products by Lokey and co-workers revealed that Nα-methylation and/or intramolecular hydrogen bonding are present in virtually all compounds of significant passive membrane permeability.92
Figure 4.
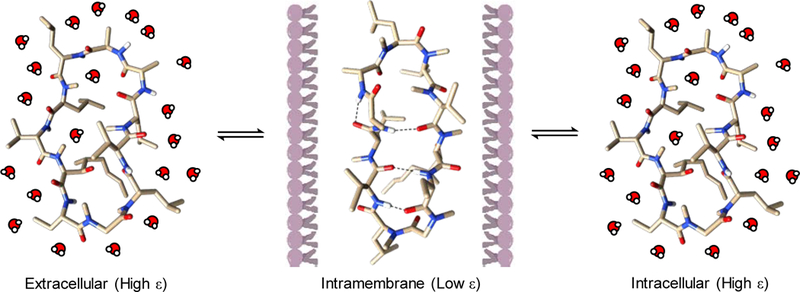
Dynamic conformations of CsA when transitioning between high and low ε environments. CsA exists in an open comformation inside the aqueous extra- and intracellular environments, but adopts a closed conformation containing three intramolecular hydrogen bonds upon entering the lipid bilayer. Nitrogen atoms are shown in blue color, oxygen atoms in red, and hydrogen bonds shown as dotted lines.
Price et al. generated a synthetic CsA analog by replacing a key unnatural amino acid required for biological activity [(2S,3R,4R,6E)-3-hydroxy-4-methyl-2-methylamino-6-octenoic acid] with valine and evaluated its impact on cell-permeability and pharmacokinetic properties.93 Interestingly, this structural change resulted in significant alterations in the compound’s low-dielectric conformation as revealed by NMR and in silico modeling, as well as a loss of most of the intramolecular hydrogen bonds and membrane permeability. Bockus et al. incorporated γ-amino acids (e.g. statines), another moiety commonly found in membrane-permeable macrocyclic peptide natural products, into cyclic peptides to evaluate its impact on permeability.94 In general, cyclic peptides containing γ-amino acids demonstrated improved permeability as determined by PAMPA and in MDCK-LE cells. NMR studies revealed that the statine β-hydroxyls were virtually all involved in hydrogen bonds with solvent-exposed amides and a positive correlation of membrane permeability with the degree of transannular hydrogen bonding within the compound series.
To further test the validity of the conformational transition hypothesis, Lokey and co-workers systemically evaluated the impact of conformation on a series of cyclohexapeptide diastereomers based on the sequence cyclo[Leu-Leu-Leu-Leu-Pro-Tyr].95 They identified two compounds whose membrane diffusion rates differed by two orders of magnitude, thus providing a robust system for testing the effect of conformational transition on cell-permeability. Similar to the observations described above, they found that the capacity to form intramolecular hydrogen bonds is the key factor for ensuring membrane permeability, even for cyclic peptides consisting of primarily hydrophobic amino acids. Solution NMR studies in CDCl3 and hydrogen/deuterium exchange studies showed that 4 out of the 5 backbone amides are involved in intramolecular hydrogen bonding in the more permeable compound, whereas only two backbone amides in the less permeable peptide were involved in intramolecular hydrogen bonds and the remaining three were solvent accessible. Furukawa et al. further demonstrated that intramolecular hydrogen bonding, not lipophilicity, is key for ensuring cyclic peptide membrane permeability.96 Analogous to what has been observed with CsA, the rate of interconversion in cyclic decapeptides to access the transannular hydrogen-bonded “closed” conformation is a key determinant for passive permeability.97 Cyclic peptides with conformational constraints (e.g. through incorporation of Pro or Nα-methylation) increasing the population of the “closed” conformer resulted in strong positive correlation with PAMPA permeability (r2 = 0.784). Moreover, scaffolds with demonstrated membrane permeability and extensive intramolecular hydrogen bonding tolerated the incorporation of more polar sidechains, provided that 2 ≤ ALogP ≤4. Incorporation of polar amino acids can be accomplished without significant losses in membrane permeability through the formation of exocyclic hydrogen bonds, which are bonds formed between sidechain HBDs and backbone carbonyl groups. In silico simulations by extensive conformational sampling in high- and low-dielectric conditions also predicted that passive permeability may be achieved by lowering the desolvation free energy.98 A strong correlation was observed between PAMPA permeability and the computational results (R2 = 0.96), although a direct correlation between permeability and SASA or the number of intramolecular hydrogen bonds was less conclusive.
In addition to the contribution of conformational transition to membrane permeability, Pye et al. noted a non-classical relationship between molecular size and passive permeability.99 Their initial work with a series of cycloocta- to cyclodecapeptides of varying MW (797–1237) and lipophilicity (0 < AlogP < 8) demonstrated that increasing ring size led to decreased permeability at the same AlogP. Partition coefficient measurements (logKhc/w) between 1,9-decadiene and water were in excellent agreement with AlogP, suggesting that lipophilicity is the key driver for partitioning under these conditions. Note that octanol is a poor model for an exclusively hydrophobic environment (such as the interior of a lipid bilayer) due to its capacity for hydrogen bonding. Correlation between logKhc/w and solubility-adjusted permeability logP0 displayed a linear relationship with a slope significantly below 1, suggesting that an additional conformational component may be associated with the decreasing permeability with ring size. Application to other beyond the Ro5 compounds revealed the same size-permeability dependence when controlling for lipophilicity. Further work is necessary to understand the mechanistic basis of these observations, but it was hypothesized that this may be a result of membrane diffusion sharing similarities with restrictive diffusion through polymeric systems or an additional energetic cost for partitioning out of the membrane and into the cytosol.100,101
Conformational flexibility may affect membrane permeability and/or biological activity through additional mechanisms. Interestingly, enantiomeric pairs of cyclic poly-Ala peptides showed different Caco-2 permeability while having identical lipophilicity (as determined from logD) and PAMPA permeability.102 This finding indicates the potential for transporter-mediated cellular uptake and/or efflux pumps for even these hydrophobic proof-of-concept scaffolds and underscores the need for additional investigation. It is conceivable that conformational flexibility allows a cyclic peptide access to a conformation(s) that is recognized by a transporter protein(s).
3.1.3. Steric Occlusion of Backbone Amides.
Another strategy for reducing desolvation energy penalties and facilitating passive diffusion through cell membranes involves steric occlusion of backbone amides. The permeability enhancement conferred by an occluded backbone amide has been found to be even greater than that of an intramolecular hydrogen bond, reflecting the importance of TPSA for efficient diffusion.82,103 Fairlie and co-workers modified sanguinamide A (7, Figure 5), a cycloheptapeptide natural product, to optimize its physiochemical properties.104 Similar to other cyclic peptides, sanguinamide A has molecular descriptors that are beyond the ranges typically associated with passive permeability, namely high MW (721 g/mol), number of HBAs (13), and TPSA (169 Å2). To complement the two strong transannular hydrogen bonds identified by NMR, the researchers Nα-methylated a solvent-exposed amide was to remove an HBD and replaced the Ala (which contains the last accessible amide) with the bulkier tert-butyl glycine. It was anticipated that the tert-butyl group would sterically occlude the backbone amide of Ala from donating a hydrogen bond, thereby reducing solvent-accessibility/TPSA and enhancing membrane permeability. Combination of these changes led to a new derivative, danamide D (8, Figure 5), whose structure (as determined by NMR) showed considerable reduction in solvent-accessibility.104 Pharmacokinetic studies in mice demonstrated improved parameters including serum half-life (t½), clearance (CL), maximum serum concentration (Cmax), time to maximum concentration (Tmax), and impressive oral bioavailability (F = 51%). Hill et al. further showed that steric occlusion alone (in the absence of any Nα-methylation) is sufficient to render cyclopenta- and cyclohexaleucine peptides membrane permeable.105 Conformational studies by X-ray crystallography and NMR revealed that the most permeable compound (Papp ~7 × 10−6 cm/s in Caco-2 assay) had two leucine sidechains projecting over the peptide backbone, reducing solvent accessibility of the backbone amides and preventing intermolecular hydrogen bonding. Indeed, work using diastereomeric compounds indicated that projection of a single Leu side-chain over the core of a macrocycle to shield backbone amides from solvent conferred improved passive permeability.82 Steric occlusion and modulation of amide solvent accessibility has been postulated to be responsible for these observed effects on permeability, but it should be noted that the increased hydrophobicity of these compounds and a resulting propensity for aggregation may confound these observations.
Figure 5.
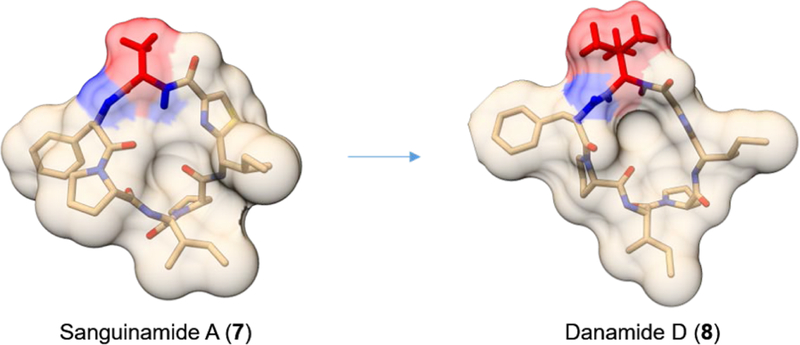
Steric occlusion of backbone amides in sanguinamide A and danamide D, represented as solvent-accessible surface colored in tan. Solvent-accessible amide bonds are colored in blue and the occluding side-chain is colored in red.
3.2. Cell-Permeability by Endocytic Uptake and Endosomal Release
3.2.1. Cyclic Cell-Penetrating Peptides.
It was discovered in the early 1990s that short, highly basic peptides from the transactivator of transcription protein of human immunodeficiency virus (Tat, GRKKRRQRRRPQ)106 and the Antennapedia homeodomain of Drosophila (penetratin, RQIKIWFQNRRMKWKK)107 were capable of entering the cytosol of mammalian cells. These findings immediately gave birth to a new research field that focuses on the discovery of additional peptides with cell-penetrating capabilities, their applications for cargo delivery, and their mechanism of action. During the ensuing three decades, nearly 2000 peptides, derived from both natural proteins and synthetic efforts, have been reported to have cell-penetrating activities.108 These peptides, typically 5–30 amino acids in length, are collectively called “cell-penetrating peptides (CPPs)”. Although their mechanism(s) of action remains a subject of debate, CPPs have been applied to deliver a wide array of cargoes (including peptides, proteins, nucleic acids, and nanoparticles) into mammalian cells in vitro and in vivo. However, for the vast majority of CPPs, the cytosolic delivery efficiency (which is defined as the ratio of cytosolic over extracellular concentration) is very poor (<5%).109 Additionally, linear CPPs consisting of primarily proteinogenic amino acids are highly susceptible to proteolytic degradation in vivo, resulting in poor pharmacokinetic properties. These and other limitations have rendered their clinical development problematic.110
The limitations of linear CPPs prompted researchers to explore cyclic peptides as CPPs, since the latter should at least have improved proteolytic stability. In 2010, Pei and co-workers observed that cyclic peptides containing an aromatic hydrophobic residue (2-naphthylalanine or Nal/Φ) and as few as three arginine residues are cell-permeable (e.g., compound 9, Figure 6).111 The cyclic peptides are relatively potent inhibitors of peptidyl-prolyl cis-trans isomerase Pin1 in vitro (KD = 30 nM) but exhibited only modest anti-proliferative activity in cell culture, indicative of moderate cell-permeability of the peptides. This was a surprising finding, because earlier studies had established that for linear CPPs, six or more arginine residues are required to have significant cellular entry activity.112 Subsequent SAR analysis and sequence optimization by the Pei group led to a family of exceptionally active cyclic CPPs, including CPP1 (10, Figure 6) [cyclo(Phe-Nal-Arg-Arg-Arg-Arg-Gln)], CPP9 (11, Figure 6) [cyclo(phe-Nal-Arg-arg-Arg-arg-Gln)], and CPP12 (12, Figure 6) [cyclo(Phe-phe-Nal-Arg-arg-Arg-arg-Gln)], which had cytosolic delivery efficiencies of 20%, 62%, and 120%, respectively (compared to 2.0% for Tat).113–115 It was found that peptide cyclization, the presence of both arginine and hydrophobic residues, and small ring sizes (≤9 aa) are critical features for high cellular entry efficiency.115
Figure 6.

Cyclic cell-penetrating peptides. Disulfide bonds in MCoTI-II (15) are shown in yellow, while the grafted arginines in ZF5.3 (16) are highlighted in pink. ZF5.3, although not a cyclic peptide, is included here because it shares the structural constraints and high cellular entry efficiency of cyclic CPPs.
Other investigators also reported that cyclization of CPPs improves their cellular entry efficiency. Cardoso and colleagues found that cyclization of Tat (13, Figure 6) and nonaarginine (R9) significantly increased their cell-penetrating activities.116,117 Parang and co-workers reported that cyclooctapeptides containing alternating arginine and tryptophan residues [e.g., cyclo(Trp-Arg-Trp-Arg-Trp-Arg-Trp-Arg) (14, Figure 6)] are active CPPs.118 Marsault and colleagues showed that a series of macrocyclic and bicyclic arginine-rich peptides of varying ring size, site of cyclization, and stereochemistry effectively entered mammalian cells.119 Horn et al. reported that cyclization of an amphipathic cationic CPP (sC18) via a triazole bridge (click chemistry) greatly improved its cellular entry.120 Meanwhile, the Craik group reported that some cyclotides (which are cyclic peptides containing multiple intramolecular disulfide bonds), e.g., MCoTI-II (15, Figure 6) are active CPPs.121 Interestingly, the Schepartz group discovered that proper spatial display of five arginine residues on a stabilized α-helix produced highly cell-permeable miniature proteins [e.g., ZF5.3 (16), Figure 6].122 Although the miniature proteins are not cyclic in nature, the α-helical structure apparently provides similar conformational constraints as does peptide cyclization (vide infra). Collectively, these observations demonstrated that structural constraints improve the activity of CPPs presumably by decreasing the entropy loss during a binding event and therefore increasing the CPPs’ binding affinity to a cell surface component(s), which in turn reduces the number of arginine residues necessary for effective binding to that component(s).
As expected, cyclic CPPs have much greater resistance to proteolytic degradation. Treatment of cyclic CPP1 (10), CPP9 (11), or CPP12 (12) in human serum for 24 h at 37 °C resulted in minimal degradation, whereas most of the linear CPPs are completely digested within the first hour.113 In fact, cyclic CPP1 and CPP9 are sufficiently stable to be orally bioavailable following PO dosing (oral gavage; without formulation).114 The cyclic CPPs have been employed to efficiently deliver a variety of cargoes (including peptides, proteins, and nucleic acids) in vitro and in vivo and it is clear they represent an exciting strategy for the effective delivery of next-generation therapeutic modalities (vide infra).
Although the focus of this review is on cyclic peptides, most of the mechanistic studies of CPPs have historically been conducted on linear peptides. Mechanistically, there is no fundamental structural difference between linear and cyclic CPPs to suggest that they might enter cells by different mechanisms. As discussed in Section 3.2.5, we believe that the difference between cyclic and linear CPPs are quantitative rather than qualitative, i.e., cyclic CPPs are more structurally constrained than their linear counterparts and when properly designed, cyclic CPPs are capable of binding to cell membranes with higher affinity and consequently achieving much greater cell-permeability. It should be pointed out that the efforts by numerous research groups over the past three decades have resulted in a vast literature on CPPs. Many of the studies and their conclusions are contradictory to each other and as a result, no consensus has been reached with regard to their molecular mechanism(s) of cell entry. It is neither our intention nor practical to cover the enormous literature on linear CPPs in its entirety. Instead, we will focus our discussion on studies performed on cyclic CPPs and mention along the way some of the most relevant studies on linear CPPs. For a comprehensive coverage of CPPs, readers are referred to several recent reviews.123, 124
There is now a general agreement that CPPs enter cells by at least two different mechanisms.125 At low concentrations, CPPs are thought to enter cells primarily by endocytosis, resulting in their initial localization inside the endosomes and lysosomes. To reach the cytosol, which is the desired destination for most of the drug delivery purposes, the CPP (or the CPP-cargo conjugate) must escape from the endosomal/lysosomal pathway by crossing the endosomal/lysosomal membrane. At high concentrations, some CPPs have been shown to cross the plasma membrane in an energy-independent fashion and enter the cytosol directly (direct translocation), thus bypassing the endosomal/lysosomal pathway altogether. For some arginine-rich CPPs (e.g., Tat and R9), the concentration threshold for direct translocation has been reported to be ~10 μM, but the threshold is likely to differ depending on the structure of the CPP, the cargo attached to the CPP, and other factors such as the cell line and extracellular media. In this section, we will discuss the mechanisms of endocytic uptake and endosomal escape as well as the experimental evidence supporting them. Section 3.3 will cover the other mechanisms of cellular entry including direct translocation.
3.2.2. Evidence for Endocytic Uptake.
Endocytosis is a natural process occurring in essentially all mammalian cells. CPPs may bind to a cell-surface component(s) and be internalized along with a physiological endocytic event (i.e., as a “bystander”). Alternatively, CPPs can bind to cell-surface components and actively trigger endocytosis. Early studies by Kaplan et al. observed that cationic CPPs enhanced the internalization of endosomal markers such as dextran,126 while Brock and colleagues reported increased TNFα and EGF receptor internalization with cationic CPP treatment.127 All known types of endocytic mechanisms have been described for the entry of the different CPPs and CPP–cargo conjugates/complexes.128 Macropinocytosis was demonstrated to be involved in the internalization of Tat126 and polyarginines.129 It was proposed that following internalization via a pore-formation mechanism, Tat interacts with the actin cytoskeleton and triggers its further uptake by macropinocytosis.129 There were also reports that after binding and clustering of proteoglycans, cationic CPPs induce activation of a small GTPase, Rac1, which in turn results in actin remodeling and induces macropinocytosis.130
The role of clathrin-mediated and caveolin-mediated endocytosis of cationic CPPs have been more controversial. While multiple studies demonstrated the role of clathrin-dependent endocytosis for the uptake of Tat peptide,131, 132 others suggested that the uptake of Tat was not affected by loss-of-function mutations in dynamin-1 which would hinder clathrin-mediated endocytosis,133 or by clathrin knockdown.134 Caveolin-mediated endocytosis has been demonstrated as an internalization pathway for Tat-protein conjugates, including Tat-GFP visualized by Ferrari et al.135 On the other hand, it was also reported that treatment of HeLa and Chinese hamster ovary (CHO) cells with nystatin and filipin III, inhibitors of caveolae-dependent endocytosis, did not affect the internalization of Tat peptide,136 and that caveolin knock-out did not affect the uptake of Tat peptide in baby hamster kidney cells.137 Finally, lipid raft-dependent endocytosis (caveolae-dependent or not) was shown to also play a role in the internalization of penetratin, Tat peptide and others.138
Pei and co-workers conducted mechanistic studies with cyclic CPPs and obtained multiple lines of evidence demonstrating endocytosis as the predominant mechanism for the uptake of cyclic CPPs under low CPP concentrations (≤10 μM).113,114 Time-lapse live-cell confocal microscopy of HeLa cells treated with fluorescein isothiocyanate (FITC)-labeled CPP9 (CPP9FITC; 3 μM) for 30 min showed predominantly punctate fluorescence in the cytoplasmic region, consistent with their initial vesicular localization (Figure 7A, B).114 Upon further incubation (60–120 min), diffuse fluorescence emerged throughout the entire cell volume (in addition to remaining punctate fluorescence), indicating that a fraction of the CPP escaped from the endosomal/lysosomal pathway into the cytosol and nucleus (Figure 7C, D). For less active cyclic CPPs (e.g., CPP1 and CPP7), their fluorescence signals remained primarily punctate even after long periods of incubation.114 Second, energy depletion by treatment of HeLa cells with sodium azide and 2-deoxyglucose reduced the amount of cyclic CPP uptake by ~80%, whereas reducing the temperature to 4 °C (which blocks endocytosis) decreased cyclic CPP uptake by 95%.113 Finally, treatment of HeLa cells with endocytosis inhibitors such as 5-(N-ethyl-N-isopropyl)amiloride (EIPA) or methyl-β-cyclodextrin (MBCD) significantly reduced the cytosolic concentration of cyclic CPP1, whereas bafilomycin A1 (a specific inhibitor of vacuolar-type H+-ATPase) or wortmannin (a potent inhibitor of phosphoinositide 3-kinase that blocks endosomal maturation) had only minor effects.113 These observations again suggest that cyclic CPPs enter cells by endocytosis and escape from the early endosome into the cytosol.
Figure 7.
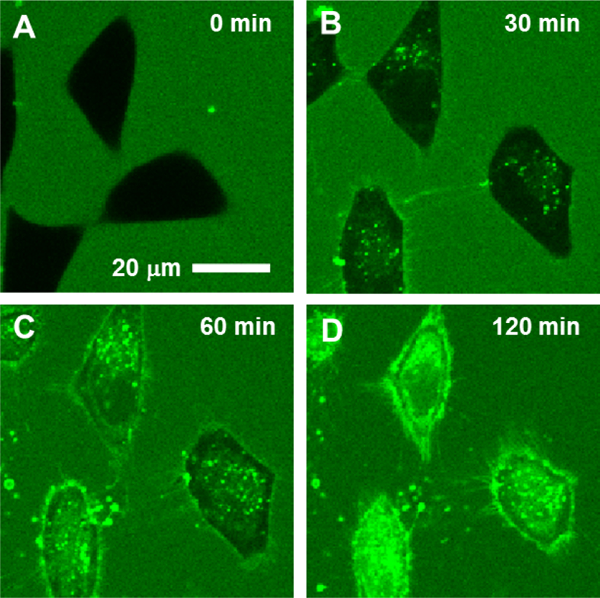
Time-lapse confocal microscopic images of HeLa cells upon treatment with 3 μM CPP9FITC (11) for 0 (A), 30 (B), 60 (C), or 120 min (D) at 37 °C. Reproduced from ref 114. Copyright 2019 American Chemical Society.
Our view is that, since CPPs can bind to various cell-surface components including the plasma membrane phospholipids, proteoglycans, and protein receptors (vide infra), any mechanism involving plasma membrane invagination may contribute to their uptake, including pinocytosis, clathrin-mediated and caveolin/lipid raft-mediated endocytosis. The relative contribution of each mechanism may differ depending on the CPP, cell type, and assay conditions. The conflicting reports in the literature may have been caused by an over-reliance on the use of chemical inhibitors and the detection of colocalization of CPPs with protein makers, neither of which are highly specific. It is also likely that inhibition of one uptake route may be compensated for by the activation of an alternative route(s), a well-known phenomenon in other fields such as cell signaling.
3.2.3. Cell Surface Receptors for CPPs.
Efficient endocytic uptake or induction of endocytosis requires association of CPP with the plasma membrane. For linear, cationic CPPs such as Tat and R9, it is well-established that charge-charge interactions are essential for establishing initial membrane-peptide contact. Guanidinium head groups effectively form bidentate hydrogen bonds with the phosphate groups of membrane phospholipids and/or the sulfate and carboxyl groups in glycosaminoglycans (GAG) such as heparan sulfate (HS) and chondroitin sulfate.139–145 HS has been well-established as an initiator for the endocytosis of macromolecular cargo.146 Both Tat and R9 bind to HS with high affinity in vitro (EC50 = 304 and 144 nM, respectively).113 Wallbrecher and colleagues evaluated the relationship between HS binding affinity and cellular uptake for disulfide-cyclized CPPs derived from human lactoferrin (hLF).147 Incorporation of four Arg residues (hLF +4R) resulted in ~50% improvement in cellular uptake and ~5-fold improvement in HS affinity (0.64 μM vs. 0.12 μM). Interestingly, guanidinium display on one face of an β-helix demonstrated optimal uptake when they were spaced ~9 Å apart, approximately corresponding to the average spacing of cell-surface proteoglycans.148 Additional support for the importance of HS in energy-independent internalization is provided by the use of HS-deficient CHO cells which demonstrate markedly reduced uptake of cationic CPPs. GAG cluster formation is also promoted by ceramide-induced lipid reorganization, supporting previous findings on the importance of ceramide formation by CPP’s on cellular uptake.149
Linear, cationic CPPs are also capable of binding directly to membrane phospholipids, although the binding affinity is dependent upon the membrane composition.150–152 In general, CPPs bind to the neutral plasma outer membrane with lower affinity than negatively charged membranes. For example, R9 bound to small unilamellar vesicles (SUVs) of plasma outer membrane composition with an EC50 value of >5 mM, while Tat showed minimal binding.114 Nevertheless, direct binding to the plasma membrane phospholipids likely contributes to the endocytic uptake of these CPPs. Several studies reported positive correlation between membrane binding affinity and the cellular uptake efficiency.114,153,154 Photocrosslinking studies using penetratin in DMPC/PCIIS membranes showed preferential association with short, unsaturated lipids that are typically found in higher disorder membrane regions.155 Furthermore, induction of membrane curvature was observed, in agreement with well-established methods for promoting endocytosis. CPP-induced membrane curvature in actin-encapsulated GUVs leads to deformations analogous to those preceding macropinocytosis, suggesting a mechanism by which direct CPP-cytoskeletal interactions lead to endocytic uptake.156
In addition to charge-charge interactions, CPPs containing hydrophobic groups may engage in hydrophobic interactions with the phospholipid membrane. For example, RW9 (RRWWRRWRR) inserted some of the hydrophobic groups into the phospholipid bilayer as suggested by molecular dynamics, with corresponding effects observed on lipid organization.157,158 By measuring the intrinsic Trp fluorescence, Jobin et al. determined that the aromatic rings of Trp residues in RW9 were inserted into the lipid bilayer to a depth of ~10 Å.159 Interestingly, incorporation of Trp into oligoarginine sequences also improves their affinity for GAG.160 Binding and crosslinking GAG by CPPs was thought to be essential for inducing endocytosis and required relatively high stoichiometry for effective internalization.
Finally, binding of cationic CPPs to cell-surface protein receptors, e.g., scavenger receptors, has been noted.161 Photocrosslinking studies with cationic linear CPPs functionalized with diazirine-containing residues identified chemokine receptor CXCR4 as a potential mediator of cellular uptake.162,163 Knockdown of CXCR4 expression reduced R12 uptake and confocal imaging revealed extensive CXCR4-R12 co-localization. However, uptake of Tat and R8 was not affected by CXCR4 knockdown, suggesting that receptor-mediated uptake is sequence-selective. Since then, additional CPP receptors have been identified, including peripheral membrane protein lanthionine synthetase component C-like protein 1 (LanCL1),164 whose overexpression increased R8 internalization.165 Syndecan-4, a ubiquitously expressed member of the syndecan family of transmembrane proteoglycans, has also been reported as a potential receptor responsible for the uptake of cationic CPPs such as Tat, penetratin and R8.165,166 Juks et al. reported that scavenger receptors (SR), a family of multifunctional receptors, are recruited to the membrane upon treatment with negatively charged CPPs.167 Two members of the class A SR family, SR-A3 and SR-A5, facilitated internalization of negatively charged CPP-nucleic acid complexes. Given the observation that a large number of cell surface receptors have been implicated in the uptake of Tat and polyarginines, it is likely that cationic CPPs bind nonspecifically to these cell surface receptors (e.g., through charge-charge interactions) and are brought into the endosomal/lysosomal pathway as a result of receptor internalization.
Compared to linear CPPs, cyclic CPPs exhibit greater binding affinity for phospholipid membranes, including the plasma outer membrane. Pei and co-workers measured the binding affinity for a panel of seven cyclic CPPs to SUVs of plasma outer membrane composition and obtained EC50 values of 0.17–2.2 mM, as compared to EC50 values of >5 mM for Tat and R9.114 Moreover, the endocytic uptake efficiency of the seven cyclic CPPs (as measured by flow cytometry) correlated well with their binding affinity for the plasma membrane. On the other hand, while Tat and R9 bound to heparin sulfate with apparent EC50 values of 304 and 144 nM, respectively, the cyclic CPPs showed minimal binding to HS under the same conditions.114 These observations suggest that the plasma membrane phospholipids may be the primary “receptors” for cyclic CPPs, although contribution from binding to other cell surface component(s) cannot be ruled out. The greater membrane-binding affinity of cyclic CPPs is likely the result of two factors: 1) the cyclic CPPs each contain two highly hydrophobic aromatic residues (Phe and Nal), which may insert into the lipid bilayer and make hydrophobic interactions with the lipids; and 2) cyclic peptides are more conformationally constrained and likely incur less entropic penalty during the binding event. Their lack of significant binding to HS is presumably because of insufficient number of positive charges (+4 for cyclic CPPs vs +9 for R9) and/or their cyclic structures not conducive to binding to the linear HS molecules.
3.2.4. Mechanism of Endosomal Escape.
Endosomal entrapment has been recognized as the key challenge for all non-viral drug delivery systems, including CPPs.168 For most of the non-viral delivery methods, the endosomal escape efficiency has been very poor, estimated to be 0.1% to a few percent.169 Lack of an efficient endosomal release system has undoubtedly contributed to the difficulty in understanding the mechanism of endosomal escape. Indeed, while there have been numerous studies on the mechanism(s) of action of CPPs over the past three decades, few dealt with endosomal escape. Further, until recently there had been no methodology available to directly measure the endosomal escape efficiency. As a result, the past attempts to improve the endosomal escape efficiency were either empirical or based on unsubstantiated mechanistic models. Fortunately, the past few years have witnessed several major advancements in the CPP field, including CPPs of vastly improved endosomal escape efficiencies as well as novel methods for directly assessing the efficiency of endosomal escape. These advancements have led to a novel mechanism for endosomal escape, as described below. The methods for evaluating CPP cytosolic delivery efficiency are described in detail under Section 5.
Several different mechanistic hypotheses have been put forward for the endosomal escape of non-viral delivery systems.170 A “proton sponge” hypothesis (Figure 8A)171–173 has often been invoked to explain the endosomal release of cationic polymer-nucleic acids complexes and is based on the buffering capacity of polyamines at the endosomal/lysosomal pH (4.5–6.5). A membrane fusion mechanism has been proposed for intracellular delivery of nucleic acids and proteins encapsulated inside lipid vesicles (Figure 8B).174,175 These two mechanisms are not applicable to CPPs, because the arginine sidechain (pKa~12) does not have buffering capacity at pH≤7.4 and CPPs are not encapsulated within lipid vesicles. In a third mechanism, which has been invoked for CPPs,176 peptides are proposed to generate barrel-stave pores, toroidal pores, or transient pores on the plasma or endosomal membrane and the CPPs and CPP-cargo conjugates/complexes passively diffuse through the pores (Figure 8C, D). This mechanism, however, cannot explain how macromolecular cargos such as large proteins covalently attached to or noncovalently associated with a CPP are delivered across the endosomal membrane. Furthermore, CPPs such as dfTat are capable of inducing the endosomal release of cargos (including proteins) that are not physically associated with the CPP in any manner.177 A fourth mechanism, which has gained increasing popularity in recent years, proposes that the delivery vector alone (e.g., CPPs) or the vector-cargo conjugate/complex interacts with and disrupts the endosomal membrane locally, thus allowing the cargo or vector/cargo complex to exit the endosome without complete destruction of the endosome (Figure 8E). A variation of this mechanism, termed “leaky endosomal membrane fusion”, has also been proposed.178–180 It states that CPPs induce membrane fusion between intraluminal vesicles or between intraluminal vesicles and the endosomal limiting membrane, generating membrane defects at the fusion junction. CPPs then pass through these membrane defects either directly into the cytosol or into the intraluminal vesicles first, followed by back fusion with the limiting membrane and cargo release into the cytosol. Unfortunately, the nature of disrupted membranes or membrane defects has not been defined, and it remains unclear how macromolecular cargos (e.g., those that are much larger in dimensions than that of the lipid bilayer) pass through the defects. Nor do they reconcile the observation that the endosomal release of nucleic acids formulated as lipoplexes or polyplexes occurs as multiple instantaneous “bursts”.181,182
Figure 8.

