

Abstract
DNA N6-methyldeoxyadenosine (6 mA) modifications were first found more than 60 years ago but were thought to be only widespread in prokaryotes and unicellular eukaryotes. With the development of high-throughput sequencing technology, 6 mA modifications were found in different multicellular eukaryotes by using experimental methods. However, the experimental methods were time-consuming and costly, which makes it is very necessary to develop computational methods instead. In this study, a machine learning-based prediction tool, named csDMA, was developed for predicting 6 mA modifications. Firstly, three feature encoding schemes, Motif, Kmer, and Binary, were used to generate the feature matrix. Secondly, different algorithms were selected into the prediction model and the ExtraTrees model received the best AUC of 0.878 by using 5-fold cross-validation on the training dataset. Besides, the ExtraTrees model also received the best AUC of 0.893 on the independent testing dataset. Finally, we compared our method with state-of-the-art predictors and the results shown that our model achieved better performance than existing tools.
Subject terms: Computational models, Machine learning
Introduction
DNA N6-methyldeoxyadenosine (6 mA) modifications were first discovered in Bacteria in 19551. However, it had not received much attention as 5-methylcytosine (5mC) did. One important reason is that 6 mA modifications were thought to be only widespread in prokaryotes and unicellular eukaryotes, but rarely in multicellular eukaryotes2,3. Researchers have proposed several experimental methods to identify 6 mA modifications in the past few decades. The first method, developed by Dunn et al. in 1955, is a combination of ultraviolet absorption spectra, electrophoretic mobility, and paper chromatographic movement, but this method is relatively insensitive and cannot be used to detect 6 mA modifications in animals1. Then a restriction enzyme method was used to discover 6 mA modifications in 1978. However, this method can only find modified adenosines that occurred in the restriction enzyme target motifs4. With the development of high-throughput sequencing technology, thousands of 6 mA modifications were found in different multicellular eukaryotes. In 2015, Fu et al. found 6 mA modifications in 84% genes of Chlamydomonas by using 6 mA immunoprecipitation sequencing (6mA-IP-Seq)5. In 2016, Koziol et al. used dot blots, HPLC, and methyl DNA immunoprecipitation followed by sequencing (MeDIP-seq) to detect 6 mA modifications in vertebrates including Xenopus laevis, mouse and human6. In 2017, Mondo et al. observed that up to 2.8% of all adenines were methylated in early-diverging fungi by using single-molecule real-time (SMRT) sequencing7. In 2018, Zhou et al. found that about 0.2% of adenines in the rice genome were 6 mA methylated by using mass spectrometry, immunoprecipitation, and SMRT, and Zhang et al. observed that the 6 mA distribution in the rice and Arabidopsis genome were very similar by using 6mA-IP-seq8,9. As the experimental methods are time-consuming and costly, researchers are trying to predict DNA 6 mA modifications by using computational methods. Two prediction tools are reported up to now, i.e., iDNA6mA-PseKNC10 and i6mA-Pred11. iDNA6mA-PseKNC is the first prediction tool for predicting 6 mA modifications in the Mus musculus genome and i6mA-Pred is the first identification method in the rice genome.
Predicting DNA 6 mA modifications based on computational algorithms is still in the infancy. However, in the parallel study of prediction of post-translational modification (PTM) sites, there are many PTM-predicting papers published by the previous researchers12–22. Although there is some detailed difference for each of the individual PTMs, the basic core is about the same. Thus, the feature extraction and classification methods proposed in these studies provide a valuable basis for the prediction of DNA 6 mA modifications. In this research, we aim to develop a prediction tool that can be used to predict DNA 6 mA modifications across species. The benchmark datasets created in the iDNA6mA-PseKNC and i6mA-Pred predictors were used and different algorithms were implemented to generate the final optimized model. 5-fold cross-validation was performed and the prediction results demonstrated that our model achieved a better performance than existing 6 mA prediction tools.
As demonstrated by a series of recent publications10,13–19 and summarized in two comprehensive review papers23,24, to develop a really useful predictor for a biological system, one needs to follow Chou’s 5-steps rule (more detailed description can be found in https://en.wikipedia.org/wiki/5-step_rules.) to go through the following five steps: (1) construct a gold standard dataset to train and test the model; (2) encode samples with effective formulations; (3) conduct the prediction model with a powerful classifier; (4) evaluate model performance by using cross-validation tests and standard measures; (5) establish a user-friendly web-server for the predictor that can be accessible to the public. Below, we are to address these points one by one, making them crystal clear in logic development and completely transparent in operation.
