

SUMMARY
Cholesterol serves as a building material for cellular membranes and plays an important role in cellular metabolism. The brain relies on its own cholesterol biosynthesis, which starts during embryonic development. Cholesterol is synthesized from two immediate precursors, desmosterol and 7-DHC. Mutations in the DHCR24 enzyme, which converts desmosterol into cholesterol, lead to desmosterolosis, an autosomal recessive developmental disorder. In this study we assessed brain content of desmosterol, 7-DHCand cholesterol from development to adulthood, and analyzed the biochemical, molecular and anatomical consequences of Dhcr24 mutations on the sterol profile in a mouse model of desmosterolosis and heterozygous Dhcr24+/− carriers. Our HPLC-MS/MS studies revealed that by P0 desmosterol almost entirely replaced cholesterol in the Dhcr24-KO brain. The greatly elevated desmosterol levels were also present in the Dhcr24-Het brains irrespective of maternal genotype, persisting into adulthood. Furthermore, Dhcr24-KO mice brains showed complex changes in expression of lipid and sterol transcripts, nuclear receptors and synaptic plasticity transcripts. Cultured Dhcr24-KO neurons showed increased arborization, which was also present in the Dhcr24-KO mouse brains. Finally, we observed a shared pathophysiological mechanism between the mouse models of desmosterolosis and Smith-Lemli-Opitz syndrome (a genetic disorder of conversion of 7-DHC to cholesterol).
Keywords: desmosterol, Dhcr24, cholesterol, 7-dehydrocholesterol, Dhcr7
INTRODUCTION
Cholesterol is essential for proper brain development and both neuronal differentiation and synaptogenesis are cholesterol-dependent processes (Dietschy 2009). It is a key component of cell membranes and it helps control the fluidity and function of the membranes (Tulenko, Boeze-Battaglia et al. 2006, Ren, Jacob et al. 2011, Chang, Ren et al. 2014). Cholesterol is the immediate precursor for the synthesis of steroid hormones and bile acids. Furthermore, it influences the activity in the sonic hedgehog pathway, an important signaling cascade for morphogenesis in the brain (Cooper, Wassif et al. 2003). Defects in cholesterol biosynthesis present with severe abnormalities and developmental disability (Kelley 2000, Porter 2002, Tierney, Bukelis et al. 2006, Porter and Herman 2011, Herman and Kratz 2012). In the cholesterol biosynthetic pathway, the two immediate precursors for cholesterol are desmosterol and 7-dehydrocholesterol (7-DHC). An inability to convert these precursors to cholesterol results in two distinct, but related disorders: desmosterolosis and Smith-Lemli-Opitz Syndrome (SLOS), respectively (FitzPatrick, Keeling et al. 1998, Porter 2008).
Desmosterolosis is a rare disorder caused by mutations in the Dhcr24 gene that encodes the enzyme 24-dehydrocholesterol reductase. Homozygous or heterozygous Dhcr24 mutations result in a systemic elevation of desmosterol and decreased levels of total cholesterol (Zolotushko, Flusser et al. 2011). The Dhcr24 gene is mapped to chromosome 1p33-p31.1 and eight missense mutations have been described in the coding region of 1550 bp (NM_014762.3) to date (Waterham, Koster et al. 2001, Schaaf, Koster et al. 2011). The observed clinical findings of desmosterolosis include shortening and flexion of extremities, brain malformations, dysmorphic facial features, syndactyly, developmental delay and diaphragmatic eventration.
In contrast, Smith-Lemli-Optiz Syndrome is caused by mutations in the Dhcr7 gene that encodes the enzyme 7-dehydrocholesterol reductase (Porter 2000). The result of these mutations is systemic elevation of 7-DHC and reduced cholesterol levels (DeBarber, Eroglu et al. 2011), leading to complex dysmorphology and intellectual and developmental disability. The clinical symptoms vary and may include hyperactivity, irritability and ritualistic behavior (Svoboda, Christie et al. 2012).
Although distinct disorders, they both arise from cholesterol biosynthesis errors, and share a number of similarities. SLOS and desmosterolosis have overlapping clinical findings and mouse models of both disorders lead to early postnatal lethality (Wassif, Zhu et al. 2001, Mirza, Hayasaka et al. 2006, Mirza, Hayasaka et al. 2008). It seems that the accumulation of desmosterol is more detrimental than the accumulation of 7-DHC, as Dhcr24 mutations in human patients are most often incompatible with life (Cross, Iben et al. 2015).
Similarly, Dhcr24-KO mice die shortly after the birth, and their sterol profile was not analyzed previously. To gain further understanding of the developmental trajectory of sterol biosynthesis, we analyzed cholesterol, desmosterol and 7-DHC levels in wild-type mouse brains across multiple developmental stages and adulthood. In addition, we assessed the biochemical, molecular and cellular changes in the brain of the Dhcr24-KO mouse model of desmosterolosis at birth, followed by a comparison to the sterol profile in a transgenic mouse model of SLOS.
MATERIALS AND METHODS
Chemicals.
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich Co (St. Louis, MO). HPLC grade solvents were purchased from VWR International (Radnor, PA). All cell culture reagents were from Mediatech (Manassas, VA), Life Technologies (Grand Island, NY) and Greiner Bio-One GmBH (Monroe, NC).
Mouse studies.