Proposed mechanisms of endosomal escape. (A) Proton sponge effect and osmotic lysis of the endosome/lysosome. (B) Membrane fusion mechanism for liposome-based delivery systems. (C) Barrel-stave pore formation. (D) Toroidal pore formation. (E) Membrane destabilization mechanism for polymer-based delivery systems; and (F) Vesicle budding and collapse mechanism.
Finally, a vesicle budding and collapse mechanism (Figure 8F) was recently proposed by Pei and colleagues, based on experimental evidence generated with highly active cyclic CPPs and giant unilamellar vesicles (GUVs) of late endosomal membrane composition.114 In this mechanism, CPPs bind to the intraluminal membrane of endosomes and gradually cluster into CPP-enriched lipid rafts, which subsequently bud off as small vesicles. For reasons that are not yet clear, the budded vesicle then disintegrates into an amorphous lipid/CPP aggregate shortly after budding off, or more frequently, as the vesicle buds off the endosomal membrane. Dissolution of the aggregate by the cytosolic milieu results in complete release of the vesicular contents into the cytosol. The budding and collapse hypothesis appears to provide a unifying mechanism for different endosomal escape events, of either natural (e.g., bacterial toxins and nonenveloped viruses) or unnatural origin (e.g., CPPs and various other non-viral delivery systems).170 Most importantly, the mechanism immediately clarifies many of the previously confusing and/or conflicting observations. For example, it readily resolves the dilemma of how large cargos such as proteins, plasmid DNA, and intact viral particles are transported across the endosomal membrane. It predicts that a small volume of the endosomal contents (i.e., volume inside the budded vesicle) would be released into the cytosol, whereas the bulk of the endosome would remain intact during and after each budding and collapse event. This is in agreement with previous observations that endosomal release does not cause the destruction of the endosome,181,182 while providing a simple explanation for how CPPs facilitate the endosomal release of cargos that are neither covalently attached to nor physically associated with the CPP.183,184 Multiple budding and collapse events may occur on the same endosome, either simultaneously or sequentially, until the endosome is depleted of the CPP.114 This is consistent with the reported endosomal release as multiple instantaneous bursts.181,182 It also explains how some molecules are nearly quantitatively released from the endosome without compromising the integrity of the endosome (vide infra).
3.2.5. What Causes the Superior Cell-Permeability of Cyclic CPPs?
As discussed in Section 3.2.3, cyclic CPPs have cytosolic entry efficiencies that are almost two orders of magnitude higher than that of linear CPPs. Schepartz and colleagues discovered that proper spatial arrangement of five arginine residues across the surface of an α-helix generated miniature proteins (5.3 and ZF5.3 (16)) of exceptional cytosolic delivery efficiency.122 Pellois et al. found that dimerization of a TMR-labeled Tat resulted in a linear CPP, dfTat (17, Figure 9) with greatly improved cell-permeability.185 Recently, Appiah Kubi et al. reported a family of non-peptidic cell-penetrating motifs (CPMs; e.g., compound 18, Figure 9) with ~5-fold higher cytosolic entry efficiency than cyclic CPP9 (11), making them the most efficacious CPPs/CPMs reported to date.186 Moreover, upon cytosolic entry, the CPMs and CPM-cargo conjugates are almost exclusively localized inside the mitochondrial matrix, suggesting nearly quantitative endosomal escape by the CPMs. These observations indicate that neither the peptidic structure nor cyclization is necessary for high cytosolic delivery efficiency.
Figure 9.
Structures of dfTat (17) and non-peptidic CPM3 (18).
For systems that enter cells by endocytosis, their cytosolic entry efficiency is governed by both the initial endocytic uptake and the efficiency of endosomal escape. Conventional linear CPPs such as Tat and R9 are limited by poor endosomal escape efficiency.109,114 The highly active cyclic CPPs (e.g., CPP9 (11) and CPP12 (12)), on the other hand, have moderately more efficient endocytic uptake than Tat (by ~3-fold), but greatly improved endosomal escape efficiency (by 9- and 18-fold, respectively, relative to Tat).114 Similarly, miniature protein 5.3, dfTat, and CPMs all have efficient endosomal escape.114,122,185,186 We conclude that efficient endosomal release is a critical parameter for successful cytosolic delivery, as it both improves the overall cytosolic delivery efficiency and minimizes any potential toxicity associated with endosomal entrapment.
What structural features, then, dictate the efficiency of endosomal release? During a vesicle budding event, the budding neck has the highest potential energy, akin to the transition state of a chemical reaction, because the saddle-shaped neck region involves drastic membrane distortion, away from the preferred lamellar structure (Figure 10A,B). The membrane curvatures at the neck have been described as the negative Gaussian curvature, which features simultaneous positive and negative curvatures in orthogonal directions. To “catalyze” the budding event (and endosomal release), a CPP (or other delivery vectors) must bind selectively to the budding neck and stabilize its structure (the “transition state”) relative to the lamellar structure (the “ground state”) (Figure 10B). To do so, the CPP needs to induce both positive and negative membrane curvatures at the same time, but in orthogonal directions. It is well established that insertion of hydrophobic group in between phospholipids, near the bilayer surface, generates positive membrane curvature (Figure 10C).187 The guanidinium group of arginine has been proposed to form bidentate hydrogen bonds with the phosphate groups of adjacent phospholipids, thereby bringing the head groups together and generating negative membrane curvature.188–190 Cyclic CPPs and CPMs are amphipathic and their guanidinium and hydrophobic groups are both required for high CPP activity.115,186 Cyclization in the case of cyclic CPPs and the rigid scaffold in CPMs (benzene) both create structural constraints that increase their binding affinity to the endosomal membrane. It is also possible that the guanidinium and hydrophobic groups in cyclic CPPs and CPMs are spatially presented in such a way that facilitates the formation of simultaneous positive and negative curvatures in orthogonal directions. When tested on GUVs that mimic the late endosome, CPP12 was indeed found to be concentrated at the budding neck (Figure 10B).114 Presumably, insertion of the aromatic side chain(s) of CPP12 into the luminal leaflet generates positive curvature in one direction (vertical direction in Figure 10A), while positioning the arginine side chains for simultaneous interactions with lipid head groups in the orthogonal direction (horizontal direction in Figure 10A). A linear correlation between endosomal escape efficiency and endosomal membrane binding affinity was also observed for a panel of 10 different CPPs (including cyclic CPPs, Tat, R9, and miniature protein 5.3).114
Figure 10.

Effect of negative Gaussian curvature at the budding neck on endosomal escape efficiency. (A) Selective binding of CPPs (indicated by small red cricles) to the saddle-shaped membrane surface at the budding neck and the associated negative Gaussian curvature (as shown, positive and negative curvatures in the vertical and horizontal directions, respectively, on the intraluminal leaflet). (B) Energy diagrams for the vesicle budding event in the absence and presence of CPP. (C) Scheme showing the induction of positive membrane curvature by insertion of hydrophobic groups in between phospholipids, whereas hydrogen bonding interactions between a guanidinium group of a CPP and two adjacent lipid molecules generate negative curvature by bringing the lipid head groups together.
3.3. Cell-Permeability by Direct Translocation
In addition to passive diffusion and endocytosis-mediated processes, a third well-documented route for the internalization of cyclic peptides is direct translocation. Most of the mechanistic studies have been carried out with linear CPPs and two terms, “direct translocation” and “direct transduction” have been coined by different researchers to describe the events occurring at low and high CPP concentrations, respectively.191 Since in neither case has the molecular mechanism been well defined, we will use the term “direct translocation” to describe both processes. Key features of direct translocation include: 1) it occurs on the plasma membrane (instead of endosomal membrane), resulting in the immediate availability of a CPP (or a CPP-cargo conjugate) inside the cytosol; 2) the CPPs are usually polycationic and highly hydrophilic (e.g., Tat and R9) and are not expected to passively diffuse through the lipid bilayer; 3) the process is energy-independent and can occur at 4° C (when endocytosis is largely shut down); and 4) it takes place much faster than endocytosis, usually occurring within a few minutes upon exposure of cells to the CPP. Below we summarize some of the experimental evidence for their presence and how they contribute to the internalization of cyclic CPPs.
Direct translocation becomes dominant when the CPP concentration exceeds certain thresholds, typically 10 μM. Duchardt et al. observed that treatment of HeLa cells with high concentrations of FAM-labeled R9 (20 μM) in the presence of chlorpromazine (CPZ), an endocytosis inhibitor, resulted in direct translocation.192 Time-lapse confocal imaging revealed that R9 formed regions of high fluorescent intensity at the plasma membrane, termed nucleation zones (NZ), from which fluorescent signal rapidly spread across the cell (Figure 11A). Internalization preceded by NZ’s had significant cell-to-cell variations with respect to the time necessary for intracellular signal to plateau. Membrane blebbing was observed, followed by the pinching-off of a highly fluorescent vesicle containing transiently confined CPP concomitant with rapid increases in cellular fluorescence. Electron microscopy uncovered that the NZ was not vesicle-associated, pointing to a non-endocytic pathway. By using Alexa 488-labeled R12 and confocal microscopy, Futaki and co-workers observed similar structures on the plasma membrane immediately before cytosolic fluorescence was visible.190 Co-staining with annexin-V revealed the presence of phosphatidylserine exclusively at the NZ sites, which has been interpreted as the result of highly localized membrane inversion events before internalization.
Figure 11.

Cellular entry of CPPs by direct translocation. (A) Scheme showing the formation of nucleation zones on the plasma membrane of a cell, followed by direct translocation across the membrane. (B) Confocal microscopic images of Hela cells upon treatment with 25 μM FITC-labeled cyclic CPP12 for 0, 5, and 10 min. CPP12 forms nucleation zones (bright fluorescent spots) on the cell surface prior to direct translocation.
Brock and colleagues showed that direct translocation of cationic CPPs required the activation of acid sphingomyelinase, which converts sphingomyelin into ceramide.193 Further work by Wallbrecher et al. confirmed that high membrane concentrations of sphingomyelin must be converted into ceramide before direct translocation could occur, whereas in sphingomyelin-deficient cell lines translocation readily occurred at CPP concentrations lower than expected.194 Ceramides generate negative curvature when present in the outer membrane, in agreement with the observed formation of membrane buds in the presence of cationic CPPs.190 SEM imaging of these budding regions, which are enriched in CPPs, revealed the presence of particles consisting of many multilamellar vesicles. Wong and colleagues also observed formation of membrane curvature as a pre-requisite for translocation and pointed out the unique capability of guanidinium groups to simultaneously form bidentate hydrogen bonds with multiple phospholipids and cause membrane deformation.189 Other investigators had observed a dynamin-dependence for translocation,192 which can be rationalized as direct translocation requiring considerable membrane restructuring. Further support of this concept was provided by the formation of extensive membrane heterogeneity upon CPP treatment, through restructuring of shorter-chain lipids.195 Takechi-Haraya et al. employed real-time in-cell NMR spectroscopy and R8 N-terminally modified with 19F-4-trifluoromethyl-L-phenylalanine (19F-R8) as a probe to monitor direct translocation into HL60 myeloid leukemia cells at 4 °C.196 Rapid non-endocytic internalization was observed at 80–100 μM concentrations, which proceeded through initial association with cell-surface GAGs, followed by permeation through the membrane into the cytosol. Time-dependent measurements indicated that extracellular R8 rapidly reached an equilibrium concentration in ~5 min, with a transient increase in GAG-bound peptide that then rapidly diminished and gradual increases in membrane- and cytosolic peptide until an equilibrium was reach in ~10 min. A prerequisite of GAG-clustering for direct translocation was also implicated in another study using WR9 at concentrations above 5 μM.197 Taken together, these results strongly support a unique, direct translocation mechanism that is inherent to cationic CPPs, requires high local membrane concentrations, and proceeds through membrane inversion/remodeling events prior to internalization.
Researchers have also explored the use of hydrophobic counter-ions to promote direct translocation of cationic CPPs at concentrations below the typical threshold. Pyrenebutyrate, a hydrophobic anion, was first used by Takeuchi et al. to promote direct translocation of polyarginine CPPs.198 Application to multiple CPPs by Guterstam et al. showed that anion pairing is a potentially general strategy for improving translocation efficiency, with the caveat that uptake is still cargo-dependent, as pyrenebutyrate/CPP/siRNA complexes did not demonstrate significant improvements.199
Interestingly, other researchers have also reported direct translocation processes at low CPP concentrations. For example, 1 μM fluorescently labeled Tat was reported to cause diffuse cytosolic fluorescence on time scales that suggest direct translocation.200 Initial reports demonstrated that Tat could induce biological responses at nanomolar concentrations, though in the presence of chloroquine which enhances endosomal escape.201 Sagan and co-workers observed differential rates of translocation and endocytosis using a MS-based quantification method, where translocation (presumed from 4 °C and proteoglycan-deficient cells) predominated at low concentration, converting to endocytosis as higher concentrations.202 This translocation process was insensitive to temperature and glycosaminoglycan identity,203 and appeared to occur within narrow time constraints, suggestive of interaction with a depletable membrane component. Further support for energy-independent direct translocation was provided by Pooga’s study with giant plasma membrane vesicles (GPMV).204 Cell-derived GPMVs have native-like membrane compositions but lack energy-dependent internalization mechanisms, making them an excellent platform for exploring direct translocation.205 Incubation of GPMVs with 1 μM fluorophore-labeled Tat showed intravesicular accumulation of Tat within minutes, suggestive of direct translocation. Greater accumulation was observed in GPMVs with reduced cholesterol content, suggesting that direct translocation relies mechanistically on dynamic, disordered membrane regions. Trypsin digestion of membrane-associated proteins suppressed accumulation, indicating involvement of cellular proteins. siRNA knockdown studies suggest nucleolin as a likely candidate for enhancing direct translocation. Other studies have reported reduced direct translocation at lower temperature as a function of declining membrane fluidity, but deconvolution from uptake by endocytosis is challenging. Tünnemann et al. showed that Tat conjugates accumulated in nucleoli on minute timescales and accumulation was inhibited by excess K+, indicating a role of membrane potential for energy-independent uptake.200 Wender and co-workers proposed that direct translocation occurred as cationic CPPs migrated across the membrane at a rate related to the overall membrane potential.206 Hyperpolarization with valinomycin and incubation with isotonic buffers raised and lowered uptake, respectively, commensurate with changes in membrane potential, although further model membrane studies have called the importance of membrane potential into question.207 Alternative mechanisms, discussed in later sections (e.g. pores, inverted micelles) are also candidates for rationalizing observed direct translocation events.
There have been relatively few studies on the direct translocation of cyclic CPPs. Parang and colleagues reported that cyclo(Trp-Arg)4 enters cells by direct translocation, based on the observation that reduction of temperature to 4 ᵒC or treatment with sodium azide did not eliminate uptake.118 Cardoso and co-workers also reported direct translocation by cyclic Tat and R10 peptides.116,117 They observed several-fold improvement in cellular uptake over linear Tat and R10 and rationalized the improvement as a result of greater distances between arginine side chains leading to faster and denser membrane association. The investigators later reported that attachment of cyclic Tat to GFP resulted in direct translocation of the CPP-protein conjugate into HeLa cells, although high concentrations (150 μM) were required.117 Pei and co-workers also observed that treatment of HeLa cells with 25 μM FITC-labeled cyclic CPP12 resulted in rapid appearance of diffuse fluorescence throughout the cell volume within minutes (Z. Qian and D.P., unpublished results). Nucleation zones were visible on the plasma membrane shortly after the addition of CPP12 and became numerous at 5 min (Figure 11B). At 10 min, intense, diffuse fluorescence was observed throughout the cell interior, without the intermediacy of punctate fluorescence observed at 3 μM concentration (Figure 7). These observations suggest that CPP12 too can enter cells through direct translocation.
Taken together, the above observations clearly demonstrate direct translocation as a third cellular entry mechanism for peptides, in addition to passive diffusion and endocytosis/endosomal escape. Direct translocation apparently occurs at both low and high CPP concentrations as well as different temperatures. We hypothesize that at low CPP concentrations, direct translocation is less robust and can only be observed when endocytic uptake is largely inhibited (e.g., at 4 ᵒC); at high CPP concentrations (>10 μM), direct translocation overtakes endocytosis and is readily observable. Future studies should focus on elucidating the molecular details of the nucleation zone and the membrane translocation process.
3.4. Cell-Permeability by Active Transport
A number of naturally occurring bicyclic peptides exert their biological activities by acting against intracellular targets (Figure 12). Analogous to their small-molecule counterparts, uptake of cyclic peptides may be facilitated by cell-surface protein transporters. Phalloidin (19, Figure 12), the first of seven bicyclic heptapeptidyl phallotoxins isolated from the death cap mushroom (Amanita phalloides), inhibits cytokinesis and cytotaxis by preventing the depolarization of actin filaments.208 Phalloidin has been demonstrated to enter hepatocytes through interaction with transporters from the organic anion transporter polypeptide (OATP) family and this is a potential mechanism for their cellular uptake.209 Similarly, the amatoxins, a family of bicyclic octapeptides which exert their cytotoxicity by selectively inhibiting mammalian RNA polymerase II [LD50 = 0.1 mg/kg in rats for α-amanitin (20, Figure 12)], have also been shown to undergo internalization in hepatocytes through the OATP family.210,211 Several other naturally occurring bicyclic peptides also exert their biologically activities by acting against intracellular targets, suggesting that they must be at least marginally cell-permeable, although their mechanisms of cellular entry have not been established. Moroidin (21, Figure 12) is a bicyclic octapeptide isolated from the seeds of Celosia argentea, which prevents eukaryotic cell division by inhibiting tubulin polymerization (IC50 = 3 μM).212 Celogentins A-K are a related class of bicyclic octapeptides, which are also derived from C. argentea and possess anti-mitotic activity.213 Among these compounds, Celogentin C (22, Figure 12) is most potent and inhibits tubulin polymerization with an IC50 value of 0.8 μM.214 Theonellamide F (23, Figure 12) induces damage to cellular membranes and stimulates Rho1 mediated 1,3-β-d-glucan synthesis.215 These examples demonstrate the potential for a diversity of cellular uptake mechanisms for naturally-occuring cyclic peptides.
Figure 12.

Examples of cyclic peptides that likely enter cells by active transport.
3.5. Cell-Permeability by Other Mechanisms
In addition to passive diffusion, direct translocation, active transport, and endocytosis-mediated processes, several alternative mechanisms have been proposed for the internalization of peptides. These processes have conventionally been examined using linear CPPs, but their mechanistic principles may be generalizable to other peptides including cyclic peptides. Experimental support for these pathways is largely confined to in vitro studies using giant unilamellar vesicles (GUVs) or in silico techniques. They may occur concomitantly with the four mechanisms described above or simply represent the underlying mechanism(s) for the observed direct translocation. In the sections below, a survey of other mechanisms that have been proposed in the literature is presented.
3.5.1. Membrane Thinning and Pore Formation.
Formation of transient pores on the plasma membrane has been proposed as an alternative, energy-independent pathway for membrane permeability. Since arginine-rich, cationic peptides have been well documented to form pores in bacterial and artificial membranes,216–218 it is not unreasonable to assume similar activity with mammalian cell membranes.219,220 Two types of membrane pores have been proposed: the barrel-stave model and the toroidal model. In barrel-stave pores, CPPs oligomerize to form a cylindrical barrel that inserts into the bilayer and forms a transmembrane channel (Figure 8C).221 In toroidal pores, the CPP-lipid head group interactions cause the outer and inner membrane leaflets to bend toward each other and become connected at the pore surface (Figure 8D).222,223 Molecular dynamics simulations employing model membranes combined with enhanced, non-physiological sampling techniques predicted pore formation for both Tat and penetratin.224,225 In these examples, pores arise due to high local CPP concentrations on the membrane leading to multiple co-localized insertion events that nucleate the formation of small toroidal-style pores. The experimental observation that cationic CPPs (e.g. Tat and R9) induce saddle-splay membrane curvatures has been used as evidence for pore formation,189 although alternative interpretations exist (e.g., vesicle budding and collapse114). Subsequent molecular dynamics simulations and in vitro studies showed that incubation with 7 μM R9, a concentration at which direct translocation is known to occur, led to permeabilization of model membranes.176 Sharmin et al. also reported rapid pore formation in GUVs on the order of a few minutes, which was promoted by high R9 concentrations.226
Concentration dependency observed for pore formation has been rationalized by the concept of CPP-induced membrane thinning. Sun et al. observed that a hydrocarbon stapled HIV capsid assembly inhibitor penetrated GUVs most effectively at a peptide : lipid ratio of 1:12 to 1:15.227 Further support of pore formation was provided by the observation that the inhibitor caused dye leakage from GUVs without apparent perturbation of the membrane integrity. Interestingly, other CPPs such as transportan 10 can translocate across the bilayer of a GUV without causing dye leakage from the GUV. To reconcile the latter observation, Yamazaki and colleagues also proposed the concept of transient pre-pores, a type of toroidal pores where hydrophilic lipid surfaces are in direct contact with water, in membranes under mechanical tension.228–230 It was proposed that CPPs act as stabilizers of these pre-pores, induce their formation, and then translocate through them. Further in vitro studies with GUVs provided support for peptide-induced pore formation,231–233 including estimation of the pore size,234,235 the kinetics of pore formation,236–238 and the concentration dependence.239 A mechanistic variant of pore formation, the sinking raft model, has also been proposed.240–242 In this model, internalization occurs as increasing local CPP concentration results in the formation of a peptide “raft”, which then sinks through the membrane, during which hydrophobic residues engage in lipid acyl chain-peptide interactions and hydrophilic residues coat the exterior of the raft. Sinking raft translocation has not seen significant experimental support and is likely a minor, if extant, contributor to energy-independent membrane translocation.
3.5.2. Inverted Micelles.
The formation of inverted micelles has been proposed as a potential mechanism for energy-independent membrane translocation of CPPs.243,244 Internalization proceeds through initial electrostatic membrane association leading to membrane lipid segregation and membrane thickening.245,246 Further increase in local peptide concentration in the segregated regions results in negative curvature and the formation of inverted micelles. Peptides entrapped inside the micelles travel through the membrane, fuse with the inner membrane leaflet and are subsequently released into the cytosol. Lamazière and colleagues observed that penetratin resulted in the formation of highly-curved membrane structures analogous to inverted micelles in model vesicles using NMR, X-ray diffraction and cryo-EM.247 Cationic CPPs (e.g. Tat, R8) were also demonstrated to form rod-like structures resembling inverted micelles in DMPC membranes by 31P NMR and TEM.248 Maniti et al. reported that penetratin interacts with varied lipid types resulting in membrane structures in which the peptide is completely engulfed by lipids.249 Coarse-grained molecular dynamics studies by Kawamoto et al. suggested the formation of multiple membrane deformation structures, including inverted micelles.250,251 Other supporting evidence include the observations that CPPs enhance lipid flipping between the inner and outer leaflets of membrane vesicles and CPP-dependent localization of anionic lipids on the inner leaflet.252
4. APPLICATION OF MECHANISTIC UNDERSTANDING TO DESIGN CELL-PERMEABLE CYCLIC PEPTIDES
Despite our incomplete understanding of the molecular mechanisms by which cyclic peptides enter the cell, rational design of cell-permeable cyclic peptides has been a flourishing area of research over the past decade. One objective is obviously to generate biologically active cyclic peptides against intracellular targets as therapeutic agents and chemical probes. An equally important goal has also been to gain further mechanistic understanding of cellular entry through the design of functional cyclic peptides. The reported cell-permeable cyclic peptides are classified according to their mechanisms of cell entry: passive diffusion vs endocytic uptake. Other macrocyclic peptides whose mechanisms of cellular entry are currently unknown, e.g., stapled peptides and cyclotides, are listed as a third category, although their actual mechanism may well be passive diffusion and/or endocytosis.
4.1. Designing Cell-Permeable Cyclic Peptides by Passive Diffusion
4.1.1. Natural Product Analogs.
Modification of natural products is a promising strategy for generating cell-permeable macrocycles with improved or new biological activities. The repurposing of cell-permeable natural product FR235222 (24, Figure 13) by Hilario et al. generated a cyclotetrapeptide scaffold (compound 25, Figure 13) that effectively internalized an aminocoumarin dye and had antiproliferative effects (IC50 = 0.27 μM against HEK293 cells), presumably by inhibiting the HDAC activity (IC50 = 35.8 nM).253 Another family of cell-permeable macrocycles, the orbitides, comprise a diverse family of ribosomally synthesized and post-translationally modified (RiPP) cyclic peptides.254,255 They tend to be rich in Leu and aromatic residues and highly hydrophobic, lacking Nα-methylation but occasionally containing backbone constraining motifs in the form of proline. Orbitides have demonstrated pro-apoptotic [e.g. (1–9-NaC)-crourorb A1256], immunosuppressive [e.g. labaditin257], antimalarial activities [e.g. ribifolin258]. Kaneda et al. prepared a series of analogs of odoamide (26, Figure 13), to evaluate the impact of changes in the peptidyl ring on passive permeability and potency and concluded that PAMPA permeability was robust even with varied structural changes (e.g., IC50 = 1.9 nM against A549 cells for compound 27, Figure 13).259 Through strategic incorporation of moieties to enhance the pharmacokinetic profile of anthelmintic natural product, cyclooctadepsipeptide PF1022A (28, Figure 13), Ohyama et al. obtained emodepside (29, Figure 13), which has been approved for veterinary applications.260
Figure 13.

Examples of cell-permeable cyclic peptide natural products that have been subjected to analog synthesis, with structural modifications highlighted in red.
4.1.2. Cyclic Peptide Scaffolds.
Improved mechanistic understanding of the processes underlying passively diffusible cyclic peptides has enabled the development of scaffold moieties with innate passive membrane permeability. Key strategies include elimination of solvent-accessible amide protons (e.g., through Nα-methylation) to reduce desolvation penalties on membrane partitioning and formation of transannular hydrogen bonding. Extensive libraries of scaffolds have been generated to better understand the impact of Nα-methylation patterns. For example, White et al. employed an on-resin strategy to synthesize large numbers of Nα-methylated cyclic peptides in a combinatorial fashion and systematically evaluated their passive permeability.261 Beck et al. reported specific patterns of Nα-methylation that conferred intestinal permeability, providing key insights on the SAR between Nα-methylation/conformation and permeability (e.g., compound 30, Figure 14).262 Recognizing the importance of intramolecular hydrogen bonding for membrane permeability, Yudin and co-workers cyclized peptides with a oxadiazole moiety to form conformationally rigid macrocycles with extensive intramolecular hydrogen bonding and correspondingly high passive permeability (e.g., compound 31, Figure 14).103 Similarly, Matsui et al. introduced conformation-restricting cyclopropane tethers (CPT) into cyclic peptides, resulting in large gains in permeability regardless of the peptide sequence, with some of the cyclic peptides demonstrating permeability superior to that of CsA (e.g., LLC-PK1 PE of 12.5 × 10−6 cm/s for compound 32, Figure 14).263
Figure 14.
Examples of synthetic, membrane-permeable cyclic peptides by the formation of intramolecular hydrogen bonds (highlighted in red).
To avoid the synthetic challenges associated with Nα-methylated residues, Kodadek and colleagues employed cycloalanine residues to eliminate backbone amides as well as introduce conformational rigidity, producing passively diffusible cyclic peptides with promising proof-of-concept biological activity.264 As reported by Rezai et al., varying the side-chain stereochemistry provides another avenue to optimize backbone geometry of a model cyclic peptide, with one scaffold demonstrating permeability rivaling that of CsA.95 Fouché et al. recently reported larger passively diffusible macrocycles of similar size to CsA, as opposed to prototypic macrocyclic scaffolds of ~5–7 residues.265 Extensive in vitro and in vivo characterization and derivatization of cyclic decapeptides of the general sequence AA-(NMe)AA-AA-(NMe)-d-AA-AA-(NMe)-AA yielded compounds that possessed extensive low-dielectric intramolecular hydrogen bonding as determined by NMR and correspondingly low polar surface areas. Further incorporation of proline to promote a β-hairpin conformation for backbone rigidification and transannular hydrogen bonding resulted in a macrocycle (compound 33, Figure 14) with 48% oral bioavailability. Although these sequences lacked target-binding activity, further development showed robust PK profiles even with the incorporation of hydrophilic residues (e.g. Thr, Tyr, [3-pyridinyl]Ala), suggesting that it might be possible to engineer target-binding activities into these scaffolds.266 In sum, while the aforementioned scaffolds generally lacked biological activity, they have served as valuable platforms for pushing the limits of side-chain diversity, backbone composition, and molecular size to facilitate understanding and design of a new generation of passively diffusible macrocycles.
4.1.3. Cyclic Peptidomimetics.
The passive permeability of cyclic peptides has also been enhanced by replacing the peptide bonds with the corresponding peptoids (Nα-substituted glycines), which has the same effect as Nα-methylation for reducing the number of backbone HBDs. Cyclization strategies for peptoids can influence cellular uptake, as demonstrated by Shin et al. with a series of amphipathic, neutral peptoids prepared to evaluate the differences in cellular uptake between linear and 1,3,5-triazine cyclized congeners.267 Lokey and colleagues generated a peptide-peptoid hybrid based on a previously identified membrane permeable cyclic peptide scaffold, termed a “peptomer”, which displayed 3-fold improvement in permeability.268 Peptoid substitutions including N-morpholines, N-phenols, and N-pyridines are well tolerated without compromising passive diffusion through MDCK cells. A follow-up study by Furukawa et al. further expanded the set of potential peptoid building blocks by preparing a library of ~400 distinct peptomer variants.269 Of the 400 compounds evaluated, 197 displayed good PAMPA permeability (Papp ≥ 1 × 10−6 cm/s), with the majority of them (154 total) having an ALogP in the range of 2–4, whereas less correlation was observed between permeability and molecular weight. These results, together with the observations described in Section 2.2, help establish hydrophobicity (as determined by LogP) as a new criterion for designing passively permeable macrocyclic peptides.