Method
Dataset generation
Feng et al. created a DNA 6 mA benchmark dataset of the M. musculus genome in 201810. The benchmark dataset includes 1,934 positive samples and 1,934 negative samples. Chen et al. launched a 6 mA benchmark dataset of the rice genome in 201911. The benchmark dataset consists of 880 positive samples and 880 negative samples. The above two benchmark datasets were used to create the cross-species dataset and the CD-HIT-EST software25 with different threshold was used to reduce sequence redundancy in the original datasets (Table 1). Finally, the cross-species dataset consists of 2,768 positive samples and 2,716 negative samples with the most rigorous threshold at 0.80, and the length of each sample is 41nt. To build a cross-species 6 mA prediction model, the stratified selection method was used and we random selected 80% samples for model training and the left 20% for independent testing. Finally, the training dataset consists of 2,214 positive samples and 2,214 negative samples, while the independent testing dataset includes 554 positive samples and 502 negative samples.
Table 1.
Reduce sequence redundancy in the different datasets by using the CD-HIT-EST software.
| Species | Dataset | Sequence identity threshold | |||
|---|---|---|---|---|---|
| 0.95 | 0.90 | 0.85 | 0.80 | ||
| Mouse | Positive | 1,931 | 1,924 | 1,914 | 1,892 |
| Negative | 1,885 | 1,866 | 1,844 | 1,836 | |
| Rice | Positive | 880 | 879 | 878 | 876 |
| Negative | 880 | 880 | 880 | 880 | |
| cross-species | Positive | 2,811 | 2,803 | 2,792 | 2,768 |
| Negative | 2,767 | 2,746 | 2,724 | 2,716 | |
Feature encoding scheme
To construct a DNA 6 mA predictor, one of the most important but also most difficult issue is how to encode feature vector for each sequence, yet still retains most of the key patterns. The pseudo amino acid composition (PseAAC) was proposed by Chou et al. and has been widely used in nearly all the areas of computational proteomics26,27. Based on the PseAAC, four powerful software, such as ‘PseAAC’28, ‘PseAAC-Builder’29, ‘propy’30, and ‘PseAAC-General’31, were established: the former three are for generating various modes of Chou’s special PseAAC32; while the 4th one for those of Chou’s general PseAAC23. Encouraged by the successes of using PseAAC to deal with protein/peptide sequences, the concept of Pseudo K-tuple Nucleotide Composition (PseKNC)33 was developed for encoding features of DNA/RNA sequences34–36 that have proved very useful as well. Particularly, recently a very powerful web-server called ‘Pse-in-One’37 and its updated version ‘Pse-in-One2.0’38 have been established that can be used to generate any desired feature vectors for protein/peptide and DNA/RNA sequences according to the need of users’ studies.
K-mer pattern
K monomeric units (k-mers), are simply patterns of k consecutive nucleic acids37 and have a total of 4k kinds of nucleotide patterns for DNA/RNA. Such as 1-mer has 4 and 2-mer has 16 kinds of nucleotide patterns. To calculate the frequencies of k-mer nucleotide patterns, the length range L of the scanning region must be determined at first, and then the absolute frequencies of the k-mer nucleotide patterns are calculated from the start position to the L-k-1 position. Finally, the relative frequencies of k-mer patterns are calculated for each region. In this study, we set k as 2, 3, 4, and extracted 42 + 43 + 44 = 336 kinds of k-mer nucleotide patterns for feature encoding.
KSNPF frequency
The KSNPF frequencies are nucleotide pairs separated by k arbitrary nucleotides and have been successfully employed for the prediction of mucin-type O-glycosylation sites39 and phosphotase-specific dephosphorylation sites40. The KSNPF can be calculated using the following equation:
| 1 |
where n1 and n2 represent a pair of sequence elements. For nucleotide, n stands for any one of A, C, G, T/U. Thus, there are 42 = 16 combinations in each pair. Gap(k) stands for k arbitrary elements at intervals and S(n1Gap(k)n2) indicates the number of occurrences of the element pair. In this study, L represents the length of the nucleotide sequence, and the k was set as 1, 2, 3, 4, and the dimension of the KSNPF can be calculated by 42 × 4 = 64.
