

Abstract
Aldosterone-producing adenomas (APA) with somatic mutations in the KCNJ5 potassium channel are a cause of primary aldosteronism. These mutations drive aldosterone excess but their role in cell growth is undefined. Our objective was to determine the role of KCNJ5 mutations in adrenal cell proliferation and apoptosis. The Ki67 proliferative index was positively correlated with adenoma diameter in APAs with a KCNJ5 mutation (r=0.435, P=0.007), a negative correlation was noted in adenomas with no mutation detected (NMD) (r=−0.548, P=0.023). Human adrenocortical cell lines were established with stable expression of cumate-inducible wild type or mutated KCNJ5. Increased cell proliferation was induced by low-level induction of KCNJ5-T158A expression compared with control cells (P=0.009) but increased induction ablated this difference. KCNJ5-G151R displayed no apparent proliferative effect but KCNJ5-G151E and L168R mutations each resulted in decreased cell proliferation (difference P<0.0001 from control cells, both comparisons). Under conditions tested, T158A had no effect on apoptosis but apoptosis increased with expression of G151R (P<0.0001), G151E (P=0.008) and L168R (P<0.0001). We generated a specific KCNJ5 monoclonal antibody which was used in immunohistochemistry to demonstrate strong KCNJ5 expression in adenomas without a KCNJ5 mutation and in the zona glomerulosa adjacent to adenomas irrespective of genotype as well as in aldosterone-producing cell clusters. KCNJ5-CYP11B2 double immunofluorescence showed markedly decreased KCNJ5 immunostaining in CYP11B2-positive cells compared with CYP11B2-negative cells in APAs with a KCNJ5 mutation. Together these findings support the concept that cell growth effects of KCNJ5 mutations are determined by the expression level of the mutated channel.
Keywords: aldosterone-producing adenoma, adrenal gland, primary aldosteronism, KCNJ5, growth and apoptosis
Introduction
Unilateral primary aldosteronism (PA) is the most prevalent surgically-correctable form of hypertension. The constitutive production of aldosterone mainly originates from a unilateral aldosterone-producing adenoma (APA) and less often from unilateral hyperplasia (30% and 2% of cases of PA, respectively).1 Major breakthroughs in understanding the pathophysiology of sporadic APAs have been made since the identification by Choi et al.2 of somatic mutations in the KCNJ5 gene (causing KCNJ5-G151R or KCNJ5-L168R missense mutations) in a high proportion of these tumors.2–4 KCNJ5 is an inwardly rectifying potassium channel (also called GIRK4, G protein coupled inwardly rectifying potassium channel) and the described mutations cause sodium ion conductance due to the loss of selectivity for potassium ions by the channel pore. In adrenocortical cells, the consequent membrane depolarization triggers opening of voltage-gated calcium channels and calcium ion influx ultimately activates aldosterone production.2, 5
The identification of additional APA somatic mutations in the Cav1.3 calcium channel (CACNA1D) and in the Na+/K+-ATPase and Ca2+-ATPase ion transporters (ATP1A1 and ATP2B3, respectively) highlighted the importance of intracellular ion homeostasis and calcium signaling in aldosterone production6, 7 and, together with somatic mutations in β-catenin (CTNNB1), these mutations can be detected in almost 90% of APAs.8 In most populations, a predominance of KCNJ5 mutations in APAs over other genotypes is reported3, 4, 9–11 with a global prevalence of 43%.12
A role for KCNJ5 mutations in adrenal cell growth has not been defined. When initially described, KCNJ5 mutations were proposed to result in both constitutive aldosterone production and cell proliferation2 due to the established role of calcium signaling in both processes.13, 14 A function in driving aldosterone excess has been demonstrated by expression of mutated forms of KCNJ5 in human adrenocortical cells in vitro5 but a decrease in cell proliferation resulted from expression of KCNJ5-T158A.5 This mutation (KCNJ5-T158A) has been identified in both sporadic APAs and a familial form of PA (called familial hyperaldosteronism type III)2, 15 and the absence of an effect on cell proliferation in vitro is seemingly paradoxical to the massive cortical hyperplasia observed in a patient carrying the germline variant.2, 16
Aldosterone-producing cell clusters (APCC) are a histopathologic feature often found beneath the adrenal capsule under normal and pathologic conditions.17 APCCs comprise tight nests of predominantly zona glomerulosa cells with intense immunohistochemistry staining for CYP11B2 (aldosterone synthase). A notable proportion of APCCs carry mutations in CACNA1D, ATP1A1 and ATP2B3 but KCNJ5 mutations are curiously absent.17, 18 Our objective was to establish the effects of KCNJ5 mutations on cell growth in human adrenocortical cells by specifically addressing their roles in cell proliferation and apoptosis.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Patient samples
The study included 72 surgically resected adrenals from patients diagnosed with unilateral PA according to the Endocrine Society Guideline.19 Patients were screened for PA using the plasma aldosterone-to-direct renin concentration ratio and diagnosis was confirmed by the intravenous saline load test according to local criteria.20 Adenoma size was assessed from the diameter of the largest nodule at pathology and CYP11B2 immunohistochemistry was done on all adrenals and any without a well circumscribed CYP11B2-positive adenoma were excluded. All participants gave written informed consent and the protocol was approved by the local ethics committee.