Cyclic peptide-peptoid hybrids consisting of predominantly peptoid units have been prepared. Schneider et al. reported a family of peptide-peptoid macrocycles that target the β-catenin-TCF interaction.87 Peptoid substituents consisted of predominately hydrophobic aromatic residues containing an ether (e.g. N-(isopropoxy-propyl)-glycine and N-(3,5-dimethoxy-phenyl)-glycine) at 5 peptoid positions and an N-terminal d-Ala for cyclization. The resulting compounds displayed robust biological activity against multiple cell lines for inhibition of β-catenin mediated signaling. One compound (34, Figure 15) demonstrated anti-proliferative effects against LNCaP-abl cells (IC50 = 0.195 μM) and reduced the volume of a model prostate tumor spheroid. Cai and co-workers synthesized and screened a OBTC library of macrocyclic peptidomimetics γ-AApeptides against the receptor tyrosine kinase EphA2 and discovered a lead compound (35, Figure 15), which bound to EphA2 with a KD value of 81 nM.270 Remarkably, the macrocyclic peptidomimetic is cell-permeable and potently antagonized EphA2-mediated signaling, although its mechanism of cellular entry was not investigated.
Figure 15.
Examples of synthetic, membrane-permeable cyclic peptidomimetics.
4.1.4. Cyclic Lipopeptides.
Lipidation is another common strategy for optimization of peptidyl leads into potential therapeutics. Incorporation of aliphatic groups increases the compound lipophilicity and can dramatically improve passive membrane permeability depending on the chain length and anchor position.271 Incorporation of lipophilic substituents can sometimes adversely affect the target binding affinity, and therefore alternative strategies that mitigate potential impact while retaining permeability enhancement is desirable. Schumacher-Klinger et al. employed a lipophilic prodrug charge masking (LPCM) strategy to convert Cilengitide, an integrin-targeting cyclic RGD pentapeptide [cyclo-(RGDf(NMe)V] containing multiple charged residues, into an aliphatically protected prodrug.272 Esterification of both guanidinium and carboxylate sidechains with hexyloxycarbonyl yielded protected groups that are readily removed by cytosolic esterases to yield the active compound. The protected prodrug demonstrated a >10-fold improvement in Caco-2 permeability (Papp from 0.07 to 0.82 × 10−6 cm/s) and an improved mouse PK profile. Mechanistic studies using these model peptides confirmed bioavailability through a transcellular process, demonstrating the effectiveness of this strategy for conferring improved membrane permeability.
4.1.5. Combinatorial Synthesis and Screening.
Hewitt et al. employed a combinatorial approach to rapidly generate structurally diverse natural product-inspired cyclic peptides by solid-phase peptide synthesis (SPPS), followed by screening to identify scaffolds that possess significant passive membrane permeability.82 Starting from a generic sequence Pro-X2–5-Tyr, they incorporated both l- and d-Leu and MeLeu at all four random positions, as well as Gly and MeGly at the X2 and X5 positions, to evaluate the effect of backbone flexibility on permeability. Passive permeability was assayed by PAMPA and contents of the acceptor well were structurally characterized and resynthesized. They observed a strong correlation between the 3D polar solvent accessible surface area, ΔGdesolv., and Caco-2 permeability, in agreement with the commonly accepted predictors of passive membrane permeability. NMR studies revealed effective intramolecular hydrogen bonding in most of the compounds. Interestingly, one of the compound series lacked any intramolecular hydrogen bond; rather, their backbone amides were occluded by the uniquely positions sidechains resulting in improved permeability.
Due to the reduced nucleophilicity of secondary amines, incorporation of multiple N-methylated amino acids into a cyclic peptide by SPPS using can be challenging. To combat this issue, investigators have developed multiple strategies for on-resin Nα-methylation. White et al. deprotonated the peptide backbone amides by treatment with LiOtBu followed by treatment with methyl iodide.261 Regioselectivity was observed as a function of backbone stereochemistry, offering a potential route to differentially Nα-methylated cyclic peptide isomers.
4.2. Designing Cell-Permeable Cyclic Peptides by Endocytic Uptake
Compared to passively diffusible cyclic peptides, a more general method for designing cell-permeable and biologically active cyclic peptides is through the integration of CPP motifs into macrocyclic peptides and by leveraging the endocytic mechanisms of the cell. Cyclic CPPs have provided a flexible platform that can be conjugated with cargo molecules in at least five different ways: exocyclic delivery, endocyclic delivery, bicyclic delivery, reversible cyclization, and noncovalent complexation (Figure 16). All of the resulting compounds (or complexes) can be considered as “cyclic peptides”, because they all contain a cyclic CPP. These cyclic peptides are expected to enter cells primarily by endocytosis and endosomal escape, as described above, but may also undergo direct translocation, especially at high concentrations. An added benefit of this method is that the resulting cyclic peptides are usually positively charged and have good aqueous solubility.
Figure 16.
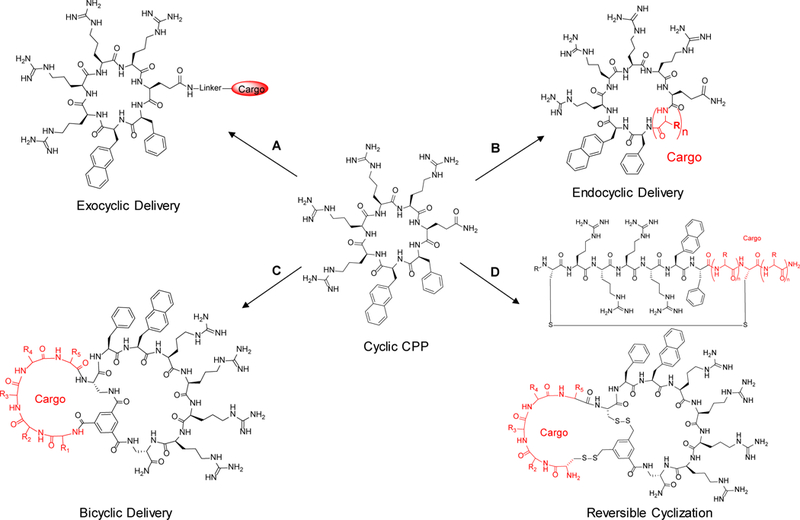
Different cargo delivery modes of cyclic CPPs. (A) Cargo conjugation to a cyclic CPP using the exocyclic strategy. (B) Incorporation of a cell-penetrating motif into a macrocycle for endocyclic delivery. (C) Incorproation of a second transport ring using the bicyclic delivery strategy. (D) Reversible cyclization of CPP-cargo fusion peptides into mono- or bicyclic peptides via disulfide bonds.
4.2.1. Exocyclic Delivery.
Exocyclic delivery involves covalent attachment of a cargo molecule to one of the side chains of a cyclic CPP. Typically, the cargo is attached to the side chain of the glutamic acid, which is not required for cellular entry, either directly or through a flexible linker (e.g., miniPEG, Figure 16A). This is the most versatile and cargo-tolerant conjugation method that has been applied to deliver a wide range of cargo molecules into the cytosol of mammalian cells. The cargo can potentially affect the cell-penetrating activity of the CPP but usually does not prevent cytosolic delivery. For example, a cargo (e.g., a highly negatively charged one) may reduce the CPP activity by physically interacting with the CPP in an intramolecular fashion and rendering the CPP less available for binding to the cell membranes. Other cargos (e.g., hydrophobic ones) may enhance the delivery efficiency by binding to the plasma and/or endosomal membrane. In such cases, experimentation with different linkers can minimize the cargo influence.274 Karpurapu et al. conjugated cyclic CPP9 to the N-terminus of a calcineurin-binding peptide, GPHPZIZITGPHEE (ZIZIT, where Z is tert-leucine) to generate a highly effective immunosuppressant, CPP9-ZIZIT (36, Figure 17), which binds to the NFAT-docking site on calcineurin and selectively blocks the dephosphorylation of NFAT and a small number of other substrates.275 Unlike cyclosporine A and FK506, CPP9-ZIZIT does not engage any other protein; as such, CPP9-ZIZIT is expected to be less toxic than cyclosporine A and FK506. In a mouse model of acute lung injury/acute respiratory distress syndrome induced by lipopolysaccharide (LPS), intranasal administration of CPP9-ZIZIT at 1 mg/kg effectively blocked the production of inflammatory cytokines (TNFα and IL6) and protected the animals against lethal doses of LPS.275
Figure 17.
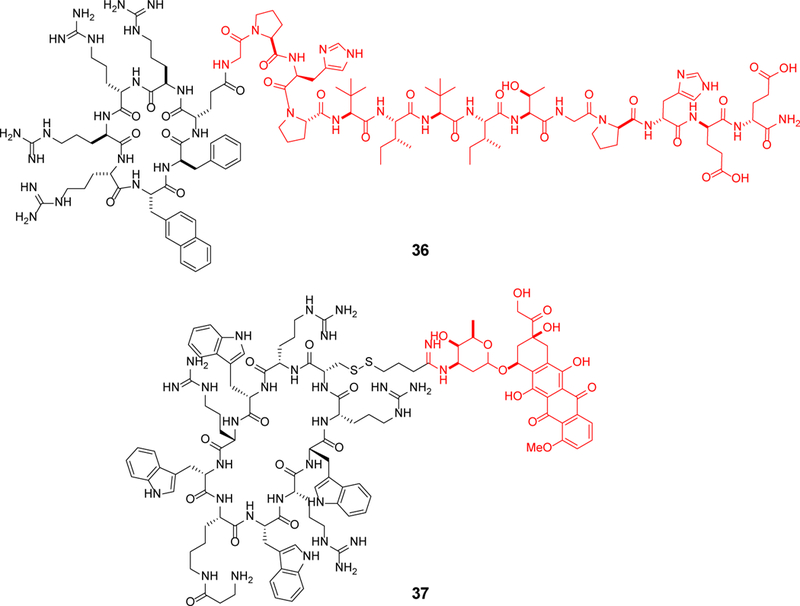
Examples of exocyclic CPP-cargo conjugates.
In addition to peptidyl cargos, cyclic CPPs have been employed to deliver small-molecule drugs and macromolecules (e.g., proteins) into the cytosol of mammalian cells. For example, Parang and co-workers attached the anticancer drug doxorubicin (Dox) to Cys-cyclo(WR)4-Lys through a disulfide bond and found that the conjugate, Dox-SS-[C(WR)4K] (37, Figure 17), was significantly less toxic in mouse myoblast cells compared to Dox at the same concentration while demonstrating enhanced cytotoxicity in a panel of cancer cell lines evaluated.276 Fluorescence microscopy exhibited that Dox-SS-[C(WR)4K] efficiently entered four different cancer cell lines and were localized in the nucleus. Further mechanistic study demonstrated that the level of intracellular reactive oxygen species in myoblast cells exposed to Dox-SS-[C(WR)4K] was reduced in comparison to Dox when co-treated with FeCl2, suggesting that Dox-SS-[C(WR)4K] has the potential to be used as Dox alternatives for anticancer treatment.
An exciting application of cyclic CPPs is intracellular delivery of proteins. Qian et al. used cyclic CPP1 to deliver green fluorescent protein (GFP) and protein tyrosine phosphatase 1B (PTP1B) into cultured mammalian cells.113 The internalized GFP was localized predominantly inside the nucleus, indicating efficient endosomal escape. Cytosolic delivery of PTP1B was also highly effective, as addition of low nanomolar concentrations of CPP1-PTP1B (but not PTP1B alone) dose-dependently reduced the phosphotyrosine levels of intracellular proteins. Nischan et al. conjugated cyclic Tat to GFP through site-specific copper-catalyzed azide-alkyne reaction.117 At high concentrations (50 – 150 μM), the cyclo-Tat-GFP conjugate demonstrated significant internalization in comparison to the linear counterpart, likely through direct translocation. Application to biologically active cargo was also demonstrated by Herce and Hackenberger who conjugated cyclic CPPs to nanobodies, which are camelid-derived single-chain VHH antibody fragments.277 Cyclic variants of Tat and R10 were prepared through side-chain macrolactamization before conjugation to GFP-binding nanobodies though expressed protein ligation. Incubation of 3T3 cells stably expressing GFP in the presence of 20 μM cyclic CPP-nanobody conjugates resulted in nucleolar redistribution of GFP. Interestingly, the cyclic CPP-nanobodies were able to bind extracellular antigens and efficiently internalize them into the cell. Further, conjugation of cyclic CPP to nanobodies through a disulfide bond allowed the Cy5-labeled nanobodies to be released inside the cell to perform intracellular immunostaining. For applications where a covalent linkage to a protein cargo is undesirable, this reversible disulfide-bond linkage can be employed, as was demonstrated by Schneider et al. in their delivery of mCherry by cyclo-R10. Upon entry to the cytosol, the disulfide bond is reduced by intracellular GSH and the cargo is liberated from the cyclic CPP to provide “traceless” delivery.278 These results demonstrate that cyclic CPPs are capable of efficiently delivering protein cargos into the cytosol of mammalian cells in their native forms. This opens the door to a wide range of therapeutic applications, e.g., intracellular enzyme replacement therapy (IC-ERT) for treating rare genetic diseases.
4.2.2. Endocyclic Delivery.
The cargo (especially when it is a peptide) may be inserted into the ring of a cyclic CPP to generate a larger cyclic peptide containing both the CPP and cargo motifs (endocyclic delivery; Figure 16B). Qian et al. systematically assessed the effect of sequence, ring size, and the stereochemistry of amino acid residues on the cellular entry efficiency of cyclic CPPs. They found that cyclohexa- to cyclooctapeptides represent optimal ring sizes; further increase in the ring size resulted in progressive and dramatic decrease in the cellular entry efficiency.115 They also found that amphipathicity is necessary for high CPP activity. Cyclic peptides containing two aromatic hydrophobic residues (e.g., Phe and Nal) and at least three (and preferably four) arginine residues are optimal, whereas cyclic R6 had little activity. These studies led to the discovery of cyclo(Phe-Nal-Arg-Arg-Arg-Arg-Gln) (CPP1) as a highly efficient CPP, with a cytosolic delivery efficiency 3- to 12-fold higher than that of linear CPPs such as Tat, penetratin, and R9.113 Next, by varying the stereochemistry of the amino acids, they discovered two exceptionally active CPPs: cyclo(phe-Nal-Arg-arg-Arg-arg-Gln) (CPP9, compound 11, where arg is d-arginine and phe is d-phenylalanine) and cyclo(Phe-phe-Nal-Arg-arg-Arg-arg-Gln) (CPP12, compound 12), which have 31- and 60-fold higher cytosolic delivery efficiencies, respectively, than Tat.114
Although limited to relatively short peptidyl cargos (1–5 aa), endocyclic delivery has the advantage (over exocyclic delivery) that the structural components may be designed to perform dual functions, i.e., the same building block is utilized for both cellular entry and target binding. For example, Bedewy et al. designed a cycloheptapeptide, cyclo(dpT-Pip-Nal-Arg-Arg-arg-arg) (38, Figure 18), as a cell-permeable and relatively potent inhibitor of peptidyl-prolyl cis-trans isomerase Pin1 (IC50 = 220 nM).279 This is, to our knowledge, the smallest cyclic peptide known to date that is cell-permeable by endocytosis and biologically active. In this case, Pip and Nal residues are critical for both cellular uptake and Pin1 binding. When added to cell culture, the cyclic peptide dose-dependently inhibited the intracellular Pin1 activity and reduced the viability of cancer (HeLa) cells but not that of non-cancerous cells. Upadhyaya et al. discovered a cycloundecapeptide as a cell-permeable direct K-Ras inhibitor, cyclorasin 9A5 (39, Figure 18), by screening a rationally designed one-bead-two-compound cyclic peptide library.280 Cyclorasin 9A5 orthosterically inhibited the Ras-effector protein interaction in vitro with an IC50 value of 120 nM and induced apoptosis in H1299 lung cancer cells (EC50 = 3 μM). SAR studies revealed that the same set of amino acids in cyclorasin 9A5 are required for both cellular entry and K-Ras binding. Parang and colleagues reported that some of their cyclic CPPs, e.g., cyclo[RW]5, are cell-permeable, moderately potent Src kinase inhibitors (IC50 = 2.8 μM).281 They also reported that a combination of acylation by long chain fatty acids and cyclization of short arginine-containing peptides results in cell-permeable cyclic peptides.282
Figure 18.
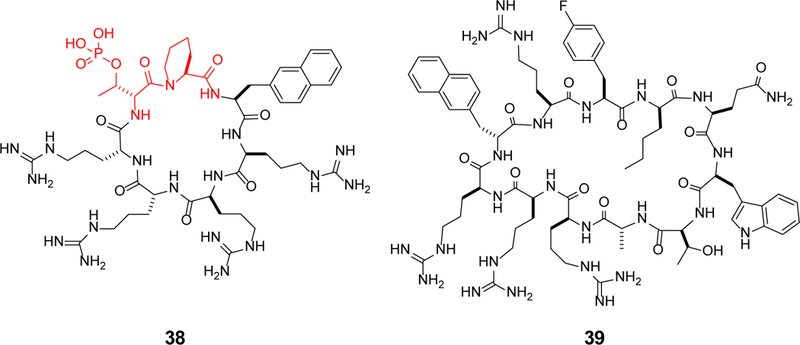
Examples of endocyclic CPP-cargo conjugates.
4.2.3. Bicyclic Delivery.
When the cargo is itself a cyclic peptide, it may be fused to a cyclic CPP to form a bicycle, in which one ring facilitates cellular uptake while the other binds to an intracellular target of interest (Figure 16C). Unlike endocyclic delivery, bicyclic delivery in principle can accommodate cargos of any size, because the cargo is confined in a separate ring and does not alter the size of the CPP ring. Lian et al. first demonstrated the validity of this strategy by fusing cyclic CPP1 to an impermeable, (phosphonodifluoromethyl)phenylalanine (F2Pmp)-containing cyclic peptidyl inhibitor against PTP1B, which was previously identified from a combinatorial library screening.283 Remarkably, the resulting bicyclic peptide (compound 40, Figure 19) retained the full inhibitory activity against PTP1B (KD = 37 nM), but became readily cell-permeable. Furthermore, the macrocyclic inhibitor was metabolically stable and highly selective for PTP1B, and potentiated insulin receptor signaling in HepG2 cells at nanomolar concentrations. Similarly, fusion of a previously discovered, membrane-impermeable cyclic peptidyl inhibitor of Pin1 with cyclic CPP1 produced a potent bicyclic Pin1 inhibitor that was highly active in cell culture assays (compound 41, Figure 19).284 Trinh et al. further demonstrated the generality of the bicyclic delivery strategy by synthesizing a combinatorial library of 5.7 million cell-permeable bicyclic peptides.285 Screening of the bicyclic peptide library against the G12V mutant K-Ras protein identified a K-Ras inhibitor that blocked the Ras-Raf interaction with an IC50 value of 3.4 μM, inhibited MEK and AKT phosphorylation, and induced apoptosis of lung cancer cells at low μM concentrations (compound 42, Figure 19). Rhodes et al. reported an interesting bicyclic peptide design that allowed the CPP ring to perform dual functions of cellular entry as well as targeting binding, thus substantially reducing the overall size of the compound.286 Briefly, the researchers synthesized a bicyclic peptide library in the OBTC format, in which the N-terminal ring consisted of a random peptide sequence of 3–6 amino acid residues, while the C-terminal ring contained a small library of different CPP sequences (12 total). The library was screened for binding to NF-κB essential modulator (NEMO), a protein required for canonical NF-κB signaling. One of the library hits, after limited optimization by conventional medicinal chemistry approach, inhibited the NEMO-IKKβ interaction with an IC50 value of 1 μM (compound 43, Figure 19). In silico modeling and alanine scan analysis confirmed that the CPP ring residues make intimate interactions with the NEMO protein surface, with the positively charged arginines interacting with an acidic patch next to the canonical IKKβ-binding site. Compound 43 inhibited NF-κB activation in a cell-based luciferase assay (IC50 = 10 μM) and reduced the viability of cisplatin-resistant ovarian cancer cells overexpressing NF-κB but not normal ovarian surface epithelial cells.
Figure 19.

Examples of bicyclic CPP-cargo conjugates.
Brock and colleagues evaluated the cell-permeability of bicyclic peptides containing different arginine compositions.287 Incorporation of an increasing number of arginine residues progressively increased heparan sulfate binding and the amount of cellular uptake. Synergy between cationic and hydrophobic aromatic residues was observed for cellular uptake, with Trp- and His-containing peptides demonstrating increased cellular uptake without varying overall charge. Perfluoroaryl-cyclized and arginine-rich bi- and tricyclic peptides possessing exceptional serum stability have been designed for the effective covalent delivery of antisense oligonucleotides, further demonstrating the benefits of structural rigidification.288
4.2.4. Reversible Cyclization.
For ligands that must be in their extended conformations for target binding, Qian et al. developed a reversible cyclization to enhance their cellular entry and metabolic stability (Figure 16D).289 The peptidyl ligand is fused with a short CPP motif at its N- or C-terminus and the fusion peptide is then cyclized by forming an intramolecular disulfide. Cyclization increases the peptide’s proteolytic stability as well as endocytic uptake; upon entering the mammalian cytosol (where the target protein is localized), the disulfide is reduced by intracellular glutathione to release the biologically active linear peptide for binding to the intended target. As a proof of principle, the investigators designed a peptidyl inhibitor against the PDZ domain of CFTR-associated ligand (CAL-PDZ), a protein that binds to the C-terminus of CFTR and chaperones CFTR to the lysosome for degradation (compound 44, Figure 20). Treatment of lung epithelial cells harboring a defective CFTR mutant (ΔF508) with the peptide inhibitor reduced lysosomal degradation, thereby increasing the amount of membrane-bound CFTR and improving the chloride ion channel activity. Formation of a monocyclic ring leaves a portion of the fusion peptide in the linear form, which remains susceptible to proteolytic degradation. Realizing this limitation, the same group later developed an improved and more general reversible bicyclization method for intracellular delivery of any linear peptidyl drug.290 Again, the cargo is fused to a short CPP motif (e.g., Phe-Nal-Arg-Arg-Arg-Arg) at its N- or C-terminus, but two cysteines are incorporated into the fusion peptide, one at the C-terminus and the other at the CPP-cargo junction. The resulting fusion peptide is converted into a bicycle by using a small-molecule scaffold, 3,5-bis(mercaptomethyl)benzoic acid. The scaffold forms an amide bond with the peptide N-terminus and two disulfide bonds with the implanted cysteine residues. Qian et al. applied the method to generate a bicyclic peptidyl inhibitor against the NEMO-IKK interaction (compound 45, Figure 20).290 The bicyclic NEMO inhibitor showed greatly improved proteolytic stability and cellular uptake efficiency. However, after cellular entry, the disulfides were reduced to release the active linear peptide, which was derived from the NEMO-binding domain of IKKβ. In a cell-based assay, the bicyclic NEMO inhibitor selectively inhibited the canonical NF-κB signaling with an IC50 value of ~20 μM.
Figure 20.
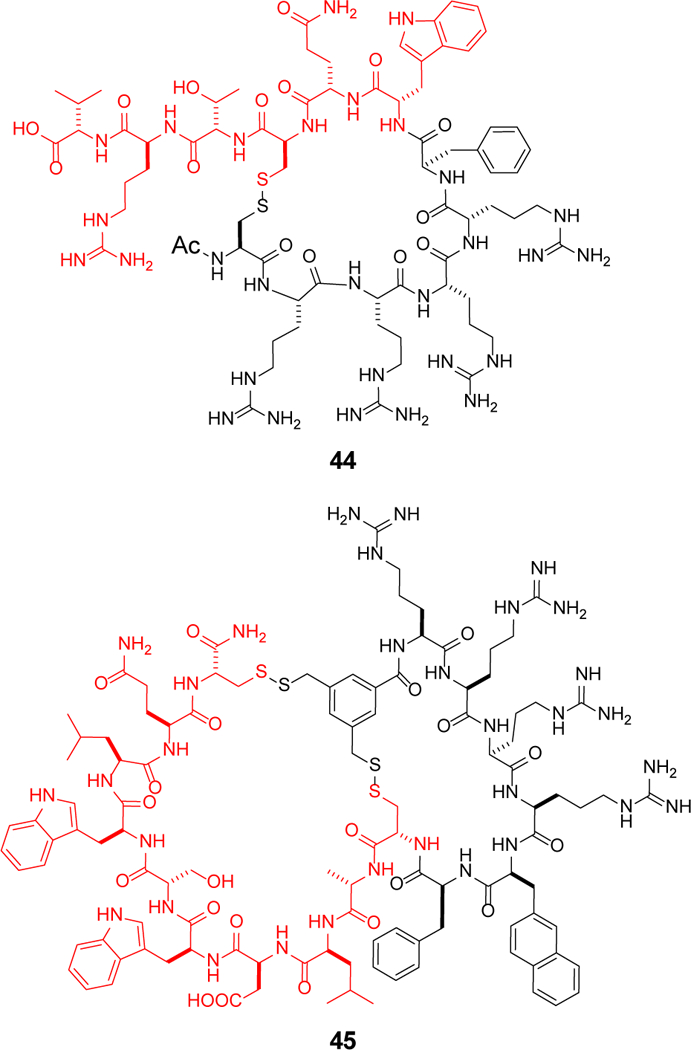
Reversibly cyclized bicyclic CPP-cargo conjugates.
4.2.5. Non-Covalent Complexation.
Parang and co-workers reported that non-covalent complexation of cyclo(WR)4 or cyclo(WR)5 with small-molecule and phosphopeptide cargos greatly increased the cellular entry of the latter.118 They later examined a number of linear and cyclic peptides containing alternating arginine and cysteine residues, e.g., cyclo(CR)4, for their capacity to deliver two molecular cargos, fluorescein-labeled cell-impermeable negatively charged phosphopeptide (F’-GpYEEI) and fluorescein-labeled lamivudine, into human leukemia cancer cells.291,292 They found that among all of the CPPs tested, cyclo(CR)4 was most effective in transporting the cargo into cells, enhancing the uptake of lamivudine and F’-GpYEEI by 16- and 20-fold, respectively. The mechanism by which the cyclic CPPs increase the uptake of non-covalently attached cargos has not yet been determined.
4.3. Cell-Permeable Cyclic Peptides by Unknown Mechanisms
4.3.1. Stapled Peptides.
Stapled peptides are α-helical peptides in which two side chains, usually at the i and i + 4 or i and i + 7 positions, are covalently crosslinked (“stapled”) to improve their helicity and proteolytic stability. They are therefore a special type of macrocyclic peptides. Hydrocarbon-stapled peptides have been developed to target a range of intracellular PPI drug targets including Bcl-2-BH3,293 MDM2-p53,3 ER-coactivator,294 protein kinase A,295 EGFR,296 and EZH2-EED,297 and have recently been reviewed.298 In particular, a stapled peptidyl inhibitor of the p53-MDM2 interaction (compound 46, Figure 21) has entered Phase 2 clinical trials, demonstrating the therapeutic potential of this class of compounds.299
Figure 21.

Examples of stapled peptides.
There have been many studies attempting to understand the mechanisms of cellular entry, the efficiency of cellular entry, as well as the parameters that govern the cellular entry efficiency of stapled peptides. With regard to the mechanism of cellular entry, fluid-phase macropinocytosis has been proposed as a possible mechanism, but how the peptides escape the endosome into the cytosol remains an open question.300 Li et al. have challenged this hypothesis by demonstrating that some of the reported MDM2-p53 inhibitors caused significant membrane disruption, implying that the peptides may enter cells through the disrupted regions of the plasma membrane and that the observed biological effects may not be pathway-mediated.301 Okamoto et al. reported similar findings for a previously reported stapled peptide inhibitor of the BH3 domain and suggested that the biological activity observed seemed to be a direct result of membrane interaction.302 Recent studies seem to suggest that stapled peptides with balanced hydrophobicity and hydrophilicity are able to partition into the lipid bilayer in significant amounts.303 When a high-affinity receptor is present inside the cell, the membrane-embedded peptides repartition back into the cytosolic side of the aqueous phase and become “entrapped” inside the cell by the protein receptors.304 This mechanism differs from classical passive diffusion in that in the absence of any protein receptor inside the cell, little peptide was found to enter the cell under otherwise identical conditions.
Recent surveys of hundreds of stapled peptide sequences by the Walensky and Verdine groups revealed that while for a small fraction of peptide sequences, hydrocarbon stapling results in significant cell-permeability, the greater majority of the stapled peptides have no to minimal cell-permeability.303,305 Understanding what structural features contribute to cell-permeability of stapled peptides is thus of paramount importance. To assess the effect of peptide sequence, stapling moiety, and other physiochemical properties on cellular entry, Bird et al. examined a family of hydrocarbon-stapled peptides mimicking the BCL-2 homology domain 3 (BH3) of pro-apoptotic protein BID and found that stapling positions that generate a hydrophobic face on the helix and enhance the helical propensity were key in ensuring cell-permeability.303 A follow-up study showed that maintaining an overall amphiphilicity resulted in the highest anti-proliferative activity against Mcl-1-dependent cells.306 Multiple studies concluded that the overall hydrophobicity of stapled peptides (as judged by HPLC retention time) is positively correlated with cellular uptake and a prerequisite when designing cell-permeable stapled peptide.307,308 Tian et al. compared different stapling strategies, including lactam, hydrocarbon, triazole, vinyl sulfide, m-xylene, and perfluoroaryl groups for their impact on the cellular uptake of a peptidyl inhibitor against the estrogen receptor-coactivator interaction.308 They also observed a clear correlation between hydrophobicity and cellular uptake, with ~3- to 5-fold increase in uptake when hydrophilic staples (e.g. lactam, triazole) were replaced with hydrophobic ones (e.g. hydrocarbon, m-xylene). These observations are consistent with the recent hypothesis that cellular entry requires partitioning of the stapled peptides into the hydrophobic region of the lipid bilayer. The challenge is to engineer optimal cellular entry efficiency while maintaining significant aqueous solubility and minimizing membrane lysis.
Zhao et al. used an N-terminal aspartic acid sidechain-to-backbone linkage to stabilize α-helical peptides that demonstrated improved cellular uptake and proof-of-concept biological activity against the estrogen receptor-coactivator interaction.309,310 Oba et al. incorporated cyclic d-amino acids into arginine-rich sequences and crosslinked their sidechains through a hydrocarbon linker, resulting in improvements in both proteolytic stability and cellular uptake over the linear counterparts.311 Fairlie and others synthesized highly constrained α-helical peptides by incorporating one, two, or three lactam (Lys/Asp) staples, generating a Bcl-xL inhibitor that showed low micromolar binding affinity and modest anti-proliferative activity in cellular assays (IC50 = 18 μM).312,313 Other investigators have incorporated positively charged residues (e.g. Arg, Lys) into stapled peptides to improve their cell-permeability. Speltz et al. appended a short cell-penetrating motif, Arg4, to the N-terminus of a membrane-impermeable hydrocarbon-stapled peptide.294 The resulting compound displayed robust biological effects against estrogen receptor α. Grossman and colleagues developed a cell-permeable stapled peptide as a β-catenin inhibitor by incorporating positively charged residues into the helical region as well as the termini of the peptide.314 Further modification of the inhibitor by replacing arginine residues with unnatural arginine analogs and adding an N-terminal nuclear localization signal resulted in 7- to 15-fold improvement in cellular uptake and substantially more potent cellular activity. Similarly, Quach et al. improved the cellular uptake of a hydrocarbon-stapled MDM2-p53 inhibitor through incorporation of cationic residues onto one face of the α-helix (compound 47, Figure 21).315 Unfortunately, the increases in cytosolic entry efficiency and anti-proliferative activity were associated with significant off-target toxicity, underscoring the challenge in balancing cellular uptake and toxicity.
