

Abstract
Endothelial-to-mesenchyme-like transition (Endo-MT) of trabecular meshwork (TM) cells is known to be associated with primary open angle glaucoma (POAG). Here, we investigated whether the prion protein (PrPC), a neuronal protein known to modulate epithelial-to-mesenchymal transition in a variety of cell types, is expressed in the TM, and plays a similar role at this site. Using a combination of primary human TM cells and human, bovine, and PrP-knock-out (PrP−/−) mouse models, we demonstrate that PrPC is expressed in the TM of all three species, including endothelial cells lining the Schlemm’s canal. Silencing of PrPC in primary human TM cells induces aggregation of β1-integrin and upregulation of α-smooth muscle actin, fibronectin, collagen 1A, vimentin, and laminin, suggestive of transition to a mesenchyme-like phenotype. Remarkably, intraocular pressure is significantly elevated in PrP−/− mice relative to wild-type controls, suggesting reduced pliability of the extracellular matrix and increased resistance to aqueous outflow in the absence of PrPC. Since PrPC is cleaved by members of the disintegrin and matrix-metalloprotease family that are increased in the aqueous humor of POAG arising from a variety of conditions, it is likely that concomitant cleavage of PrPC exaggerates and confounds the pathology by inducing Endo-MT-like changes in the TM.
Subject terms: Cell biology, Glaucoma
Introduction
Prion protein (PrPC) is a cell surface glycoprotein known mostly for its obligate role in the pathogenesis of prion disorders, a group of neurodegenerative conditions characterized by extensive degeneration of the brain parenchyma and the neuroretina. The key pathogenic event in all prion disorders is a change in the conformation of PrPC to a β-sheet rich PrP-scrapie (PrPSc) isoform1–3. The resulting loss of function of PrPC combined with gain of toxic function by PrPSc are believed to contribute to disease-associated pathology4. In support of the loss of function hypothesis, recent reports suggest that dysfunction of PrPC impairs its ability to fine-tune the Ras homolog gene family member A (RhoA)-associated coiled-coil containing kinase (ROCK) signaling pathway, resulting in over-activation of ROCK and signaling through the LIMK-cofilin pathway5,6. Deletion of PrP in knock-out (PrP−/−) mice or silencing in neuronal cells produces a similar outcome, supporting a key role of PrPC in regulating cytoskeletal homeostasis7,8. PrPC also interacts with several extracellular matrix (ECM) proteins9, and its absence or altered function induces cell-ECM dyshomeostasis, resulting in loss of neuronal polarity and axonal degeneration in diseased brains5,7,10.
One of the principal outcomes of RhoA-ROCK activation is a shift from cell-cell interactions to cell-substrate interactions, a key event in PrPC-mediated epithelial to mesenchymal transition (EMT) in several cell types8,11. Trabecular meshwork cells also respond to RhoA-ROCK activation, and upregulate fibrillogenic proteins that deposit in the extracellular matrix (ECM) and increase its stiffness12–14. This compromises the response of ECM meshwork to fluctuations in intraocular pressure (IOP) and increases resistance to aqueous outflow, the hallmark of primary open angle glaucoma (POAG). Although TM cells do not show the phenotypic changes typical of EMT because of their endothelial nature, the endothelial to mesenchyme-like transition (Endo-MT-like) is sufficient to elevate the IOP and precipitate glaucoma15. It is encouraging to note that ROCK inhibitors have provided significant benefit in reversing this change, and are promising therapeutic agents for the management of POAG16–18.
While the emphasis on inhibitors of ROCK activation for the therapeutic management of POAG is well-placed, it is equally important to identify pathways upstream or parallel to RhoA-ROCK, and cross-talk between different pathways. Examples include signaling through integrins19–21, TGFβ221,22, autotaxin-LPA23, JNK-paxillin24, and cross-talk between TGFβ, integrins, and the ECM25 to name a few. An additional possible player is PrPC, a documented inducer of EMT through β1-integrin and RhoA-ROCK activation5,7,11,26. This question deserves attention because PrPC is cleaved by members of the disintegrin and matrix-metalloprotease family of enzymes that are upregulated in the aqueous humor (AH) of glaucomatous eyes27,28, and is likely to confound the pathogenesis and therapeutic management of POAG through cross-talk with other pathways29.