Adult mice, B6.129S5-Dhcr24tm1lex/SbpaJ, stock no: 012564 and B6.129P2(Cg)-Dhcr7tm1Gst/J, stock no: 007453 were purchased from the Jackson Laboratory (Bar Harbor, ME). The mice were housed under a 12-h light-dark cycle at constant temperature (25 °C) and humidity with ad libitum access to food and water in Comparative Medicine at UNMC, Omaha, NE. Embryonic and newborn mice from Het (Dhcr24+/−) x Het (Dhcr24+/−) Het (Dhcr7+/−) x Het (Dhcr7+/−) matings were used for the study. The whole brain was dissected then bisected along the longitudinal fissure prior to freezing in 2-methyl butane pre-chilled on dry ice and stored at −80°C. Tail clips were collected for genotype confirmation. Half of the brain was used for sterol analysis and the other half was used for either total RNA extraction and qPCR or protein extraction and western blotting. All procedures were performed in accordance with the Guide for the Humane Use and Care of Laboratory Animals. The use of mice in this study was approved by the Institutional Animal Care and Use Committee of UNMC. The brief history of the mouse model can be found in the Supplemental Materials and Methods.
Primary neuronal cultures, immunocytochemistry and imaging.
Primary cortical neuronal cultures were prepared from E15 mice as previously described (Korade, Mi et al. 2007, Xu, Mirnics et al. 2012). The neurite outgrowth was evaluated on the ImageXpress Pico Automated Cell Imaging System using the 20X Plan Fluor objective and Cy3 channel. Images were acquired with CellReporterXpress software. The acquisition of images was done by stitching 49 individual fields within one well. The images were processed using the neurite tracing algorithm.
RNA isolation and cleanup.
This method was previously described (Korade, Xu et al. 2010).
cDNA synthesis and RT2 Profiler™ PCR Mouse Arrays.
500 ng of total RNA was reverse transcribed using the first strand cDNA synthesis kit (Qiagen, Germantown, MD) following the manufacturer’s protocol. All data from the PCR was collected by the StepOne Software v2.3 (Thermo Fisher Scientific) and analyzed by Qiagen’s PCR Array Data Analysis Web Portal. We used several ways to normalize gene expression changes, including the best fit from the panel of housekeeping genes and geometric mean from the full plate. Only gene expression changes that are statistically significant with all different normalization protocols are presented in the Results section.
Sterol extraction from tissue and LC-MS/MS (SRM) analyses were previously described (Korade, Xu et al. 2010, Korade, Xu et al. 2013, Liu, Xu et al. 2014, Genaro-Mattos, Tallman et al. 2018).
Western blotting.
Details of the method along the list of antibodies used are shown in the Supplement.
Statistical analyses.
Data are presented as the mean ± standard error of the mean (SEM). Differences in means were tested using unpaired two-tailed t-tests, employing Welch’s correction when the variances between the two groups was significantly different. The p values for statistically significant differences are highlighted in Figure Legends. Statistical analyses were performed using GraphPad Prism version 8 (GraphPad Software, Inc, La Jolla, CA) for Windows and Microsoft Excel 2016.
Supplemental Materials and Methods contain detailed description of all methods.
RESULTS
Cholesterol, desmosterol and 7-DHC levels show a developmental trajectory.
Cholesterol is the major sterol in brain tissue and starts accumulating early in embryonic development. However, the temporal production of cholesterol and its two immediate precursors (desmosterol and 7-DHC) in the brain is not well understood to date. To gain insight into this process, we compared the brain levels of cholesterol and its immediate precursors starting from embryonic stages (E9) to adulthood (P240) (Supplementary Figure 1A-C). We found that compared to cholesterol, the absolute levels of its precursors were significantly lower at all stages of development: cholesterol was an order of magnitude more abundant than desmosterol, while desmosterol was an order of magnitude higher than 7-DHC. Cholesterol and its precursors showed a developmental trajectory, which was distinct. The most significant increase in cholesterol levels was observed from E17 to P30, with levels plateauing thereafter. Desmosterol levels increased linearly from E9 through P0 and after P30 stayed at low levels. In contrast, 7-DHC was present at very low levels in the mouse brains, peaking at P7 and declining thereafter.
Cholesterol deficiency leads to early postnatal death.
Desmosterolosis is an autosomal recessive disorder with Mendelian inheritance, thus the expected frequency of knockouts is approximately 25%. In our mouse colony we found a slightly lower than expected number of Dhcr24-KO mice (Figure 1A): in 12 litters with total 96 embryos (average number of embryos per litter = 8) we obtained 21% of KOs. At P0, Dhcr24-KO pups could be visually identified by lack of wrinkled skin, (Figure 1B), shorter limbs and a missing a milk spot. Western blot analyses of P0 brains revealed DHCR24 protein was completely absent in Dhcr24-KO pups, while Dhcr24-Het mice brains revealed a 50% reduction of DHCR24 when compared to WT littermates (Figure 1C). At P0, in addition to commonly observed KO stillborn pups (9 stillborn out of 27 born), all live-born KO pups died shortly after birth (Figure 1D). Furthermore, KO pups weighed approximately 10% less than WT littermates (p <0.0001), while WT and Dhcr24-Het P0 weights were comparable (Figure 1D).
Figure 1. Desmosterol is greatly elevated in the mouse model of desmosterolosis.