While cation grafting is a valid approach to optimizing stapled peptide permeability, deleterious effects on target binding affinity and off-target toxicity may arise. Since stapled peptides do not require an overall positive charge for internalization, alternative strategies focused on optimizing overall physiochemical properties have also been explored.316 For example, Spiegel et al. identified hydrocarbon-stapled peptides that inhibit the Rab GTPase-effector protein interactions in the μM range, but the compounds displayed no biological activity as they were cell-impermeable.317 To render the peptide (StRIP3) biologically active, they installed a double hydrocarbon staple for proteolytic stability and carried out arginine scanning to enhance cellular uptake.318 However, incorporation of arginines resulted in large losses in binding affinity. The researchers therefore replaced the negatively charged residues with neutral analogs and incorporated additional hydrophobic residues (e.g., Trp). The resulting peptides were charge neutral and demonstrated modest cellular uptake. Kritzer and colleagues reported a series of autophagy-inducing stapled peptides which, despite carrying no net charge, possessed intrinsic cytosolic entry capability.319 Starting from a previously known autophagy-inducing peptide, Tat-Beclin 1, the investigators crosslinked two free Cys residues in the peptide with a library of bis-alkylating agents and optimized the peptide sequence at the same time. These efforts yielded an 11-mer peptide, DD5-o containing an i to i+3 o-xylene staple between two d-cysteine residues. DD5-o entered cells effectively and induced autophagy in vitro and in vivo (compound 48, Figure 21).
4.3.2. Cyclotides.
Cyclotides are a family of cysteine-rich polycyclic peptides containing both an N-to-C backbone cyclization and multiple intramolecular disulfide bonds.320 Multiple cyclotides and related cyclic peptides have been identified as plant-derived natural products, such as kalata B1,321 sunflower trypsin inhibitor 1 (SFTI-1),322 Momordica cochinchinensis trypsin inhibitor I and II (MCoTI-I, -II),323 and numerous others.324 Cyclotides have been shown to enter mammalian cells, likely by multiple forms of endocytic mechanisms (e.g. macropinocytosis, clathrin-mediated endocytosis), although endocytosis inhibitors only resulted in modest reduction in their uptake (~40%).325–327 Additional mechanisms contributing to cellular uptake, potentially including curvature-induced direct translocation,328 have been proposed and efforts to fully understand cyclotide internalization are currently ongoing.121 Efforts to optimize cellular uptake by D’Souza et al. through arginine incorporation and replacement resulted in modest, sequence-specific gains over the parent compounds.329 Engineered sequences demonstrated negligible toxicity, strong membrane binding, and internalization efficiencies of ~70% relative to Tat. Innate cell-penetrating activity and robust proteolytic stability have made cyclotides an attractive framework upon which biologically active peptidyl binding domains may be grafted. Ji et al. incorporated a phage-derived peptidyl inhibitor of the p53-MDM2 interaction into the MCoTI-I scaffold.330 The resulting engineered cyclotide, MCo-PMI, retained potent binding affinity to MDM2 (KD = 2.3 nM), robust human serum stability (t½ = 55 h), anti-proliferative activity in HCT116 cells (EC50 = 2 μM), and suppressed tumor growth in vivo through reactivation of the p53 pathway.
The extraordinary metabolic stability exhibited by cyclotides also allows them to be engineered into useful tumor imaging agents. Directed mutagenesis of an agouti-related protein (AgRP) resulted in a high-affinity ligand of αvβ3 integrin. Conjugation of the engineered cyclotide with chelating agent DOTA and 64Cu generated a tumor-specific imaging agent, which revealed wide tissue distribution and in vitro cellular uptake animal PET studies.331
Thell et al. reported that cyclotide kalata B1 (kB1), specifically [T20K]kB1, effectively inhibited T-cell proliferation via an IL-2-dependent mechanism in vivo.332 When tested in a mouse model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE), treatment with 10 mg/kg [T20K]kB1 7 days before immunization significantly reduced axon demyelination and inflammation. Impressively, an oral dose of [T20K]kB1 (at 20 mg/kg) resulted in significant systemic exposure and limited toxicity. A shortened dosing regimen (20 mg/kg, three times in 3 d intervals) resulted in statistically significant reduction in EAE progression.
4.3.3. Other Cyclic Peptides.
Many other methods have been explored to endow cell-permeability to cyclic peptides. Mechanistic studies were generally lacking or inconclusive to determine their internalization mechanism(s). Early on, researchers directly conjugated known CPP sequences (e.g., Tat, R9, or penetratin) to an exocyclic position of cyclic peptides, as demonstrated for a cyclic peptidyl inhibitor of Pin1111 and disulfide-cyclized DOCK2 inhibitors.333 As expected, the cellular entry efficiency is dictated by the CPP motif and generally low. Other investigators explored various spatial displays of guanidinium groups to optimize the cellular uptake of cyclic peptides.112,334 Traboulsi et al. explored various oligoarginine sequences in the context of linear, cyclic, and bicyclic scaffolds, as well as different combinations of endo- and exocyclic Arg residues.335 They reported that monocyclic oligoarginine peptides had superior uptake relative to their linear congeners of ≥7 residues, while bicyclic peptides had generally lower uptake, presumably because they are unable to adopt favorable membrane-binding conformations.
Other researchers have found that dimerization of certain impermeable sequences resulted in membrane permeability. Jang et al. designed dimeric α-helical peptidyl inhibitors against the HIV Tat-TAR interaction.336 They dimerized an amphipathic α-helical peptide consisting of leucine and lysine residues through a pair of disulfide bonds and the dimer, LK-4, showed potent binding to the TAR RNA sequence (KD = 0.059 nM) and robust cellular uptake (70–90% fluorescently positive cells at 10 nM concentration). Endocytosis was proposed as the predominant uptake mechanism at low concentrations (10 nM), as treatment with wortmannin, amiloride, or at 4 °C virtually eliminated uptake. At higher concentrations (e.g., 500 nM), the endocytosis inhibitors had negligible effect, suggesting alternate uptake mechanisms. Based on the peptides identified by Jang, Hyun et al. synthesized a series of hydrocarbon-stapled, Leu- and Lys-rich amphipathic peptides for siRNA delivery both in vitro and in vivo.337 Mechanistic studies supported multiple mechanisms of internalization for the most active peptide, stEK, as inhibition of macropinocytosis with EIPA reduced the uptake by 36% while inhibition of proteoglycan-mediated endocytosis by NaClO3 reduced uptake by 25%. Incorporation of multiple His residues further improved the endosomal escape efficiency and siRNA delivery as determined by target gene knockdown.338 The resulting His-containing siRNA-peptide complex, siRNA-LKH-stEK, resulted in 49–58% repression of connective tissue growth factor (CTGF) in mice following intradermal injection (100 nM).
Cell surface thiols have been exploited to confer cell-permeability to compounds that are otherwise impermeable to the cell membrane. Sagan and colleagues utilized an activated disulfide to deliver an impermeable peptidyl inhibitor of protein kinase C into the cell.339 Meng et al. developed a Cys-X-Cys motif (where X is any amino acid) motif as a general peptide delivery vehicle.340 Further application of these strategies has led to the development of a range of disulfide-containing CPPs.341–344 Apparently, the activated disulfides undergo exchange reactions with cell surface thiols, resulting in covalent attachment of the cargo to the cell surface. How the attached cargo subsequently crosses the plasma membrane is currently unknown. It was thought that internalization occurs through an endocytosis-independent pathway, providing a method for direct cytosolic cargo delivery and avoiding the pitfalls associated with endosomal entrapment.345
4.3.4. Formulation.
Optimizing pharmacokinetic profiles through selection of the correct formulation to ensure consistent biodistribution is an essential component of the small molecule development process. As most peptidyl therapeutics have traditionally been delivered via intravenous administration, inclusion of novel excipients is uncommon. As cyclic peptide clinical candidates emerge, formulation strategies to enhance their oral bioavailability have been developed. Specifically, self-emulsifying drug delivery systems (SEDDS) – mixtures of surfactants, oils, and co-solvents – have been successfully employed, for example in both the initial characterization of Circle Pharma’s CXCR786 inhibitors and in the clinic with CsA (trade name Neoral® by Sandimmune).346 In general, these formulations do not alter the inherent cell-permeability of a given compound, but instead ensure consistent and reliable biodistribution and are therefore beyond the scope of this review. Utilization of a formulation to directly enhance cell-permeability or endosomal escape is an intriguing area for further research as the field continues to mature. For example, Thean et al. screened several commercially available cationic lipid-based formulations in combination with stapled peptide MDM2-p53 inhibitors (e.g. ATSP-7041) and found that a proprietary formulation (Saint PhD) improved the EC50 for p53 activation by 2- to 6-fold.347 Confocal imaging studies with unformulated FAM-ATSP-7041 showed predominantly punctate fluorescence, whereas the formulated compound demonstrated more diffuse cytosolic fluorescence, suggesting that improved endosomal escape was a potential contributor to the enhanced cellular activity, although an alternative mechanism of cellular entry is possible.
5. METHODS FOR ASSESSING CELL-PERMEABILITY
Further mechanistic investigation of CPPs or improvement of their cellular entry efficiencies would be greatly facilitated by the availability of robust, convenient methods to quantitatively assess the efficiency of each step along the cellular entry pathway(s). At the minimum, methods are needed to quantify total cellular uptake efficiency (i.e., the total amount of cargo in all intracellular compartments), the cytosolic entry efficiency (defined as the ratio of cytosolic/extracellular cargo concentration), and the endosomal escape efficiency (i.e., the fraction of endocytosed cargo that exits the endosome/lysosome into the cytosol). Many different methods have already been developed to study the cellular entry of CPPs and can be broadly classified into label-based and label-free categories (Table 1). In the following sections, we will briefly discuss the methodologies and their advantages and disadvantages. It is clear that further improvement of the current methods and/or development of novel methods are highly desirable.
Table 1.
Methods for evaluating the cellular permeabilty of cyclic CPPs.
| Method | Label Probe | Readout | Section Reference |
|---|---|---|---|
| Confocal microscopy | Fluorophore | Fluorescence from labeled peptide | 5.1/ [114] |
| Flow cytometry | Fluorophore | Fluorescence from labeled peptide | 5.2/[355] |
| Mass spectrometry | Biotin, heteroatom, or no label | Mass Spectrometry | 5.3/[361,364,367] |
| Fluorescence correlation spectroscopy (FCS) | Fluorophore | Fluorescence from labeled peptide | 5.4/[369,370] |
| FACS-FCS | Fluorophore | Fluorescence from labeled peptide | 5.4/[371] |
| Glucocorticoid receptor-based assays | Dexamethasone | Luminescence from transcriptionally-controlled reporter or fluorescence from GFP | 5.5/[372,373] |
| Protein complementation assay | Split GFP fragment | Fluorescence after complementation of split protein fragments | 5.6/[374,376] |
| β-lactamase complementation assay | N-terminal fragment of β-lactamase | Shift in fluorescence from green to blue | 5.6/[376] |
| Enzymatic dephosphorylation | pCAP | Fluorescence from dephosphorylatedpCAP | 5.7/[113] |
| Kinase activity | Peptide-based substrates for respective kinase | Phosphorylated substrate peptide concentration | 5.7/[378] |
| Biotin ligase | Avi tag. HA tag | Western blot of biotinylated peptide | 5.7/[109] |
| β-galactosidase | Fluorescein di-β-D-galactopyranoside | Fluorescence from unmasked fluorescein | 5.7/[379] |
| Luciferin release | Luciferin | Luminescence | 5.7/[380] |
| Chloroalkane penetration assay (CAPA) | Chloroalkane | Fluorescence from dye-labeled chloroalkane | 5.7/[319] |
| mRNA splicing assay | Oligonucleotide | Fluorescence from GFP | 5.8/[288] |
| Radioactivity | Radionuclide | Radioactivity | 5.9/[383,384] |
| FRET | Fluorophore | Fluorescence | 5.10/[385,386] |
| Intrinsic fluorescence | None (for tryptophan-containing peptides) | Fluorescence | 5.10/[388] |
| Apoptosis | Doxorubicin | Cell viability | 5.10/[389] |
| PAMPA & Caco-2 | None | LC MS identification | 5.11/[92,391] |
5.1. Confocal Microscopy
Confocal microscopy is often the first experiment of choice to assess the cellular entry capabilities of a new CPP, because of the ease of experimental setup and its capacity to reveal the subcellular localization of the CPP. CPPs labeled with a fluorescent dye are incubated with primary cells, cultured cells or three-dimensional organoids followed by live-cell microscopy. Because CPP uptake is sensitive to factors such as temperature, number of cells, media serum content and imaging settings (e.g., laser exposure), it is of utmost importance to keep such experimental variables constant when comparing the uptake of different CPPs. It should be noted that fixation of cells using methanol or paraformaldehyde prior to imaging may lead to redistribution of membrane-bound CPPs to intracellular structures.348 Live cell confocal microscopy is therefore preferred to avoid these artifacts arising from fixation.
One of the primary advantages of confocal microscopy is the capacity to visualize the subcellular localization of the peptide by carrying out co-localization studies with various organelle-specific fluorescent markers. Such studies have allowed the identification of peptides that target specific subcellular structures, such as the mitochondria.186,349 Co-localization with GFP-fused Rab5 (early endosome), Rab7 (late endosome), and/or Lamp1 (lysosome) has been frequently employed to follow the intracellular trafficking of CPPs.350 Confocal microscopy has also been used to monitor the cellular entry of CPPs in real time, contributing significantly to the mechanistic studies of cyclic CPPs.113,114 However, confocal microscopy also has significant limitations. First, it is primarily a qualitative method, although high-content epifluorescence imaging coupled with custom image analysis algorithms has been used to semi-quantify fluorescence signals inside cells.303 Second, the fluorescent tag may interact with cellular contents and alter the cellular uptake efficiency of a CPP, either positively or negatively.351–353 Third, because fluorescent dyes are often highly sensitive to the environment, a number of factors may render fluorescence intensity an unreliable measure of the intracellular cargo concentration. For example, binding to intracellular components may either enhance the fluorescence intensity or result in fluorescence quenching.354 CPPs may undergo proteolytic degradation while outside the cell, during endosomal/lysosomal trafficking, or inside the cytosol, leaving the cleaved fluorescent tags in these compartments and resulting in overestimation of the CPP concentration.
5.2. Flow Cytometry
Flow cytometry provides an efficient method to rapidly assess the cell-penetrating activity of fluorescently labeled cyclic peptides.355 Typically, a fluorescently-labeled cyclic peptide of interest is incubated with ~10 × 104 primary or cultured cells for 1–4 h, and the cells are then suspended in a proper buffer solution and analyzed by using a fluorescence-activated flow cytometer.356 Flow cytometry provides a quantitative measurement of the total fluorescence in a cell population, is rapid, and has high throughput. By labeling peptides with different dyes of non-overlapping spectral properties, multiple peptides may be analyzed simultaneously and specific cells populations in a heterogeneous cell mixture may be isolated for further analysis. Additionally, flow cytometry is relatively tolerant to the dye properties, e.g., dyes that are not bright enough for other techniques can still be effective for flow cytometry analysis (vide infra). A major limitation of flow cytometry is that it does not differentiate peptides associated with the cell surface vs peptides internalized by the cells or peptides inside the cytosol vs peptides still entrapped inside the endosomal/lysosomal compartments. Whereas cell surface-bound peptides can be largely removed by exhaustive washing procedures (e.g., incubation with trypsin), the latter limitation has been particularly problematic, since conventional CPPs such as Tat and R9 are mostly entrapped inside the endosomes and lysosomes. Additionally, because flow cytometry requires dye labeling, it suffers from the same problem as confocal microscopy, in that the fluorescence label may significantly perturb the cellular entry properties of the CPP. Therefore, selection of a proper dye label should be a key consideration when using flow cytometry for assessing CPP uptake and the use of appropriate controls is essential.
Researchers have devised a number of strategies to differentiate CPPs inside the cytosol (and nucleus) from those entrapped inside the endosome/lysosome and quantify cytosolic delivery efficiency of CPPs by flow cytometry. Langel and others attached a disulfide-linked fluorescence donor-quencher pair to CPPs; upon entry into the cytoplasm, the disulfide bond is cleaved to release the quencher, resulting in an increase in the fluorescence yield of the donor.357 Qian et al. took advantage of the acidic pH inside the endosomes/lysosomes and employed a pH-sensitive fluorophore, naphthofluorescein (NF), as the reporter.358 NF has a pKa of ~7.8 and is nearly completely protonated and non-fluorescent (when excited at ≥590 nm) inside the acidic endosomes/lysosomes (pH<6) but becomes highly fluorescent once escaping into the cytosol (pH 7.4). Thus, the fluorescence intensity of NF-labeled peptides as measured by flow cytometry reflects the amount of peptides inside the cytosol and nucleus.
Besides specialty dyes such as NF, flow cytometry is amenable to a wide range of commercially-available dyes excited with multiple laser lines and assays can employ virtually any cell type, including stem and primary cells, lymphocytes, and monocytes.358,359 Mechanistic and kinetic studies can be readily performed using flow cytometry. Multiple investigators have evaluated cellular pathways responsible for uptake by pretreating the cells with endocytosis inhibitors, ATP-depleting conditions, and low temperatures to suppress active transport.200–203, 338–345
5.3. Mass Spectrometry
Burlina and colleagues employed MALDI-TOF mass spectrometry to quantify cellular uptake of Tat, R9, and penetratin.361 Cells were treated with biotinylated CPP, washed, and digested with trypsin to remove membrane-associated CPPs. Immediately before lysis, a known concentration of deuterated, biotinylated CPP was added to serve as an internal standard for MS quantification. Cells were lysed and the lysate was incubated with streptavidin-coated magnetic beads to recover the peptides, which were then washed and analyzed by MALDI-TOF. Quantification was achieved by comparing the peak intensity of the non-deuterated peptide (sample) to that of the deuterated peptide control (internal standard). This assay is highly reproducible; incubation of CHO cells with 7.5 μM peptide revealed final cellular concentrations of 4.5 ± 1 μM for R9, 3.5 ± 0.8 μM for penetratin, and 0.7 ± 0.2 μM for Tat. This method can also be applied to investigate intracellular peptide degradation by identifying proteolytic fragments in the lysate.362,363
An alternative MS-based method employed inductively coupled plasma-MS and CPPs labeled with low-abundance heteroatoms [e.g. Se in selenomethionine (MSe)].364 This method has wide dynamic ranges and low detection limits, and replacement of a CPP residue with MSe should have less impact on the physiochemical properties of the CPP. Møller et al. applied this strategy to analyze the uptake of penetratin containing a MSe (PenMSe), as compared to its fluorophore-labeled analog, TAMRA-Pen.365,366 MSe was incorporated during SPPS, displayed limited cytotoxicity, and was stable to the experimental conditions. Treatment of HeLa cells with PenMSe, TAMRA-PenMSe, and TAMRA-Pen followed by ICP-MS or LC-fluorimetry analysis showed cellular uptake of 4%, 15%, and 18%, respectively. These results demonstrate that while the incorporation of MSe has limited effect on uptake, the addition of TAMRA dye dramatically enhances cellular uptake efficiency.
A label-free MS-based method was reported by Rakowska et al.367 Adherent cells (e.g., human dermal fibroblasts) were incubated with a CPP, washed, trypsinized, and lysed. The lysate was fractionated by size-exclusion chromatography and then reversed-phase HPLC to isolate the CPP. Pure fractions were spiked with a known concentration of deuterated CPP analog and quantified using MALDI-TOF MS. They reported an intracellular concentration of 1.8 ± 0.24 μM for Tat following 3 h treatment of cells with 10 μM peptide, in reasonable agreement with previous findings. Note that while the MS-based methods provide an accurate measurement of the amount of CPPs internalized by cells without the interference of a fluorescent dye, they do not differentiate cytosolic from endosomally entrapped CPPs.
5.4. Fluorescence Correlation Spectroscopy
Fluorescence correlation spectroscopy (FCS) has emerged as a powerful technique for directly measuring the cytosolic concentration of fluorescently labeled CPPs. Briefly, FCS measures the fluorescence intensity in a small volume of a sample that is at equilibrium. The observed intensity fluctuates as a function of time as fluorescent molecules diffuse into and out of the small volume, giving rise to an autocorrelation function. This autocorrelation function relates to the duration of intensity fluctuations and can be correlated to the sample concentration by calculating the number of molecules that entered the viewing area.368 Weizenegger et al. employed FCS to evaluate the uptake efficiency of penetratin and an FGF-derived membrane translocating sequence (MTS) at low nanomolar concentrations and found that penetratin was internalized ~3-fold better than MTS.369 Schepartz and colleagues further expanded the study to a larger set of cationic CPPs and stapled peptides.370 Incubation of HeLa cells with 500 nM rhodamine-labeled peptides resulted in intracellular concentrations of 350 nM for cationic miniprotein ZF5.3 (16) and 253 nM for hydrocarbon-stapled peptide SAH-p53–8.
Rezgui et al. used a combination of FCS and flow cytometry to determine multiple parameters related to the cellular uptake of CPPs, such as cytosolic concentration, delivery efficiency, toxicity, internalization mechanism, and the extent of intracellular degradation.371 Cells were incubated with 1 μM fluorescently labeled CPP for 2 h at 37 °C and subjected to fluorescence-activated cell sorting (FACS) to obtain cells that had internalized the peptide and remained healthy. Lysates prepared from this cell population were then analyzed by FCS to determine the internalized CPP concentration. This procedure showed an uptake efficiency of ~5% for penetratin.
5.5. Nuclear Translocation
Kodadek and co-workers developed a high-throughput luciferase assay to measure the cell-permeability of peptides and peptoids based on the activation of glucocorticoid receptor (GR).372 They transfected cells with genes coding for firefly luciferase, Renilla luciferase, and a fusion protein consisting of the DNA-binding and dimerization domains of Gal4, the ligand binding domain of GR, and VP16 transactivation domain, Gal4-GR-VP16. In the absence of any GR agonist, this fusion protein is tightly bound to heat shock protein 90 (Hsp90) in the cytosol through the GR ligand binding domain. Treatment with a GR agonist, such as dexamethasone or a dexamethasone-CPP conjugate, disrupts this interaction in a dose-dependent manner and releases the fusion protein from Hsp90, allowing it to translocate into the nucleus. This leads to the activation of Gal4-responsive element and expression of firefly luciferase, while Renilla luciferase serves as a transfection control. Therefore, the firefly luciferase activity reflects the cytosolic concentration of dexamethasone or the dexamethasone-CPP conjugate.
Kodadek and co-workers used this assay to determine cell-permeability of a library of peptides and peptidomimetics. They first showed that conjugation with dexamethasone does not affect the relative cell-permeability of the test compounds. They then used it to compare the cell-permeability of a library of peptoid tetramers versus octamers, cyclic versus linear peptides,84 and peptides versus peptoids.85 Schepartz and co-workers later made two modifications to the Kodadek assay.373 In the first modification, termed “glucocorticoid-induced eGFP induction (GIGI)”, the firefly luciferase reporter was replaced by eGFP. The resulting eGFP fluorescence can be assessed by confocal microscopy, by flow cytometry, or on a plate reader. In the second modification, termed “glucocorticoid-induced eGFP translocation (GIGT)”, the Gal4 DBD-VP16 domains of the fusion protein were replaced by eGFP and the nuclear translocation of GR-eGFP was monitored. The relative cell-permeability of a dexamethasone-CPP conjugate is reflected by the extent of GR-eGFP nuclear translocation, which is in turn determined by calculating the ratio of the mean GFP signal in the nucleus to that of the surrounding cytosol. GIGT is particularly useful for applications during which a fast readout is desired (e.g., in RNA interference screening), which may not be possible for transcriptionally activated reporters. Compared to the original Kodadek assay, the modified methods are less costly since they do not require the expensive luciferase substrate and avoid potential false positive results caused by luciferase stabilization by allosteric inhibitors.
The above assays can be used in a high-throughput manner to screen compound libraries of moderate size as well as evaluating poorly permeable compounds due to signal amplification during transcription and translation. Most importantly, they provide functional assays for cytosolic delivery efficiency. However, these assays depend on the binding affinity of the dexamethasone-CPP conjugate to GR and conjugation with a CPP may affect dexamethasone binding to GR. As such, these assays are well suited for comparing the cytosolic entry efficiency of the same dexamethasone-CPP conjugate under different conditions, but caution should be exercised when comparing the cytosolic entry efficiency of different dexamethasone-CPP conjugates. These assays do not provide information about the mechanism by which compounds enter the cell and cannot be adapted easily for live-cell confocal microscopy.
5.6. Protein Complementation
Protein complementation provides a robust, convenient assay for assessing functional delivery of cargo to the cytosol. The assay involves splitting a protein into two fragments, neither of which is biologically active. Typically, the larger fragment is overexpressed inside the cytosol of target cells, whereas the small fragment (usually a short peptide) is chemically synthesized and fused to a CPP to be evaluated. Successful delivery of the CPP-peptide fusion into the cytosol results in reassembly of the two fragments into a functional protein, whose biological activity is readily quantified. Milech and co-workers developed a GFP-based assay, termed the “split-complementation endosomal escape (SEE)” assay.374 It is based on a prior observation that GFP can be split into two non-fluorescent fragments, GFP1–10 and GFP11, which can reassemble into a functional GFP.375 A CPP of interest is fused to GFP11 and cells overexpressing GFP1–10 are incubated with the CPP-GFP11 fusion. Successful cytosolic delivery of CPP-GFP11 leads to the reassembly of a functional GFP, whose fluorescent signal can be quantified by flow cytometry. Dowdy and co-workers have used this assay to screen a panel of hydrophobic peptides for their capacity to enhance endosomal escape.376 Stone et al. developed a similar complementation assay by using TEM-1 β-lactamase and a FRET-based substrate CCF2-AM.375 A CPP of interest is fused to the N-terminal fragment of β-lactamase (N-BLA_SpyT) while the C-terminal fragment (SpyC_C-BLA) is expressed inside cells. If the CPP successfully delivers the N-terminal fragment into the cytosol, the SpyTag/SpyCatcher pair brings together the two fragments to form a functional β-lactamase, which cleaves CCF2-AM and shifts its fluorescence from green to blue. The dose-dependent increase in the blue signal is monitored by flow cytometry and the extent of blue fluorescence increase reflects the relative cytosolic delivery efficiency of the CPP.
5.7. Enzymatic Activation
The cytosolic delivery efficiency of CPPs can be assessed by conjugating the CPPs to an enzyme substrate, which upon cytosolic entry undergoes a chemical modification by either an endogenous enzyme or a heterologous enzyme overexpressed inside the cytosol of target cells. Pei and co-workers labeled CPPs with phosphocoumaryl aminopropionic acid (pCAP) as a reporter for cytosolic and nuclear delivery.113 pCAP is non-fluorescent, but is rapidly dephosphorylated by the endogenous protein tyrosine phosphatases in the cytosol of mammalian cells to produce coumaryl aminopropionic acid as a fluorescent product, which was quantified by flow cytometry. Soughayer et al. exploited the activity of endogenous protein kinase B and Ca2+/calmodulin-activated kinase II to measure the cytosolic delivery of CPPs.378 Verdurmen et al. introduced a biotin ligase (BirA)-based assay to quantify the total cellular uptake as well as the cytosolic entry efficiencies of CPPs.109 This method exploits the capabilities of the avi tag, a 15-amino acid peptide, to act as a substrate for prokaryotic BirA (but not the eukaryotic biotin ligase). A peptide to be evaluated for cell-permeability was modified to contain a C-terminal avi tag and HA tag. Successful delivery of the avi tag into the cytosol rendered the peptide a substrate of BirA, which had been overexpressed inside the cells. The cell lysate containing biotinylated CPP construct was next subjected to western blotting with streptavidin and anti-HA antibody, the signals of which represent the cytosolic and total cellular uptake efficiencies, respectively. Similarly, Chao and Raines devised a fluorescence turn-on assay by employing an enzyme-substrate pair orthogonal to human cells.379 They expressed E. coli β-d-galactosidase in the cytosol of HeLa cells and used CPPs labeled with fluorescein di-β-d-galactopyranoside (FDG) as substrate. Successful delivery of FDG into the cytosol results in its hydrolysis by the expressed β-d-galactosidase and activation of fluorescein fluorescence. Wender and co-workers developed a luciferin-release assay using a novel releasable disulfide-carbonate linker system to tether the transporter peptide and luciferin (reporter cargo).380 Their design allows for the release of active luciferin in the reducing environment of the cytosol as a measure of peptide uptake and cargo release. Using cells transfected with luciferase, they implemented a real time assay to monitor luciferase signal that can possibly be used in transgenic mice to monitor drug uptake. Wadia et al. used a Cre recombinase-based assay and a loxP-STOP-loxP eGFP reporter to evaluate the internalization efficiency of Tat under a range of conditions.133
Kritzer and colleagues recently developed a quantitative, high-throughput HaloTag-based assay to assess cytosolic delivery efficiency, termed the “chloroalkane penetration assay (CAPA)”.319 HaloTags are modified bacterial haloalkane dehalogenases which can be stably expressed in mammalian cells and react irreversibly with halogenated substrates.381 In essence, CAPA is a “pulse-chase” assay, where HaloTag-expressing cells are incubated (“pulsed”) with chloroalkane-bearing CPPs, followed by incubation with a cell-permeable chloroalkane-containing dye (“chase”). Alkylation of HaloTag by CPPs that reach the cytosol prevents it from reacting with the dye, which is normalized and quantified as a dose-dependent reduction in fluorescent intensity. Differential cellular expression of HaloTags conjugated with mitochondrial- or nuclear-targeting sequences enables exclusive quantification of cytosolic or nuclear delivery. While the chloroalkane tag has relatively small effect on the physiochemical properties of a CPP, it was found that the linker between the CPP and HaloTag can have a profound effect on the as measured cytosolic entry efficiency. CAPA represents a functional assay for cytosolic delivery, is high throughput, and requires low sample volumes, making it an attractive option for cyclic CPP profiling. CAPA has been employed to compare the cellular uptake of linear peptoids and their cyclic counterparts,382 as well as for thioether-stapled autophagy-inducing peptides.319
5.8. mRNA Splicing
Wolfe and co-workers reported an exon-skipping assay to assess the uptake of antisense oligonucleotides.288 They used HeLa-654 cells stably transfected with a split eGFP construct containing a mutant intron responsible for the expression of a truncated, non-fluorescent eGFP. In the presence of cell-permeable phosphorodiamidate morpholino oligonucleotide (PMO) IVS2–654, which binds to the mutant intron, alternative splicing occurs resulting in the production of a functional (fluorescent) eGFP. The resulting eGFP signal was quantified by flow cytometry. The investigators used this assay to compare the cell-permeability of several CPP-PMO conjugates.