Here, we explored the expression and functional significance of PrPC in the TM of human, bovine, and mouse eyes. We demonstrate that PrPC is expressed in the TM of all three species, and plays a significant role in maintaining the ECM structure at this site. In addition, significant amounts of soluble PrPC are present in the AH, suggesting constitutive or regulated shedding of membrane-bound PrPC in the anterior segment30.
Results
Expression of PrPC in human, bovine, and murine trabecular meshwork
Immunoreaction of human TM sections for PrPC showed strong reaction in all layers of the TM (Fig. 1a, panels 1 & 2). Reaction with mouse IgG and H&E staining of serial sections confirmed the specificity of the immunoreaction and accurate identification of the TM region (Fig. 1a, panels 3 & 4). A similar evaluation of non-permeabilized primary human TM cells revealed expression of PrPC on the plasma membrane as in neuronal and other cells (Fig. 1b, panel 1, arrowheads)31. No reaction was detected with non-specific mouse IgG processed in parallel (Fig. 1b, panel 2).
Figure 1.
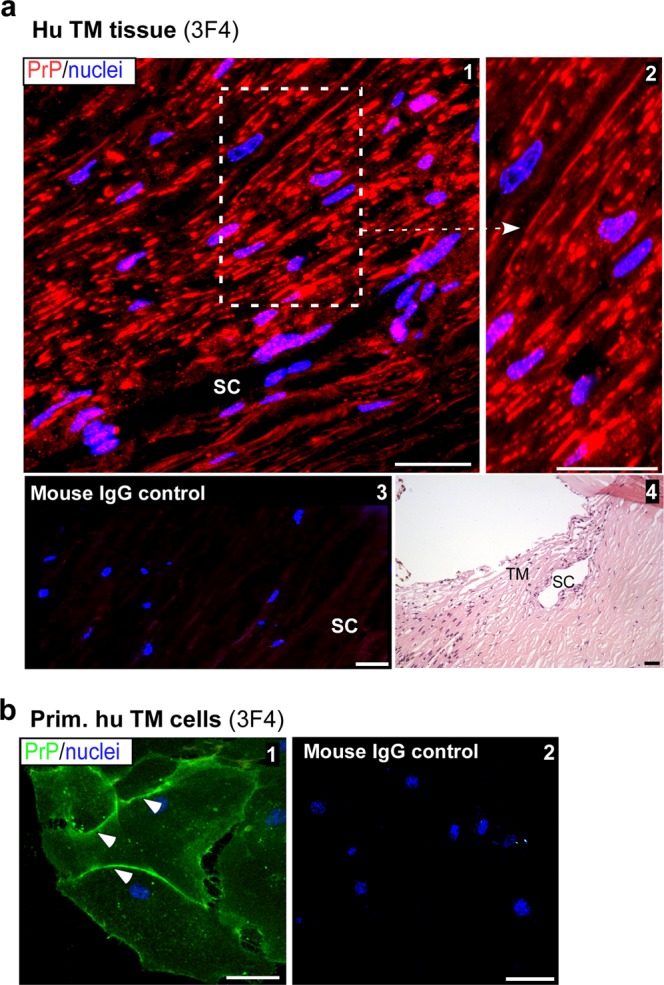
Distribution of PrPC in the human trabecular meshwork. (a) Immunoreaction of human TM section with PrP-specific antibody 3F4 followed by Alexa fluor 546-conjugated secondary antibody shows strong reactivity in all layers of the TM (panel 1). High magnification image demonstrates expression of PrPC on the plasma membrane of TM cells (panel 2). A serial section reacted with mouse IgG and Alexa fluor 546-conjugated secondary antibody shows no reaction (panel 3). H&E staining of a serial section confirms the TM region and Schlemm’s canal (SC) (panel 4). Scale bar: 25 µm. (b) Non-permeabilized primary human TM cells reacted with 3F4 followed by Alexa Fluor 488-conjugated secondary antibody show expression of PrPC on the plasma membrane (panel 1). No reaction is detected in control cells exposed to mouse IgG followed by the same secondary antibody (panel 2). Scale bar: 25 µm.