DNA sequencing
Genomic DNA was extracted from dissected nodules from fresh frozen adrenal tissues, and DNA fragments were amplified using primers flanking mutation hot spot regions in KCNJ5, ATP1A1, ATP2B3, and CACNA1D before DNA sequencing as described elsewhere.21
Production of HAC15 stable cells lines with inducible KCNJ5 expression
cDNAs encoding mutated and wild type forms of KCNJ5 were prepared by Gateway cloning (ThermoFisher Scientific) in cumate inducible PiggyBac vectors (System Biosciences, Palo Alto, CA). Stable cell lines were established by co-transfection of human adrenocortical cells (HAC15 cells, a kind gift from Professor William E. Rainey, University of Michigan, Ann Arbor, USA) with the PiggyBac vector (carrying the human KCNJ5 cDNA) and the Super PiggyBac transposase according to the manufactureŕs instructions (System Biosciences, Palo Alto, CA). Transfected cells were selected with puromycin (4 μg/mL) in the presence of verapamil (10 μM) to inhibit the P glycoprotein.22 The macrolide antibiotic roxithromycin (20 μM) was also included to inhibit any potential effects on cell growth of mutant KCNJ5 channels23 in the absence of the cumate inducer. Total RNA was extracted from stable cell lines after induction with cumate (10 μg/mL) for 72 h, reverse transcribed and the KCNJ5 gene was sequenced to confirm the mutated or wild-type KCNJ5 genotype of all cell lines.21
Cell proliferation and apoptosis assays
HAC15 cells (2.5 × 104 cells/ well) stably transfected with wild type or mutated forms of KCNJ5 (T158A, G151R, G151E or L168R) or empty vector were plated in 96-well plates and transcription was induced with 1 μg/mL or 10 μg/mL cumate in the absence of roxithromycin for 24 hours. Cell proliferation was determined with a WST-1 assay (Roche), and apoptosis was quantified by an Annexin V apoptosis assay (Promega).
Generation of monoclonal antibodies against human KCNJ5
A peptide corresponding to the N-terminal portion of human KCNJ5 (acetyl-36-ATDRTRLLAEGKKP-49-C) with the addition of a cysteine at the C-terminal end was synthesized by LifeTein LLC (Hillsborough, NJ) and conjugated to 5 mg of ImjectTM Blue CarrierTM Protein (ThermoFisher Scientific) using Succinimidyl-6-(iodoacetyl)aminocaproate (Molecular Biosciences (Boulder, CO). Four Swiss Webster Female mice were immunized initially with 10 μg of immunogen with Complete Freund’s Adjuvant (Millipore-Sigma) followed by immunization using incomplete Freund’s adjuvant every two weeks. After 2 months of biweekly immunizations, the mice received the immunogen in saline intraperitoneally and 3 days later were sacrificed using isofluorane anesthesia, blood was withdrawn and spleens removed under aseptic conditions. Spleen cells were then obtained and frozen in liquid nitrogen using DMEM media containing 20% newborn calf serum, 5% dimethylsulfoxide and 2.5% of polyethylene glycol 1,000.
After titers were performed on the serum, the spleen from the mouse with the higher titer was fused with PEG 1450 (ATCC.org) to the mouse myeloma SP2-mIL6-hIL21-hTERT cells and plated into 10 × 96 well plates. After 10 days the wells were screened by ELISA on plates coated with the acetyl-36-ATDRTRLLAEGKKP-49-C conjugated to chicken ovalbumin. Positive clones were then screened by Western blotting of cell lysates from HEK 293T cells transduced with a tetracycline-inducible lentivirus containing the human KCNJ5 sequence.5 Clones which gave single bands of the appropriate molecular mass for KCNJ5 on Western blots were subcloned using high density methyl cellulose24 and were isotyped. The use of mice for the generation of monocloncal antibodies was approved by the University of Mississippi Medical Center IACUC.
Immunohistochemistry and immunofluorescence
Formalin-fixed paraffin-embedded (FFPE) adrenal tissue sections (3 μm) were used for CYP11B2 immunohistochemistry to detect aldosterone synthase expression with a monoclonal antibody (clone 17B) diluted 1:200 as described25 and KCNJ5 immunohistochemistry was performed using the KCNJ5 monoclonal antibody generated herein (clone # 36–33-5, dilution 1:2000). Double immunofluorescence CYP11B2 and KCNJ5 staining used an anti-mouse IgG1 Alexa Fluor 488 secondary antibody (to detect CYP11B2 primary antibody) and anti-mouse IgG2B Alexa Fluor 594 (to detect KCNJ5 antibodies) both diluted 1:200 (Invitrogen). A rabbit anti-PARP monoclonal antibody diluted 1:2000 (Cell Signaling) was used for immunofluorescence staining of cleaved PARP (poly-ADP ribose polymerase) with an anti-rabbit Alexa Fluor 594 secondary antibody diluted 1:200 (Invitrogen).
Scoring adrenals for Ki67 proliferation index and KCNJ5 immunostaining
Ki67 immunohistochemistry was performed on FFPE adrenal sections (3 μm) using a rabbit monoclonal antibody (clone # SP6 1:200 dilution, Sigma-Aldrich). The Ki67 proliferation index was assessed as the percentage of the manual count of intense Ki67 stained nuclei relative to the total hematoxylin stained nuclei which were quantified by color segmentation using ImageJ software. Three separate fields of view were used for scoring and the final proliferation index was calculated as the average of the 3 Ki67 scores.26 To score KCNJ5 immunostaining intensity in adenomas and paired adjacent cortical tissue, a semi-quantitative score system was used in which intensity of immunohistochemistry staining was graded 0 to 4 for undetectable, low, moderate or high27 from a field of view at x 20 magnification acquired from each adrenal sample. Both the Ki67 proliferation index and H scores for CYP11B2 were evaluated by researchers blinded to mutational status and pathological reports of the assessed adrenals (HS and TAW). Adenoma sizes (to determine correlations with Ki67 index) were determined by the pathologist (TK) as the diameter of the largest nodule.
Statistical analyses
Statistical analyses were performed using SPSS, version 25.0 and Graphpad Prism version 7.0. Comparisons between two groups were determined using a t test or a Mann-Whitney test, multiple comparisons were analyzed by ANOVA with a Bonferroni test or Kruskal-Wallis tests with pairwise comparisons. Pearsońs correlation coefficients were used to analyze univariate correlations. P<0.05 was considered significant.