5.9. Radiolabeling
Radiolabeling is a relatively common biochemical technique and has been successfully applied to explore intracellular concentrations across a range of different moieties. For CPPs, Gammon et al. used a radioactivity-based assay to quantify the absolute intracellular concentration in cells free from the interference of fluorescent tags.383 They synthesized [99mTc] labeled peptides and measured the gamma activity of cell extracts to determine the concentration of intracellular CPP. The specific activity of the extracellular solution was also measured to normalize extracellular concentration of the [Tc]peptide to cell-associated activity. This assay allowed them to quantify the total cellular uptake of different peptides, but it did not distinguish cytosolic from endosomal peptides. This drawback was addressed by Zaro and Shen, who employed a similar radionuclide-based assay with 125I-labelled oligopeptides but conducted subcellular fractionation to separate peptides inside the cytosol from those located in intracellular vesicles.384 They also excluded membrane-bound CPPs by carrying out trypsin digest and heparin washes prior to fractionation and radioactivity measurement. However, radioactivity-based assays have yet to be used for CPPs conjugated with cargos and operational concerns with the use of radioactive isotopes may render them undesirable to some investigators.
5.10. Other Methods
Pellois and co-workers employed a FRET assay to study the CPP-assisted transduction of proteins.385 They labeled a CPP with fluorescein (donor) and conjugated the peptide to mCherry (quencher) via a disulfide linkage. CPPs localized in the extracellular space or endosomes emitted red mCherry fluorescence. After cytosolic entry, the disulfide is reduced and the CPP produced the green fluorescence of fluorescein. The investigators used confocal microscopy to quantify donor and FRET activation to study Tat-mediated protein delivery. Tsien and co-workers used FRET to study the cytosolic entry of oligoarginine CPPs by exploiting the highly specific interaction between FlAsH, a fluorescein biarsenical, and a tetracysteine-containing peptide.386,387 They expressed a tetracysteine-containing cyan fluorescent protein (CFP) in the cytosol of HeLa cells and incubated the cells with oligoarginine-FlAsH conjugates. Delivery of the peptide conjugates to the cytosol results in FRET-mediated quenching of the CFP signal. Jobin and Alves carried out label-free quantification of Trp-containing CPPs by measuring the intrinsic fluorescence of tryptophan.388 Aroui et al. evaluated the cellular entry of CPPs by conjugating them to doxorubicin and monitoring apoptotic cell death induced by doxorubicin.389
5.11. Methods for Determining Passive Permeability
To evaluate the in vitro permeability of peptides, two assays have been commonly used - parallel artificial membrane permeability assay (PAMPA) and Caco-2 cell-permeability. PAMPA is a cell-free assay that measures the passive permeability of peptides (and other drugs) across an artificial membrane composed of a lipid membrane and a porous hydrophobic support.390 Permeability coefficients can be determined in a high-throughput manner, as the assay depends on LC-MS (liquid chromatography-mass spectrometry) or UV detection of the peptides. Lokey et al. recently used this assay to study the effect of conformation on membrane permeability for 39 cyclic peptide natural products.92 Yudin et al. also used PAMPA to study the structure-permeability relationship of cyclic peptides with exocyclic amide bonds.80 Morimoto et al. introduced a novel fluorogenic Click label in order to improve peptide detection and tested their modified PAMPA assay with cyclic peptide derivatives.391
Caco-2 cell monolayers (a human colon epithelial cancer cell line) have been used as a model system to examine the absorption of peptides across the human intestinal epithelium.392 The flux of peptides across the monolayer is measured in order to calculate their permeability coefficients. Radiolabeled peptides were used in the original Caco-2 monolayer assays, while recent studies employ LC-MS, UV or fluorescence readouts. This assy has been used to study the mechanism of CPP uptake.393 Kessler et al. also used this assay to show that enantiomeric cyclic peptide pairs have different permeability coefficients.102
6. SUMMARY AND OUTLOOK
6.1. Summary
Since the beginning of this millennium, several major breakthroughs have been made in both peptide science and technology. The development of several powerful combinatorial library technologies has enabled the rapid discovery of potent, specific, and in many cases metabolically stable cyclic peptide ligands against essentially any protein target. The discovery of highly active CPPs (including cyclic CPPs and non-peptidic CPMs) allows these cyclic peptide ligands to be effectively delivered into the cell interior to engage intracellular targets. Great strides have also been made in elucidating the molecular mechanisms by which cyclic peptides enter the cell and the structural features conducive to cellular entry by different mechanisms. Cyclic peptides may enter the mammalian cell cytosol by at least four different mechanisms (Figure 22). Cyclic peptides that enter cells by passive diffusion across the plasma membrane tend to be relatively small (≤10 aa), contain predominantly hydrophobic side chains, and possess structural features that effectively reduce the number of exposed HBDs and HBAs when inside hydrophobic environments (e.g., Nα-methylation, formation of intramolecular hydrogen bonds, and/or steric occlusion of polar groups). To enter cells effectively by endocytic uptake and endosomal escape, a cyclic peptide should have a small ring size (≤9 aa) and contain 3 or 4 arginine residues and at least one but preferably two hydrophobic residues (especially aromatic hydrophobic residues such as Nal and Phe). The constrained amphipathic structures bind effectively to the plasma and endosomal membranes, enhancing the endocytic uptake and endosomal escape efficiencies, respectively. At high concentrations, these amphipathic cyclic peptides can also directly translocate across the plasma membrane, although the molecular mechanism(s) of direct translocation remains to be elucidated. Still other cyclic peptides have been demonstrated to enter the cell via active transport, by binding nonspecifically to and acting as substrates of cell-surface protein transporters (e.g., OATP). Finally, innovative assay methods capable of quantitatively assessing the individual steps of cellular entry (i.e., total cellular uptake, endosomal escape efficiency, and cytosolic delivery efficiency) have begun to emerge. These scientific and technical advancements have now made it possible to rationally design cell-permeable, biologically active cyclic peptides that efficiently enter the cell by either passive diffusion or endocytic mechanisms and engage specific intracellular targets (e.g., PPIs). This has in turn rendered cyclic peptides an exciting drug modality, which may provide the ultimate solution to the “undruggable” intracellular PPI targets.
Figure 22.

Overview of pathways utilized by cyclic CPPs to enter the cell. Left, some hydrophobic cyclic peptides have demonstrated ability to cross the plasma membrane by passive diffusion. Middle, other peptides can directly cross the plasma membrane by yet poorly defined mechanisms. A number of putative mechanisms including pore formation, carpet model, and inverted micelle have been put forward in the literature. Right, cyclic peptides are brough into the early endosome by various endocytic and pinocytic mechanisms and some of them efficiently escape the endosomal/lysosomal pathway into the cytosol.
6.2. Outlook
We expect a potentially explosive growth in cyclic peptide-related research and development during the coming decades, in both academia and industry. All of the major obstacles that confronted peptide drugs in the past – proteolytic liability, impermeability, and inaccessibility (in the case of cyclic peptides) have now been largely removed. Properly designed cyclic peptides (e.g., by incorporation of d-amino acids) and in our hands, most of the bicyclic peptides, are highly stable against proteolytic degradation. There are now numerous examples of synthetic cyclic and bicyclic peptides that bind to intended protein targets with antibody-like affinity and specificity.6–8 In this review, we show that cyclic peptides can now be engineered to be cell-permeable by passive diffusion or endocytic mechanisms. Therefore, the time seems to be ripe for a major effort in developing cyclic peptide-based therapeutics as well as for other applications (e.g., as targeting and imaging agents). In our opinion, the greatest potential of cyclic peptides lies in their capacity to target intracellular proteins that are currently undruggable by small molecules or monoclonal antibodies. A second area of opportunity is orally bioavailable cyclic peptides, which would have significant advantage over the current biologics with respect to convenience and patient compliance. A large number of predominantly hydrophobic cyclic peptides have already demonstrated good and even excellent oral bioavailability. Cyclic CPPs have also been shown to be orally bioavailable.114 A particularly exciting prospect is the possibility that conjugation to an orally bioavailable cyclic CPP may render an otherwise membrane-impermeable cargo molecule orally active. Finally, a third area of opportunity is intracellular biologics, i.e., the use of cyclic CPPs to deliver proteins (e.g., antibodies), peptides of various kinds, nucleic acids (e.g., siRNA), and protein-nucleic acid complexes (e.g., CRISPR/Cas9) into the cell. The capacity of cyclic CPPs to effectively deliver a wide range of biologically active cargos into the cell should also open up numerous opportunities in biomedical research.
ACKNOWLEDGEMENTS
We thank the Pei group members (past and present) for their contributions to the studies covered in this review and all of the reviewers for their constructive comments. Work in the Pei laboratory is supported by grants from the NIH (GM122459 and A139600), Cystic Fibrosis Foundation (PEI18G0), and Kimberly Professorship from The Ohio State University.
ABBREVIATIONS
- aa
amino acid
- AgRP
agouti-related protein
- BBB
blood brain barrier
- BH3
B-cell lymphoma-2 homology domain 3
- Cmax
maximum serum concentration
- CAPA
chloroalkane penetration assay
- CAL
CFTR-associated ligand
- CFP
cyan fluorescent protein
- CHO
Chinese hamster ovary
- CL
clearance
- CPP
cell-penetrating peptide
- CPM
cell-penetrating motif
- CPT
cyclopropane tether
- CsA
cyclosporine A
- CTGF
connective tissue growth factor
- CXCR
CXC chemokine receptor
- Dap
2,3-diaminopropionic acid
- dfTat
dimeric fluorescent Tat
- DOCK2
dedicator of cytokinesis 2
- DOS
diversity-oriented synthesis
- Dox
doxorubicin
- DPMC
1,2-dimyristoyl-sn-glycero-3- phosphocholine
- DTS
DNA-templated synthesis
- EAE
experimental autoimmune encephalomyelitis
- EGF
epidermal growth factor
- eGFP
enhanced green fluorescent protein
- EIPA
5-(N-ethyl-N-isopropyl) amiloride
- EphA2
ephrin type-A receptor 2
- ER
estrogen receptor
- EZH2-EED
enhancer of zeste homolog 2 - embryonic ectoderm development complex
- F2Pmp
(phosphonodifluoromethyl)phenylalanine
- FACS
fluorescence-activated cell sorting
- FAM
fluorescein amidite
- FCS
fluorescence correlation spectroscopy
- FDG
fluorescein di-β-d-galactopyranoside
- FITC
fluorescein isothiocyanate
- FRET
Fӧrster resonance energy transfer
- GAG
glycosaminoglycans
- GFP
green fluorescent protein
- GIGI
glucocorticoid-induced eGFP induction
- GIGT
glucocorticoid-induced eGFP translocation
- GPMV
giant plasma membrane vesicle
- GR
glucocorticoid receptor
- GUV
giant unilamellar vesicle
- HA
human influenza hemagglutinin (tag)
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- HDAC
histone deacetylase
- HIF
hypoxia inducible factor
- HIV
human immunodeficiency virus
- hLF
human lactoferrin
- HPLC
high performance liquid chromatography
- HS
heparan sulfate
- Hsp90
heat shock protein 90
- IC-ERT
intracellular enzyme replacement therapy
- ICP-MS
inductively coupled plasma - mass spectrometry
- IKK
IκB kinase
- IL6
interleukin-6
- Lamp 1
lysosome-associated membrane protein 1
- LanCL1
lanthionine synthetase component C-like protein 1
- LPCM
lipophilic prodrug charge masking
- LPS
lipopolysaccharide
- MALDI-TOF
matrix assisted laser desorption/ionization – time of flight
- MBCD
methyl-β-cyclodextrin
- Mcl-1
myeloid cell leukemia-1
- MCoTI-I, -II
Momordica cochinchinensis trypsin inhibitor I and II
- MDM2
mouse double minute 2 homolog
- MTS
membrane translocating sequence
- MW
molecular weight
- Nal/Φ
l-2-naphthylalanine
- NEMO
NF-κB essential modulator
- NF
naphthofluorescein
- NZ
nucleation zone
- OATP
organic anion transporting polypeptide
- OBOC
one-bead-one-compound
- OBTC
one-bead-two-compound
- PAMPA
parallel artificial membrane permeability assay
- pCAP
phosphocoumaryl aminopropionic acid
- PET
positron emission tomography
- Pip
l-piperidine-2-carboxylic acid
- PK
pharmacokinetics
- PMI
p53–Mdm2/MdmX inhibitor
- PMO
phosphorodiamidate morpholino oligonucleotide
- PPI
protein-protein interaction
- PSA
polar surface area
- PTP-1B
protein tyrosine phosphatase 1B
- pY
phosphotyrosine
- RaPID
Random non-standard Peptides Integrated Discovery
- RiPP
Ribosomally synthesized and Post-translationally modified Peptides
- Ro5
Rule of Five
- SAR
structure activity relationship
- SLCO2B1
solute carrier organic anion transporter
- SEE
split-complementation endosomal escape
- SEM
scanning electron microscopy
- SFTI-1
sunflower trypsin inhibitor 1
- SICLOPPS
split intein circular ligation of peptides and proteins
- SLC
solute carrier
- SLCO
solute carrier organic anion
- SPPS
solid-phase peptide synthesis
- SR
scavenger receptor
- SUV
small unilamellar vesicle
- Tmax
time to maximum concentration
- TAR
transactivation response element
- Tat
transactivator of transcription
- TCF
T-cell factor
- TEM
transmission electron microscopy
- TMR (TAMRA)
tetramethylrhodamine
- TNFα
tumor necrosis factor-α
- TPSA
total polar surface area
- ZF
zinc finger
- ΔGdesolv
free energy of desolvation
- t½
half-time
- EC50
effective concentration 50
- IC50
inhibitory concentration 50
- KD
dissociation constant
- kf
membrane-water partition coefficient
- LD50
lethal dose 50
- LogP
octanol-water partition coefficient
Biographies
Patrick G. Dougherty obtained a B.S in Chemistry and Biochemistry from Florida State University (2013). He then continued to The Ohio State University to pursue his Ph.D. in Professor Dehua Pei’s group. His research interests are focused on the discovery and optimization of peptidyl therapeutics through combinatorial, medicinal, and computational chemistry.
Ashweta Sahni obtained her B.Sc. (Hons.) in Chemistry (2014), and subsequently her M.Sc. in Chemistry (2016) from the University of Delhi, India. She then joined Prof. Dehua Pei’s research group at the Ohio State University in 2016 to pursue her Ph.D. in Biochemistry. Her current research focuses on elucidating the mechanism of endosomal escape of cell penetrating peptides.
Dehua Pei is the Charles H. Kimberly Professor of Chemistry and Biochemistry at The Ohio State University. He received his PhD degree in organic chemistry from University of California, Berkeley (1991), and was a Damon Runyon-Winchell Walter Cancer Fund postdoctoral fellow at Harvard Medical School (1992–1995) before joining the faculty at The Ohio State University (1995). He was promoted to Associate Professor in 2001 and Professor in 2004. His research group is currently developing new methodologies for combinatorial synthesis and screening of macrocyclic peptides, cell-penetrating molecules for drug delivery, and macrocyclic inhibitors against previously undruggable targets, such as intracellular protein-protein interactions. He is a co-Founder and the Chief Scientific Advisor to Entrada Therapeutics Inc. (Boston, MA), a privately owned biotechnology company specializing in the development of intracellular biologic drugs.
Footnotes
The authors declare the following competing financial interests: D.P. is a co-founder and shareholder of Entrada Therapeutics.
REFERENCES
- (1).Verdine GL; Walensky LD The Challenge of Drugging Undruggable Targets in Cancer: Lessons Learned from Targeting BCL-2 Family Members. Clin. Cancer Res. 2007, 13, 7264–7270. [DOI] [PubMed] [Google Scholar]
- (2).Gentilucci L; De Marco R; Cerisoli L Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Psuedo-Peptide Bonds, and Cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [DOI] [PubMed] [Google Scholar]
- (3).Chang YS; Graves B; Guerlavais V; Tovar C; Packman K; To K; Olson KA; Kesavan K; Gangurde P; Mukherjee A; et al. Stapled α-Helical Peptide Drug Development: A Potent Dual Inhibitor of MDM2 and MDMX for p53-Dependent Cancer Therapy. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Guillen Schlippe YV; Hartman MCT; Joesphson K; Szotsak JW In Vitro Selection of Highly Modified Cyclic Peptides that Act as Tight Binding Inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Morimoto J; Hayashi Y; Suga H Discovery of Macrocyclic Peptides Armed with a Mechanism-Based Warhead: Isoform-Selective Inhibition of Human Deacetylase SIRT2. Angew. Chem. Int. Ed. Engl. 2012, 51, 3423–3427. [DOI] [PubMed] [Google Scholar]
- (6).Yamagishi Y; Shoji I; Miyagawa S; Kawakami T; Katoh T; Goto Y, Suga H Natural Product-Like Macrocyclic N-Methyl-Peptide Inhibitors Against a Ubiquitin Ligase Uncovered from a Ribosome-Expressed De Novo Library. Chem. Biol. 2011, 18, 1562–1570. [DOI] [PubMed] [Google Scholar]
- (7).Abdalla MA; McGaw LJ Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review. Molecules 2018, 23, 2080–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zorzi A; Deyle K; Heinis C; Cyclic Peptide Therapeutics: Past, Present and Future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]
- (9).Thorstholm L; Craik DJ Discovery and Applications of Naturally Occurring Cyclic Peptides. Drug Discov. Today 2012, 9, 13–21. [DOI] [PubMed] [Google Scholar]
- (10).White CJ; Yudin AK Contemporary Strategies for Peptide Macrocyclization. Nat. Chem. 2011, 3, 509–524. [DOI] [PubMed] [Google Scholar]
- (11).Zorzi A; Deyle K; Heinis C; Cyclic Peptide Therapeutics: Past, Present and Future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]
- (12).Driggers EM; Hale SP; Lee J; Terrett NK The Exploration of Macrocycles for Drug Discovery – An Underexploited Structural Class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [DOI] [PubMed] [Google Scholar]
- (13).Hillman RA; Nadraws JW; Bertucci MA The Hydrocarbon Staple & Beyond: Recent Advances towards Stapled Peptide Therapeutics that Target Protein-Protein Interactions. Curr. Top. Med. Chem. 2018, 18, 611–624. [DOI] [PubMed] [Google Scholar]
- (14).Hosseinzadeh P; Bhardwaj G; Mulligan VK; Shortridge MD; Craven TW; Pardo-Avila F; Rettie SA; Kim DE; Silva D-A; Ibrahim YM; et al. Comprehensive Computational Design of Ordered Peptide Macrocycles. Science 2017, 358, 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Foster AD; Ingram JD; Leitch EK; Lennard KR; Osher EK; Tavassoli A Methods for the Creation of Cyclic Peptide Libraries for Use in Lead Discovery. SLAS Discov. 2015, 20, 563–576. [DOI] [PubMed] [Google Scholar]
- (16).Wada A Development of Next-Generation Peptide Binders Using In Vitro Display Technologies and Their Potential Applications. Front. Immunol. 2013, 4, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Huang Y, Wiedmann MM; Suga H RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem Rev. 2019, ASAP. [DOI] [PubMed] [Google Scholar]
- (18).Smith GP; Scott JK Libraries of Peptides and Proteins Displayed on Filamentous Phage. Methods Enzymol. 1993, 217, 228–257. [DOI] [PubMed] [Google Scholar]
- (19).Heinis C; Rutherford T; Freund S; Winter G Phage-Encoded Combinatorial Chemical Libraries Based on Bicyclic Peptides. Nat. Chem. Biol. 2009, 5, 502–507. [DOI] [PubMed] [Google Scholar]
- (20).Mund T; Lewis MJ; Maslen S; Pelham HR Peptide and Small Molecule Inhibitors of HECT-type Ubiquitin Ligase. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 16736–16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wrighton NC; Farrell FX; Chang R; Kashyap AK; Barbone FP; Mulcahy LS; Johnson DL; Barrett RW; Jolliffe LK; Dower WJ Small Peptides as Potent Mimetics of the Protein Hormone Erythropoietin. Science 1996, 273, 458–463. [DOI] [PubMed] [Google Scholar]
- (22).Scott CP; Abel-Santos E; Wall M; Wahnon DC; Benkovic SJ Production of Cyclic Peptides and Proteins In Vivo. Prc. Natl. Acad. Sci. U. S. A. 1999, 96, 13638–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wu H; Hu Z; Liu X Protein Trans-Splicing by a Split Intein Encoded in a Split DnaE Gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U. S. A. 1999, 95, 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tavassoli A; Benkovic SJ Genetically Selected Cyclic-Peptide Inhibitors of AICAR Transformylase Homodimerization. Angew. Chem. Int. Ed. Engl. 2005, 44, 2760–2763. [DOI] [PubMed] [Google Scholar]
- (25).Tavassoli A; Lu Q; Gam J; Pan H; Benkovic SJ; Cohen SN Inhibition of HIV Budding by a Genetically Selected Cyclic Peptide Targeting the Gag-TSG101 Interaction. ACS Chem. Biol. 2008, 3, 757–764. [DOI] [PubMed] [Google Scholar]
- (26).Miranda E; Nordgren IK; Male AL; Lawrence CE; Hoakwie F; Cuda F; Court W; Fox KR; Townsend PA; Packham GK; et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Roberts RW; Szostak JW RNA-Peptide Fusions for the In Vitro Selection of Peptides and Proteins. Proc. Natl Acad. Sci. U.S.A. 1997, 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Millward SW; Takahashi TT; Roberts RW A General Route for Post-Translational Cyclization of mRNA Display Libraries. J. Am. Chem. Soc. 2005, 127, 14142–14143 [DOI] [PubMed] [Google Scholar]
- (29).Goto Y; Katoh T; Suga H Flexizymes for Genetic Code Reprogramming. Nat. Protoc. 2011, 6, 779–790. [DOI] [PubMed] [Google Scholar]
- (30).Iwasaki K; Goto Y; Katoh T; Suga H Selective Thioether Macrocyclization of Peptides Having the N-Terminal 2-Chloroacetyl Group and Competing Two or Three Cysteine Residues in Translation. Org. Biomol. Chem. 2012, 10, 5783–5786. [DOI] [PubMed] [Google Scholar]
- (31).Lam KS; Salmon SE; Hersh EM; Hruby VJ; Kazmierski WM; Knapp RJ A New Type of Synthetic Peptide Library for Identifying Ligand-Binding Activity. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- (32).Houghten RA; Pinilla C; Blondelle SE; Appel JR; Dooley CT; Cuervo JH Generation and Use of Synthetic Peptide Combinatorial Libraries for Basic Research and Drug Discovery. Nature, 1991, 354, 84–86. [DOI] [PubMed] [Google Scholar]
- (33).Redman JE; Wilcoxen KM; Ghadiri MR Automated Mass Spectrometric Sequence Determination of Cyclic Peptide Library Members.” J. Comb. Chem. 2003, 5, 33–40. [DOI] [PubMed] [Google Scholar]
- (34).Siegel M; Huang J; Lin B; Tsao R; Edmonds CG Structures of Bacitracin A and Isolated Congeners: Sequencing of Cyclic Peptides with Blocked Linear Side Chains by Electrospray Ionization Mass Spectrometry. Bio. Mass Spectrom. 1994, 23, 186–204. [DOI] [PubMed] [Google Scholar]
- (35).Joo SH; Xiao Q; Ling Y; Gopishetty B; Pei D High-Throughput Sequence Determination of Cyclic Peptide Library Members by Partial Edman Degradation/Mass Spectrometry. J. Am. Chem. Soc. 2006, 128, 13000–13009. [DOI] [PubMed] [Google Scholar]
- (36).Wang P; Arabaci G; Pei D Rapid Sequencing of Library-Derived Peptides by Partial Edman Degradation and Mass Spectrometry. J. Comb. Chem. 2001, 3, 251–254 [DOI] [PubMed] [Google Scholar]
- (37).Lian W; Upadhyaya P; Rhodes CA; Liu Y; Pei D Screening Bicyclic Peptide Libraries for Protein–Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-α Antagonist. J. Am. Chem. Soc. 2013, 135, 11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gartner ZJ; Tse BN; Grubina R; Doyon JB; Snyder TM; Liu DR DNA-Templated Organic Synthesis and Selection of a Library of Macrocycles. Science 2004, 305, 1601–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Usanov DL; Chan AI; Maianti JP; Liu DR Second-Generation DNA-Templated Macrocycle Libraries for the Discovery of Bioactive Small Molecules. Nat. Chem. 2018, 10, 704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nielsen DS; Shepherd NE; Xu W; Lucke AJ; Stoermer MJ; Fairlie DP Orally Absorbed Cyclic Peptides, Chem. Rev. 2017, 117, 8094–8128. [DOI] [PubMed] [Google Scholar]
- (41).Wang CK; Craik DJ Cyclic Peptide Oral Bioavailability: Lessons from the Past. Biopolymers, 2016, 106, 901–909. [DOI] [PubMed] [Google Scholar]
- (42).Räder AFB; Weinmüller M; Reichart F; Schumacher-Klinger A; Merzbach S; Gilon C; Hoffman A; Kessler H Orally Active Peptides: Is There a Magic Bullet? Angew. Chem. Int. Ed. Engl. 2018, 57, 14414–14438. [DOI] [PubMed] [Google Scholar]
- (43).Avdeef A; Kansy M; Bendels S; Tsinman K Absorption-Excipient-pH Classification Gradient Maps: Sparingly Soluble Drugs and the pH Partition. Eur. J. Pharm. Sci. 2008, 33, 29–41. [DOI] [PubMed] [Google Scholar]
- (44).Dickson CJ; Hornak V; Pearlstein RA; Duca JS Structure-Kinetic Relationships of Passive Membrane Permeation from Multiscale Modeling. J. Am. Chem. Soc. 2017, 139, 442–452. [DOI] [PubMed] [Google Scholar]
- (45).Frézard F; Garnier-Suillerot A Permeability of Lipid Bilayer to Anthracycline Derivatives. Role of the Bilayer Composition and of the Temperature. Biochim. Biophys. Acta. 1998, 1389, 13–22. [DOI] [PubMed] [Google Scholar]
- (46).Mann SC; Andrews PA; Howell SB Comparison of Lipid Content, Surface Membrane Fluidity, and Temperature Dependence of cis-diamminedichloroplatinum(II) Accumulation in Sensitive and Resistant Human Ovarian Carcinoma Cells. Anticancer Res. 1988, 8, 1211–1215. [PubMed] [Google Scholar]
- (47).Geldenhuys WJ; Mohammad AS; Adkins CE; Lockman PR Molecular Determinants of Blood-Brain Barrier Permeation. Ther. Deliv. 2015, 6, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Mikitsh JL; Chacko A Pathways for Small Molecule Delivery to the Central Nervous System across the Blood-Brain Barrier. Perspect. Medicin. Chem. 2014, 6, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kell DB; Dobson PD; Oliver SG Pharmaceutical Drug Transport: The Issues and the Implications that It Is Essentially Carrier-Mediated Only. Drug Discov. Today 2011, 16, 704–714. [DOI] [PubMed] [Google Scholar]
- (50).Dobson PD; Kell DB Carrier-Mediated Cellular Uptake of Pharmaceutical Drugs: An Exception or the Rule? Nat. Rev. Drug. Discov. 2008, 7, 205–220. [DOI] [PubMed] [Google Scholar]
- (51).Galinis-Luciani D; Nguyen L; Yazdanian M Is PAMPA a Useful Tool for Discovery? J. Pharm. Sci. 2007, 96, 2886–2892. [DOI] [PubMed] [Google Scholar]