Further confirmation of the above results was obtained by performing a similar analysis on bovine and mouse TM tissue (Fig. 2). Immunoreaction of fixed bovine TM sections for PrPC revealed strong reactivity on the plasma membrane of TM cells and endothelial cells of the aqueous plexus (AP) (Fig. 2a panel 1, arrowheads)32. No reaction was detected in a serial section reacted with non-specific mouse IgG (Fig. 2a, panel 2). A similar evaluation of mouse TM sections showed strong reactivity for PrPC in PrP+/+, and complete absence in PrP−/− sections as expected (Fig. 2b, panels 1 & 2). Serial sections from the same block reacted with mouse IgG showed no reactivity (Fig. 2b panel 3). H&E staining confirmed accurate identification of the TM region in PrP+/+ sections of the anterior segment (Fig. 2b, panel 4).
Figure 2.

Expression of PrPC in bovine and murine TM. (a) Immunoreaction of bovine TM section with PrP-specific antibody SAF32 followed by Alexa fluor 546-conjugated secondary antibody shows strong reactivity for PrPC on the plasma membrane of TM cells and endothelial cells lining the aqueous plexus (AP) (panel 1, arrowhead). No reaction is detected in a serial section exposed to mouse IgG followed by the same secondary antibody (panel 2). Scale bar: 25 µm. (b) Immunoreaction of the anterior segment of PrP+/+ mouse eye with PrP-specific antibody 8H4 followed by Alexa fluor 546-conjugated secondary antibody shows expression of PrPC in all layers of the TM (panel 1). No reaction is noted in PrP−/− mouse sample processed in parallel (panel 2). Reaction of PrP+/+ sample with mouse IgG followed by the same secondary antibody shows no reaction (panel 3). H&E staining of a serial section confirms accurate identification of the TM region (panel 4). Scale bar: 25 µm.
Processing of PrPC in human ocular tissues
To evaluate the expression and processing of PrPC in different regions of the eye, antibodies spanning the entire sequence of PrPC were used (Fig. 3a). Thus, lysates of primary human TM cells were evaluated as such, or deglycosylated before processing for Western blotting. Probing of lysates with 3F4 that reacts with full-length (FL) and β-cleaved PrPC showed glycosylated and unglycosylated forms as expected (Fig. 3b, lanes 1–3). However, deglycosylation with PNGase-F revealed that 30–50% of FL PrPC (#, 27 kDa) was cleaved at the β-site (C2, white arrowhead, 20 kDa) (Fig. 3b, lanes 4–6; Fig. 3f). No reaction was detected in samples from TM cells transfected with PrPC-specific siRNA (Fig. 3b, lane 7). Lysate from human brain was fractionated in parallel as a positive control (Fig. 3b, lane 8). Re-probing with 8H4 showed α-cleaved 18 kDa band (C1, *), representing a minor fraction of the total (Fig. 3b, lanes 12–14). No reactivity was detected in samples treated with PrPC-specific siRNA (Fig. 3b, lane 15).
Figure 3.

Processing of PrPC in human ocular tissue. (a) Schematic representation of full length (FL), α-cleaved (C1, 18 kDa), β-cleaved (C2, 20 kDa), and ~19 kDa forms of PrPC. Antibody 8B4 reacts with FL and N-terminal fragments of PrPC, 3F4 reacts with FL and C2, and 8H4, G-12, and 2301 react with FL, C1, and C2. (b) Probing of lysates from primary human TM cells cultured from three different cases with 3F4 and 8H4 shows FL and mainly C2 fragment of PrPC. C1 represents a small fraction of total PrPC. Human brain lysate provides a positive control, and lysates from cells transfected with PrP-siRNA serve as a negative control (lanes 1–15). (c) Probing of lysates from the TM, retina (Ret), optic nerve (ON), and CB with 8H4 shows glycosylated PrPC in all samples (lanes 1–4), and a ~19 kDa fragment in lysates from the ON (lane 3,?) (d) Probing of human TM and CB lysates with 8B4, 3F4, and G-12 shows FL glycosylated and deglycosylated PrPC in all samples as in human brain. The TM shows significantly more C2 relative to FL and C1, the CB shows mainly C1, while the brain shows mainly FL and a small amount of C1 (lanes 1–18) (lighter exposures are shown for lanes 11 and 12. Complete membrane is shown in Supplementary Fig. S2). TM and CB lysates probed with 2301 antibody mimicked the G-12 probing data (Supplementary Fig. S3) (e) Probing of lysates from the ON and retina with 8B4, 3F4, and 2301 shows FL PrPC as in human brain, and a ~19 kDa fragment in deglycosylated ON sample. The retina has significantly more C2 relative to FL and C1, while the ON has more C1 relative to C2. The ~19 kDa fragment does not react with 2301 (lanes 15-18). (FL: #; C1: star; C2: white arrowhead;?: ~19 kDa). All membranes were re-probed for β-actin to control for loading. (f) The relative abundance of FL, C1, C2, and the ~19 kDa fragment is shown graphically. Figures 3c-e are from tissue harvested from the same eye. Similar results were obtained from two other eye globes.