Results
Clinical characteristics of patients with APA according to genotype
Genotyping of 72 resected adrenals from patients with an APA, determined 39 APAs with a KCNJ5 mutation (L168R, n=22; G151R, n=16, and T158A, n=1), 5 with a CACNA1D mutation, and 3 and 2 APAs with ATP1A1 or ATP2B3 mutations, respectively. The remaining 23 APAs did not carry a mutation in known hotspots of target genes and were referred to as tumors with no mutation detected (NMD).
Patients with a KCNJ5-mutated APA were younger than patients with an NMD-APA (47.2 years ± 10.4 versus 57.7 years ± 11.0, P= 0.001) with a higher proportion of women than patients with an NMD-APA (82.1% of 39 patients versus 30.4% of 23, P<0.001) or relative to the small group of patients with other somatic APA mutations (10.0 % of 10 patients, P<0.001). The largest adenoma diameter at pathology was greater in KCNJ5-mutated APAs (17.0 mm [14.0–24.0]) compared with both NMD-APAs and APAs with other mutations combined (12.0 mm [8.0–25.0], P=0.019 and 9.0 mm [7.8–15.3], P=0.003, respectively). We noted a lower PAC in KCNJ5-mutated APAs compared with the group of APAs with a mutation in ATP1A1, ATP2B3 and CACNA1D combined (979 pmol/L [500–1470] compared with 1989 pmol/L [1624–3346], P=0.006) (Table S1).
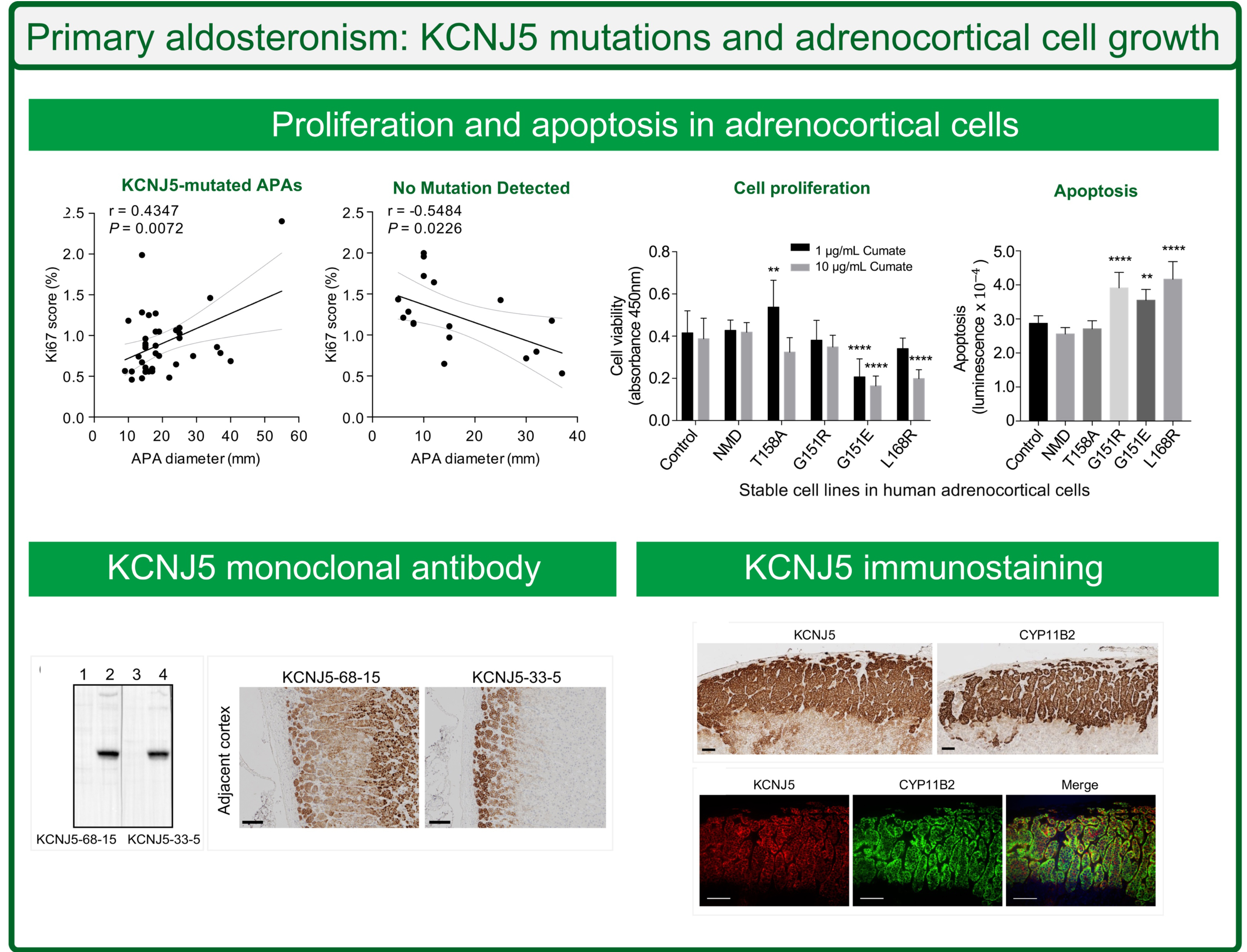
Diverse proliferation in adenomas with or without a KCNJ5 mutation
Ki67 proliferation index was assessed in a subset of adrenals (37 APAs with KCNJ5 mutations; 17 designated NMD and 10 with either a CACNA1D, ATP1A1 or ATP2B3 mutation). Adenoma size was larger in APAs with a KCNJ5 mutation compared with NMD (17.0 mm [14.5–24.5] versus 12.0 mm [8.0–27.5], P=0.0327). APAs with a KCNJ5 mutation had a lower proliferation index relative to APAs with NMD (0.9% ± 0.4 versus 1.2% ± 0.4, P=0.011). The Ki67 proliferation index was positively correlated with adenoma diameter in KCNJ5-mutated APAs (r=0.4347, P=0.0072) in contrast to the negative linear correlation noted in NMD-APAs (r=−0.5484, P=0.0226) (Figure 1). There was no correlation of adenoma diameter with Ki67 index in the small group of APAs with a CACNA1D, ATP1A1 or ATP2B3 mutation combined. There was no significant difference in adenoma diameter between APAs with a L168R or a G151R mutation (L168R, 16.0 mm [15.0–27.3] versus G151R, 18.0 mm [14.0–22.0], P=0.636) or in Ki67 score (L168R, 1.0 % ± 0.4 versus G151R, 0.8 % ± 0.4, P=0.339).