Nucleic shift density
Nucleic shift density encoding can be used to calculate the density of any nucleotide at the current position in its prefix string and has been used to encode nucleotide sequences in the iDNA6mA-PseKNC predictor10. A nucleic shift density feature at any position can be defined as follows:
| 2 |
where q represents of any nucleotide at current position i, Ni is the length of the ith prefix string in the sequence. For example, the DNA sequence “CAGCTG”. The Nucleic shift density of ‘C’ at the position 1, 2, 3, 4, 5 or 6 is 1/1 = 1, 1/2 = 0.5, 1/3 ≈ 0.33, 2/4 = 0.5, 2/5 = 0.4 or 2/6 ≈ 0.33, respectively. In this study, the length of each sample is 41nt. Thus, 41 Nucleic shift density features were generated for each sample.
Binary code
Binary encoding scheme is used to predict 6 mA modifications in the iDNA6mA-PseKNC predictor10. For the nucleotide in position i, the Binary features can be defined as following:
| 3 |
In this research, the Binary encoding scheme generates a vector with 3 × 41 = 123 elements by characterizing each nucleotide, “A”, “C”, “G”, or “T”, with (1, 1, 1), (0, 0, 1), (1, 0, 0), or (0, 1, 0), respectively.
Motif score matrix
The MEME Suite (http://meme-suite.org/) consists of several motif-based sequence analysis tools. In this study, the MEME tool with differential enrichment mode was used and the maximum number of motifs was set to 10. The most enriched motifs were selected based on E-value and the motif matrixes were used for generating motif scores of each sample.
Performance evaluation
Five different classifiers, Random Forest, GradientBoosting, AdaBoost, ExtraTrees and SVM, were implemented by using Python. For Random Forest, GradientBoosting, AdaBoost, ExtraTrees Classifiers, 1,000 trees were selected for each of them. For SVM, grid research was used to search the best combination of C and gamma parameters. 5-fold cross-validation was used to evaluate the performance of our model. In a different fold of cross-validation, each subset was iteratively selected as a testing set, while the left 4 subsets were used to train the model. The mean results of the five experiments were finally used as the performance estimates of the algorithms.
Based on the Chou’s symbols introduced for studying signal peptides41,42, Four standard measures were derived and have been adopted by several recent publications43–45. The measures can be defined as follows:
| 4 |
where N+ and N− refer to the number of positive samples or negative samples, respectively. stands for the number of positive samples that were predicted to be negatives, refers to the number of negative samples that were predicted to be positives. However, these measures are valid only for single-label learning issues. For the multi-label learning problems, whose appearances are more common in system biology46, system medicine47 and biomedicine16, a completely different set of standard measures is needed48. Besides, the receiver operating characteristic curve (ROC) combined with the area under the ROC curve (AUC), the Precision-Recall curve combined with the average precision (AP), and the F1 score49 were also used to evaluate the performance of different classifiers.
Using graphic approaches to study biological and medical systems can provide an intuitive vision and useful insights for helping analyze complicated relations therein as shown in the systems of enzyme fast reaction50, graphical rules in molecular biology51, and low-frequency internal motion in biomacromolecules (such as protein and DNA)52. Particularly, what happened is that this kind of insightful implication has also been demonstrated in53 and many follow-up publications54–56. The framework of csDMA is shown in Fig. 1.
Figure 1.

The framework of csDMA.
As pointed out by Chou et al.57 and demonstrated in a series of recent publications16–18, publicly accessible web-servers or online bioinformatics tools have significantly increased the impacts of bioinformatics on medical science58, driving medicinal chemistry into an unprecedented revolution59. Accordingly, the datasets and online tool involved in this paper are all available at https://github.com/liuze-nwafu/csDMA.
Results
Differential enrichment motifs discovery
To find the enriched motifs in the flank of 6 mA sites, the MEME tool with differential enrichment mode was used and the maximum number of motifs was set to 10. We used the positive samples in the cross-species dataset as the input and treated the negative samples as the control sequences. The detailed information of the enriched motifs can be found in the supplementary materials. Consider the statistical significance of the motifs, the E-value lower than 0.05 was used to find the most statistically significant motifs and two motifs were selected. The first motif, NNNNNNNHHNHHNHWNTNTNWNNNWNYNNNNNNNNNNNNNN, with an E-value of 3.3e-18 was the most statistically significant. And the third motif ACCGATCSA, with an E-value of 2.9e-2, was also selected. The probability matrixes can also be downloaded from the MEME website which can be used to build motif score matrixes in the training process.