- (52).Hsiang B; Zhu Y; Wang Z; Wu Y; Sasseville V; Yang WP; Kirchgessner TG A Novel Human Hepatic Organic Anion Transporting Polypeptide (OATP2). Identification of a Liver-Specific Human Organic Anion Transporting Polypeptide and Identification of Rat and Human Hydroxymethylglutaryl-CoA Reductase Inhibitor Transporters. J. Biol. Chem. 1999, 274, 37161–37168. [DOI] [PubMed] [Google Scholar]
- (53).Busch AE; Karbach U; Miska D; Gorboulev B; Akhoundova A; Volk C; Arndt P; Ulzheimer JC; Sonders MS; Baumann C; et al. Human Neurons Express the Polyspecific Cation Transporter hOCT2, which Translocates Monoamine Neurotransmitters, Amantadine, and Memantine. Mol. Pharmacol. 1998, 54, 342–352. [DOI] [PubMed] [Google Scholar]
- (54).Dudley AJ; Bleasby K; Brown CD The Organic Cation Transporter OCT2 Mediates the Uptake of β-Adrenoceptor Antagonists across the Apical Membrane of Renal LLC-PK(1) Cell Monolayers. Br. J. Pharmacol. 1999, 56, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Grundemann D; Liebich G; Kiefer N; Koster S; Schomig E; Selective Substrates for Non-Neuronal Monoamine Transporters. Mol. Pharmacol. 1999, 56, 1–10. [DOI] [PubMed] [Google Scholar]
- (56).Lin L; Yee SW; Kim RB; Giacomini KM SLC Transporters as Therapeutic Targets: Emerging Opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yang NY; Hinner MJ Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods. Mol. Biol. 2016, 1266, 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mueckler M; Thorens B The SLC2 (GLUT) Family of Membrane Transporters. Mol. Aspects Med. 2013, 34, 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Colas C; Ung PM; Sclessinger A SLC Transporters: Structure, Function, and Drug Discovery. MedChemComm. 2016, 7, 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computation Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (61).Xiang TX; Anderson BD. The Relationship between Permeant Size and Permeability in Lipid Bilayer Membranes. J. Membrane Biol. 1994, 140, 111–122. [DOI] [PubMed] [Google Scholar]
- (62).Palm K; Stenberg P; Luthman K; Artursson P Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [DOI] [PubMed] [Google Scholar]
- (63).Goetz GH; Philippe L; Shapiro MJ EPSA: A Novel Supercritical Fluid Chromatography Technique Enabling the Design of Permeable Cyclic Peptides. ACS Med. Chem. Lett. 2014, 5, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Rossi Sebastiano M.; Doak BC; Backlund M; Poongavanam V; Over B; Ermondi G; Caron G; Matsson P; Kihlberg J Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. [DOI] [PubMed] [Google Scholar]
- (65).Leung SS; Mijalkovic J; Borrelli K; Jacobson MP Testing Physical Models of Passive Membrane Permeation. J. Chem. Inf. Model. 2012, 52, 1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Veber DF; Johnson SR; Cheng HY; Smith BR; Ward KW; Kopple KD Molecular Properties that Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- (67).Ritchie TJ; Macdonald SJF The Impact of Aromatic Ring Count on Compound Developability – Are Too Many Aromatic Rings a Liability in Drug Design? Drug Discov. Today 2009, 14, 1011–1020. [DOI] [PubMed] [Google Scholar]
- (68).Riviere JE; Brooks JD Predicting Skin Permeability from Complex Chemical Mixtures. Toxicol. Appl. Pharmacol. 2005, 208, 99–110. [DOI] [PubMed] [Google Scholar]
- (69).Burton PS; Conradi RA; Ho NF; Hilgers AR; Borchardt RT How Structural Features Influence the Biomembrane Permeability of Peptides. J. Pharm. Sci. 1996, 82, 1336–1340. [DOI] [PubMed] [Google Scholar]
- (70).Lau JL; Dunn MK Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [DOI] [PubMed] [Google Scholar]
- (71).Smolewski P; Robak T The Discovery and Development of Romidepsin for the Treatment of T-Cell Lymphoma. Expert Opin. Drug Discov. 2017, 12, 859–873. [DOI] [PubMed] [Google Scholar]
- (72).Faulds D; Goa KL; Benfield P Cyclosporin. A Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Use in Immunoregulatory Disorders. Drugs 1993, 45, 953–1040. [DOI] [PubMed] [Google Scholar]
- (73).Biron E; Chatterjee J; Ovadia O; Langenegger D; Brueggen J; Hoyer D; Schmid HA; Jelinek R; Gilon C; Hoffman A; Kessler H Improving Oral Bioavailability of Peptides by Multiple N-Methylation: Somatostatin Analogues. Angew. Chem. Int. Ed. Engl. 2008, 47, 2595–2599. [DOI] [PubMed] [Google Scholar]
- (74).Veber DF; Friedlinger RM; Perlow DS; Paleveda WJ; Holly FW; Strachan RG; Nutt RF; Arison BH; Homnick C; et al. A Potent Cyclic Hexapeptide Analogue of Somatostatin. Nature 1981, 292, 55–58. [DOI] [PubMed] [Google Scholar]
- (75).Mas-Moruno C; Rechenmacher F; Kessler H Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate Design, Synthesis, and Clinical Evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Ovadia O; Greenberg S; Chatterjee J; Laufer B; Opperer F; Kessler H; Gilon C; Hoffman A The Effect of Multiple N-Methylation on Intestinal Permeability of Cyclic Hexapeptides. Mol. Pharmaceutics 2011, 8, 479–487. [DOI] [PubMed] [Google Scholar]
- (77).Weinmüller M; Rechenmacher F; Kiran Marelli U.; Reichart F; Kapp TG; Räder AFB; Di Leva FS; Marinelli L; Novellino E; et al. Overcoming the Lack of Oral Availability of Cyclic Hexapeptides: Design of a Selective and Orally Available Ligand for the Integrin αvβ3. Angew. Chem. Int. Ed. Engl. 2017, 56, 16405–16409. [DOI] [PubMed] [Google Scholar]
- (78).Laufer B; Chatterjee J; Frank AO; Kessler H Can N-Methylated Amino Acids Serve as Substitutes for Prolines in Conformational Design of Cyclic Pentapeptides? J. Pept. Sci. 2009, 15, 141–146. [DOI] [PubMed] [Google Scholar]
- (79).Chatterjee J; Mierke D; Kessler H N-Methylated Cyclic Pentaalanine Peptides as Template Structures. J. Am. Chem. Soc. 2006, 128, 15164–15172. [DOI] [PubMed] [Google Scholar]
- (80).Hickey JL; Zaretsky S; St. Denis MA; Chakka SK; Monzur Morshed M.; Scully CCG; Roughton AL; Yudin AK Passive Membrane Permeability of Macrocycles Can Be Controlled by Exocyclic Amide Bonds. J. Med. Chem. 2016, 59, 5368–5376. [DOI] [PubMed] [Google Scholar]
- (81).Doedens L; Opperer F; Cai M; Beck JG; Dedek M; Palmer E; Hruby VJ; Kessler H Multiple N-Methylation of MT-II Backbone Amide Bonds Leads to Melanocortin Receptor Subtype hMC1R Selectivity: Pharmacological and Conformational Studies. J. Am. Chem. Soc. 2010, 132, 8115–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Hewitt WM; Leung SS; Pye CR; Ponkey AR; Bednarek M; Jacobson MP; Lokey RS Cell-Permeable Cyclic Peptides from Synthetic Libraries Inspired by Natural Products. J. Am. Chem. Soc. 2015, 137, 715–721. [DOI] [PubMed] [Google Scholar]
- (83).Bockus AT; Schwochert JA; Pye CR; Townsend CE; Sok V; Bednarek MA; Lokey RS Going out on a Limb: Delineating the Effects of β-Branching, N-Methylation, and Side Chain Size on the Passive Permeability, Solubility, and Flexibility of Sanguinamide A Analogues. J. Med. Chem. 2015, 58, 7409–7418. [DOI] [PubMed] [Google Scholar]
- (84).Kwon YU; Kodadek T Quantitative Comparison of the Relative Cell-permeability of Cyclic and Linear Peptides. Chem. Biol. 2007, 14, 671–677. [DOI] [PubMed] [Google Scholar]
- (85).Tan NC; Yu P; Kwon YU; Kodadek T High-Throughput Evaluation of Relative Cell-permeability between Peptoids and Peptides. Bioorg. Med. Chem. 2008, 16, 5853–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Boehm M; Beaumont K; Jones R; Kalgutkar AS; Zhang L; Atkinson K; Bai G; Brown JA; Eng H; Goetz GH; et al. Discovery of Potent and Orally Bioavailable Macrocyclic Peptide-Peptoid Hybrid CXCR7 Modulators. J. Med. Chem. 2017, 60, 9653–9663. [DOI] [PubMed] [Google Scholar]
- (87).Schneider JA; Craven TW; Kasper AC; Yun C; Haugbro M; Briggs EM; Svetlov V; Nudler E; Knaut H; Bonneau R; et al. Design of Peptoid-Peptide Macrocycles to Inhibit the β-Catenin TCF Interaction in Prostate Cancer. Nat. Commun. 2018, 9, 4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Wang CK; Swedberg JE; Harvey PJ; Kaas Q; Craik DJ Conformational Flexibility Is a Determinant of Permeability for Cyclosporin. J. Phys. Chem. B. 2018, 122, 2261–2276. [DOI] [PubMed] [Google Scholar]
- (89).Witek J; Keller BG; Blatter M; Messiner A; Wagner T; Riniker S Kinetic Models of Cyclosporin A in Polar and Apolar Environments Reveal Multiple Congruent Conformational States. J. Chem. Inf. Model. 2016, 56, 1547–1562. [DOI] [PubMed] [Google Scholar]
- (90).Witek J; Mühlbauer M; Keller BG; Blatter M; Meissner A; Wagner T; Riniker S Interconversion Rates between Conformational States as Rationale for the Membrane Permeability of Cyclosporines. ChemPhysChem 2017, 18, 3309–3314. [DOI] [PubMed] [Google Scholar]
- (91).Masuda Y; Tanaka R; Ganesan A; Doi T Systematic Analysis of the Relationship among 3D Structure, Bioactivity, and Membrane Permeability of PF1171F, a Cyclic Hexapeptide with Paralyzing Effects on Silkworms. J. Org. Chem. 2017, 82, 11447–11463. [DOI] [PubMed] [Google Scholar]
- (92).Ahlbach CL; Lexa KW; Bockus AT; Chen V; Crews P; Jacobsen MP; Lokey RS Beyond Cyclosporine A: Conformation-Dependent Passive Membrane Permeabilities of Cyclic Peptide Natural Products. Future Med. Chem. 2015, 7, 2121–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Price DA; Eng H; Farley KA; Goetz GH; Huang Y; Jiao Z; Kalgutkar AS; Kablaoui NM; Khunte B; Liras S; et al. Comparative Pharmacokinetic Profile of Cyclosporine (CsA) with a Decapeptide and a Linear Analogue. Org. Biomol. Chem. 2017, 15, 2501–2506. [DOI] [PubMed] [Google Scholar]
- (94).Bockus AT; Lexa KW; Pye CR; Kalgutkar AS; Gardner JW; Hund KC; Hewitt WM; Schwochert JA; Glassey E; Price DA; et al. Probing the Physicochemical Boundaries of Cell-permeability and Oral Bioavailability in Lipophilic Macrocycles Inspired by Natural Products. J. Med. Chem. 2015, 58, 4581–4589. [DOI] [PubMed] [Google Scholar]
- (95).Rezai T; Yu B; Millhauser GL; Jacobson MP; Lokey RS Testing the Conformational Hypothesis of Passive Membrane Permeability Using Synthetic Cyclic Peptide Diastereomers. J. Am. Chem. Soc. 2006, 128, 2510–2511. [DOI] [PubMed] [Google Scholar]
- (96).Furukawa A; Townsend CE; Schwochert J; Pye CR; Bednarek MA; Lokey RS Passive Membrane Permeability in Cyclic Peptomer Scaffolds Is Robust to Extensive Variation in Side Chain Functionality and Backbone Geometry. J. Med. Chem. 2016, 59, 9503–9512. [DOI] [PubMed] [Google Scholar]
- (97).Witek J; Wang S; Schroeder B; Lingwood R; Dounas A; Roth H-J; Fouché M; Blatter M; Lemke O; Keller B; Riniker S Rationalization of the Membrane Permeability Differences in a Series of Analogue Cyclic Decapeptides. J. Chem. Inf. Model. 2019, 59, 294–308 [DOI] [PubMed] [Google Scholar]
- (98).Rezai T; Bock JE; Zhou MV; Kalyanaraman C; Lokey RS; Jacobson MP Conformational Flexibility, Internal Hydrogen Bonding, and Passive Membrane Permeability: Successful In Silico Prediction of the Relative Permeabilities of Cyclic Peptides. J. Am. Chem. Soc. 2006, 128, 14073–14080. [DOI] [PubMed] [Google Scholar]
- (99).Pye CR; Hewitt WM; Schwochert J; Haddad TD; Townsend CE; Etienne L; Lao Y; Limberakis C; Furukawa A; Mathiowetz AM; et al. Nonclassical Size Dependence of Permeation Defines Bounds for Passive Adsorption of Large Drug Molecules. J. Med. Chem. 2017, 60, 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Lieb WR; Stein WD Implications of Two Different Types of Diffusion for Biological Membranes. Nature 1971, 234, 220–222. [DOI] [PubMed] [Google Scholar]
- (101).Filipe HAL; Salvador A; Silvestre JM; Vaz WLC; Moreno MJ Beyond Overton’s Rule: Quantitative Modeling of Passive Permeation through Tight Cell Monolayers. Mol. Pharmaceutics 2014, 11, 3696–3706. [DOI] [PubMed] [Google Scholar]
- (102).Marelli UK; Bezençon J; Puig E; Ernst B; Kessler H Enantiomeric Cyclic Peptides with Different Caco-2 Permeability Suggest Carrier-Mediated Transport. Chemistry 2015, 21, 8023–8027. [DOI] [PubMed] [Google Scholar]
- (103).Frost JR; Scully CCG; Yudin AK Oxadiazole Grafts in Peptide Macrocycles. Nat. Chem. 2016, 8, 1105–1111. [DOI] [PubMed] [Google Scholar]
- (104).Nielsen DS; Hoang HN; Lohman RJ; Hill TA; Lucke AJ; Craik DJ; Edmonds DJ; Griffith DA; Rotter CJ; Ruggeri RB et al. Improving on Nature: Making a Cyclic Heptapeptide Orally Bioavailable. Angew. Chem. Int. Ed. Engl. 2014, 53, 12059–12063. [DOI] [PubMed] [Google Scholar]
- (105).Hill TA; Lohman RJ; Hoang HN; Nielsen DS; Scully CC; Kok WM; Liu L; Lucke AJ; Stoermer MJ; Schroeder C,I et al. Cyclic Penta- and Hexaleucine Peptides without N Methylation Are Orally Absorbed. ACS Med. Chem. Lett. 2014, 5, 1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Mann DA; Frankel AD Endocytosis and Targeting of Exogenous HIV-1 Tat Protein. EMBO J. 1991, 10, 1733–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Derossi D; Joliot AH; Chassaing G; Prochiantz A The Third Helix of the Antennapedia Homeodomain Translocates through Biological Membranes. J. Biol. Chem. 1994, 269, 10444–10450. [PubMed] [Google Scholar]
- (108).Milletti F Cell-Penetrating Peptides: Classes, Origin, and Current Landscape. Drug Discov. Today 2012, 17, 850–860. [DOI] [PubMed] [Google Scholar]
- (109).Verdurmen WPR; Mazlami M; Plückthun A A Quantitative Comparison of Cytosolic Delivery via Different Protein Uptake Systems. Sci. Rep. 2017, 7, 13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Bechara C; Sagan S Cell-Penetrating Peptides: 20 Years Later, Where Do We Stand? FEBS Lett. 2013, 587, 1693–1702. [DOI] [PubMed] [Google Scholar]
- (111).Liu T; Liu Y; Kao H; Pei D Membrane Permeable Cyclic Peptidyl Inhibitors against Human Peptidylprolyl Isomerase Pin1. J. Med. Chem. 2010, 53, 2494–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Mitchell DJ; Kim DT; Steinman L; Fathman CG; Rothbard JB Polyarginine Enters Cells More Efficiently Than Other Polycationic Homopolymers. J. Pept. Res. 2000, 56, 318–325. [DOI] [PubMed] [Google Scholar]
- (113).Qian Z; LaRochelle JR; Jiang B; Lian W; Hard RL; Selner NG; Luechapanichkul R; Barrios AM; Pei D Early Endosomal Escape of a Cyclic Cell-Penetrating Peptide Allows Effective Cytosolic Cargo Delivery. Biochemistry 2014, 53, 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Qian Z; Liu T; Liu Y-Y; Briesewitz R; Barrios AM; Jhiang SM; Pei D Efficient Delivery of Cyclic Peptides into Mammalian Cells with Short Sequence Motifs. ACS. Chem. Biol. 2013, 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Lättig-Tünnemann G; Prinz M; Hoffmann D; Behlke J; Palm-Apergi C; Morano I; Herce HD; Cardoso MC Backbone Rigidity and Static Presentation of Guanidinium Groups Increases Cellular Uptake of Arginine-Rich Cell-Penetrating Peptides. Nat. Commun. 2011, 2, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Nischan N; Herce HD; Natale F; Bohlke N; Budisa N; Cardoso MC; Hackenberger CP Covalent Attachment of Peptides to GFP Results in Protein Delivery into Live Cells with Immediate Bioavailability. Angew. Chem. Int. Ed. Engl. 2015, 54, 1950–1953. [DOI] [PubMed] [Google Scholar]
- (118).Mandal D; Nasrolahi Shirazi A.; Parang K Cell-Penetrating Homochiral Cyclic Peptides as Nuclear-Targeting Molecular Transporters. Angew. Chem. Int. Ed. Engl. 2011, 50, 9633–9637. [DOI] [PubMed] [Google Scholar]
- (119).Traboulsi H; Larkin H; Bonin MA; Volkov L; Lavoie CL; Marsault É Macrocyclic cell penetrating peptides: a study of structure-penetration properties. Bioconjugate Chem. 2015, 26, 405–411. [DOI] [PubMed] [Google Scholar]
- (120).Horn M; Reichart F; Natividad-Tietz S; Diaz D; Neundorf I Tuning the Properties of a Novel Short Cell-Penetrating Peptide by Intramolecular Cyclization with a Triazole Bridge. Chem. Comm. 2016, 52, 2261–2264. [DOI] [PubMed] [Google Scholar]
- (121).Cascales L; Henriques ST; Kerr MC; Huang YH; Sweet MJ; Daly NL; Craik DJ Identification and Characterization of a New Family of Cell-penetrating Peptides: Cyclic Cell-Penetrating Peptides. J. Biol. Chem. 2011, 286, 36932–36943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Appelbaum JS; LaRochelle JR; Smith BA; Balkin DM; Holub JM Schepartz A Arginine Topology Controls Escape of Minimally Cationic Proteins from Early Endosomes to the Cytoplasm. Chem. Biol. 2012, 19, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Gallo M; Defaus S; Andreu D 1988–2018: Thirty Years of Drug Smuggling at the Nano Scale. Challenges and Opportunities of Cell-Penetrating Peptides in Biomedical Research. Arch. Biochem. Biophys. 2018, 661, 74–86. [DOI] [PubMed] [Google Scholar]
- (124).Guidotti G; Brambilla L; Rossi D Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [DOI] [PubMed] [Google Scholar]
- (125).Kauffman WB Fuselier T; He J; Wimley WC Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Kaplan IM; Wadia JS; Dowdy SF Cationic TAT Peptide Transduction Domain Enters Cells by Macropinocytosis. J. Control. Release 2005, 102, 247–253. [DOI] [PubMed] [Google Scholar]
- (127).Fotin-Mleczek M; Welte S; Mader O; Duchardt F; Fischer R; Hufnagel H; Scheurich P; Brock R Cationic Cell-Penetrating Peptides Interfere with TNF Signaling by Induction of TNF Receptor Internalization. J. Cell. Sci. 2005, 118, 3339–3351. [DOI] [PubMed] [Google Scholar]
- (128).Madani F; Lindberg S; Langel U; Futaki S; Gräslund A Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, ID 414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Nakase I; Tadokoro A; Kawabata N; Takeuchi T; Katoh H; Hiramoto K; Negishi M; Nomizu M; Sugiura Y; Futaki S Interaction of Arginine-Rich Peptides with Membrane-Associated Proteoglycans is Crucial for Induction of Actin Organization and Macropinocytosis. Biochemistry 2007, 46, 492–501. [DOI] [PubMed] [Google Scholar]
- (130).Gerbal-Chaloin S; Gondeau C; Aldrian-Herrada G; Heitz F; Gauthier-Rouvière C; Divita G; First Step of the Cell-Penetrating Peptide Mechanism Involves Rac1 GTPase-Dependent Actin-Network Remodeling. Biol. Cell. 2007, 99, 223–238. [DOI] [PubMed] [Google Scholar]
- (131).Mäger I; Eiríksdóttir E; Langel K, El Andaloussi S; Langel U Assessing the Uptake Kinetics and Internalization Mechanisms of Cell-Penetrating Peptides Using a Quenched Fluorescence Assay. Biochim. Biophys. Acta. 2010, 1798, 338–343. [DOI] [PubMed] [Google Scholar]
- (132).Drin G; Cottin S; Blanc E; Rees AR; Temsamani J Studies on the Internalization Mechanism of Cationic Cell-penetrating Peptides. J. Biol. Chem. 2003, 278, 31192–31201. [DOI] [PubMed] [Google Scholar]
- (133).Wadia JS; Stan RV; Dowdy SF Transducible TAT-HA Fusogenic Peptide Enhances Escape of TAT-Fusion Proteins after Lipid Raft Macropinocytosis. Nat. Med. 2004, 10, 310–315. [DOI] [PubMed] [Google Scholar]
- (134).Säälik P; Padari K; Niinep A; Lorents A; Hansen M; Jokitalo E; Langel U; Pooga M Protein Delivery with Transportans is Mediated by Caveolae Rather than Flotillin-Dependent Pathways. Bioconjugate Chem. 2009, 20, 877–887. [DOI] [PubMed] [Google Scholar]
- (135).Ferrari A; Pellegrini V; Arcangeli C; Fittipaldi A; Giacca M; Beltram F Caveolae-Mediated Internalization of Extracellular HIV-1 Tat Fusion Proteins Visualized in Real Time. Mol. Ther. 2003, 8, 284–294. [DOI] [PubMed] [Google Scholar]
- (136).Richard JP; Melikov K; Brook H; Prevot P; Lebleu B; Chernomordik LV Cellular Uptake of Unconjugated TAT Peptide Involves Clathrin Dependent Endocytosis and Heparan Sulfate Receptors. J. Biol. Chem. 2005, 280, 15300–15306. [DOI] [PubMed] [Google Scholar]
- (137).Ter-Avetisyan G; Tunnemann G; Nowak D; Nitschke M; Herrmann A; Drab M; Cardoso MC Cell Entry of Arginine-Rich Peptides is Independent of Endocytosis. J. Biol. Chem. 2009, 284, 3370–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Fittipaldi A; Ferrari A; Zoppé M; Arcangeli C; Pellegrini V; Beltram F; Giacca M Cell Membrane Lipid Rafts Mediate Caveolar Endocytosis of HIV-1 Tat Fusion Proteins. J. Biol. Chem. 2003, 278, 34141–34149. [DOI] [PubMed] [Google Scholar]
- (139).Morris MC; Deshayes S; Heitz F; Divita G Cell-Penetrating Peptides: From Molecular Mechanisms to Therapeutics. Biol. Cell. 2008, 100, 201–217. [DOI] [PubMed] [Google Scholar]
- (140).Su Y; Doherty T; Waring AJ; Ruchala P; Hong M Roles of Arginine and Lysine Residues in the Translocation of a Cell-Penetrating Peptide from 13C, 31P, and 19F Solid-State NMR. Biochemistry 2009, 48, 4587–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Fuchs SM; Raines RT Pathway for Polyarginine Entry into Mammalian Cells. Biochemistry 2004, 43, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Leifert JA; Whitton JL “Translocatory Proteins” and “Protein Transduction Domains”: A Critical Analysis of Their Biological Effects and Underlying Mechanisms. Mol. Ther. 2003, 8, 13–20. [DOI] [PubMed] [Google Scholar]
- (143).Belting M Heparan Sulfate Proteoglycan as a Plasma Membrane Carrier. Trends Biochem. Sci. 2003, 28, 145–151. [DOI] [PubMed] [Google Scholar]
- (144).Hakansson S; Caffrey M Structural and Dynamic Properties of the HIV-1 TAT Transduction Domain in the Free and Heparin-Bound States. Biochemistry 2003, 42, 8999–9006. [DOI] [PubMed] [Google Scholar]
- (145).Ziegler A; Blatter XL; Seelig A; Seelig J Protein Transduction Domains of HIV-1 and SIV TAT Interact with Charged Lipid Vesicles. Binding Mechanism and Thermodynamic Analysis. Biochemistry 2003, 42, 9185–9194. [DOI] [PubMed] [Google Scholar]
- (146).Christianson HC; Belting M Heparan Sulfate Proteoglycan as a Cell-Surface Endocytosis Receptor. Matrix Biol. 2014, 35, 51–55. [DOI] [PubMed] [Google Scholar]
- (147).Wallbrecher R; Verdurmen WP; Schmidt S; Bovee-Geurts PH; Broecker F; Reinhardt A; van Kuppevelt TH; Seeberger PH; Brock R The Stoichiometry of Peptide-Heparan Sulfate Binding as a Determinant of Uptake Efficiency of Cell-Penetrating Peptides. Cell Mol. Life Sci. 2014, 71, 2717–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Nagel YA; Raschle PS; Wennemers H Effect of Preorganized Charge-Display on the Cell-Penetrating Properties of Cationic Peptides. Angew. Chem. Int. Ed. Engl. 2017, 56, 122–126. [DOI] [PubMed] [Google Scholar]
- (149).Bechara C; Pallerla M; Burlina F; Illien F; Cribier S; Sagan S Massive Glycosaminoglycan-Dependent Entry of Trp-Containing Cell-Penetrating Peptides Induced by Exogenous Sphingomyelinase or Cholesterol Depletion. Cell. Mol. Life Sci. 2015, 72, 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Arsov Z; Nemec M; Schara M; Johansson H; Langel U; Zorko M Cholesterol Prevents Interaction of the Cell-Penetrating Peptide Transportan with Model Lipid Membranes. J. Pept. Sci. 2008, 14, 1303–1308. [DOI] [PubMed] [Google Scholar]
- (151).Hallock KJ; Lee DK; Omnaas J; Mosberg HI; Ramamoorthy A Membrane Composition Determines Pardaxin’s Mechanism of Lipid Bilayer Disruption. Biophys. J. 2002, 83, 1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Mano M; Henrigues A; Paiva A; Prieto M; Gavilanes F; Simões S; de Lima MC Interaction of S413-PV Cell Penetrating Peptide with Model Membranes: Relevance to Peptide Translocation across Biological Membranes. J. Pept. Sci. 2007, 13, 301–313. [DOI] [PubMed] [Google Scholar]
- (153).Walrant A; Correia I, Jiao CY; Leguin O; Bent EH; Goasdoué N; Lacombe C; Chassaing G; Sagan S; Alves ID Different Membrane Behaviour and Cellular Uptake of Three Basic Arginine-Rich Peptides. Biochim. Biophys. Acta. 2011, 1808, 382–393. [DOI] [PubMed] [Google Scholar]
- (154).Amand HL; Boström CL; Lincoln P; Nordén B; Esbjörner EK Binding of Cell-Penetrating Penetratin Peptides to Plasma Membrane Vesicles Correlates Directly with Cellular Uptake. Biochim. Biophys. Acta. 2011, 1808, 1860–1867. [DOI] [PubMed] [Google Scholar]
- (155).Jiao C; Sachon E; Alves ID; Chassaing G; Bolbach G; Sagan S Exploiting Benzophenone Photoreactivity to Probe the Phospholipid Environment and Insertion Depth of the Cell‐Penetrating Peptide Penetratin in Model Membranes. Angew. Chem. Int. Ed. Engl. 2017, 56, 8226–8230. [DOI] [PubMed] [Google Scholar]
- (156).Mishra A; Lai GH; Schmidt NW; Sun VZ; Rodriguez AR; Tong R; Tang L; Cheng J Deming TJ; Kamei DT; Wong GCL Translocation of HIV TAT Peptide and Analogues Induced by Multiplexed Membrane and Cytoskeletal Interactions. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16883–16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Walrant A; Vogel A; Correia I; Lequin O; Olausson BE; Desbat B; Sagan S; Alves ID Membrane Interactions of Two Arginine-Rich Peptides with Different Cell Internalization Capacities. Biochim. Biophys. Acta. 2012, 1818, 1755–1763. [DOI] [PubMed] [Google Scholar]
- (158).Witte K; Olausson BE; Walrant A; Alves ID; Vogel A Structure and Dynamics of the Two Amphipathic Arginine-Rich Peptides RW9 and RL9 in a Lipid Environment Investigated by Solid-State NMR and MD Simulations. Biochim. Biophys. Acta. 2013, 1828, 824–833. [DOI] [PubMed] [Google Scholar]
- (159).Jobin ML; Blanchet M; Henry S; Chaignepain S; Manigand C; Castano S; Lecomte S; Burlina F; Sagan S; Alves ID The Role of Tryptophans on the Cellular Uptake and Membrane Interaction of Arginine-Rich Cell Penetrating Peptides. Biochim. Biophys. Acta. 2015, 1848, 593–602. [DOI] [PubMed] [Google Scholar]
- (160).Bechara C; Pallerla M; Zaltsman Y; Burlina F; Alves ID; Lequin O; Sagan S Tryptophan within Basic Peptide Sequences Triggers Glycosaminoglycan-Dependent Endocytosis. FASEB J. 2013, 27, 738–739. [DOI] [PubMed] [Google Scholar]
- (161).Juks C; Lorents A; Arukuusk P; Langel U; Pooga M Cell-Penetrating Peptides Recruit Type A Scavenger Receptors to the Plasma Membrane for Cellular Delivery of Nucleic Acids. FASEB J. 2017, 31, 975–988. [DOI] [PubMed] [Google Scholar]
- (162).Tanaka G; Nakase I; Fukuda Y; Masuda R; Oishi S; Shimura K; Kawaguchi Y; Takatani-Nakase T; Langel U; Gräslund A; et al. CXCR4 Stimulates Macropinocytosis: Implications for Cellular Uptake of Arginine-Rich Cell-Penetrating Peptides and HIV. Chem. Biol. 2012, 19, 1437–1446. [DOI] [PubMed] [Google Scholar]
- (163).Nakashima H; Hashimoto M; Sadakane Y; Tomohiro T; Hatanaka Y Simple and Versatile Method for Tagging Phenyldiazirine Photophores. J. Am. Chem. Soc. 2006, 128, 15092–1509. [DOI] [PubMed] [Google Scholar]
- (164).Mayer H; Bauer H; Breuss J; Ziegler S; Prohaska R Characterization of Rat LANCL1, a Novel Member of the Lanthionine Synthetase C-like Protein Family, Highly Expressed in Testis and Brain. Gene 2001, 269, 73–80. [DOI] [PubMed] [Google Scholar]
- (165).Kawaguchi Y; Tanaka G; Nakase I; Imanishi M; Chiba J; Hatanaka Y; Futaki S Identification of Cellular Proteins Interacting with Octaarginine (R8) Cell-Penetrating Peptide by Photo-Crosslinking. Bioorg. Med. Chem. Lett. 2013, 23, 3738–3740. [DOI] [PubMed] [Google Scholar]
- (166).Letoha T; Keller-Pintér A; Kusz E; Kolozsi C; Bozsó Z; Tóth G; Vizler C; Oláh Z, Szilák L Cell-Penetrating Peptide Exploited Syndecans. Biochim. Biophys. Acta 2010, 1798, 2258–2265 [DOI] [PubMed] [Google Scholar]
- (167).Juks C; Lorents A; Arukuusk P; Langel U; Pooga M Cell-Penetrating Peptides Recruit Type A Scavenger Receptors to the Plasma Membrane for Cellular Delivery of Nucleic Acids. FASEB J. 2017, 31, 975–988. [DOI] [PubMed] [Google Scholar]