Figure 1: Correlation of Ki67 score with nodule diameter according to genotype.

Ki67 score was positively linearly correlated with APA diameter in KCNJ5-mutated APAs (r=0.4347, P=0.0072) (Panel A), whereas a linear negative correlation was observed in the group of tumors with no mutation detected (NMD) (r=−0.5484, P=0.0226) (Panel B). Ki67 index was not correlated with adenoma diameter in the small group of APAs with a CACNA1D, ATP1A1 or ATP2B3 mutation combined (Panel C). Ki67 score was derived using ImageJ software and calculated from the average intense Ki67 nuclei staining count divided by the total nuclei hematoxylin staining count from 3 fields of view. Lines represent the Pearson correlation (thick black line) and 95% CI (thin grey line). When the outlier in panel A is omitted, a positive linear correlation between Ki67 index and APA diameter is still observed (r=0.5454, P=0.0006).
Effects of KCNJ5 mutations on cell growth in adrenocortical cells
Stable HAC15 cell lines expressing KCNJ5 with different genotypes were established using the selection marker puromycin. Sensitivity to puromycin was increased in the presence of verapamil (10 μM) (Figure S1) and the presence of KCNJ5 mutations was confirmed by Sanger sequencing. The cell viability of the KCNJ5-T158A HAC15 cell line was significantly higher compared with control cells (transfected with empty vector) after 24-hour induction with 1 μg/mL cumate (P=0.0094). This effect on cell proliferation was absent in cells with increased transcriptional induction of KCNJ5-T158A (10 μg/mL cumate). KCNJ5-G151R had no apparent effect on adrenocortical proliferation in vitro, whereas decreased proliferation was observed in HAC15 cells with KCNJ5-G151E and L168R mutations (P<0.0001 versus control cells, both comparisons) (Figure 2A).
Figure 2: Effects of KCNJ5 mutants on cell growth in adrenocortical cells.

HAC15 cells stably transfected with wild type or mutated forms of KCNJ5 (T158A, G151R, G151E or L168R) or empty vector (control) were used to measure cell viability (Panel A) or apoptosis (Panel B). Cell viability was measured using a WST-1 assay after 24-hour incubation with either 1 μg/mL or 10 μg/mL cumate (black and grey bars, respectively) to induce expression of KCNJ5 (Panel A). Apoptosis was measured using an Annexin V assay after 24-hour incubation with 1 μg/mL cumate (Panel B). Bars represent means of 6 separate experiments, error bars indicate SD. P values were calculated by ANOVA with a post hoc Bonferroni test, **difference (P<0.01) from control, **** difference (P<0.0001) from control. NMD, no mutation detected
Higher levels of cell death by apoptosis were observed in cells with KCNJ5-G151R, G151E and L168R mutations (P<0.0001, P=0.0078, and P<0.0001 versus control cells, respectively) under the conditions tested (24-hour incubation with 1 μg/mL cumate). Cells carrying the KCNJ5-T158A mutation did not induce apoptosis under the same conditions (Figure 2B). These observations were consistent with immunofluorescence detection of cleaved poly-ADP ribose polymerase (PARP), a hallmark of apoptosis, which showed increased numbers cells with positive cleaved PARP staining in the nuclei of KCNJ5-G151E and L168R transfected cells compared with control cells (Figure S2). HAC15 cells with KCNJ5-T158A and G151R mutations displayed a similar proportion of cleaved-PARP positive cells compared with control cells (Figure S2).
Generation of monoclonal antibodies against human KCNJ5
There were 100 positive clones from the ELISA screen and of these, 2 clones (#33 and #68) displayed specific binding to KCNJ5 on Western blots of HEK 293T cell lysates transduced with a lentivirus carrying the human KCNJ5 sequence. Clones #33 and #68 were subcloned to produce antibodies KCNJ5–33-5 and KCNJ5–68-15 and their specificity was validated by Western blotting (Figure 3A). The two clones were isotyped, clone KCNJ5–33-5 was IgG2b and the KCNJ5–68-15 was IgG2c. Both antibodies were used for immunohistochemistry of FFPE sections of resected adrenals from patients with an APA. Analysis of the cortical tissue adjacent to an adenoma demonstrated membrane and cytoplasmic staining with #68–15 quite diffuse throughout the cortex compared with predominant plasma membrane staining of zona glomerulosa cells with #33–5 (Figure 3B, C). Clone #33–5 was selected for further immunohistochemistry and immunofluorescence staining.
Figure 3: Generation of KCNJ5 monoclonal antibodies.

Monoclonal antibodies against KCNJ5 were produced by injection of mice with a synthetic peptide corresponding to the N-terminal portion of KCNJ5 (acetyl-36-ATDRTRLLAEGKKP-49-C). See methods for details. The specificity of antibodies KCNJ5–68-15 and KCNJ5–33-11 was validated by Western blotting of cell lysates of HEK 293T cells transduced with a tetracycline-inducible lentivirus containing the human KCNJ5 sequence (Panel A, uninduced [lanes 1 and 3] and tetracycline-induced [lanes 2 and 4]). KCNJ5 immunohistochemistry of adrenal cortex adjacent to an APA using KCNJ5–68-15 and KCNJ5–33-5 (Panel B). KCNJ5–68-15 resulted in staining of most of the cortical tissue with evident staining of nuclei (Panel B, left). KCNJ5–33-5 produced intense staining of the zone glomerulosa with clear localization to the plasma membrane (Panel B, right). Panel B scale bar = 100 μm.