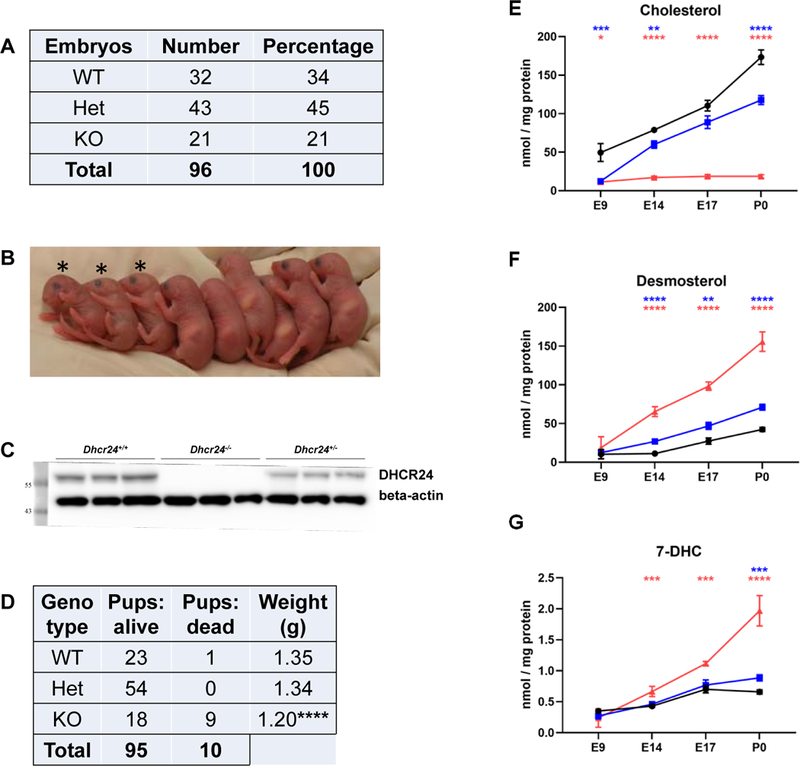
A) Number of embryos by genotype, E9-E18, recovered from 12 litters. B) An example of a litter with 3 KO pups (marked with stars). The KO pups are missing milk spots, have shorter legs and skin without wrinkles. C) DHCR24 protein expression is missing in Dhcr24-KO brains and is greatly decreased in Dhcr24-Het mouse brain. D) Number of P0 pups obtained from 12 litters which contained pups with all three genotypes. Comparison of number of alive and dead pups and their weight. E-G) Comparison of E) cholesterol, F) desmosterol and G) 7-DHC levels in mouse brain from WT (black), Het (blue) and KO (red) mice during embryonic development. Statistical significance and N values for each stage is shown in Supplemental Figure 2. Statistical significance (two-tailed t test): *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Desmosterol is greatly elevated in the brains of Dhcr24-deficient mice.
Next, we compared cholesterol, desmosterol and 7-DHC levels in brains of WT, Het and Dhcr24-KO littermates during embryonic development and shortly after birth (Figure 1E-G). While there was a steady increase of cholesterol and desmosterol in the WT and Het brains during embryonic stages, no accumulation of cholesterol in Dhcr24-KOs could be observed. Instead, desmosterol progressively accumulated in embryonic KO brains, reaching comparable levels to that of cholesterol seen in the WT brains by P0. Interestingly, the difference among WT, Het and Dhcr24-KO sterol levels was already present at E14, with the most pronounced differences at P0. Detailed statistical analyses are shown in Supplementary Figure 2. Our results also confirmed previously reported data that endogenous brain cholesterol synthesis in the developing brain starts around E12 (Tint, Yu et al. 2006, Lamberson, Xu et al. 2014).
Desmosterol is elevated in Dhcr24-Het mice compared to WT littermates.
Since we found that desmosterol is significantly elevated in P0 brain of Dhcr24-Het mice, we hypothesized that in these mice i) elevated desmosterol levels will persist into adulthood, and ii) maternal Het genotype will by itself have an effect on the developing pups. The effect of the maternal genotype on the pup’s brain sterol levels is shown in Figure 2A. We found decreased cholesterol (p < 0.001) and increased desmosterol (p < 0.01) levels in the brain tissue of Dhcr24-Het pups, which was independent of the maternal genotype (e.g. Dhcr24-Het pups originating from Dhcr24-Het vs WT mothers). In addition, we observed that 7-DHC levels were elevated in Het pups compared to their WT littermates in litters from Het mothers only (p =0.0280).
Figure 2. Desmosterol is elevated in the brain of Dhcr24-Het mice compared to WT littermates.