- (168).Allen JK; Brock DJ; Kondow-McConaghy HM; Pellois J Efficient Delivery of Macromolecules into Human Cells by improving the Endosomal Escape Activity of Cell-Penetrating Peptides: Lessons Learned from dfTAT and its Analogs. Biomolecules 2018, 8, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Erazo-Oliveras A; Muthukrishnan N; Baker R; Wang T-Y; Pellois J-P Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges. Pharmaceuticals 2012, 5, 1177–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Pei D; Buyanova M Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem. 2019, 30, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Behr J-P The Proton Sponge: A Trick to Enter Cells the Viruses Did Not Exploit. CHIMIA Int. J. Chem. 1997, 51, 34–36. [Google Scholar]
- (172).Sonawane ND; Szoka FC Jr.; Verkman AS Chloride Accumulation and Swelling in Endosomes Enhances DNA Transfer by Polyamine-DNA Polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [DOI] [PubMed] [Google Scholar]
- (173).Haensler J; Szoka FC Polyamidoamine Cascade Polymers Mediate Efficient Transfection of Cells in Culture. Bioconjugate Chem. 1993, 4, 372–379. [DOI] [PubMed] [Google Scholar]
- (174).White JM; Whittaker GR Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Zelphati O; Szoka FC Mechanism of Oligonucleotide Release from Cationic Liposomes. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 11493–11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Herce HD; Garcia AE; Litt J; Kane RS; Martin P; Enrique N; Rebolledo A; Milesi V Arginine-Rich Peptides Destabilize the Plasma Membrane, Consistent with a Pore Formation Translocation Mechanism of Cell-Penetrating Peptides. Biophys. J. 2009, 97, 1917–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Allen JK; Brock DJ; Knodow-McConaghy HM; Pellois JP Efficient Delivery of Macromolecules into Human Cells by Improving the Endosomal Escape Activity of Cell-Penetrating Peptides: Lessons Learned from dfTAT and its Analogs. Biomolecules 2018, 8, E50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Yang S; Zaitseva E; Chernomordik LV; Melikov K Cell-Penetrating Peptide Induces Leaky Fusion of Liposomes Containing Late Endosome-Specific Anionic Lipid. Biophys. J. 2010, 99, 2525–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Vermeulen LMP; Brans T; Samal SK; Dubruel P; Demeester J; De Smedt SC; Remaut K; Braeckmans K Endosomal Size and Membrane Leakiness Influence Proton Sponge-Based Rupture of Endosomal Vesicles. ACS Nano 2018, 12, 2332–2345. [DOI] [PubMed] [Google Scholar]
- (180).Brock DJ; Kustigian L; Jiang M; Graham M; Wang T; Erazo-Oliveras A; Najjar K; Zhang J; Rye H; Pellois JP Efficient Cell Delivery Mediated by Lipid‐Specific Endosomal Escape of Supercharged Branched Peptides. Traffic 2018, 19, 421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).ur Rehman Z; Hoekstra D; Zuhorn IS Mechanism of Polyplex- and Lipoplex-Mediated Delivery of Nucleic Acids: Real-Time Visualization of Transient Membrane Destabilization Without Endosomal Lysis. ACS Nano 2013, 7, 3767–3777. [DOI] [PubMed] [Google Scholar]
- (182).Wittrup A; Ai A; Liu X; Hamar P; Trifonova R; Charisse K; Manoharan M; Kirchhausen T; Lieberman J Visualizing Lipid-Formulated siRNA Release from Endosomes and Target Gene Knockdown. Nat. Biotechnol. 2015, 33, 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Erazo Oliveras A; Najjar K; Dayani L; Wang TY; Johnson G; Pellois J‐P Protein Delivery into Live Cells by Incubation with an Endosomolytic Agent. Nat Methods 2014, 11, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Akishiba M; Takeuchi T; Kawaguchi Y; Sakamoto K; Yu H-H; Nakase I; Takatani-Nakase T; Madani F; Gräslund A; Futaki S Cytosolic Antibody Delivery by Lipid-Sensitive Endosomolytic Peptide. Nat. Chem. 2017, 9, 751–761. [DOI] [PubMed] [Google Scholar]
- (185).Erazo Oliveras A; Najjar K; Dayani L; Wang TY; Johnson G; Pellois J‐P Protein Delivery into Live Cells by Incubation with an Endosomolytic Agent. Nat Methods 2014, 11, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Appiah Kubi G.; Qian Z; Amiar S; Sahni A; Stahelin RV; Pei D Non-Peptidic Cell-Penetrating Motifs for Mitochondrion-Specific Cargo Delivery. Angew. Chem. Int. Ed. Engl. 2018, 57, 17183–17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Campelo F; McMahon HT; Kozlov MM The Hydrophobic Insertion Mechanism of Membrane Curvature Generation by Proteins. Biophys. J. 2008, 95, 2325–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Alves ID; Goasdoué N; Correia I; Aubry S; Galanth C; Sagan S; Lavielle S; Chassaing G Membrane Interaction and Perturbation Mechanisms Induced by Two Cationic Cell Penetrating Peptides with Distinct Charge Distribution. Biochim. Biophys. Acta. 2008, 1780, 948–959. [DOI] [PubMed] [Google Scholar]
- (189).Mishra A; Gordon VD; Yang L; Coridan R; Wong GC HIV TAT Forms Pores in Membranes by Inducing Saddle-Splay Curvature: Potential Role of Bidentate Hydrogen Bonding. Angew. Chem. Int. Ed. Engl. 2008, 47, 2986–2989. [DOI] [PubMed] [Google Scholar]
- (190).Hirose S; Takeuchi T; Osakada H; Pujals S; Katayama S; Nakase I; Kobayashi S; Haraguchi T; Futaki S Transient Focal Membrane Deformation Induced by Arginine-Rich Peptides Leads to Their Direct Penetration into Cells. Mol. Ther. 2012, 20, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Brock R The Uptake of Arginine-Rich Cell-Penetrating Peptides: Putting the Puzzle Together. Bioconjugate Chem. 2014, 25, 863–868. [DOI] [PubMed] [Google Scholar]
- (192).Duchardt F; Fotin-Mleczek M; Schwarz H; Fischer R; Brock R A Comprehensive Model for the Cellular Uptake of Cationic Cell‐penetrating Peptides. Traffic 2007, 8, 848–866. [DOI] [PubMed] [Google Scholar]
- (193).Verdurmen WP; Thanos M; Ruttekolk IR; Gulbins E; Brock R Cationic Cell-Penetrating Peptides Induce Ceramide Formation via Acid Sphingomyelinase: Implications for Uptake. J. Control. Release 2010, 147, 171–179. [DOI] [PubMed] [Google Scholar]
- (194).Wallbrecher R; Ackels T; Olea RA; Klein MJ; Caillon L; Schiller J; Bovée-Geurts PH; van Kuppevelt TH; Ulrich AS; Spehr M; et al. Membrane Permeation of Arginine-Rich Cell-Penetrating Peptides Independent of Transmembrane Potential as a Function of Lipid Composition and Membrane Fluidity. J. Control. Release 2017, 256, 68–78. [DOI] [PubMed] [Google Scholar]
- (195).Joanne P; Galanth C; Goasdoué N; Nicolas P; Sagan S; Lavielle S; Chassaing G; El Amri C; Alves ID Lipid Reorganization Induced by Membrane-Active Peptides Probed Using Differential Scanning Calorimetry. Biochim. Biophys. Acta. 2009, 1788, 1772–1781. [DOI] [PubMed] [Google Scholar]
- (196).Takechi-Haraya Y; Aki K; Tohyama Y; Harano Y; Kawakami T; Saito H; Okamura E Glycosaminoglycan Binding and Non-Endocytic Membrane Translocation of Cell-Permeable Octaarginine Monitored by Real-Time In-Cell NMR Spectroscopy. Pharmaceuticals 2017, 10, E42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Ziegler A; Seelig J Contributions of Glycosaminoglycan Binding and Clustering to the Biological Uptake of the Nonamphipathic Cell-Penetrating Peptide WR9. Biochemistry 2011, 50, 4650–4664. [DOI] [PubMed] [Google Scholar]
- (198).Takeuchi T; Kosuge M; Tadokoro A; Sugiura Y; Nishi M; Kawata M; Sakai N, Matile S; Futaki S Direct and Rapid Cytosolic Delivery Using Cell-Penetrating Peptides Mediated by Pyrenebutyrate. ACS Chem. Biol. 2006, 1, 299–303. [DOI] [PubMed] [Google Scholar]
- (199).Guterstam P; Madani F; Hirose H; Takeuchi T; Futaki S; El Andalossui S; Gräslund A; Langel U Elucidating Cell-Penetrating Peptide Mechanisms of Action for Membrane Interaction, Cellular Uptake, and Translocation Utilizing the Hydrophobic Counter-Anion Pyrenebutyrate. Biochim. Biophys. Acta. 2009, 1788, 2509–2517. [DOI] [PubMed] [Google Scholar]
- (200).Tünnemann G; Martin RM; Haupt S; Patsch C; Edenhofer F; Cardoso MC Cargo-Dependent Mode of Uptake and Bioavailability of TAT-Containing Proteins and Peptides in Living Cells. FASEB J. 2006, 20, 1775–1784. [DOI] [PubMed] [Google Scholar]
- (201).Vivès E; Brodin P; Lebleu B A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [DOI] [PubMed] [Google Scholar]
- (202).Jiao CY; Delaroche D; Burlina F; Alves ID; Chassaing G; Sagan S Translocation and Endocytosis for Cell-Penetrating Peptide Internalization. J. Biol. Chem. 2009, 284, 33957–33965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Pae J; Liivamägi L; Lubenets D; Arukuusk P, Langel U; Pooga M Glycosaminoglycans Are Required for Translocation of Amphipathic Cell-Penetrating Peptides across Membranes. Biochim. Biophys. Acta. 2016, 1858, 1860–1870. [DOI] [PubMed] [Google Scholar]
- (204).Lorents A; Säälik P; Langel U; Pooga M Arginine-Rich Cell-Penetrating Peptides Require Nucleolin and Cholesterol-Poor Subdomains for Translocation across Membranes. Bioconjugate Chem. 2018, 29, 1168–1177. [DOI] [PubMed] [Google Scholar]
- (205).Säälik P; Niinep A; Pae J; Hansen M; Lubenets D; Langel U; Pooga M Penetration Without Cells: Membrane Translocation of Cell-Penetrating Peptides in the Model Giant Plasma Membrane Vesicles. J. Control. Release 2011, 153, 117–125. [DOI] [PubMed] [Google Scholar]
- (206).Rothbard JB; Jessop TC; Lewis RS; Murray BA; Wender PA Role of Membrane Potential and Hydrogen Bonding in the Mechanism of Translocation of Guanidinium-Rich Peptides into Cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [DOI] [PubMed] [Google Scholar]
- (207).Copolovici DM; Langel K; Eriste E; Langel U Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano, 2014, 8, 1972–1994. [DOI] [PubMed] [Google Scholar]
- (208).Cooper JA Effects of Cytochalasin and Phalloidin on Actin. J. Cell. Biol. 1987, 105, 1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Meier-Abt F; Faulstich H; Hagenbuch B Identification of Phalloidin Uptake Systems of Rat and Human Liver. Biochim. Biophys. Acta. 2004, 1664, 64–69. [DOI] [PubMed] [Google Scholar]
- (210).Rudd MD; Luse DS Amanitin Greatly Reduces the Rate of Transcription by RNA Polymerase II Ternary Complexes but Fails to Inhibit Some Transcript Cleavage Modes. J. Biol. Chem. 1996, 271, 21549–21558. [DOI] [PubMed] [Google Scholar]
- (211).Letschert K; Faulstich H; Keller D; Keppler D Molecular Characterization and Inhibition of Amanitin Uptake into Human Hepatocytes. Toxicol. Sci. 2006, 91, 140–149. [DOI] [PubMed] [Google Scholar]
- (212).Morita H; Shimbo K; Shigemori H; Kobayashi J Antimitotic Activity of Moroidin, a Bicyclic Peptide from the Seeds of Celosia argentea. Bioorganic Med. Chem. Lett. 2000, 10, 469–471. [DOI] [PubMed] [Google Scholar]
- (213).Kobayashi J; Suzuki H; Shimbo K; Takeya K; Morita H Celogentins A-C, New Antimitotic Bicyclic Peptides from the Seeds of Celosia argentea. J. Org. Chem. 2001, 66, 6626–6633. [DOI] [PubMed] [Google Scholar]
- (214).Ma B; Banerjee B; Litvinov DN; He L; Castle SL Total Synthesis of the Antimitotic Bicyclic Peptide Celogentin C. J. Am. Chem. Soc. 2010, 132, 1159–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Nishimura S; Arita Y; Honda M; Iwamoto K; Matsuyama A; Shirai A; Kawasaki H; Kobayashi T; Matsunaga S; Yoshida M Marine Antifungal Theonellamides Target 3-β-Hydroxysterol to Activate Rho1 Signaling. Nat. Chem. Biol. 2010, 6, 519–526. [DOI] [PubMed] [Google Scholar]
- (216).Oren Z; Shai Y Mode of Action of Linear Amphipathic Alpha-Helical Antimicrobial Peptides. Biopolymer 1998, 47, 451–463. [DOI] [PubMed] [Google Scholar]
- (217).Matsuzaki K Why and How are Peptide-Lipid Interactions Utilized for Self-Defense? Magainins and Tachyplensins as Archetypes. Biochim. Biophys. Acta. 1999, 1462, 1–10. [DOI] [PubMed] [Google Scholar]
- (218).Tang M; Waring AJ; Hong M Phosphate-Mediated Arginine Insertion into Lipid Membranes and Pore Formation by a Cationic Membrane Peptide from Solid-State NMR. J. Am. Chem. Soc. 2007, 129, 11438–11446. [DOI] [PubMed] [Google Scholar]
- (219).Matsuzaki K; Yoneyama S; Murase O Miyajima K Transbilayer Transport of Ions and Lipids Coupled with Mastoparan X Translocation. Biochemistry 1996, 35, 8450–8456. [DOI] [PubMed] [Google Scholar]
- (220).Pouny Y; Rapaport D; Mor A; Nicolas P; Shai Y Interaction of Antimicrobial Dermaseptin and its Fluorescently Labeled Analogs with Phospholipid Membranes. Biochemistry 1992, 31, 12416–12423. [DOI] [PubMed] [Google Scholar]
- (221).Shai Y Mode of Action of Membrane Active Antimicrobial Peptides. Biopolymers 2002, 66, 236–248. [DOI] [PubMed] [Google Scholar]
- (222).Matsuzaki K; Murase O; Fuiji N; Miyajima K An Antimicrobial Peptide, Magainin 2, Induced Rapid Flip-Flop of Phospholipids Coupled with Pore Formation and Peptide Translocation. Biochemistry 1996, 35, 11361–11368. [DOI] [PubMed] [Google Scholar]
- (223).Allende D; Simon SA; McIntosh TJ Melittin-Induced Bilayer Leakage Depends on Lipid Material Properties; Evidence for Toroidal Pores. Biophys. J. 2005, 88, 1828–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Herce HD; Garcia AE; Molecular Dynamics Simulations Suggest a Mechanism for Translocation of the HIV-1 TAT Peptide across Lipid Membranes. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 20805–20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Yesylevskyy S; Marrink S; Mark AE Alternative Mechanisms for the Interaction of the Cell-Penetrating Peptides Penetratin and the TAT Peptide with Lipid Bilayers. Biophys. J. 2009, 97, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Sharmin S; Islam MZ; Karal MAS; Shibly SUA; Dohra H; Yamazaki M Effects of Lipid Composition on the Entry of Cell-Penetrating Peptide Oligoarginine into Single Vesicles. Biochemistry 2016, 55, 4154–4165. [DOI] [PubMed] [Google Scholar]
- (227).Sun TL; Sun Y; Lee CC; Huang HW Membrane Permeability of Hydrocarbon-Cross-Linked Peptides. Biophys. J. 2013, 104, 1923–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Levadny V; Tsuboi T; Belaya M; Yamazaki M Rate Constant of Tension-Induced Pore Formation in Lipid Membranes. Langmuir 2013, 29, 3848–3852. [DOI] [PubMed] [Google Scholar]
- (229).Karal MAS; Alam JM; Takahashi T; Levadny V; Yamazaki M Stretch-Activated Pore of Antimicrobial Peptide Magainin 2. Langmuir 2015, 31, 3391–3401. [DOI] [PubMed] [Google Scholar]
- (230).Islam MZ; Sharmin S; Levadnyy V; Alam Shibly SU; Yamazaki M Effects of Mechanical Properties of Lipid Bilayers on the Entry of Cell-Penetrating Peptides into Single Vesicles. Langmuir 2017, 33, 2433–2443. [DOI] [PubMed] [Google Scholar]
- (231).Islam MZ; Ariyama H; Alam JM; Yamazaki M Entry of Cell-Penetrating Peptide Transportan 10 into a Single Vesicle by Translocating Across Lipid Membrane and its Induced Pores. Biochemistry 2014, 53, 386–396 [DOI] [PubMed] [Google Scholar]
- (232).Lamazière A; Burlina F; Wolf C; Chassaing G; Trugnan G; Ayala-Sanmartin J Non-Metabolic membrane Tubulation and Permeability Induced by Bioactive Peptides. PLoS One 2007, 2, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Via MA; Del Pópolo MG; Wilke N Negative Dipole Potentials and Carboxylic Polar Head Groups Foster the Insertion of Cell-Penetrating Peptides into Lipid Monolayers. Langmuir 2018, 34, 3102–3111. [DOI] [PubMed] [Google Scholar]
- (234).Tamba Y; Yamazaki M Single Giant Unilamellar Vesicle Method Reveals Effect of Antimicrobial Peptide, Magainin 2, on Membrane Permeability. Biochemistry 2005, 44, 15823–15833. [DOI] [PubMed] [Google Scholar]
- (235).Tamba Y; Yamazaki M Magainin 2-Induced Pore Formation in Membrane Depends on its Concentration in Membrane Interface. J. Phys. Chem. B 2009, 113, 4846–4852. [DOI] [PubMed] [Google Scholar]
- (236).Tamba Y; Ariyama H; Levadny V; Yamazaki M Kinetic Pathway of Antimicrobial Peptide Magainin 2-Induced Pore Formation in Lipid Membranes. J. Phys. Chem. B 2010, 114, 12018–12026. [DOI] [PubMed] [Google Scholar]
- (237).Alam JM; Kobayashi T; Yamazaki M The Single Giant Unilamellar Vesicle Method Reveals Lysenin-Induced Pore Formation in Lipid Membranes Containing Sphingomyelin. Biochemistry 2012, 51, 5160–5172. [DOI] [PubMed] [Google Scholar]
- (238).Moniruzzaman M; Alam JM; Dohra H; Yamazaki M Antimicrobial Peptide Lactoferricin B-Induced Rapid Leakage of Internal Contents from Single Giant Unilamellar Vesicles. Biochemistry 2015, 54, 5802–5814. [DOI] [PubMed] [Google Scholar]
- (239).Chen X; Liu S; Deme B; Cristiglio V; Marquardt D; Weller R; Rao P; Wang Y; Bradshaw J Efficient Internalization of TAT Peptide in Zwitterionic DOPC Phospholipid Membrane Revealed by Neutron Diffraction. Biochim. Biophys. Acta. Biomembr. 2017, 1859, 910–916. [DOI] [PubMed] [Google Scholar]
- (240).Pokorny A; Birkbeck H; Almeida PFF Mechanism and Kinetics of δ-lysin Interaction with Phospholipid Vesicles, Biochemistry 2002, 41, 11044–11056. [DOI] [PubMed] [Google Scholar]
- (241).Silvestro L; Axelsen PH Membrane-Induced Folding of Cecropin A. Biophys. J. 2000, 79, 1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Pokorny A; Almeida PF Kinetics of Dye Efflux and Lipid Flip-Flop Induced by δ-Lysin in Phosphatidylcholine Vesicles and the Mechanism of Graded Release by Amphipathic, α-Helical Peptides. Biochemistry 2004, 43, 8846–8857. [DOI] [PubMed] [Google Scholar]
- (243).Derossi D; Calvet S; Trembleau A; Brunissen A; Chassaing G; Prochiantz A Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-Independent. J. Biol. Chem. 1996, 271, 1811–18193. [DOI] [PubMed] [Google Scholar]
- (244).Derossi D; Joliot AH; Chassaing G; Prochiantz A The Third Helix of the Antennapedia Homeodomain Translocates through Biological Membranes. J. Biol. Chem. 1994, 269, 10444–10450. [PubMed] [Google Scholar]
- (245).Prochiantz A Getting Hydrophilic Compounds into Cells: Lessons from Homeopeptides. Curr. Opin. Neurobiol. 1996, 6, 629–634. [DOI] [PubMed] [Google Scholar]
- (246).Almeida C; Lamazière A; Filleau A; Corvis Y; Espeau P; Ayala-Sanmartin J Membrane Re-Arrangements and Rippled Phase Stabilisation by the Cell Penetrating Peptide Penetratin. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2584–2591. [DOI] [PubMed] [Google Scholar]
- (247).Lamazière A; Maniti O; Lambert O; Chassaing G; Trugnan G; Ayala-Sanmartin J Lipid Domain Separation, Bilayer Thickening and Pearling Induced by the Cell Penetrating Peptide Penetratin. Biochim. Biophys. Acta 2010, 1798, 2223–2230. [DOI] [PubMed] [Google Scholar]
- (248).Afonin S; Frey A; Bayerl S; Fischer D; Wadhwani P; Weinkauf S; Ulrich AS The Cell-Penetrating Peptide TAT(48–60) Induces a Non-Lamellar Phase in DMPC Membranes. ChemPhysChem. 2006, 7, 2134–2142. [DOI] [PubMed] [Google Scholar]
- (249).Maniti O; Alves I; Trugnan G; Ayala-Sanmartin J Distinct Behaviour of the Homeodomain Derived Cell Penetrating Peptide Penetratin in Interaction with Different Phospholipids. PLoS One 2010, 5, e15819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Kawamoto S; Miyakawa T; Takasu M; Morikawa R; Tatsuki O; Saito H; Futaki S; Hidemi N Cell-Penetrating Peptide Induces Various Deformations of Lipid Bilayer Membrane: Inverted Micelle, Double Bilayer, and Transmembrane. Int. J. Quantum Chem. 2012, 112, 178–183. [Google Scholar]
- (251).Kawamoto S; Takasu M; Miyakawa T; Morikawa R; Oda T; Futaki S; Nagao H Inverted Micelle Formation of Cell-Penetrating Peptide Studied by Coarse-Grained Simulation: Importance of Attractive Force between Cell-Penetrating Peptides and Lipid Head Group. J. Chem. Phys. 2011, 134, 095103. [DOI] [PubMed] [Google Scholar]
- (252).Swiecicki JM; Bartsch A; Tailhades J; Di Pisa M; Heller B; Chassaing G; Mansuy C; Burlina F; Lavielle S The Efficacies of Cell-Penetrating Peptides in Accumulating in Large Unilamellar Vesicles Depend on Their Ability to Form Inverted Micelles. Chembiochem. 2014, 15, 884–891. [DOI] [PubMed] [Google Scholar]
- (253).Francisco Hilário F.; Traoré MD; Zwick V; Berry L; Simões-Pires CA; Cuendet M; Fantozzi N; Pereira de Freitas R; Maynadier M; Wein S; et al. Synthesis of an Uncharged Tetra-Cyclopeptide Acting as a Transmembrane Carrier: Enhanced Cellular and Nuclear Uptake. Org. Lett. 2017, 19, 612–615. [DOI] [PubMed] [Google Scholar]
- (254).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camaero JA; Campopiano DJ; Challis GL; Clardy J et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Ramalho SD; Pinto MEF; Ferreira D; Bolzani VS; Biologically Active Orbitides from the Euphorbiaceae Family. Planta Med. 2018, 84, 558–567. [DOI] [PubMed] [Google Scholar]
- (256).Candido-Bacani PM; Figueiredo PO; Matos MF; Garcez FR; Garcez WS; Cytotoxic Orbitide from the Latex of Croton urucurana. J. Nat. Prod. 2015, 78, 2754–2760. [DOI] [PubMed] [Google Scholar]
- (257).Kosasi S; Van der Sluis WG; Boelens R; Hart LA; Labadie RP Labaditin, a Novel Cyclic Decapeptide from the Latex of Jatropha multifida L. (Euphorbiaceae). FEBS Lett. 1989, 256, 91–96. [Google Scholar]
- (258).Pinto ME; Batista JMJ; Koehbach J; Gaur P; Sharma A; Nakabashi M; Cilli EM; Giesel GM; Verli H; Gruber CW; et al. Ribifolin, an Orbitide from Jatropha ribifolia, and its Potential Antimalarial Activity. J. Nat. Prod. 2015, 78, 374–380. [DOI] [PubMed] [Google Scholar]
- (259).Kaneda M; Kawaguchi S; Fujii N; Ohno H; Oishi S SAR Study on Odoamide: Insights into the Bioactivities of Aurilide-Family Hybrid Peptide-Polyketides. ACS Med. Chem. Lett. 2018, 9, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Ohyama M; Okada Y; Takahashi M; Sakanaka O; Matsumoto M; Atsumi K Structure-Activity Relationship of Anthelmintic Cyclooctadepsipeptides. Biosci. Biotechnol. Biochem. 2011, 75, 1354–1363. [DOI] [PubMed] [Google Scholar]
- (261).White TR; Renzelman CM; Rand AC; Rezai T; McEwen CM; Gelev VM; Turner RA; Linington RG; Leung SSF; Kalgutkar AS; et al. On-Resin N-Methylation of Cyclic Peptides for Discovery of Orally Bioavailable Scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Beck JG; Chatterjee J; Laufer B; Kiran MY; Frank AO; Neubauer S; Ovadia O; Greenberg S; Gilon C; Hoffman A; Kessler H Intestinal Permeability of Cyclic Peptides: Common Key Backbone Motifs Identified. J. Am. Chem. Soc. 2012, 134, 12125–12133. [DOI] [PubMed] [Google Scholar]
- (263).Matsui K; Kido Y; Watari R; Kashima Y; Yoshida Y Shuto S Highly Conformationally Restricted Cyclopropane Tethers with Three‐Dimensional Structural Diversity Drastically Enhance the Cell-permeability of Cyclic Peptides. Chem. Eur. J. 2016, 23, 3034–3041. [DOI] [PubMed] [Google Scholar]
- (264).Wu H; Mousseau G; Mediouni S; Valente ST; Kodadek T Cell‐Permeable Peptides Containing Cycloalanine Residues. Angew. Chem. Int. Ed. Engl. 2016, 55, 12637–12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Fouché M; Schäfer M; Berghausen J; Desrayaud S; Blatter M; Piéchon P; Dix I; Martin Garcia A.; Roth HJ Design and Development of a Cyclic Decapeptide Scaffold with Suitable Properties for Bioavailability and Oral Exposure. ChemMedChem. 2016, 11, 1048–1059. [DOI] [PubMed] [Google Scholar]
- (266).Fouché M; Schäfer M; Blatter M; Berghausen J; Desrayaud S; Roth HJ Pharmacokinetic Studies around the Mono- and Difunctionalization of a Bioavailable Cyclic Decapeptide Scaffold. ChemMedChem. 2016, 11, 1060–1068. [DOI] [PubMed] [Google Scholar]
- (267).Shin MK; Hyun YJ; Lee JH; Lim HS Comparison of Cell-permeability of Cyclic Peptoids and Linear Peptoids. ACS Comb. Sci. 2018, 20, 237–242. [DOI] [PubMed] [Google Scholar]
- (268).Schwochert J; Turner R; Thang M; Berkeley RF; Ponkey AR; Rodriguez KM; Leung SSF; Khunte B; Goetz G; Limberakis C; et al. Peptide to Peptoid Substitutions Increase Cell-permeability in Cyclic Hexapeptides. Org. Lett. 2015, 17, 2928–2931. [DOI] [PubMed] [Google Scholar]
- (269).Furukawa A; Townsend CE; Schwochert J; Pye CR; Bednarek MA; Lokey RS Passive Membrane Permeability in Cyclic Peptomer Scaffolds Is Robust to Extensive Variation in Side Chain Functionality and Backbone Geometry. J. Med. Chem. 2016, 59, 9503–9512. [DOI] [PubMed] [Google Scholar]
- (270).Shi Y; Challa S; Sang P; She F; Li C; Gray GM; Nimmagadda A; Teng P; Odom T; Wang Y; et al. One-Bead–Two-Compound Thioether Bridged Macrocyclic γ-AApeptide Screening Library against EphA2. J. Med. Chem. 2017, 60, 9290–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Zhang L; Bulaj G Converting Peptides into Drug Leads by Lipidation. Curr. Med. Chem. 2012, 19, 1602–1618. [DOI] [PubMed] [Google Scholar]
- (272).Schumacher-Klinger A; Fanous J; Merzbach S; Weinmüller M; Reichart F; Räder AFB; Gitlin-Domagalska A; Gilon C; Kessler H; Hoffman A Enhancing Oral Bioavailability of Cyclic RGD Hexa-peptides by the Lipophilic Prodrug Charge Masking Approach: Redirection of Peptide Intestinal Permeability from a Paracellular to Transcellular Pathway. Mol. Pharm. 2018, 15, 3468–3477. [DOI] [PubMed] [Google Scholar]
- (273).Slough DP; McHugh SM; Lin YS Understanding and Designing Head-to-Tail Cyclic Peptides. Biopolymers 2018, 109, e23113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Dinca A; Chien W Chin MT Intracellular Delivery of Proteins with Cell-Penetrating Peptides for Therapeutic Uses in Human Disease. Int. J. Mol. Sci. 2016, 17, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Karpurapu M; Lee YG; Qian Z; Wen J; Ballinger MN; Rusu L; Chung S; Deng J; Qian F; Reader BF; et al. Inhibition of Nuclear Factor of Activated T Cells (NFAT) c3 Activation Attenuates Acute Lung Injury and Pulmonary Edema in Murine Models of Sepsis. Oncotarget 2018, 9, 10606–10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Darwish S; Sadehhiani N; Fong S; Mozaffari S; Hamidi P; Withana T; Yang S; Tiwari RK; Parang K Synthesis and Antiproliferative Activities of Doxorubicin Thiol Conjugates and Doxorubicin-SS-Cyclic Peptide. Eur. J. Med. Chem. 2019, 161, 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Herce HD; Schumacher D; Schnider AFL; Ludwig AK; Mann FA; Filies M; Reinke S; Krause E; Leonhardt H; Cardoso MC; Hackenberger CPR Cell-Permeable Nanobodies for Targeting Immunolabelling and Antigen Manipulation in Living Cells. Nat. Chem. 2017, 9, 762–771. [DOI] [PubMed] [Google Scholar]
- (278).Schneider AFL; Wallabregue ALD; Franz L; Hackenberger PR Targeted Subcellular Protein Delivery Using Cleavable Cyclic Cell-Penetrating Peptides. Biconjugate Chem. 2019, 30, 400–404. [DOI] [PubMed] [Google Scholar]