KCNJ5 expression in APAs varies according to genotype
Immunohistochemistry using the KCNJ5 #33–5 monoclonal antibody was performed on 33 adrenal samples with various APA genotypes (KCNJ5, n=13; WT, n=10; CACNA1D, n=5; ATP1A1, n=3; ATP2B3, n=2). Adenomas of all adrenals showed positive-immunostaining for KCNJ5 and CYP11B2 (Figure 4, Figure S3) with decreased intensity of KCNJ5 immunostaining in APAs with KCNJ5 mutations compared with other adenomas (Figure 4). Semi-quantitative H score assessment of KCNJ5 immunostaining highlighted the decreased KCNJ5 expression in APAs with a KCNJ5 mutation (Figure 5A, difference P<0.0001 for KCNJ5-mutated APAs versus NMD-APAs and APAs with ATP1A1, ATP2B3, CACNA1D mutations combined). There were no apparent differences in KCNJ5 immunostaining intensity between NMD-APAs versus APAs with CACNA1D, ATP1A1, ATP2B3 mutations (Figure 4, Figure 5A). No differences in intensity of KCNJ5 immunostaining were apparent between APAs with different KCNJ5 mutations (KCNJ5-G151R, L168R or T158A) (Figure S3).
Figure 4: Heterogeneous immunostaining of KCNJ5 in APA according to genotype.

Immunohistochemical staining of KCNJ5 and CYP11B2 in an APA with a KCNJ5 mutation or in an APA with no mutation detected (NMD) showing decreased KCNJ5 immunostaining in the adenoma with a KCNJ5 mutation (Panel A, Panel B). APAs with ATP1A1, ATP2B3 or CACNA1D mutations displayed intense KCNJ5 immunostaining (Panel C). Double immunofluorescence staining of KCNJ5 and CYP11B2 in an APA with a KCNJ5 mutation compared with a NMD-APA (Panel D). KCNJ5 was intensely expressed in CYP11B2-negative cells in KCNJ5-mutated adenoma but markedly decreased KCNJ5 immunofluorescence was observed in CYP11B2-positive cells (Panel D, upper panel). In wild type APAs, KCNJ5 and CYP11B2 were co-localized to the same cells (Panel D, lower panel). DAPI staining (blue) was only included in the merged image. Panels A - C scale bar = 2 μm; panel D scale bar = 100 μm.
Figure 5: KCNJ5 immunostaining in APAs according to genotype.

Semi-quantitative H score of KCNJ5 immunohistochemistry in adenomas according to genotype is shown (Panel A). Horizontal Lines represent median, vertical lines represent range (Panel A). P value was calculated by the Mann-Whitney test, ****difference (P<0.0001) from NMD or from other mutations combined. Relatively lower KCNJ5 immunostaining was noted in all adenomas with a KCNJ5 mutation compared with paired adjacent cortex, whereas 75% of 20 adenomas with other genotypes combined (NMD, n=10; ATP1A1, n=3; ATP2B2, n=2; CACNA1D, n=5) showed either increased or similar expression in APAs compared with paired adjacent cortex (Panel B). NMD, no mutation detected; other, adenomas with ATP1A1, ATP2B3 and CACNA1D mutation.
KCNJ5 immunostaining was lower in all 13 tumors with KCNJ5 mutations compared with the paired adjacent cortex (Figure 4A and B, Figure 5B). In contrast, the majority of APAs with other genotypes showed either increased or similar KCNJ5 immunostaining intensity in adenomas (75% of 20 adrenals) (Figure 4C, Figure 5B).
Double KCNJ5-CYP11B2 immunofluorescence was performed on APAs of different genotypes. Co-localization of KCNJ5 with CYP11B2 was demonstrated in all adrenals but a decrease of KCNJ5 immunostaining was evident in CYP11B2-positive cells relative to CYP11B2-negative cells of the same adenoma carrying a KCNJ5 mutation (Figure 4D, Figure S4). This difference of KCNJ5 immunostaining intensity was absent in APAs of other genotypes (Figure S4).
Expression of KCNJ5 in aldosterone-producing cell clusters
KCNJ5 and CYP11B2 immunohistochemistry and double KCNJ5-CYP11B2 immunofluoresence of APCCs showed moderate to high expression of KCNJ5 in APCCs (n=11) (Figure 6A) and the co-localization of the high-level KCNJ5 and CYP11B2 immunostaining (Figure 6B).
Figure 6: KCNJ5 and CYP11B2 immunostaining of aldosterone-producing cell clusters.

Immunohistochemistry (Panel A) and double immunofluorescence (Panel B) showed intense KCNJ5 staining in aldosterone-producing cell clusters and co-localization with CYP11B2. DAPI (blue) was only included in the merged image. Scale bar = 100 μm.