Model training with different feature subsets
To find the best combination of feature subsets, different feature subsets were selected into the Random Forest classifier and 5-fold cross-validation was used on the training dataset to evaluate the performance of our model. As shown in Fig. 2, the classifier received an AUC value of 0.866 only by using the Binary code features, which means that the Binary code features were the most significant features that can be used to distinguish positive samples from negative samples. Interestingly, this result was even slightly higher than using combined feature subsets, such as Motif and Binary, Ksnpf and Binary, which achieved an AUC value of 0.861 and 0.862, respectively. Besides, the model achieved the best AUC value of 0.871 when three feature subsets Motif, Kmer, and Binary feature subsets were selected into the classifier. This result was even a little better than the model performance by using all feature subsets. Thus, we used the Motif, Kmer, and Binary encoding scheme to generate the optimized feature matrix.
Figure 2.

Model performance based on the different feature subsets. 1,000 decision trees were selected into the Random Forest classifier and 5-fold cross-validation was used to evaluate the performance of csDMA.
Performance evaluation with different classifiers
Five different algorithms were implemented in this research. For the Random Forest, GradientBoosting, AdaBoost, ExtraTrees Classifiers, 1,000 trees were selected for each of them. For the SVM classifier, grid research was used to search the best combination of C and gamma parameters and the SVM classifier achieved the best performance with C of 0.98 and gamma of 0.01. To compare the performance of different classifiers, 5-fold cross-validation was used and each classifier was trained with the same fold. As shown in Fig. 3, the ExtraTrees classifier received the best ACC of 0.799 and Sn of 0.864, while the AdaBoost got the lowest ACC of 0.715, Sn of 0.713, Sp of 0.718. However, the ExtraTrees classifier performed not very well for predicting negative samples and received an Sp of 0.735, but it is only a little lower than those of other methods. A more detailed comparison of different classifiers is also shown in Table 2. What’s more, the ExtraTrees classifier also achieved the highest MCC of 0.603, AUC of 0.878 and F1 of 0.811. Thus, we used the ExtraTrees algorithm to train the optimized model.
Figure 3.
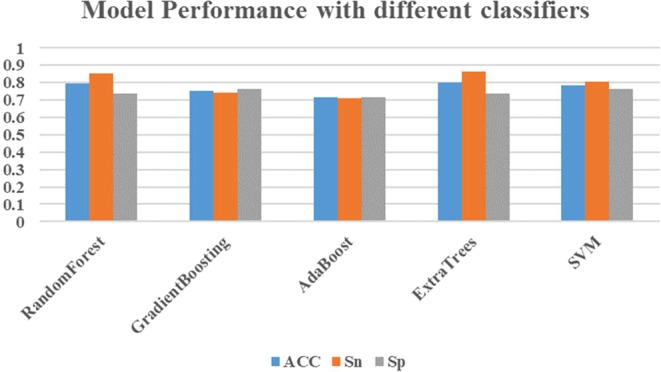
The model performance of different classifiers. The Motif, Kmer, and Binary feature subsets were selected into each classifier and the optimized parameters were used for model training. To evaluate the performance of each classifier, 5-fold cross-validation was used and Standard measures such as ACC, Sn and Sp were used to evaluate the performance of our model.
Table 2.
Model performance of each algorithm on the training dataset.
| Algorithm | Sn | Sp | ACC | MCC | AUC | F1 |
|---|---|---|---|---|---|---|
| RandomForest | 0.853 | 0.735 | 0.794 | 0.593 | 0.871 | 0.806 |
| GradientBoosting | 0.743 | 0.762 | 0.752 | 0.506 | 0.818 | 0.750 |
| AdaBoost | 0.713 | 0.718 | 0.715 | 0.431 | 0.777 | 0.715 |
| ExtraTrees | 0.864 | 0.735 | 0.799 | 0.603 | 0.878 | 0.811 |
| SVM | 0.807 | 0.764 | 0.785 | 0.572 | 0.858 | 0.790 |
The highest value of each column is marked in bold.