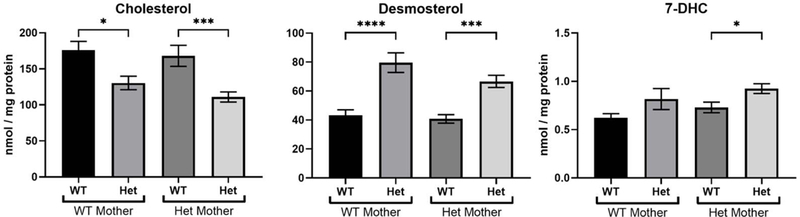
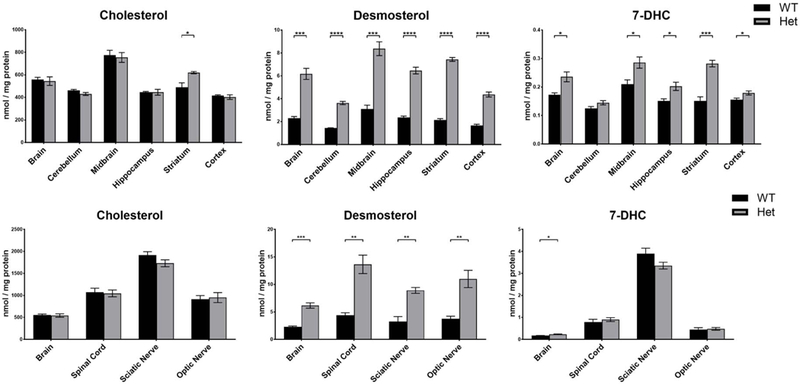
A) Sterols were analyzed in the brains from WT and Dhcr24-Het pups derived from either WT or Dhcr24-Het mothers at P0. The maternal phenotype did not affect cholesterol and desmosterol levels in the brain of newborn pups. N for maternal WT genotype: 16 WT pups and 14 Het pups; N for maternal Het genotype: 8 WT pups and 18 Het pups. B) Cholesterol, desmosterol and 7-DHC in different brain regions in WT and Het adult mice at P60. N=4 per group, 2 males and 2 females for each genotype. No sex differences were observed, so males and females of the same genotype were grouped and analyzed together. C) Cholesterol, desmosterol and 7-DHC in brain, spinal cord, sciatic nerve and optic nerve. N=4 per group. Statistical significance for A, B and C: groupwise t-test with Welch’s correction; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Next, desmosterol, 7-DHC and cholesterol levels were compared across different brain regions and the whole brain of two-month-old WT and Dhcr24-Het mice (Figure 2B). While there was significant difference in cholesterol levels between WT and Het brains at P0, no difference was detected at P60 across the tested brain regions, except in the striatum, which showed elevated cholesterol levels. In contrast, desmosterol levels remained greatly elevated at P60 in all brain regions, with the most pronounced increase in the striatum. Surprisingly, the largest difference in 7-DHC levels was observed at P60, with the most pronounced changes in the striatum. In addition to brain tissue, several other tissues were analyzed at P60. These include spinal cord, sciatic and optic nerve (Figure 2C) and liver (not shown). Compared to the brain, the liver had the lowest levels of sterols, while the spinal cord, sciatic and optic nerves had larger amount of sterols. With the exception of liver (data not shown), desmosterol was greatly elevated in all tissues originating from Dhcr24-Het mice compared to their WT littermates.
Elevated desmosterol levels result in complex transcriptional changes.
Since sterols are potent regulators of gene expression (Korade, Kenworthy et al. 2009, Korade, Xu et al. 2010), we screened P0 brain tissue using specific PCR arrays (Qiagen) to determine if elevated desmosterol changes gene expression. The pathway-focused panels used in this study include lipoprotein signaling and cholesterol metabolism, nuclear receptors and coregulators and synaptic plasticity arrays. The screening of both Dhcr24-KO and Dhcr7-KO brain tissues was done to compare similarities and differences. The full list of differentially regulated transcripts and their function is shown in Figure 3. Six lipid transcripts were downregulated and four were upregulated in Dhcr24-KO brain. Among the lipid transcripts, Dhcr24 was undetectable in Dhcr24-KO brains, and Dhcr7 transcript was undetectable in Dhcr7-KO brains. Among lipid transcripts with altered expression in Dhcr24-KO brains, three were also changed in the brains of Dhcr7-KO mice, albeit in the opposing direction. These three included Srebf1, a transcription factor and two sterol transporters, Abca1 and Abcg1. These findings are in agreement with the elegant studies by Yang et al. (Yang, McDonald et al. 2006) showing that desmosterol regulates the expression of Abca1 through the activation of LXRs.
Figure 3. Elevated desmosterol changes lipoprotein signaling, cholesterol biosynthesis transcripts, nuclear receptors and coregulators, and synaptic plasticity transcripts.
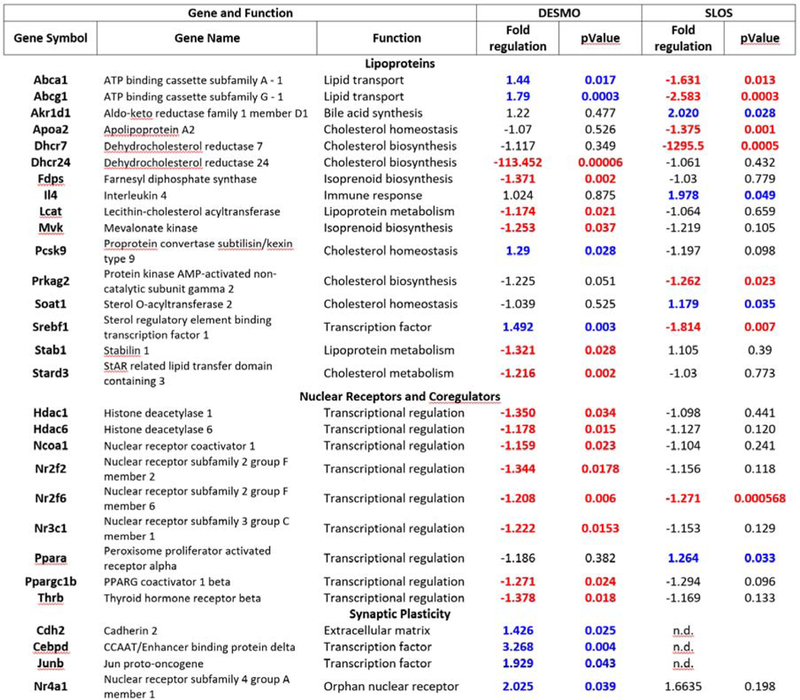
Table shows gene symbol, gene name and biological function, fold regulation and p-values. The upregulated transcripts are blue, downregulated transcripts are red and all statistically significant changes are shown in bold. p<0.05.