Discussion
We demonstrate the diverse effects of KCNJ5 mutations on adrenocortical cell growth. We show an increase in adrenocortical cell proliferation with low-level transcriptional induction of KCNJ5-T158A and, under similar conditions, stimulation of apoptosis with KCNJ5-G151R, L168R and G151E. In adenomas with KCNJ5 mutations, CYP11B2-positive cells display strikingly reduced levels of KCNJ5 expression compared with CYP11B2-negative cells of the same tumor and compared with CYP11B2-positive cells in APAs of other genotypes. We found decreased KCNJ5 immunostaining in KCNJ5-mutated APAs compared with paired adjacent cortical tissue in agreement with a previous study which also showed the absence of KCNJ5 mutations in the adjacent cortex.28
These observations indicate that only low-level expression of KCNJ5 mutations is compatible with adrenocortical cell survival. KCNJ5 mutations are absent (or at least rarely found) in APCCs which comprise tight nests of zona glomerulosa cells.17 The cell toxicity of KCNJ5 mutations combined with the high KCNJ5 expression in the zona glomerulosa layer is consistent with the absence of KCNJ5 mutations in APCCs and the particular phenotype of KCNJ5-mutated APAs with a predominance of zona fasciculata cells over zona glomerulosa cells.29–31
It is unlikely that the differences in intensity of KCNJ5 immunostaining are due to diminished antibody binding to mutated KCNJ5 because the monoclonal antibody was raised against a peptide corresponding to an extracellular N-terminal sequence at positions 36–49 (ATDRTRLLAEGKKP), far removed from the KCNJ5 mutations which are located in or near the channel pore region. Further, KCNJ5 immunohistochemistry with a polyclonal antibody (binding to multiple epitopes) shows a similar reduction of KCNJ5 immunostaining compared with the adjacent cortex.28
As reported in other studies,4, 12 APAs with KCNJ5 mutations were larger than other APAs and we show a positive correlation between nodule diameter of tumors with a KCNJ5-G151R or L168R mutation with cell proliferation. The pro-apoptotic effects of G151R and L168R and the relatively larger adenoma diameter of tumors carrying these mutations suggests a selective pressure to override apoptosis in these tumors. KCNJ5-mutated APAs have distinct transcriptional profiles compared with other APAs32–34 which may result in the expression of specific pro-survival factors to counteract the pro-apoptotic effects of KCNJ5-G151R and L168R.35–37 Conversely, in NMD-APAs, a decreased Ki67 index was noted with increasing tumor diameter such that NMD-APAs with large tumor diameters displayed relatively lower Ki67 indices. This is probably due to a decline in proliferation rate during the lifespan of the tumor, as described previously for sporadic parathyroid adenomas,38 and potentially explained by the activation of anti-proliferation and pro-apoptotic mechanisms to self-regulate tumor growth.
KCNJ5 potassium channel mutations associated with PA display a loss of selectivity for potassium ions and aberrant sodium ion conductance.2, 5 This disturbance in channel conductance appears less severe in with KCNJ5-T158A because human embryonic kidney (HEK) cells expressing this mutant display an increased permeability ratio for potassium relative to sodium ions compared with cells expressing G151R or L168R.2 Transduction of human adrenocortical cells with a lentivirus carrying the cDNA encoding the KCNJ5-T158A channel resulted in a decrease in cell proliferation compared with control cells.5 Our data with the higher level of transcriptional induction of KCNJ5 concord with the observations of Oki et al.5 but we did observe an increase in cell proliferation when the level of induction of KCNJ5-T158A gene expression was decreased.
Germline variants of KCNJ5 cause a familial form of PA called familial hyperaldosteronism type III (FH type III).39 Patients with germline KCNJ5-T158A or G151R mutations present with a severe form of PA with extensive adrenocortical hyperplasia requiring bilateral adrenalectomy.2, 16, 40 Patients with FH type III with a KCNJ5-G151E mutation display a relatively mild, medically-treatable clinical phenotype with apparently normal adrenals from computerized tomography scan results.41 Patch clamp electrophysiology of HEK 293T cells transfected with KCNJ5-G151E and G151R, demonstrated the increased sodium ion conductance of the G151E mutated channel and cell survival assays established the greater cell lethality induced by G151E relative to G151R.41 Our study supports this suggestion because KCNJ5-G151E, but not G151R, caused a highly significant reduction in the viability of human adrenocortical cells. The increased cell toxicity associated with KCNJ5-G151E was inferred to limit adrenocortical cell mass and account for the milder phenotype of carriers of this germline variant41 probably because only a subset of cells expressing low-levels of the mutated channel can survive and produce excess aldosterone.
Strengths and limitations of the study
The strength of our study is the production of stable human adrenocortical cell lines with inducible expression of KCNJ5 mutations to study the cell growth effects of sporadic and germline KCNJ5 mutations. A further strength is the analysis of the proliferative status of a large cohort of APAs with genotype data that were homogeneously selected for surgery according to a stringent diagnostic flow chart that included adrenal venous sampling. Finally, we used highly specific monoclonal antibodies to demonstrate by immunohistochemistry and double immunofluorescence the variance in KCNJ5 and CYP11B2 expression in APAs according to genotype. A limitation of our study is that genotyping was performed on dissected pieces of adrenal nodule rather than targeted to CYP11B2 expressing regions. However, we minimized the potential genotyping of a non-functional nodule because we performed CYP11B2 immunochemistry of all adrenals included in the study and those with non-functional nodules were excluded.
Perspectives
KCNJ5 mutations cause cell lethality to a variable degree according to genotype and expression level. The proliferative function of KCNJ5 mutations in vivo is challenging to reproduce in vitro because any long-term chronic effects of potential survival factors is difficult to replicate in adrenal cell cultures. Transcriptome studies are planned to identify genes and signaling pathways which enable cell proliferation of adenomas with KCNJ5 mutations, despite the increased cell lethality caused by their expression, and which limit growth rates of tumors with no mutation detected.
Supplementary Material
{kind=link}
Novelty and Significance.
What is New?
Ki67 proliferation index is positively correlated with adenoma diameter in KCNJ5-mutated APAs, a negative correlation was noted in tumors with no mutation detected
Adrenocortical cell expression of the sporadic and germline KCNJ5-T158A mutation caused cell proliferation at low induction of expression, other KCNJ5 mutations induced apoptosis
The zona glomerulosa layer and aldosterone-producing cell clusters adjacent to adenomas show intense KCNJ5 immunostaining
KCNJ5-mutated adenomas comprise CYP11B2-positive cells with a marked reduction of KCNJ5 immunostaining compared with CYP11B2-negative cells and APAs of other genotype
What is relevant?