The independent testing dataset was also used to further evaluate the performance of each classifier. Each classifier was trained on the whole training dataset and evaluated on the independent testing dataset. As shown in Table 3, the ExtraTrees classifier received the best Sn of 0.888, AUC of 0.893 and F1 of 0.832, while the SVM model got the highest Sp of 0.761. Interestingly, the performance of each classifier on the independent testing dataset was even a little higher than that on the training dataset, which suggests that the classifier will receive better performance with a larger training dataset.
Table 3.
Model performance of the different algorithms on the independent testing dataset.
| Algorithm | Sn | Sp | ACC | MCC | AUC | F1 |
|---|---|---|---|---|---|---|
| RandomForest | 0.875 | 0.747 | 0.814 | 0.630 | 0.884 | 0.832 |
| GradientBoosting | 0.765 | 0.757 | 0.761 | 0.522 | 0.854 | 0.771 |
| AdaBoost | 0.776 | 0.719 | 0.749 | 0.496 | 0.814 | 0.764 |
| ExtraTrees | 0.888 | 0.729 | 0.813 | 0.628 | 0.893 | 0.832 |
| SVM | 0.843 | 0.761 | 0.804 | 0.607 | 0.875 | 0.819 |
The highest value of each column is marked in bold.
Comparison with existing 6 mA predictors
The SVM-based tool iDNA6mA-PseKNC was also implemented in this research. Grid research was used to find the best C and gamma, and the iDNA6mA-PseKNC achieved the best performance with C of 0.336 and gamma of 0.02. The same fold used for training csDMA was also used for training iDNA6mA-PseKNC. The iDNA6mA-PseKNC predictor received Sn of 0.767, Sp of 0.769, ACC of 0.767, MCC of 0.536, and F1 of 0.767. Most of the measures were lower except Sp is higher than our model with the ExtraTrees classifier. To further compare the performance of the two algorithms. The ROC and Precision-Recall curves were also plotted in Fig. 4. Our model received an AUC of 0.893, while iDNA6mA-PseKNC got an AUC of 0.840, which also demonstrates that our model achieved better performance than the iDNA6mA-PseKNC predictor.
Figure 4.

Performance comparison of csDMA and iDNA6mA-PseKNC. (A) The ROC curves of csDMA and iDNA6mA-PseKNC. (B) The Precision-Recall curves of csDMA and iDNA6mA-PseKNC.
To test the performance of our model across species, we compared the performance of csDMA and iDNA6mA-PseKNC on the different datasets, i.e., Cross-species, rice, and M. musculus datasets. For each dataset, 5-fold cross-validation was performed and the previously optimized parameters were used. We used the same fold for training on different datasets. The five-round results of each measure were averaged and shown in Table 4. For the Cross-species dataset, iDNA6mA-PseKNC got an AUC of 0.844, while our model received a higher AUC of 0.879. For the rice dataset, iDNA6mA-PseKNC received an AUC of 0.896, while our model achieved a higher AUC of 0.923. For the M. musculus dataset, both models got the same AUC values, but our model also received higher MCC and F1 than those of iDNA6mA-PseKNC. All these results show that the proposed method is very accurate and can be used to predict 6 mA sites in different species.
Table 4.
Model performance of each algorithm across species.
| Algorithm | Species | Sn | Sp | ACC | MCC | AUC | F1 |
|---|---|---|---|---|---|---|---|
| csDMA | Cross-species | 0.863 | 0.735 | 0.799 | 0.603 | 0.879 | 0.811 |
| Rice | 0.842 | 0.880 | 0.861 | 0.723 | 0.923 | 0.858 | |
| M. musculus | 0.932 | 1 | 0.966 | 0.935 | 0.974 | 0.965 | |
| iDNA6mA-PseKNC | Cross-species | 0.762 | 0.769 | 0.765 | 0.531 | 0.844 | 0.764 |
| Rice | 0.569 | 0.721 | 0.641 | 0.394 | 0.896 | 0.543 | |
| M. musculus | 0.869 | 1 | 0.935 | 0.877 | 0.974 | 0.930 |
Discussion
Unlike the prediction of m6A modifications in mRNA, the identification of 6 mA modifications in DNA is still at the beginning. In this study, we developed an improved tool, called csDMA, for predicting 6 mA modifications in different species. Three feature encoding strategies were used to generate the feature matrix and different algorithms were selected into the model. For performance evaluation, 5-fold cross-validation and independent test were used and the ExtraTrees classifier received the best performance on the training and independent test datasets. We also compared the performance of our tool with that of iDNA6mA-PseKNC. And the results showed that our model improved the recognition performance of DNA 6 mA modifications effectively.