Evaluation of tissue from different regions of the human eye with 8H4 showed strong reactivity for FL PrPC in the TM, retina (Ret), optic nerve (ON), and ciliary body (CB) (Fig. 3c, lanes 1–4, #)). However, the presence of a unique 19 kDa band in the ON was surprising (Fig. 3c, lane 3), prompting additional exploration.
To achieve this goal, three pairs of human eyes were dissected to isolate the TM, CB, retina, and ON, and tissue homogenates were evaluated as such or deglycosylated before Western blotting. Lysate from human brain was fractionated in parallel as a positive control. To avoid experimental artifacts, membranes probed once with a particular PrPC-specific antibody were re-probed only for β-actin, not with another PrP antibody.
Probing of lysates from the TM and CB with 8B4 that reacts with FL and N-terminal fragments revealed mainly FL PrPC in all samples (Fig. 3d, lanes 1–6, #). N-terminal fragments of α- and β-cleaved PrP were not detected because these are soluble and unlikely to be present in significant amounts in tissue lysates33. Probing with 3F4 showed mainly β-cleaved PrPC (Fig. 3d, lanes 7–10, white arrowhead). Reaction with G-12, a C-terminal antibody that reacts with FL, β-cleaved, α-cleaved, and probably Ɣ-cleaved soluble FL PrPC showed mainly β-cleaved PrPC in the TM, and α-cleaved fragment in the CB and brain samples (Fig. 3d, lanes 13–18, white arrowhead, *). Similar results were obtained with 2301, validating the results with G-12 (Supplementary Fig S3). In bovine CB, however, PrPC showed equivalent representation of α- and β-cleaved forms30, suggesting species-specific differences in its proteolytic processing.
Lysates from ON and retina showed FL and α-cleaved PrPC (Fig. 3e, lanes 1–18, #, *). The retina also showed β-cleaved PrPC (Fig. 3e, lanes 6, 12, and 18, white arrowhead). Surprisingly, the optic nerve showed a unique fragment of ~19 kDa that reacted with the N-terminal antibody 8B4 (Fig. 3e, lane 2,?). This fragment also reacted with 8H4 (Fig. 3c, lane 3) and 3F4 (Fig. 3e, lane 14), but not with 2301 (Fig. 3e, lane 16). These observations suggest that this fragment is distinct from α, β, and Ɣ-cleaved PrPC for the following reasons; (1) it is glycosylated and hence includes at least one N-glycan (amino acid 175) (Fig. 3e, lanes 1 & 2, lanes 13 & 14), (2) it reacts with 8B4, suggesting the presence of N-terminal amino acid 35 (Fig. 3a), (3) it does not react with 2301 (Fig. 3a), and (4) migrates at ~19 kDa, a molecular mass that is inconsistent with Ɣ-cleaved or FL PrPC. Further exploration is necessary to characterize this fragment fully and understand the conditions that precipitate its cleavage.
Quantification of relative abundance of FL, C1 (α-cleaved), and C2 (β-cleaved) forms of PrPC in different tissues showed a ratio of 32:6:62 for TM cells, 9:27:64 for TM tissue, 20:60:20 for CB, 30:35:5:30 (unique) for optic nerve, and 14:45:41 for the retina (Fig. 3f). These ratios are an approximation at best because of the efficiency of reactivity of different antibodies. However, it is clear that unlike brain and neuronal cells where 60–70% of PrPC is cleaved at the α-site, processing of PrPC in ocular tissues is distinct, and is mainly at the β-site in human TM tissue.