KCNJ5 mutations in APAs are associated with increased adrenal cell proliferation
KCNJ5 mutations may be absent from aldosterone-producing cell clusters due to the high level of KCNJ5 expression in the zona glomerulosa
Summary
KCNJ5 mutations induce cell toxicity and their effects on adrenocortical cell growth are determined in part by the expression level of the mutated KCNJ5 potassium channel
Acknowledgments
Sources of Funding
This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No [694913] to M Reincke) and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Projektnummer: 314061271-TRR 205 to F Beuschlein, M Reincke and TA Williams; and by DFG grant RE 752/20–1 to M Reincke. This work was also supported by the Else Kröner-Fresenius Stiftung in support of the German Conns Registry-Else-Kröner Hyperaldosteronism Registry (2013_A182 and 2015_A171 to M Reincke). CE Gomez-Sanchez is supported by National Heart, Lung and Blood Institute grant R01 HL27255, the National Institute of General Medical Sciences grant U54 GM115428. Y. Yang is supported by a fellowship from the China Scholarship Council.
Footnotes
Conflicts of Interest/Disclosure
None
References
- 1.Young WF Jr. Diagnosis and treatment of primary aldosteronism: Practical clinical perspectives. J Intern Med. 2019;285:126–148. doi: 10.1111/joim.12831. [DOI] [PubMed] [Google Scholar]
- 2.Choi M, Scholl UI, Yue P. et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulkroun S, Beuschlein F, Rossi GP. et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension. 2012;59:592–598. doi: 10.1161/HYPERTENSIONAHA.111.186478. [DOI] [PubMed] [Google Scholar]
- 4.Akerstrom T, Crona J, Delgado Verdugo A. et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS One. 2012;7:e41926. doi: 10.1371/journal.pone.0041926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology. 2012;153:1774–1782. doi: 10.1210/en.2011-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prada ETA, Burrello J, Reincke M, Williams TA. Old and new concepts in the molecular pathogenesis of primary aldosteronism. Hypertension. 2017;70:875–881. doi: 10.1161/HYPERTENSIONAHA.117.10111. [DOI] [PubMed] [Google Scholar]
- 7.Perez-Rivas LG, Williams TA, Reincke M. Inherited forms of primary hyperaldosteronism: New genes, new phenotypes and proposition of a new classification. Exp Clin Endocrinol Diabetes. 2019;127:93–99. doi: 10.1055/a-0713-0629. [DOI] [PubMed] [Google Scholar]
- 8.Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, Miller BS, Giordano TJ, Tomlins SA, Rainey WE. Targeted molecular characterization of aldosterone-producing adenomas in white americans. J Clin Endocrinol Metab. 2018;103:3869–3876. doi: 10.1210/jc.2018-01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams TA, Monticone S, Schack VR et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension. 2014;63:188–195. doi: 10.1161/HYPERTENSIONAHA.113.01733. [DOI] [PubMed] [Google Scholar]
- 10.Fernandes-Rosa FL, Williams TA, Riester A et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension. 2014;64:354–361. doi: 10.1161/HYPERTENSIONAHA.114.03419. [DOI] [PubMed] [Google Scholar]
- 11.Zheng FF, Zhu LM, Nie AF, Li XY, Lin JR, Zhang K, Chen J, Zhou WL, Shen ZJ, Zhu YC, Wang JG, Zhu DL, Gao PJ. Clinical characteristics of somatic mutations in chinese patients with aldosterone-producing adenoma. Hypertension. 2015;65:622–628. doi: 10.1161/HYPERTENSIONAHA.114.03346. [DOI] [PubMed] [Google Scholar]
- 12.Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. A meta-analysis of somatic KCNJ5 K(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab. 2015;100:E1089–1095. doi: 10.1210/jc.2015-2149. [DOI] [PubMed] [Google Scholar]
- 13.Berridge MJ. Calcium signalling and cell proliferation. Bioessays. 1995;17:491–500. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- 14.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulatero P, Tauber P, Zennaro MC. et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension. 2012;59:235–240. doi: 10.1161/HYPERTENSIONAHA.111.183996. [DOI] [PubMed] [Google Scholar]
- 16.Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 2008;93:3117–3123. doi: 10.1210/jc.2008-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez-Sanchez CE, Nanba K, Rainey WE. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A. 2015;112:E4591–4599. doi: 10.1073/pnas.1505529112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamazaki Y, Nakamura Y, Omata K, Ise K, Tezuka Y, Ono Y, Morimoto R, Nozawa Y, Gomez-Sanchez CE, Tomlins SA, Rainey WE, Ito S, Satoh F, Sasano H. Histopathological classification of cross-sectional image-negative hyperaldosteronism. J Clin Endocrinol Metab. 2017;102:1182–1192. doi: 10.1210/jc.2016-2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, Stowasser M, Young WF Jr. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2016;101:1889–1916. doi: 10.1210/jc.2015-4061. [DOI] [PubMed] [Google Scholar]
- 20.Williams TA, Reincke M. MANAGEMENT OF ENDOCRINE DISEASE: Diagnosis and management of primary aldosteronism: The endocrine society guideline 2016 revisited. Eur J Endocrinol. 2018;179:R19–R29. doi: 10.1530/EJE-17-0990. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Burrello J, Burrello A, Eisenhofer G, Peitzsch M, Tetti M, Knosel T, Beuschlein F, Lenders JWM, Mulatero P, Reincke M, Williams TA. Classification of microadenomas in patients with primary aldosteronism by steroid profiling. J Steroid Biochem Mol Biol. 2019;189:274–282. doi: 10.1016/j.jsbmb.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bello-Reuss E, Ernest S, Holland OB, Hellmich MR. Role of multidrug resistance P-glycoprotein in the secretion of aldosterone by human adrenal NCI-H295 cells. Am J Physiol Cell Physiol. 2000;278:C1256–1265. doi: 10.1152/ajpcell.2000.278.6.C1256. [DOI] [PubMed] [Google Scholar]