The i6mA-Pred predictor is another of the two existing tools for DNA 6 mA prediction. However, the research paper is still in the corrected proof phase and their method cannot be reached until our work finished. Fortunately, we acknowledge from their online abstract that the method received an ACC of 0.831 by using a jackknife test. As jackknife test will generate a fixed ACC on the same dataset and their dataset was also downloaded as the rice dataset in this study. Thus, we also evaluated the performance of our model on the rice dataset by using a jackknife test and our model received an ACC of 0.859, which is also higher than that of i6mA-Pred.
Although our model received a high performance on the M. musculus dataset, the performance on the rice and cross-species datasets were relatively low. In the future, more feature encoding schemes, such as genomic and structural features, will be used to improve the performance of csDMA. And also we will extend csDMA to other species, such as human and Arabidopsis thaliana.
Supplementary information
Acknowledgements
This work was supported by the Start-up foundation of Northwest A&F University (Z109021809), the National Natural Science Foundation of China (51809218), and the Postdoctoral Research Foundation of China (2018M643744).
Author Contributions
Z.L. participated in conceiving and performing the experiments. W.D. and W.J. participated in analyzing the data. All authors contributed to the writing of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-49430-4.
References
- 1.Dunn DB, Smith JD. Occurrence of a new base in the deoxyribonucleic acid of a strain of bacterium coli. Nature. 1955;175:336–337. doi: 10.1038/175336a0. [DOI] [PubMed] [Google Scholar]
- 2.Vanyushin BF, Belozersky AN, Kokurina NA, Kadirova DX. 5-Methylcytosine and 6-Methylaminopurine in Bacterial DNA. Nature. 1968;218:1066–1067. doi: 10.1038/2181066a0. [DOI] [PubMed] [Google Scholar]
- 3.Casadesus J, Low D. Epigenetic gene regulation in the bacterial world. Microbiol and Molecular Biology Reviews. 2006;70:830. doi: 10.1128/MMBR.00016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird A. Use of restriction enzymes to study eukaryotic DNA methylation: II. The symmetry of methylated sites supports semi-conservative copying of the methylation pattern. Journal of Molecular Biology. 1978;118:49–60. doi: 10.1016/0022-2836(78)90243-7. [DOI] [PubMed] [Google Scholar]
- 5.Fu Y, et al. N6-Methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell. 2015;161:879–892. doi: 10.1016/j.cell.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koziol MJ, et al. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nature Structural & Molecular Biology. 2016;23:24–30. doi: 10.1038/nsmb.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mondo, S. et al. Widespread adenine N6-methylation of active genes in fungi. Nature Genetics. 49 (2017). [DOI] [PubMed]
- 8.Zhou C, et al. Identification and analysis of adenine N6-methylation sites in the rice genome. Nature Plants. 2018;4:554–563. doi: 10.1038/s41477-018-0214-x. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Q, et al. N(6)-Methyladenine DNA methylation in Japonica and Indica rice genomes and its association with gene expression, Plant Development, and Stress Responses. Molecular Plant. 2018;11:1492–1508. doi: 10.1016/j.molp.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Feng PM, et al. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics. 2018;111:96–102. doi: 10.1016/j.ygeno.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Chen, W., Lv, H., Nie, F. & Lin, H. i6mA-Pred: identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics. btz015 (2019). [DOI] [PubMed]
- 12.Xu Y, et al. iNitro-Tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. Plos One. 2014;9:e105018. doi: 10.1371/journal.pone.0105018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Feng P, Ding H, Lin H, Chou KC. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Analytical Biochemistry. 2015;490:26–33. doi: 10.1016/j.ab.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Tang H, Ye J, Lin H, Chou KC. iRNA-PseU: Identifying RNA pseudouridine sites. Molecular Therapy-Nucleic Acids. 2016;5:e332. doi: 10.1038/mtna.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia J, Zhang LX, Liu Z, Xiao X, Chou KC. pSumo-CD: Predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics. 2016;32:3133–3141. doi: 10.1093/bioinformatics/btw387. [DOI] [PubMed] [Google Scholar]