Silencing of PrPC induces mesenchyme-like transition in the TM
Absence of PrPC has been reported to induce aggregation of β1-integrin in neuronal cells8, triggering the Rho/ROCK pathway. To evaluate if a similar process occurs in the TM, primary human TM cells were transfected with PrP-specific siRNA, and non-permeabilized cells were immunoreacted with antibody specific for activated β1-integrin. Downregulation of PrPC resulted in clustering of β1-integrin on the plasma membrane as opposed to control cells that showed uniform distribution (Fig. 4a, panels 1–4, arrowheads). Control cells where the primary antibody was omitted did not show any reaction (Fig. 4a, panel 5). Western blotting of lysates did not show a significant difference in β1-integrin expression levels as reported in a previous study5 (Supplementary Fig. S4).
Figure 4.

Downregulation of PrPC aggregates β1-integrin and upregulates laminin and its receptor. (a) Silencing of PrPC in human TM cells followed by immunostaining with antibody specific for activated β1-integrin shows clustering of activated β1-integrin on the plasma membrane in the absence of PrPC (arrowheads) (panels 1–4). Cells transfected with PrP-siRNA and reacted with mouse IgG and respective secondary antibody do not show any reaction (panel 5). Scale bar: 25 µm. (b) Probing of TM cell lysates for PrPC shows the expected glycoforms in control cells transfected with scrambled siRNA, and minimal reaction in cells exposed to PrP-siRNA (lanes 1 & 2). Probing for laminin and laminin receptor (LR) shows significant upregulation in the absence of PrPC relative to control (lanes 1 & 2). (c) Quantification by densitometry after normalization with β-actin shows 2-fold upregulation of laminin and LR due to downregulation of PrPC. Values are mean ± SEM of the indicated n. *p < 0.05. Full-length blots are included in the Supplementary Fig. S2.
To evaluate whether clustering of β1-integrin activates downstream pathways resulting in Endo-MT-like transition, primary human TM cells were transfected with PrP-specific siRNA to downregulate PrPC, and the expression and distribution of fibrillogenic proteins indicative of Endo-MT-like transition was evaluated by Western blotting.
Probing for PrPC revealed the expected glycoforms in cells transfected with scrambled siRNA, and >98% downregulation by PrP-specific siRNA (Fig. 4b, lanes 1 & 2). Probing for laminin and laminin receptor (LR) showed significant upregulation in the absence of PrPC relative to controls (Fig. 4b, lanes 1 & 2; Fig. 4c). Immunoreaction of control and experimental cells for laminin and laminin-receptor confirmed the immunoblotting results (Supplementary Fig. S5).
A similar evaluation of duplicate cultures showed significant downregulation of PrPC by siRNA treatment as above (Fig. 5a, lanes 1 & 2). Notably, downregulation of PrPC resulted in significant upregulation of α-smooth muscle actin (α-SMA) and fibronectin relative to controls (Fig. 5b, lanes 1 & 2; Fig. 5c). Immunostaining of sections from PrP−/− and PrP+/+ mouse eyes for α-SMA and fibronectin showed more reaction in the TM of PrP−/− relative to PrP+/+ controls (Fig. 5d & 5e, panels 1–4), supporting the above results. Immunoreaction with mouse IgG showed no reaction (Fig. 5d, panels 5 & 6).
Figure 5.

Downregulation of PrPC upregulates α-SMA and fibronectin in the TM. (a) Probing of TM cell lysates treated with scrambled and PrP-siRNA for PrPC shows the expected glycoforms in the scrambled control, and minimal reactivity for PrPC in the experimental sample (lanes 1 & 2). (b) Probing of the same lysates for α-SMA and fibronectin shows upregulation in the absence of PrPC relative to controls (lanes 1 & 2). (c) Quantification of protein expression by densitometry after normalization with β-actin shows 6.1-fold and 5.9-fold upregulation of α-SMA and fibronectin respectively. Values are mean ± SEM of the indicated n. *p < 0.05, **p < 0.01. Full-length blots (Supplementary Fig. S2). (d & e) Immunoreaction of fixed sections from the anterior segment of PrP+/+ and PrP−/− mice shows stronger reactivity for α-SMA and fibronectin in the TM of PrP−/− relative to PrP+/+ samples (panel 1 vs. 2). Scale bar: 25 µm. No reaction was detected in samples reacted with mouse IgG followed by Alexa 546-conjugated secondary antibody (panels 5 & 6).