Among transcripts encoding nuclear receptors and co-regulators we identified eight transcripts with downregulated expression in Dhcr24-KO mice and two transcripts altered in Dhcr7-KO brains (one downregulated and one upregulated). Common for two disorders was downregulation of Nr2f2 transcription factor. While all nuclear receptors were downregulated in Dhcr24-KO brain, synaptic plasticity transcripts were all upregulated. Of these four upregulated genes, one encodes neural cell adhesion molecule, while the remaining three are transcription factors with expression patterns ranging from ubiquitous to highly region-specific expression in the brain. To determine if there is a common dysregulated pathway and visualize the interaction among various transcripts, all genes with altered expression were uploaded into the STRING database (https://string-db.org/). The visualization of interactions is shown in Supplementary Figure 3. This expanded network analysis revealed that these genes had common interaction partners, suggesting that Dhcr24- and Dhcr7-deficiency might affect the same nuclear transcriptional network.
High desmosterol levels alter neuronal outgrowth.
Synaptic development is a cholesterol-dependent process and altered sterol composition of neuronal membranes affects neuronal arborization. For example, in a SLOS mouse model, elevated 7-DHC increases dendritic and axonal length of hippocampal neurons (Jiang, Backlund et al. 2010) and 7-DHC derived oxysterols greatly increase neuronal arborization (Xu, Mirnics et al. 2012). To test if elevated desmosterol has an effect on neuronal morphology, we prepared cortical neuronal cultures from embryonic day 15 (E15) Dhcr24-KO and WT brains. Neurons were cultured for up to six days and their morphology was analyzed using the neuron-specific marker MAP2 (Figure 4). Imaging and quantification of total neuronal outgrowth revealed that arborization of Dhcr24-KO neurons was significantly increased compared to the neuronal outgrowth seen in samples originating from WT brains (p < 0.0001). To corroborate these results, protein was extracted from E18 and P0 brains, followed by western blotting for MAP2 (Figure 4). This experiment revealed that MAP2 was also greatly elevated in Dhcr24-KO brains at both E18 and P0, confirming the initially observed in vitro data.
Figure 4. Increased neuronal arborization in Dhcr24-KO brains.
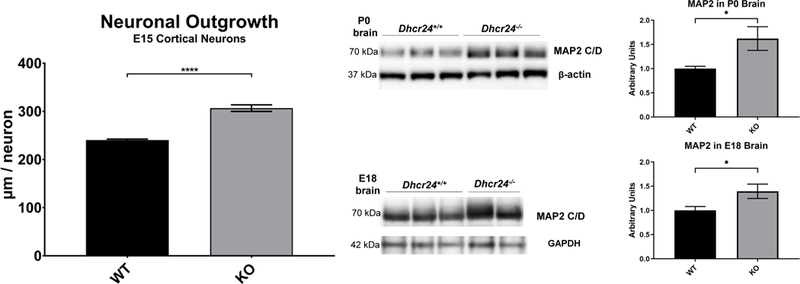
A) Total neuronal outgrowth was compared in WT and KO cultures from E15 cortex. Neuronal cultures in 96-well plates were stained with MAP2. The total outgrowth was assessed with the ImageXpress Pico Automated Cell Imaging System, using CellReporterXpress software and the Neurite Tracing analysis protocol. The total outgrowth was normalized to total number of cells. Neuronal cultures were prepared from 2 WT embryos and 4 KO embryos. For each culture condition, 8 wells with 49 images per well were analyzed at 20X. ****p<0.0001. B) Western blots for MAP2 in whole brain extracts from WT and Dhcr24-KO P0 pups at P0 and E18. Normalizers were Beta-Actin and Gapdh. C) Acquired TIFF images on Azure C300 instrument were analyzed with AzureSpot Software and quantification of band intensities performed in GraphPad Prism 8. *p<0.05.
DISCUSSION
The outcome of our studies can be summarized as follows: A) Cholesterol deficiency leads to early postnatal death or stillborn pups. B) Desmosterol is the second most abundant sterol at P0 during normal development, with a decreasing developmental trajectory into adulthood. C) In Dhcr24-KO mice, while desmosterol and 7-DHC are significantly elevated, cholesterol is greatly decreased. D) Greatly elevated desmosterol in Het brain at P0 continues to stay elevated in the adult brain. E) Changes in brain sterol composition lead to altered transcriptome response, affecting synaptic, lipid and nuclear receptor transcripts in a complex fashion. F) Total arborization of cultured Dhcr24-KO neurons is increased compared to WT neurons.
Studies in the SLOS mouse models were informative and provided a new insight into the human disorder (Waage-Baudet, Lauder et al. 2003, Korade, Xu et al. 2014, Sparks, Wassif et al. 2014, Sharif, Korade et al. 2017), as the sterol biosynthesis pathway is conserved across the two species. SLOS is the most frequent human disorder caused by mutation in cholesterol biosynthesis enzymes, and SLOS transgenic mouse models recapitulate molecular and biochemical changes seen in SLOS patients (Porter 2000, Porter 2002, Porter 2003, Porter 2008, Porter and Herman 2011). Similarly, DHCR24 mutations in desmosterolosis patients share the same fundamental phenotype with the Dhcr24 knockout mice – they have highly elevated desmosterol, and diminished levels of cholesterol. Thus, the highly conserved cholesterol biosynthesis pathways, and similarity of biochemical profile between human patients and transgenic mouse models suggest that the current model is a clinically relevant representation of pathophysiological processes that take place in the brain of human desmosterolosis patients.