- (279).Bedewy W; Liao H; Abou-Taleb NA; Hammad SF; Nasr T; Pei D Generation of a Cell-Permeable Cycloheptapeptidyl Inhibitor against Peptidyl-Prolyl Isomerase Pin1. Org. Biomol. Chem. 2017, 15, 4540–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Upadhyaya P; Qian Z; Selner NG; Clippinger SR; Wu Z; Briesewitz R; Pei D Inhibition of Ras Signaling by Blocking Ras-Effector Interactions with Cyclic Peptides. Angew. Chem. Int. Ed. Engl. 2015, 54, 7602–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Shirazi AN; Tiwari RK; Brown A; Mandal D; Sun G; Parang K Cyclic Peptides Containing Tryptophan and Arginine as Src Kinase Inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 3230–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Oh D; Shirazi AN; Northup K; Sullivan B; Tiwari RK; Bisoffi M; Parang K Enhanced Cellular Uptake of Short Polyarginine Peptides through Fatty Acylation and Cyclization. Mol. Pharmaceutics 2014, 11, 2845–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Lian W; Jiang B; Qian Z; Pei D Cell-Permeable Bicyclic Peptide Inhibitors against Intracellular Proteins. J. Am. Chem. Soc. 2014, 136, 9830–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Jiang B; Pei D A Selective, Cell-Permeable Nonphosphorylated Bicyclic Peptidyl Inhibitor against Peptidyl-Prolyl Isomerase Pin1. J. Med. Chem. 2015, 58, 6306–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Trinh TB; Upadhyaya P; Qian Z; Pei D Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci. 2016, 18, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Rhodes CA; Dougherty PG; Cooper JK; Qian Z; Lindert S; Wang Q; Pei D Cell-Permeable Bicyclic Peptidyl Inhibitors against NEMO-IκB Kinase Interaction Directly from a Combinatorial Library. J. Am. Chem. Soc. 2018, 140, 12102–12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Wallbrecher R; Depré L; Verdurmen WPR; Bovée PH; van Duinkerken RH; Zekveld MJ; Timmerman P Brock R Exploration of the Design Principles of a Cell-Penetrating Bicylic Peptide Scaffold. Bioconjugate Chem. 2014, 25, 955–964. [DOI] [PubMed] [Google Scholar]
- (288).Wolfe JM; Fadzen CM; Holden RL; Yao M; Hanson GJ; Pentelute BL Perfluoroaryl Bicyclic Cell-Penetrating Peptides for Delivery of Antisense Oligonucleotides. Angew. Chem. Int. Ed. Engl. 2018, 57, 4756–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Qian Z; Xu X; Amacher JF; Madden DR; Cormet-Boyaka E; Pei D Intracellular Delivery of Peptidyl Ligands by Reversible Cyclization: Discovery of a PDZ Domain Inhibitor that Rescues CFTR Activity. Angew. Chem. Int. Ed. Engl. 2015, 54, 5874–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Qian Z; Rhodes CA; McCroskey LC; Wen J; Appiah-Kubi G; Wang DJ; Guttridge DC; Pei D Enhancing the Cell-permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chem. Int. Ed. Engl. 2017, 56, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Shirazi AN; El-Sayed NS; Mandal D; Tiwari RK; Tavakoli K; Etesham M; Parang K Cysteine and Arginine-Rich Peptides as Molecular Carriers. Bioorg. Med. Chem. Lett. 2016, 26, 656–661. [DOI] [PubMed] [Google Scholar]
- (292).Shirazi AN; Tiwari RK; Oh D; Banerjee A; Yadav A; Parang K Efficient Delivery of Cell Impermeable Phosphopeptides by a Cyclic Peptide Amphiphile Containing Tryptophan and Arginine. Mol. Pharm. 2013, 10, 2008–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Walensky LD; Pitter K; Morash J; Oh KJ; Barbuto S; Fisher J; Smith E; Verdine GL; Korsmeyer SJ A Stapled BID BH3 Helix Directly Binds and Activates BAX. Mol. Cell 2006, 24, 199–210. [DOI] [PubMed] [Google Scholar]
- (294).Speltz TE; Danes JM; Stender JD; Frasor J; Moore TW A Cell-Permeable Stapled Peptide Inhibitor of the Estrogen Receptor/Coactivator Interaction. ACS Chem. Biol. 2018, 13, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Wang Y; Ho TG; Bertinetti D; Neddermann M; Franz E; Mo GC; Schendowich LP; Sukhu A; Spelts RC; Zhang J; et al. Isoform-Selective Disruption of AKAP-Localized PKA Using Hydrocarbon Stapled Peptides. ACS Chem. Biol. 2014, 9, 635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Ahsan A; Ramanand SG; Bergin IL; Zhao L; Whitehead CE; Rehemtulla A; Ray D; Pratt WB; Lawrence TS; Nyati MK Efficacy of an EGFR-Specific Peptide against EGFR-Dependent Cancer Cell Lines and Tumor Xenografts. Neoplasia, 2014, 16, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Kim W; Bird GH; Neff T; Guo G; Kerenyi MA; Walensky LD; Orkin SH Targeted Disruption of the EZH2–EED Complex Inhibits EZH2-Dependent Cancer. Nat. Chem. Biol. 2013, 9, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Hillman RA; Nadraws JW; Bertucci MA The Hydrocarbon Staple & Beyond: Recent Advances towards Stapled Peptide Therapeutics that Target Protein-Protein Interactions. Curr. Top. Med. Chem. 2018, 18, 611–624. [DOI] [PubMed] [Google Scholar]
- (299).Meric-Bernstam F; Saleh MN; Infante JR; Goel S; Falchook GS; Shapiro G; Chung KY; Conry RM; Hong DS; Wang JS-Z; et al. Phase I Trial of a Novel Stapled Peptide ALRN-6924 Disrupting MDMX- and MDM2-Mediated Inhibition of WT p53 in Patients with Solid Tumors and Lymphomas. J. Clin. Oncology 2017, 25, 2505. [Google Scholar]
- (300).Walensky LD; Kung AL; Escher I; Malia TJ; Barbuto S; Wright RD; Wanger G; Verdine GL; Korsmeyer SJ Activation of Apoptosis In Vivo by a Hydrocarbon-Stapled BH3 Helix. Science 2004, 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Li YC; Rodewald LW; Hoppmann C; Wong ET; Lebreton S; Safar P; Patek M; Wertman KF; Wahl GM A Versatile Platform to Analyze Low-Affinity and Transient Protein-Protein Interactions in Living Cells in Real Time. Cell Rep. 2014, 9, 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Okamoto T; Zobel K; Fedorova A; Quan C; Yang H; Fairbrother WJ; Huang DC; Smith BJ; Deshayes K; Czabotar PE Stabilizing the Pro-Apoptotic BimBH3 Helix (BimSAHB) Does Not Necessarily Enhance Affinity or Biological Activity. ACS Chem. Biol. 2013, 8, 297–302. [DOI] [PubMed] [Google Scholar]
- (303).Bird GH; Mazzola E; Opoku-Nsiah K; Lammert MA; Godes M; Neuberg DS; Walensky LD Biophysical Determinants for Cellular Uptake of Hydrocarbon-Stapled Peptide Helices. Nat. Chem. Biol. 2016, 12, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Cromm PM; Spiegel J; Grossman TN Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS. Chem. Biol. 2015, 10, 1362–1375. [DOI] [PubMed] [Google Scholar]
- (305).Chu Q; Moellering RE; Hilinski GJ; Kim Y; Grossmna TN; Yeh JT-H Verdine GL Towards Understanding Cell Penetration by Stapled Peptides. Med. Chem. Commun. 2015, 6, 111–119. [Google Scholar]
- (306).Rezaei Araghi R.; Bird GH; Ryan JA; Jenson JM; Godes M; Pritz JR; Grant RA; Letai A; Walensky LD; Keating AE Iterative Optimization Yields Mcl-1-Targeting Stapled Peptides with Selective Cytotoxicity to Mcl-1-Dependent Cancer Cells. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 886–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Sakagami K; Masuda T; Kawano K Futaki S Importance of Net Hydrophobicity in the Cellular Uptake of All-Hydrocarbon Stapled Peptides. Mol. Pharm. 2018, 15, 1332–1340. [DOI] [PubMed] [Google Scholar]
- (308).Tian Y; Jiang Y; Li J; Wang D; Zhao H; Li Z Effect of Stapling Architecture on Physiochemical Properties and Cell-permeability of Stapled α-Helical Peptides: A Comparative Study. Chembiochem. 2017, 18, 2087–2093. [DOI] [PubMed] [Google Scholar]
- (309).Zhao H; Liu QS; Geng H; Tian Y; Cheng M; Jiang YH; Xie MS; Niu XG; Jiang F; Zhang YO; et al. Crosslinked Aspartic Acids as Helix-Nucleating Templates. Angew. Chem. Int. Ed. Engl. 2016, 55, 12088–12093. [DOI] [PubMed] [Google Scholar]
- (310).Zhao H; Jiang Y; Tian Y; Yang D; Qin X; Li Z Improving Cell Penetration of Helical Peptides Stabilized by N-Terminal Crosslinked Aspartic Acids. Org. Biomol. Chem. 2017, 15, 459–464. [DOI] [PubMed] [Google Scholar]
- (311).Oba M; Kunitake M; Kato T; Ueda A; Tanaka M Enhanced and Prolonged Cell-Penetrating Abilities of Arginine-Rich Peptides by Introducing Cyclic α,α-Disubstituted α-Amino Acids with Stapling. Bioconjugate Chem. 2017, 28, 1801–1806. [DOI] [PubMed] [Google Scholar]
- (312).Klein MJ; Schmidt S; Wadhwani P; Bürck J; Reichert J; Berditsch M; Schober T; Brock R; Kansy M; Ulrich AS Lactam-Stapled Cell-Penetrating Peptides: Cell Uptake and Membrane Binding Properties. J. Med. Chem. 2017, 60, 8071–8082. [DOI] [PubMed] [Google Scholar]
- (313).Shepherd NE; Harrison RS; Ruiz-Gomez G; Abbenante G; Mason JM; Fairlie DP Downsizing the BAD BH3 Peptide to Small Constrained α-Helices with Improved Ligand Efficiency. Org. Biomol. Chem. 2016, 14, 10939–10945. [DOI] [PubMed] [Google Scholar]
- (314).Dietrich L; Rathmer B; Ewan K; Bange T; Heinrichs S; Dale TC; Schade D; Grossman TN Cell Permeable Stapled Peptide Inhibitor of Wnt Signaling that Targets β-Catenin Protein-Protein Interactions. Cell Chem. Biol. 2017, 24, 958–968. [DOI] [PubMed] [Google Scholar]
- (315).Quach K; LaRochelle J; Li XH; Rhoades E; Schepartz A Unique Arginine Array Improves Cytosolic Localization of Hydrocarbon-Stapled Peptides. Bioorg. Med. Chem. 2018, 26, 1197–1202. [DOI] [PubMed] [Google Scholar]
- (316).Iegre J; Ahmen NS; Gaynord JS; Wu Y; Herlihy KM; Tan YS; Lopes-Pires ME; Jha R; Lau YH; Sore HF; et al. Stapled Peptides as a New Technology to Investigate Protein-Protein Interactions in Human Platelets. Chem. Sci. 2018, 9, 4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Spiegel J; Cromm PM; Itzen A; Goody RS; Grossmann TN; Waldmann H Direct Targeting of Rab-GTPase-Effector Interactions. Angew. Chem. Int. Ed. Engl. 2014, 53, 2498–2503. [DOI] [PubMed] [Google Scholar]
- (318).Cromm PM; Spiegel J; Küchler P; Dietrich L; Kriegsmann J; Wendt M; Goody RS; Waldmann H; Grossmann TN Protease-Resistant and Cell-Permeable Double-Stapled Peptides Targeting the Rab8a GTPase. ACS Chem. Biol. 2016, 11, 2375–2382. [DOI] [PubMed] [Google Scholar]
- (319).Peraro L; Zou Z; Makawana KM; Cummings AE; Ball HL; Yu H; Lin Y; Levin B; Kritzer JA Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy. J. Am. Chem. Soc. 2017, 139, 7792–7802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Craik DJ; Daly NL, Bond T; Waine C Plant Cyclotides: A Unique Family of Cyclic and Knotted Proteins That Defines the Cyclic Cysteine Knot Structural Motif. J. Mol. Biol. 1999, 294, 1327–1336. [DOI] [PubMed] [Google Scholar]
- (321).Saether O; Craik DJ; Campbell ID; Sletten K; Juul J; Norman DG Elucidation of the Primary and Three-Dimensional Structure of the Uterotonic Polypeptide Kalata B1. Biochemistry 1995, 34, 4147–4158. [DOI] [PubMed] [Google Scholar]
- (322).Luckett S; Garcia RS; Barker JJ; Konarev AV; Shewry PR; Clarke AR; Brady RL High-Resolution Structure of a Potent, Cyclic Proteinase Inhibitor from Sunflower Seeds. J. Mol. Biol. 1999, 290, 525–533. [DOI] [PubMed] [Google Scholar]
- (323).Felizmenio-Quimio ME; Daly NL; Craik DJ Circular Proteins in Plants: Solution Structure of a Novel Macrocyclic Trypsin Inhibitor from Momordica cochinchinensis. J. Biol. Chem. 2001, 276, 22875–22882. [DOI] [PubMed] [Google Scholar]
- (324).Craik DJ; Lee MH; Rehm FBH; Tombling B; Doffek B; Peacock H Ribosomally-Synthesized Cyclic Peptides from Plants as Drug Leads and Pharmaceutical Scaffolds. Bioorg. Med. Chem. 2018, 26, 2727–2737. [DOI] [PubMed] [Google Scholar]
- (325).Cascales L; Henrigues ST; Kerr MC; Huang Y,H; Sweet MJ; Daly NL; Craik DJ Identification and Characterization of a New Family of Cell-Penetrating Peptides: Cyclic Cell-Penetrating Peptides. J. Biol. Chem. 2011, 286, 36932–36943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Greenwood KP; Daly NL; Brown DL; Stow JL; Craik DJ The Cyclic Cysteine Knot Miniprotein MCoTI-II is Internalized into Cells by Macropinocytosis. Int. J. Biochem. Cell. Biol. 2007, 39, 2252–2264. [DOI] [PubMed] [Google Scholar]
- (327).Contreras J; Elnagar AY; Hamm-Alvarez SF; Camarero JA Cellular Uptake of Cyclotide MCoTI-I Follows Multiple Endocytic Pathways. J. Control. Release 2011, 155, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Henrigues ST; Huang YH; Chaousis S; Sani MA; Poth AG; Separovic F; Craik DJ The Prototypic Cyclotide Kalata B1 Has a Unique Mechanism of Entering Cells. Chem. Biol. 2015, 22, 1087–1097. [DOI] [PubMed] [Google Scholar]
- (329).D’Souza C; Henrigues ST; Wang CK; Craik DJ Structural Parameters Modulating the Cellular Uptake of Disulfide-Rich Cyclic Cell-Penetrating Peptides: MCoTI-II and SFTI-1. Eur. J. Med. Chem. 2014, 88, 10. [DOI] [PubMed] [Google Scholar]
- (330).Ji Y; Majumnder S; Millard M; Borra R; Bi T; Elnagar AY; Neamati N; Shekhtman A; Camarero JA In Vivo Activation of the p53 Tumor Suppressor Pathway by an Engineered Cyclotide. J. Am. Chem. Soc. 2013, 135, 11623–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Jiang L; Kimura RH; Miao Z; Silverman AP; Ren G; Liu H; Li P; Gambhir SS; Cochran JR; Cheng Z Evaluation of a 64Cu-Labeled Cystine-Knot Peptide Based on Agouti-Related Protein for PET of Tumors Expressing αvβ3 Integrin. J. Nucl. Med. 2010, 51, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Thell K; Hellinger R; Sahin E; Michenthaler P; Gold-Binder M; Haider T; Kuttke M; Liutkevičiūtė Z; Göransson U; Gründemann C; et al. Oral Activity of a Nature-Derived Cyclic Peptide for the Treatment of Multiple Sclerosis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113¸ 3960–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Adachi Y; Sakamoto K; Umemoto T; Fukuda Y; Tani A; Asami T Investigation on Cellular Uptake and Pharmacodynamics of DOCK2-Inhibitory Peptides Conjugated with Cell-Penetrating Peptides. Bioorg. Med. Chem. 2017, 25, 2148–2155. [DOI] [PubMed] [Google Scholar]
- (334).Smith BA; Daniels DS; Coplin AE; Jordan GE; McGregor LM; Schepartz A Minimally Cationic Cell-Permeable Miniature Proteins via α-Helical Arginine Display. J. Am. Chem. Soc. 2008, 130, 2948–2949. [DOI] [PubMed] [Google Scholar]
- (335).Trabolusi H; Larkin H; Bonin M; Volkov L; Lavoie CL; Marsault E Macrocyclic Cell Penetrating Peptides: A Study of Structure-Penetration Properties. Bioconjugate Chem. 2015, 26, 405–411. [DOI] [PubMed] [Google Scholar]
- (336).Jang S; Hyun S; Kim S; Lee S; Lee IS; Baba M; Lee Y; Yu J Cell-Penetrating, Dimeric α-Helical Peptides: Nanomolar Inhibitors of HIV-1 Transcription. Angew. Chem. Int. Ed. Engl. 2014, 53, 10086–10089. [DOI] [PubMed] [Google Scholar]
- (337).Hyun S; Choi Y; Lee HN; Lee C; Oh D; Lee D; Lee C; Lee Y; Yu J Construction of Histidine-Containing Hydrocarbon Stapled Cell-Penetrating Peptides for In Vitro and In Vivo Delivery of siRNAs. Chem. Sci. 2018, 9, 3820–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Chang KL; Higuchi Y; Kawakami S; Yamashita F; Hashida M Efficient Gene Transfection by Histidine-Modified Chitosan through Enhancement of Endosomal Escape. Bioconjugate Chem. 2010, 21, 1087–1095. [DOI] [PubMed] [Google Scholar]
- (339).Aubry S; Burlina F; Dupont E; Delaroche D; Joliot A; Lavielle S; Chassaing G; Sagan S Cell-Surface Thiols Affect Cell Entry of Disulfide-Conjugated Peptides. FASEB J. 2009, 23, 2956–2967. [DOI] [PubMed] [Google Scholar]
- (340).Meng X; Li T; Zhao Y; Wu C; CXC-Mediated Cellular Uptake of Miniproteins: Forsaking “Arginine Magic”. ACS Chem. Biol. 2018, 13, 3078–3086. [DOI] [PubMed] [Google Scholar]
- (341).Jha D; Mishra R; Gottschalk S; Wiesmüller KH; Ugurbil K; Maier ME; Engelmann J CyLoP-1: A Novel Cysteine-Rich Cell-Penetrating Peptide for Cytosolic Delivery of Cargoes. Bioconjugate Chem. 2011, 22, 319–328. [DOI] [PubMed] [Google Scholar]
- (342).Bode SA; Wallbrecher R; Brock R; van Hest JC; Löwik DW Activation of Cell-Penetrating Peptides by Disulfide Bridge Formation of Truncated Precursors. Chem. Commun. 2014, 50, 415–417. [DOI] [PubMed] [Google Scholar]
- (343).Kenien R; Shen WC; Zaro JL Vesicle-to-Cytosol Transport of Disulfide-Linked Cargo Mediated by an Amphipathic Cell-Penetrating Peptide. J. Drug Target. 2012, 20, 793–800. [DOI] [PubMed] [Google Scholar]
- (344).Li T; Gao W; Liang J; Zha M; Chen Y; Zhao Y; Wu C Biscysteine-Bearing Peptide Probes to Reveal Extracellular Thiol–Disulfide Exchange Reactions Promoting Cellular Uptake. Anal. Chem. 2017, 89, 8501–8508. [DOI] [PubMed] [Google Scholar]
- (345).Gasparini G; Bang EK; Molinard G; Tulumello DV; Ward S; Kelley SO; Roux A; Sakai N; Matile S Cellular Uptake of Substrate-Initiated Cell-Penetrating Poly(disulfide)s. J. Am. Chem. Soc. 2014, 136, 6069–6074. [DOI] [PubMed] [Google Scholar]
- (346).Gursoy RN; Benita S Self-Emulsifying Drug Delivery Systems (SEDDS) for Improved Oral Delivery of Lipophilic Drugs. Biomed. Pharmacother. 2004, 58, 173–182. [DOI] [PubMed] [Google Scholar]
- (347).Thean D; Ebo JS; Luxton T; Lee XC; Ferrer FJ; Johannes CW; Lane DP; Brown CJ Enhancing Specific Disruption of Intracellular Protein Complexes by Hydrocarbon Stapled Peptides Using Lipid Based Delivery. Sci. Rep. 2017, 7, 1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Lundberg M; Wikström S; Johansson M Cell Surface Adherence and Endocytosis of Protein Transduction Domains. Mol. Ther. 2003, 8, 143–150. [DOI] [PubMed] [Google Scholar]
- (349).Rotstein BH; Mourtada R; Kelley SO; Yudin AK Solvatochromic Reagents for Multicomponent Reactions and their Utility in the Development of Cell‐Permeable Macrocyclic Peptide Vectors. Chem. Eur. J. 2011, 17, 12257–12261. [DOI] [PubMed] [Google Scholar]
- (350).Majzoub RN; Chan C; Ewert KK; Silva BFB; Liang KS; Safinya CR Fluorescence Microscopy Colocalization of Lipid-Nucleic Acid Nanoparticles with Wildtype and Mutant Rab5-GFP: A Platform for Investigating Early Endosomal Events. Biochim. Biophys. Acta 2015, 1848, 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Hedegaard SF; Derbas MS; Lind TK; Kasimova MR; Christensen MV; Michaelsen MH; Campbell RA; Jorgensen L; Franzyk H; Cárdenas M et al. Fluorophore Labeling of a Cell-Penetrating Peptide Significantly Alters the Mode and Degree of Biomembrane Interaction. Sci. Rep. 2018, 8, 6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Birch D; Christensen MV; Staerk D; Franzyk H; Nielsen HM Fluorophore Labeling of a Cell-Penetrating Peptide Induces Differential Effects on its Cellular Distribution and Affects Cell Viability. Biochim. Biophys. Acta 2017, 1859, 2483–2494. [DOI] [PubMed] [Google Scholar]
- (353).Muthukrishnan N; Donovan S; Pellois JP The Photolytic Activity of Poly-Arginine Cell Penetrating Peptides Conjugated to Carboxy-Tetramethylrhodamine is Modulated by Arginine Residue Content and Fluorophore Conjugation Site. Photochem. Photobiol. 2014, 90, 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Swiecicki JM; Thiebaut F; Di Pisa M; Gourdin-Bertin S; Tailhades J; Mansuy C; Burlina F; Chwetzoff S; Trugnan G; Chassaing G; Lavielle S How to Unveil Self-Quenched Fluorophores and Subsequently Map the Subcellular Distribution of Exogenous Peptides. Sci. Rep. 2016, 6, 20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Harreither E; Rydberg HA; Amand HL; Jadhav V; Fliedl L; Benda C; Esteban MA; Pei D; Borth N; Brillari-Voglauer R; et al. Characterization of a Novel Cell Penetrating Peptide Derived from Human Oct4. Cell Regen. 2014, 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Ibrahim SF; van den Engh G; Flow Cytometry and Cell Sorting. Adv. Biochem. Eng. Biotechnol. 2007, 106, 19–39. [DOI] [PubMed] [Google Scholar]
- (357).Hällbrink M; Florén A; Elmquist A; Pooga M; Bartfai T; Langel Ü Cargo Delivery Kinetics of Cell-Penetrating Peptides. Biochim. Biophys. Acta 2006, 1515, 101–109. [DOI] [PubMed] [Google Scholar]
- (358).Qian Z; Dougherty PG; Pei D Monitoring the Cytosolic Entry of Cell-Penetrating Peptides Using a pH-Sensitive Fluorophore. Chem. Comm. 2015, 51, 2162–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (359).Manceur A; Wu A; Audet J Flow Cytometric Screening of Cell-Penetrating Peptides for Their Uptake into Embryonic and Adult Stem Cells. Anal. Biochem. 2007, 364, 51–59. [DOI] [PubMed] [Google Scholar]
- (360).Rodrigues M; de la Torre BG; Andreu D; Santos NC Kinetic Uptake Profiles of Cell Penetrating Peptides in Lymphocytes and Monocytes. Biochim. Biophys. Acta 2013, 1830, 4554–4563. [DOI] [PubMed] [Google Scholar]
- (361).Burlina F; Sagan S; Bolbach G; Chassaing G Quantification of the Cellular Uptake of Cell-Penetrating Peptides by MALDI-TOF Mass Spectrometry. Angew. Chem. Int. Ed. 2005, 44, 4244–4247. [DOI] [PubMed] [Google Scholar]
- (362).Delaroche D; Aussedat B; Aubry S; Chassaing G; Burlina F; Clodic G; Bolbach G; Lavielle S; Sagan S Tracking a New Cell-Penetrating (W/R) Nonapeptide, through an Enzyme-Stable Mass Spectrometry Reporter Tag. Anal. Chem. 2007, 79, 1932–1938. [DOI] [PubMed] [Google Scholar]
- (363).Aussedat B; Sagan S; Chassaing G; Bolbach G; Burlina F Quantification of the Efficinecy of Cargo Delivery by Peptidic and Pseudo-Peptidic Trojan Carriers using MALDI-TOF Mass Spectrometry. Biochim. Biophys. Acta 2006, 1758, 375–383. [DOI] [PubMed] [Google Scholar]
- (364).Pröfrock D; Prange A Inductively Coupled Plasma– Mass Spectrometry (ICP-MS) for Quantitative Analysis in Environmental and Life Sciences: A Review of Challenges, Solutions, and Trends. Appl. Spectrosc. 2012, 66, 834–868. [DOI] [PubMed] [Google Scholar]
- (365).Møller LH; Gabel-Jensen C; Franzyk H; Bahnsen JS; Stürup S; Gammelgaard B Quantification of Pharmaceutical Peptides Using Selenium as an Elemental Detection Label. Metallomics 2014, 6, 1639–1647. [DOI] [PubMed] [Google Scholar]
- (366).Møller LH; Bahnsen JS; Nielsen HM; Østergaard J; Stürup S; Gammelgaard B Selenium as an Alternative Peptide Label – Comparison to Fluorophore-Labeled Penetratin. Eur. J. Pharm. Sci. 2015, 67, 76–84. [DOI] [PubMed] [Google Scholar]
- (367).Rakowska PD; Lamarre B; Ryadnov MG Probing Label-Free Intracellular Quantification of Free Peptide by MALDI-ToF Mass Spectrometry. Methods 2014, 68, 331–337. [DOI] [PubMed] [Google Scholar]
- (368).Thompson NL Fluorescence Correlation Spectroscopy. Top. Fluoresc. Spectrosc. 1991, 337–378. [Google Scholar]
- (369).Waizenegger T; Fischer R; Brock R; Intracellular Concentration Measurements in Adherent Cells: A Comparison of Import Efficiencies of Cell-Permeable Peptides, Biol. Chem. 2002, 383, 291. [DOI] [PubMed] [Google Scholar]
- (370).LaRochelle JR; Cobb GB; Steinauer A; Rhoades E; Schepartz A Fluorescence Correlation Spectroscopy Reveals Highly Efficient Cytosolic Delivery of Certain Penta-Arg Proteins and Stapled Peptides. J. Am. Chem. Soc. 2015, 137, 2536–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Rezgui R; Blumer K; Yeoh-Tan G; Trexler AJ; Magzoub M Precise Quantification of Cellular Uptake of Cell-Penetrating Peptides Using Fluorescence-Activated Cell Sorting and Fluorescence Correlation Spectroscopy. Biochim. Biophys. Acta 2016, 1858, 1499–1506. [DOI] [PubMed] [Google Scholar]
- (372).Yu P; Liu B; Kodadek T A High-Throughput Assay for Assessing the Cell-permeability of Combinatorial Libraries. Nat. Biotechnol. 2005, 23, 746–751. [DOI] [PubMed] [Google Scholar]
- (373).Holub JM; LaRochelle JR; Appelbaum JS; Schepartz A Improved Assays for Determining the Cytosolic Access of Peptides, Proteins, and Their Mimetics. Biochemistry 2013, 52, 9036–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Milech N; Longville BAC; Cunningham PT; Scobie MN; Bogdawa HM; Winslow S; Anastasas M; Connor T; Ong F; Stone SR; et al. GFP-Complementation Assay to Detect Functional CPP and Protein Delivery into Living Cells. Sci. Rep. 2015, 5, 18329–18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Cabantous S; Terwilliger TC; Waldo GS Protein Tagging and Detection with Engineered Self-Assembling Fragments of Green Fluorescent Protein. Nat. Biotech. 2005, 23, 102–107. [DOI] [PubMed] [Google Scholar]
- (376).Lonn P; Kacsinta AD; Cui XS; Hamil AS; Kaulich M; Gogoi K; Dowdy SF Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6, 32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Stone SR; Heinrich T; Juraja SM; Satiaputra JN; Hall CM; Anastasas M; Mills AD; Chamberlain CA; Winslow S; Priebatsch K; et al. β-Lactamase Tools for Establishing Cell Internalization and Cytosolic Delivery of Cell Penetrating Peptides. Biomolecules 2018, 8, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Soughayer JS; Wang Y; Li H; Cheung SH; Rossi FM; Stanbridge EJ; Sims CE; Allbritton NL Characterization of TAT-Mediated Transport of Detachable Kinase Substrates. Biochemistry 2004, 43, 8528–8540. [DOI] [PubMed] [Google Scholar]
- (379).Chao TY; and Raines RT Fluorogenic Label to Quantify the Cytosolic Delivery of Macromolecules. Mol. BioSyst. 2013, 9, 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Jones LR; Goun EA; Shinde R; Rothbard JB; Contag CH; Wender PA Releasable Luciferin−Transporter Conjugates: Tools for the Real-Time Analysis of Cellular Uptake and Release. J. Am. Chem. Soc. 2006, 128, 6526–6527. [DOI] [PubMed] [Google Scholar]
- (381).Los GV; Encell LP; McDougall MG; Hartzell DD; Karassina N; Zimprich C; Wood MG; Learish R; Ohana RF; Urh M; et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- (382).Peraro L; Deprey KL; Moser MK; Zou Z; Ball HL; Levin B; Kritzer JA Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J. Am. Chem. Soc. 2018, 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Gammon ST; Villalobos VM; Prior JL; Sharma V; Piwnica-Worms D Quantitative Analysis of Permeation Peptide Complexes Labeled with Technetium-99m: Chiral and Sequence-Specific Effects on Net Cell Uptake. Bioconjugate Chem. 2003, 14, 368–376. [DOI] [PubMed] [Google Scholar]
- (384).Zaro JL; and Shen WC Quantitative Comparison of Membrane Transduction and Endocytosis of Oligopeptides. Biochem. Biophys. Res. Commun. 2003, 307, 241–247. [DOI] [PubMed] [Google Scholar]
- (385).Lee YJ; Datta S; Pellois JP Real-Time Fluorescence Detection of Protein Transduction into Live Cells. J. Am. Chem. Soc. 2008, 130, 2398–2399. [DOI] [PubMed] [Google Scholar]
- (386).Adams SR; Campbell RE; Gross LA; Martin BR; Walkup GK; Yao Y; Llopis J; Tsien RY New Biarsenical Ligands and Tetracysteine Motifs for Protein Labeling In Vitro and In Vivo: Synthesis and Biological Applications. J. Am. Chem. Soc. 2002, 124, 6063–6076. [DOI] [PubMed] [Google Scholar]
- (387).Adams SR; Tsien RY; Langel U (Ed.). Imaging the Influx of Cell-Penetrating Peptides into the Cytosol of Individual Live Cells In Handbook of Cell-Penetrating Peptides, 2007, CRC Press: Boca Raton. [Google Scholar]
- (388).Jobin ML; Alves ID Label-Free Quantification of Cell-Penetrating Peptide Translocation into Liposomes. Anal. Methods 2016, 8, 4608–4616. [Google Scholar]
- (389).Aroui S; Brahim S; De Waard M; Kenani A Cytotoxicity, Intracellular Distribution and Uptake of Doxorubicin and Doxorubicin Coupled to Cell-Penetrating Peptides in Different Cell Lines: A Comparative Study. Biochem. Biophys. Res. Commun. 2010, 391, 419–425. [DOI] [PubMed] [Google Scholar]
- (390).Kansy M; Senner F; Gubernator K Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeability Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- (391).Morimoto J; Amano R; Ono T; Sando S A Parallel Permeability Assay of Peptides across Artificial Membranes and Cell Monolayers using a Fluorogenic Reaction. Org. Biomol. Chem. 2019, 17, 2887–2891. [DOI] [PubMed] [Google Scholar]
- (392).Conradi RA; Hilgers AR; Ho NFH; Burton PS The Influence of Peptide Structure on Transport across Caco-2 Cells. Pharm. Res. 1991, 8, 1453–1460. [DOI] [PubMed] [Google Scholar]
- (393).Kamei N; Onuki Y; Takayama K; Takeda-Morishita M Mechanistic Study of the Uptake/Permeation of Cell-Penetrating Peptides across a Caco-2 Monolayer and Their Stimulatory Effect on Epithelial Insulin Transport. J. Pharm. Sci. 2013, 102, 3998–4008. [DOI] [PubMed] [Google Scholar]