Further probing of lysates from human TM cells transfected with scrambled and PrP-siRNA showed significant upregulation of vimentin and collagen 1A by downregulation of PrPC in comparison to controls (Fig. 6a, lanes 1 & 2; Fig. 6b). Immunostaining of sections from the anterior segment of PrP−/− and PrP+/+ mouse eyes for vimentin and collagen 1A showed more reactivity in PrP−/− relative to PrP+/+ samples (Fig. 6c,d, panels 1–4), supporting the results from primary human TM cells. Immunoreaction with mouse IgG (for vimentin) and rabbit IgG (for collagen 1A) showed no reaction (Fig. 6c,d, panels 5 & 6).
Figure 6.

Downregulation of PrPC upregulates vimentin and collagen 1A. (a) PrPC was silenced in human TM cells and lysates were processed as above. Probing for vimentin and collagen 1A shows significant upregulation in the absence of PrPC relative to controls (lanes 1 & 2). (b) Quantification of protein expression by densitometry after normalization with β-actin shows 1.9-fold upregulation of vimentin and 2.2-fold upregulation of collagen 1A in the absence of PrPC. Values are mean ± SEM of the indicated n. *p < 0.05, **p < 0.01. Full-length blots (Supplementary Fig. S2). (c & d) Immunoreaction of fixed sections from the anterior segment of PrP+/+ and PrP−/− mice for vimentin and collagen 1A shows stronger reaction in the TM of PrP−/− relative to PrP+/+ samples (panel 1 vs. 2). No reaction was detected when serial sections were reacted with mouse or rabbit IgG followed by Alexa 546-conjugated secondary antibody (panels 5 & 6). Scale bar: 25 µm.
Together, the above results demonstrate that absence or downregulation of PrPC in mouse models or in primary human TM cells aggregates β1-integrin and upregulates fibrillogenic proteins including α-SMA, fibronectin, vimentin, and collagen 1A, the ECM protein laminin, and surprisingly, also the receptor for laminin.
Absence of PrPC upregulates myocilin
Myocilin is a biomarker for TM cells, and is upregulated by dexamethasone treatment34. Since human TM cells are likely to change their morphology in culture, dexamethasone-mediated upregulation of myocilin, a reliable method for their validation35, was used before every experiment (Fig. 7a, lanes 1 & 2; Fig. 7b). Surprisingly, downregulation of PrPC also upregulated myocilin in primary human TM cells (Fig. 7a, lane 1 vs. 3; Fig. 7b), and blunted their response to dexamethasone (Fig. 7a, lane 1 vs 3 & 4; Fig. 7b). Probing for PrPC showed the expected glycoforms in cells treated with scrambled siRNA as expected, and almost complete absence in cells transfected with PrP-siRNA (Fig. 7a, lanes 1–4).
Figure 7.

Myocilin is upregulated by downregulation of PrPC. (a) PrPC was silenced in primary human TM cells and lysates were processed as above. Probing for PrPC shows the expected glycoforms in controls, and minimal reaction in samples treated with PrP-siRNA (lanes 1 & 2 vs. 3 & 4). Re-probing for myocilin shows significant upregulation in the absence of PrPC (lane 1 vs 3). Exposure of control and experimental cells to dexamethasone (Dex) shows upregulation of myocilin as expected (lanes 1 & 2). However, no additive effect of dexamethasone is noted in the absence of PrPC (lanes 3 & 4). (b) Quantification by densitometry after normalization with β-actin shows 8-fold upregulation of myocilin by dexamethasone, and ~5.3-fold upregulation in the absence of PrPC regardless of dexamethasone. Values are mean ± SEM of the indicated n. **p < 0.01; ns: not significant. Full-length blots are included in the Supplementary Fig. S2. (c) Immunoreaction of anterior segment of PrP+/+ and PrP−/− mice for myocilin shows upregulation in the TM of PrP−/− sections relative to controls (panels 1 & 2). No reaction was detected in a serial section reacted with mouse IgG followed by Alexa 546-conjugated secondary antibody (panel 3). Scale bar: 25 µm. (d) Measurement of IOP shows significant upregulation in PrP−/− eyes relative to PrP+/+ controls (n = 8). Values are mean ± SEM of the indicated n. **p < 0.01.