While whole body cholesterol biosynthesis and metabolism have been extensively studied in the context of energy metabolism and cardiovascular diseases, the origin, role, metabolism and regulation of the central nervous system (CNS) cholesterol pool remains understudied. As shown in this study, desmosterol is the second most abundant sterol in the brain with rapidly increasing levels prenatally, and steady levels in adulthood. Thus, this raises the interesting question: does desmosterol have a function of its own, and can it influence normal brain function? Jansen et al. proposed that transient accumulation of desmosterol during early postnatal period may serve to increase the sterol pool in the brain (Jansen, Wang et al. 2013). This could be accomplished through different mechanisms, and all due to distinct chemical properties of desmosterol. Desmosterol has decreased propensity to be esterified, it cannot be hydroxylated to generate 24S-dhydroxycholesterol, and it activates the LXR-dependent pathway (Jansen, Wang et al. 2013). Based on the effects of desmosterol on neuronal arborization and synaptic plasticity transcripts that we have observed, it is conceivable that desmosterol may have additional roles in the regulation of neuronal differentiation and synaptic remodeling and this should be further investigated.
Our studies and previous findings raise an interesting question. In recessive diseases, heterozygous parents are typically asymptomatic, with the disease phenotype being expressed only in the offspring homozygous or compound heterozygous for the mutations. This might not be true in the case of DHCR24 mutations. Namely, in the three clinical reports of desmosterolosis (FitzPatrick, Keeling et al. 1998, Andersson, Kratz et al. 2002, Zolotushko, Flusser et al. 2011), in addition to description of affected individual, the authors assessed desmosterol levels in parents (carriers of DHCR24 mutations). All six carriers had elevated plasma desmosterol levels compared to control samples. In addition, the cultured immortalized lymphocytes obtained from patient parents showed a 10-fold increase in desmosterol levels compared to control samples. This finding has potential clinical implications, raising a host of questions. At what level do sterol intermediates levels become detrimental? How wide is the “normal” range for cholesterol biosynthesis intermediates? Is the elevated level of desmosterol predisposing, protective or a modifying factor for another condition?
An interesting and important observation of our study is related to the significantly increased desmosterol levels of the Dhcr24-Het mice throughout the lifespan. With a decreased DHCR24 activity observed in the Dhcr24-Het mice, we speculate that 7-dehydrodesmosterol (7-DHD), the immediate precursor to desmosterol, would also be increased in the brain. This would not be surprising and can be extrapolated from the increased 7-DHC levels in the Dhcr24-Het brains at P0 (Figure 1G and Supplemental Figure 2). The increase in 7-DHC suggests that intermediates upstream to desmosterol on the pathway would also be affected. Since both 7-DHC and 7-DHD are more prone to oxidation due to their chemical structure (Yin, Xu et al. 2011, Lamberson, Muchalski et al. 2017), the resulting oxidative products may have cytotoxic effects in the brain. Thus, in the DHCR24+/− heterozygous human population the elevated desmosterol and 7-DHD levels have the potential to result in elevated levels of oxysterols and increased oxidative stress, which might have a long-term effect on overall health status. These potential oxidative stress effects are likely to be regional and tissue-dependent, as we observed differences in desmosterol accumulation between the various brain regions. Thus, we propose that health status of the human heterozygous DHCR24+/− carriers should be carefully assessed, as this single-copy mutation might affect more than one domain of health and lead to a predisposition to various disorders in which oxidative stress plays a pathophysiological part.
While desmosterol is greatly elevated in Dhcr24-KO and significantly decreased in Dhcr7-KO mice, 7-DHC was increased in both of these two mouse models, and both disorders are characterized by markedly decreased cholesterol levels in the brain. Thus, it is perhaps not surprising that the molecular and microanatomical changes also show some similarities between Dhcr24-KO and Dhcr7-KO mice. Namely, the affected gene expression, while not identical across the two mouse models, impact the same transcriptional networks, and increased neuronal arborization is a feature of both models. Untangling the precise origin of all these changes is challenging and we propose that the primary driver of the majority of common changes is lack of cholesterol, while the Dhcr24-KO specific changes are due to elevated desmosterol levels. However, we acknowledge that increased neural arborization can be potentially a result of increased 7-DHC observed in both models, and might be due to the previously reported toxic effect of 7-DHC derived oxysterols.
In conclusion, desmosterol is an abundant, temporally regulated precursor of cholesterol in brain cells, with putative physiological function. Its disturbances might be a significant contributor to disease states, and this should be further investigated.
Supplementary Material
Supplemental Figure 1. Ontogeny of cholesterol, desmosterol and 7-DHC in the mouse brain from E9 to P240. Note different y-axis for three sterols. Embryonic stages E13–15 and E16-E18 were combined and labeled E14 and E17, respectively. N values for each time point: E9=3; E14=13; E17=8; P0=23; P30=3; P60=3; P150=3; P240=2.
Supplemental Figure 2. Desmosterol is greatly elevated in the brains of Dhcr24-deficient mice. Statistical significance (two-tailed t test): *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Supplemental Figure 3. Interaction network of transcripts altered in Dhcr24-deficient mice. STRING database of known and predicted biological interactions was used to analyze altered transcripts.
Acknowledgement:
Special thanks to Adriana Ocon, medical student at Texas A&M University School of Medicine, who participated in this research project during her undergraduate studies at Vanderbilt University and to Qian Chen, medical student from Shanghai Jiao Tong University School of Medicine, who participated in this research during summer exchange program.
Details of funding: This work was supported by The National Institutes of Health NIMH MH110636 (KM, NAP); NIEHS ES024133 (NAP, ZK), NICHD HD064727 (NAP).
Footnotes
A concise 1 sentence take-home message:
Greatly elevated desmosterol in Dhcr24-KO brain changes gene expression and alters neuronal arborization in the embryonic brain.