- 16.Qiu WR, Sun BQ, Xiao X, Xu ZC, Chou KC. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics. 2016;32:3116–3123. doi: 10.1093/bioinformatics/btw380. [DOI] [PubMed] [Google Scholar]
- 17.Feng P, et al. iRNA-PseColl: Identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Molecular Therapy-Nucleic Acids. 2017;7:155–163. doi: 10.1016/j.omtn.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, et al. iRNA-3typeA: identifying 3-types of modification at RNA’s adenosine sites. Molecular Therapy-Nucleic Acid. 2018;11:468–474. doi: 10.1016/j.omtn.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu WR, et al. iKcr-PseEns: Identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics. 2018;110:239–246. doi: 10.1016/j.ygeno.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Li F, et al. Positive-unlabelled learning of glycosylation sites in the human proteome. BMC Bioinformatics. 2019;20:112. doi: 10.1186/s12859-019-2700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, et al. Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework. Briefings in Bioinformatics. 2018 doi: 10.1093/bib/bby079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, et al. Large-scale comparative assessment of computational predictors for lysine post-translational modification sites. Briefings in Bioinformatics. 2018 doi: 10.1093/bib/bby089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition. Journal of Theoretical Biology. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou KC. Advance in predicting subcellular localization of multi-label proteins and its implication for developing multi-target drugs. Current Medicinal Chemistry. 2019 doi: 10.2174/0929867326666190507082559. [DOI] [PubMed] [Google Scholar]
- 25.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins. 2001;43:246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 27.Chou KC. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005;21:10–19. doi: 10.1093/bioinformatics/bth466. [DOI] [PubMed] [Google Scholar]
- 28.Shen HB, Chou KC. PseAAC: a flexible web-server for generating various kinds of protein pseudo amino acid composition. Analytical Biochemistry. 2008;373:386–388. doi: 10.1016/j.ab.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Du P, Wang X, Xu C, Gao Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo amino acid compositions. Analytical Biochemistry. 2012;425:117–119. doi: 10.1016/j.ab.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 30.Cao DS, Xu QS, Liang YZ. propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics. 2013;29:960–962. doi: 10.1093/bioinformatics/btt072. [DOI] [PubMed] [Google Scholar]
- 31.Du P, Gu S, Jiao Y. PseAAC-General: Fast building various modes of general form of Chou’s pseudo amino acid composition for large-scale protein datasets. International Journal of Molecular Sciences. 2014;15:3495–3506. doi: 10.3390/ijms15033495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Current Proteomics. 2009;6:262–274. doi: 10.2174/157016409789973707. [DOI] [Google Scholar]
- 33.Chen W, Lei TY, Jin DC, Lin H, Chou KC. PseKNC: a flexible web-server for generating pseudo K-tuple nucleotide composition. Analytical Biochemistry. 2014;456:53–60. doi: 10.1016/j.ab.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Chen W, Lin H. Pseudo nucleotide composition or PseKNC: an effective formulation for analyzing genomic sequences. Molecular BioSystems. 2015;11:2620–2634. doi: 10.1039/C5MB00155B. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Yang F, Huang DS, Chou KC. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics. 2018;34:33–40. doi: 10.1093/bioinformatics/btx579. [DOI] [PubMed] [Google Scholar]
- 36.Tahir M, Tayara H, Chong KT. iRNA-PseKNC(2methyl): Identify RNA 2’-O-methylation sites by convolution neural network and Chou’s pseudo components. Journal of Theoretical Biology. 2019;465:1–6. doi: 10.1016/j.jtbi.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 37.Liu B, et al. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Research. 2015;43:W65–W71. doi: 10.1093/nar/gkv458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu B, Wu H. Pse-in-One 2.0: An improved package of web servers for generating various modes of pseudo components of DNA, RNA, and protein sequences. Natural Science. 2017;9:67–91. doi: 10.4236/ns.2017.94007. [DOI] [Google Scholar]