Immunoreaction of mouse TM sections for myocilin revealed significantly stronger reactivity in PrP−/− samples relative to PrP+/+ controls (Fig. 7c, panels 1 & 2), confirming the results from human TM cells. Immunoreaction with mouse IgG showed no reactivity (Fig. 7c panel 3).
Absence of PrPC elevates intraocular pressure
To establish the clinical relevance of our observations to POAG, age and sex-matched PrP+/+ and PrP−/− mice were anesthetized, and IOP was measured in both eyes with a tonometer. To eliminate bias, measurements were performed in separate sets of mice by three different individuals blinded to the mouse genotype. Surprisingly, IOP was significantly elevated in PrP−/− eyes relative to PrP+/+ controls (Fig. 7d). The increase in IOP in PrP−/− mice is in good agreement with upregulation of fibrillogenic proteins36,37, the hallmark of altered cell-ECM interactions in the TM.
Discussion
We report that PrPC is expressed in the TM, and modulates cell-ECM interactions at this site. Downregulation of PrPC in primary human TM cells induced aggregation of β1-integrin on the plasma membrane and upregulation of fibrillogenic proteins. Likewise, fibrillogenic proteins were upregulated in the TM of PrP−/− mice, indicating an Endo-MT-like transition. In vivo measurement of IOP revealed significant elevation in PrP−/− relative to PrP+/+ mouse eyes, implicating PrPC in the pathophysiology of POAG.
Our data demonstrate expression of PrPC in the TM of human, bovine and mouse eyes, including endothelial cells of the Schlemm’s canal and the aqueous plexus (in bovine) that modulate aqueous outflow12,38. In primary human TM cells, PrPC was detected on the plasma membrane as in neuronal and other cell types. However, unlike neurons where majority of PrPC is cleaved at the α-site, most of the PrPC in TM cells was cleaved at the β-site. Unlike α-cleavage that occurs during physiological recycling of PrPC from the plasma membrane39, β-cleavage is associated with oxidative stress40–42, iron transport30,43,44, conversion of PrPC to PrPSc1,3, and possibly other stimuli45. It is surprising that human TM, ciliary body, optic nerve, and the retina showed distinct cleavage patterns of PrPC. In TM cells, TM tissue, and ciliary body, PrPC was mostly β-cleaved, while in the retina PrPC showed almost equal representation of α- and β-cleaved forms. Full-length PrPC was minimal in all of the above ocular tissues. These observations differ from ~50% β-cleavage of PrPC human retinal pigment epithelial cells44, and almost equal representation of α- and β-cleaved PrPC forms in bovine ciliary body30. Since bovine and human eyes have different concentrations of oxalate, apo-transferrin, and possibly other anti-oxidants that determine susceptibility to light-induced oxidative stress46, it is likely that cleavage of PrPC partly depends on the exposure of a particular ocular region to light or other stimuli that increase oxidative stress. It is notable that the optic nerve showed a novel internal fragment of ~19 kDa that requires further characterization. The soluble N-terminal fragments of α-, β-, and other cleaved forms of PrPC are likely to accumulate in the AH and vitreous humor and play distinct physiological roles as in neurons45, a possibility that is currently under investigation.