A competing interest statement: None declared.
Details of ethics approval: N/A
Documentation of approval from the IACUC: All procedures were performed in accordance with the Guide for the Humane Use and Care of Laboratory Animals. The use of mice in this study was approved by the Institutional Animal Care and Use Committee of UNMC.
REFERENCES
- Qiagen Data Center: https://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/
- Chang S, Ren G, Steiner RD, Merkens L, Roullet JB, Korade Z, DiMuzio PJ and Tulenko TN (2014). “Elevated Autophagy and Mitochondrial Dysfunction in the Smith-Lemli-Opitz Syndrome.” Mol Genet Metab Rep 1: 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, Porter FD and Beachy PA (2003). “A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis.” Nat Genet 33(4): 508–513. [DOI] [PubMed] [Google Scholar]
- Cross JL, Iben J, Simpson CL, Thurm A, Swedo S, Tierney E, Bailey-Wilson JE, Biesecker LG, Porter FD and Wassif CA (2015). “Determination of the allelic frequency in Smith-Lemli-Opitz syndrome by analysis of massively parallel sequencing data sets.” Clin Genet 87(6): 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBarber AE, Eroglu Y, Merkens LS, Pappu AS and Steiner RD (2011). “Smith-Lemli-Opitz syndrome.” Expert Rev Mol Med 13: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM (2009). “Central nervous system: cholesterol turnover, brain development and neurodegeneration.” Biol Chem 390(4): 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzPatrick DR, Keeling JW, Evans MJ, Kan AE, Bell JE, Porteous ME, Mills K, Winter RM and Clayton PT (1998). “Clinical phenotype of desmosterolosis.” Am J Med Genet 75(2): 145–152. [PubMed] [Google Scholar]
- Genaro-Mattos TC, Tallman KA, Allen LB, Anderson A, Mirnics K, Korade Z and Porter NA (2018). “Dichlorophenyl piperazines, including a recently-approved atypical antipsychotic, are potent inhibitors of DHCR7, the last enzyme in cholesterol biosynthesis.” Toxicol Appl Pharmacol 349: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman GE and Kratz L (2012). “Disorders of sterol synthesis: beyond Smith-Lemli-Opitz syndrome.” Am J Med Genet C Semin Med Genet 160C(4): 301–321. [DOI] [PubMed] [Google Scholar]
- Jansen M, Wang W, Greco D, Bellenchi GC, di Porzio U, Brown AJ and Ikonen E (2013). “What dictates the accumulation of desmosterol in the developing brain?” FASEB J 27(3): 865–870. [DOI] [PubMed] [Google Scholar]
- Jiang XS, Backlund PS, Wassif CA, Yergey AL and Porter FD (2010). “Quantitative proteomics analysis of inborn errors of cholesterol synthesis: identification of altered metabolic pathways in DHCR7 and SC5D deficiency.” Mol Cell Proteomics 9(7): 1461–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RI (2000). “Inborn errors of cholesterol biosynthesis.” Adv Pediatr 47: 1–53. [PubMed] [Google Scholar]
- Korade Z, Kenworthy AK and Mirnics K (2009). “Molecular consequences of altered neuronal cholesterol biosynthesis.” J Neurosci Res 87(4): 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korade Z, Mi Z, Portugal C and Schor NF (2007). “Expression and p75 neurotrophin receptor dependence of cholesterol synthetic enzymes in adult mouse brain.” Neurobiol Aging 28(10): 1522–1531. [DOI] [PubMed] [Google Scholar]
- Korade Z, Xu L, Harrison FE, Ahsen R, Hart SE, Folkes OM, Mirnics K and Porter NA (2014). “Antioxidant supplementation ameliorates molecular deficits in Smith-Lemli-Opitz syndrome.” Biol Psychiatry 75(3): 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korade Z, Xu L, Mirnics K and Porter NA (2013). “Lipid biomarkers of oxidative stress in a genetic mouse model of Smith-Lemli-Opitz syndrome.” J Inherit Metab Dis 36(1): 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korade Z, Xu L, Shelton R and Porter NA (2010). “Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith-Lemli-Opitz syndrome.” J Lipid Res 51(11): 3259–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberson CR, Muchalski H, McDuffee KB, Tallman KA, Xu L and Porter NA (2017). “Propagation rate constants for the peroxidation of sterols on the biosynthetic pathway to cholesterol.” Chem Phys Lipids 207(Pt B): 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberson CR, Xu L, Muchalski H, Montenegro-Burke JR, Shmanai VV, Bekish AV, McLean JA, Clarke CF, Shchepinov MS and Porter NA (2014). “Unusual kinetic isotope effects of deuterium reinforced polyunsaturated fatty acids in tocopherol-mediated free radical chain oxidations.” J Am Chem Soc 136(3): 838–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Xu L, Lamberson C, Haas D, Korade Z and Porter NA (2014). “A highly sensitive method for analysis of 7-dehydrocholesterol for the study of Smith-Lemli-Opitz syndrome.” J Lipid Res 55(2): 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza R, Hayasaka S, Kambe F, Maki K, Kaji T, Murata Y and Seo H (2008). “Increased expression of aquaporin-3 in the epidermis of DHCR24 knockout mice.” Br J Dermatol 158(4): 679–684. [DOI] [PubMed] [Google Scholar]
- Mirza R, Hayasaka S, Takagishi Y, Kambe F, Ohmori S, Maki K, Yamamoto M, Murakami K, Kaji T, Zadworny D, Murata Y and Seo H (2006). “DHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermis.” J Invest Dermatol 126(3): 638–647. [DOI] [PubMed] [Google Scholar]