- 39.Chen Y, Tang Y, Sheng Z, Zhang Z. Prediction of mucin-type O-glycosylation sites in mammalian proteins using the composition of k-spaced amino acid pairs. BMC Bioinformatics. 2008;9:101. doi: 10.1186/1471-2105-9-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Yan R, Song J. DephosSite: a machine learning approach for discovering phosphotase-specific dephosphorylation sites. Scientific Reports. 2016;6:23510. doi: 10.1038/srep23510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou KC. Using subsite coupling to predict signal peptides. Protein Engineering. 2001;14:75–79. doi: 10.1093/protein/14.2.75. [DOI] [PubMed] [Google Scholar]
- 42.Chou KC. Prediction of signal peptides using scaled window. Peptides. 2001;22:1973–1979. doi: 10.1016/S0196-9781(01)00540-X. [DOI] [PubMed] [Google Scholar]
- 43.Liu B, Wang S, Long R, Chou KC. iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics. 2017;33:35–41. doi: 10.1093/bioinformatics/btw539. [DOI] [PubMed] [Google Scholar]
- 44.Cheng X, Lin WZ, Xiao X, Chou KC. pLoc_bal-mAnimal: predict subcellular localization of animal proteins by balancing training dataset and PseAAC. Bioinformatics. 2019;35:398–406. doi: 10.1093/bioinformatics/bty628. [DOI] [PubMed] [Google Scholar]
- 45.Song J, Wang Y, Li F. iProt-Sub: a comprehensive package for accurately mapping and predicting protease-specific substrates and cleavage sites. Briefings in Bioinformatics. 2018;20:638–658. doi: 10.1093/bib/bby028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng X, Zhao SG, Lin WZ, Xiao X, Chou KC. pLoc-mAnimal: predict subcellular localization of animal proteins with both single and multiple sites. Bioinformatics. 2017;33:3524–3531. doi: 10.1093/bioinformatics/btx476. [DOI] [PubMed] [Google Scholar]
- 47.Cheng X, Zhao SG, Xiao X, Chou KC. iATC-mISF: a multi-label classifier for predicting the classes of anatomical therapeutic chemicals. Bioinformatics. 2017;33:341–346. doi: 10.1093/bioinformatics/btx245. [DOI] [PubMed] [Google Scholar]
- 48.Chou KC. Some remarks on predicting multi-label attributes in molecular biosystems. Molecular Biosystems. 2013;9:1092–1100. doi: 10.1039/c3mb25555g. [DOI] [PubMed] [Google Scholar]
- 49.Song J, et al. Transcriptome-wide annotation of m5C RNA modifications using machine learning. Frontiers in Plant Science. 2018;9:519. doi: 10.3389/fpls.2018.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou KC, Forsén S. Diffusion-controlled effects in reversible enzymatic fast reaction system: Critical spherical shell and proximity rate constants. Biophysical Chemistry. 1980;12:255–263. doi: 10.1016/0301-4622(80)80002-0. [DOI] [PubMed] [Google Scholar]
- 51.Carter RE, Forsén S. A new graphical method for deriving rate equations for complicated mechanisms. Chemica Scripta. 1981;18:82–86. [Google Scholar]
- 52.Chou K, Chen N, Forsén S. The biological functions of low-frequency phonons: 2. Cooperative effects. Chemica Scripta. 1981;18:126–132. [Google Scholar]
- 53.Jiang SP, Liu WM, Fee CH. Graph theory of enzyme kinetics: 1. Steady-state reaction system. Scientia Sinica. 1979;22:341–358. [Google Scholar]
- 54.Shen HB, Song JN, Chou KC. Prediction of protein folding rates from primary sequence by fusing multiple sequential features. Journal of Biomedical Science and Engineering. 2009;2:136–143. doi: 10.4236/jbise.2009.23024. [DOI] [Google Scholar]
- 55.Chou KC. Graphic rule for drug metabolism systems. Current Drug Metabolism. 2010;11:369–378. doi: 10.2174/138920010791514261. [DOI] [PubMed] [Google Scholar]
- 56.Zhou GP. The disposition of the LZCC protein residues in wenxiang diagram provides new insights into the protein-protein interaction mechanism. Journal of Theoretical Biology. 2011;284:142–148. doi: 10.1016/j.jtbi.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chou KC, Shen HB. Recent advances in developing web-servers for predicting protein attributes. Natural Science. 2009;1:63–92. doi: 10.4236/ns.2009.12011. [DOI] [Google Scholar]
- 58.Chou KC. Impacts of bioinformatics to medicinal chemistry. Medicinal Chemistry. 2015;11:218–234. doi: 10.2174/1573406411666141229162834. [DOI] [PubMed] [Google Scholar]
- 59.Chou KC. An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Current Topics in Medicinal Chemistry. 2017;17:2337–2358. doi: 10.2174/1568026617666170414145508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.