- 23.Scholl UI, Abriola L, Zhang C, Reimer EN, Plummer M, Kazmierczak BI, Zhang J, Hoyer D, Merkel JS, Wang W, Lifton RP. Macrolides selectively inhibit mutant KCNJ5 potassium channels that cause aldosterone-producing adenoma. J Clin Invest. 2017;127:2739–2750. doi: 10.1172/JCI91733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis JM. A single-step technique for selecting and cloning hybridomas for monoclonal antibody production. Methods Enzymol. 1986;121:307–322. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, Satoh F, Maekawa T, Nakamura Y, Sasano H, Gomez-Sanchez EP. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol. 2014;383:111–117. doi: 10.1016/j.mce.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan GC, Negro G, Pinggera A. et al. Aldosterone-producing adenomas: Histopathology-genotype correlation and identification of a novel CACNA1D mutation. Hypertension. 2017;70:129–136. doi: 10.1161/HYPERTENSIONAHA.117.09057. [DOI] [PubMed] [Google Scholar]
- 27.Fernandes-Rosa FL, Amar L, Tissier F, Bertherat J, Meatchi T, Zennaro MC, Boulkroun S. Functional histopathological markers of aldosterone producing adenoma and somatic KCNJ5 mutations. Mol Cell Endocrinol. 2015;408:220–226. doi: 10.1016/j.mce.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 28.Boulkroun S, Golib Dzib JF, Samson-Couterie B, Rosa FL, Rickard AJ, Meatchi T, Amar L, Benecke A, Zennaro MC. KCNJ5 mutations in aldosterone producing adenoma and relationship with adrenal cortex remodeling. Mol Cell Endocrinol. 2013;371:221–227. doi: 10.1016/j.mce.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Azizan EA, Poulsen H, Tuluc P. et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet. 2013;45:1055–1060. doi: 10.1038/ng.2716. [DOI] [PubMed] [Google Scholar]
- 30.Dekkers T, ter Meer M, Lenders JW, Hermus AR, Schultze Kool L, Langenhuijsen JF, Nishimoto K, Ogishima T, Mukai K, Azizan EA, Tops B, Deinum J, Kusters B. Adrenal nodularity and somatic mutations in primary aldosteronism: One node is the culprit? J Clin Endocrinol Metab. 2014;99:E1341–1351. doi: 10.1210/jc.2013-4255. [DOI] [PubMed] [Google Scholar]
- 31.Monticone S, Castellano I, Versace K, Lucatello B, Veglio F, Gomez-Sanchez CE, Williams TA, Mulatero P. Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol Cell Endocrinol. 2015;411:146–154. doi: 10.1016/j.mce.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azizan EA, Lam BY, Newhouse SJ, Zhou J, Kuc RE, Clarke J, Happerfield L, Marker A, Hoffman GJ, Brown MJ. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab. 2012;97:E819–829. doi: 10.1210/jc.2011-2965. [DOI] [PubMed] [Google Scholar]
- 33.Akerstrom T, Willenberg HS, Cupisti K. et al. Novel somatic mutations and distinct molecular signature in aldosterone-producing adenomas. Endocr Relat Cancer. 2015;22:735–744. doi: 10.1530/ERC-15-0321. [DOI] [PubMed] [Google Scholar]
- 34.Backman S, Akerstrom T, Maharjan R, Cupisti K, Willenberg HS, Hellman P, Bjorklund P. RNA sequencing provides novel insights into the transcriptome of aldosterone producing adenomas. Sci Rep. 2019;9:6269. doi: 10.1038/s41598-019-41525-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams TA, Monticone S, Morello F, Liew CC, Mengozzi G, Pilon C, Asioli S, Sapino A, Veglio F, Mulatero P. Teratocarcinoma-derived growth factor-1 is upregulated in aldosterone-producing adenomas and increases aldosterone secretion and inhibits apoptosis in vitro. Hypertension. 2010;55:1468–1475. doi: 10.1161/HYPERTENSIONAHA.110.150318. [DOI] [PubMed] [Google Scholar]
- 36.Williams TA, Monticone S, Crudo V, Warth R, Veglio F, Mulatero P. Visinin-like 1 is upregulated in aldosterone-producing adenomas with KCNJ5 mutations and protects from calcium-induced apoptosis. Hypertension. 2012;59:833–839. doi: 10.1161/HYPERTENSIONAHA.111.188532. [DOI] [PubMed] [Google Scholar]
- 37.Shaikh LH, Zhou J, Teo AE, Garg S, Neogi SG, Figg N, Yeo GS, Yu H, Maguire JJ, Zhao W, Bennett MR, Azizan EA, Davenport AP, McKenzie G, Brown MJ. LGR5 activates noncanonical Wnt signaling and inhibits aldosterone production in the human adrenal. J Clin Endocrinol Metab. 2015;100:E836–844. doi: 10.1210/jc.2015-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parfitt AM, Braunstein GD, Katz A. Radiation-associated hyperparathyroidism: comparison of adenoma growth rates, inferred from weight and duration of latency, with prevalence of mitosis. J Clin Endocrinol Metab. 1993;77:1318–1322. doi: 10.1210/jcem.77.5.8077327 [DOI] [PubMed] [Google Scholar]
- 39.Monticone S, Tetti M, Burrello J, Buffolo F, De Giovanni R, Veglio F, Williams TA, Mulatero P. Familial hyperaldosteronism type III. J Hum Hypertens. 2017;31:776–781. doi: 10.1038/jhh.2017.34. [DOI] [PubMed] [Google Scholar]
- 40.Therien B, Mellinger RC, Caldwell JR, Howard PJ. Primary aldosteronism due to adrenal hyperplasia; occurrence in a boy aged 10 years. AMA J Dis Child. 1959;98:90–99. [DOI] [PubMed] [Google Scholar]
- 41.Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, Couch R, Hammer LK, Harley FL, Farhi A, Wang WH, Lifton RP. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A. 2012;109:2533–2538. doi: 10.1073/pnas.1121407109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.