- Porter FD (2000). “RSH/Smith-Lemli-Opitz syndrome: a multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis.” Mol Genet Metab 71(1–2): 163–174. [DOI] [PubMed] [Google Scholar]
- Porter FD (2002). “Malformation syndromes due to inborn errors of cholesterol synthesis.” J Clin Invest 110(6): 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter FD (2003). “Human malformation syndromes due to inborn errors of cholesterol synthesis.” Curr Opin Pediatr 15(6): 607–613. [DOI] [PubMed] [Google Scholar]
- Porter FD (2008). “Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management.” Eur J Hum Genet 16(5): 535–541. [DOI] [PubMed] [Google Scholar]
- Porter FD and Herman GE (2011). “Malformation syndromes caused by disorders of cholesterol synthesis.” J Lipid Res 52(1): 6–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Jacob RF, Kaulin Y, Dimuzio P, Xie Y, Mason RP, Tint GS, Steiner RD, Roullet JB, Merkens L, Whitaker-Menezes D, Frank PG, Lisanti MP, Cox RH and Tulenko TN (2011). “Alterations in membrane caveolae and BKCa channel activity in skin fibroblasts in Smith-Lemli-Opitz syndrome.” Mol Genet Metab 104(3): 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Koster J, Katsonis P, Kratz L, Shchelochkov OA, Scaglia F, Kelley RI, Lichtarge O, Waterham HR and Shinawi M (2011). “Desmosterolosis-phenotypic and molecular characterization of a third case and review of the literature.” Am J Med Genet A 155A(7): 1597–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif NF, Korade Z, Porter NA and Harrison FE (2017). “Oxidative stress, serotonergic changes and decreased ultrasonic vocalizations in a mouse model of Smith-Lemli-Opitz syndrome.” Genes Brain Behav 16(6): 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks SE, Wassif CA, Goodwin H, Conley SK, Lanham DC, Kratz LE, Hyland K, Gropman A, Tierney E and Porter FD (2014). “Decreased cerebral spinal fluid neurotransmitter levels in Smith-Lemli-Opitz syndrome.” J Inherit Metab Dis 37(3): 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda MD, Christie JM, Eroglu Y, Freeman KA and Steiner RD (2012). “Treatment of Smith-Lemli-Opitz syndrome and other sterol disorders.” Am J Med Genet C Semin Med Genet 160C(4): 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney E, Bukelis I, Thompson RE, Ahmed K, Aneja A, Kratz L and Kelley RI (2006). “Abnormalities of cholesterol metabolism in autism spectrum disorders.” Am J Med Genet B Neuropsychiatr Genet 141B(6): 666–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tint GS, Yu H, Shang Q, Xu G and Patel SB (2006). “The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols.” J Lipid Res 47(7): 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulenko TN, Boeze-Battaglia K, Mason RP, Tint GS, Steiner RD, Connor WE and Labelle EF (2006). “A membrane defect in the pathogenesis of the Smith-Lemli-Opitz syndrome.” J Lipid Res 47(1): 134–143. [DOI] [PubMed] [Google Scholar]
- Waage-Baudet H, Lauder JM, Dehart DB, Kluckman K, Hiller S, Tint GS and Sulik KK (2003). “Abnormal serotonergic development in a mouse model for the Smith-Lemli-Opitz syndrome: implications for autism.” Int J Dev Neurosci 21(8): 451–459. [DOI] [PubMed] [Google Scholar]
- Wassif CA, Zhu P, Kratz L, Krakowiak PA, Battaile KP, Weight FF, Grinberg A, Steiner RD, Nwokoro NA, Kelley RI, Stewart RR and Porter FD (2001). “Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith--Lemli--Opitz syndrome.” Hum Mol Genet 10(6): 555–564. [DOI] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Romeijn GJ, Hennekam RC, Vreken P, Andersson HC, FitzPatrick DR, Kelley RI and Wanders RJ (2001). “Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis.” Am J Hum Genet 69(4): 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Mirnics K, Bowman AB, Liu W, Da J, Porter NA and Korade Z (2012). “DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model.” Neurobiol Dis 45(3): 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC and Hobbs HH (2006). “Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands.” J Biol Chem 281(38): 27816–27826. [DOI] [PubMed] [Google Scholar]
- Yin H, Xu L and Porter NA (2011). “Free radical lipid peroxidation: mechanisms and analysis.” Chem Rev 111(10): 5944–5972. [DOI] [PubMed] [Google Scholar]
- Zolotushko J, Flusser H, Markus B, Shelef I, Langer Y, Heverin M, Bjorkhem I, Sivan S and Birk OS (2011). “The desmosterolosis phenotype: spasticity, microcephaly and micrognathia with agenesis of corpus callosum and loss of white matter.” Eur J Hum Genet 19(9): 942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Ontogeny of cholesterol, desmosterol and 7-DHC in the mouse brain from E9 to P240. Note different y-axis for three sterols. Embryonic stages E13–15 and E16-E18 were combined and labeled E14 and E17, respectively. N values for each time point: E9=3; E14=13; E17=8; P0=23; P30=3; P60=3; P150=3; P240=2.
Supplemental Figure 2. Desmosterol is greatly elevated in the brains of Dhcr24-deficient mice. Statistical significance (two-tailed t test): *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Supplemental Figure 3. Interaction network of transcripts altered in Dhcr24-deficient mice. STRING database of known and predicted biological interactions was used to analyze altered transcripts.