The stimuli and the identity of enzymes responsible for the mainly β-cleavage of PrPC in most ocular tissues and a unique cleavage in the optic nerve is not clear from our data. These are important unanswered questions with significant physiological and pathological implications45,47–49. In neuronal cells, PrPC undergoes at least four different proteolytic events. α-Cleavage is predominant, and the neuroprotective role of the resultant N-terminal fragment N1 has been described45. The proteases responsible for this cleavage, however, are not clear, and are arbitrarily termed α-PrPases48,50. Cleavage near the C-terminus releases almost full-length PrPC in the extracellular milieu, and is believed to protect neurons by reducing the substrate for PrP-scrapie, the disease-associated isoform of PrPC, on neuronal cells. Implications of soluble PrPC in the extracellular milieu, however, are not clear. This cleavage is mediated by the disintegrin and metalloprotease ADAM1051–53. ADAM9 influences ADAM10 activity, and is thus indirectly responsible for this event41,42,54. Additional cleavage of mainly unglycosylated PrPC near the C-terminus has been described, and is termed Ɣ-cleavage. The responsible protease is probably a member of the matrix metalloprotease family42. It is pertinent to mention here that matrix metalloproteases 2 and 9, ADAM proteases 9 and 10, and tetraspanin 6, a member of the tetraspanin family necessary for the maturation and transport of ADAM10, are increased in the AH of glaucomatous eyes of diverse etiology27,55,56. This raises the possibility that shedding of PrPC from TM cells may induce Endo-MT-like transition and altered TM-ECM interactions, contributing to the ongoing pathology. β-cleavage of PrPC is mainly associated with pathological conditions, and is mediated by calpains, lysosomal proteases, and oxidative stress. It is believed that the released N-terminal fragment N2 is an anti-oxidant and thus neuroprotective45,49,57. This raises the interesting possibility that β-cleavage of PrPC is an adaptive response, and increased levels of N2 protect the highly sensitive ocular tissues from light-induced oxidative stress. Further exploration is necessary to understand the physiological and pathological implications of this phenomenon fully.
It is remarkable that downregulation of PrPC in TM cells caused significant upregulation of several fibrillogenic proteins including α-SMA, fibronectin, and collagen 1A, suggesting transformation to a mesenchyme-like phenotype11,13,38,58. Absence of PrPC in neuronal cells induces aggregation of β1-integrin on the plasma membrane, activating signaling pathways including RhoA-ROCK that interfere with neuronal polarity and axonal growth by altering cell-ECM interactions5–8,26,59,60. Our data suggest that a similar mechanism operates in TM cells, and induces upregulation of fibrillogenic proteins typical of the glaucomatous change12,15.
PrPC is also a cell surface receptor for laminin, an extracellular matrix glycoprotein that plays a major role in neuronal differentiation10. Deletion or dysfunction of PrPC causes aggregation and accumulation of laminin in intra- and extracellular compartments and compensatory upregulation of laminin receptor in astrocytes and neuronal cells9,10. Our data show a similar response in TM cells, where upregulation of fibronectin is likely to contribute further to endo-MT-like changes12,38. Upregulation of vimentin upon downregulation of PrPC suggests loss of adherens junctions, another characteristic of such a change. Since laminin and vimentin are putative ligands of PrPC 31, these changes are likely to be independent of ROCK activation, and suggest that PrPC contributes to Endo-MT-like transition in TM cells by both ROCK-dependent and independent pathways.
Upregulation of myocilin due to silencing of PrPC is difficult to explain from our data. Since exposure to dexamethasone, a known inducer of myocilin did not cause additional upregulation of myocilin in the absence of PrPC, it is likely that both pathways intersect, perhaps through ROCK activation34. Additional studies are necessary to understand the relationship between PrPC and myocilin.
In conclusion, this study demonstrates a significant role of PrPC in maintaining cell-ECM interactions TM, and possibly as an anti-oxidant. Downregulation or absence of PrPC induces an endo-MT-like transition in the TM and elevation of IOP in PrP−/− mice, typical of POAG (Fig. 8)61. These observations underscore the significance of PrPC as a trigger for endo-TM-like transition in TM cells, and its potential to aggravate glaucomatous pathology due to shedding by ADAM and matrix-metalloproteases in the AH of glaucomatous eyes. Future exploration in additional PrP−/− and over expression mouse models and ex-vivo perfusion models of human eye where levels of PrPC have been altered experimentally are necessary to define its precise role in ocular tissues.
Figure 8.

Hypothetical representation of PrPC-mediated Endo-MT-like change in the TM. Physiological role of PrPC in the TM: (1) PrPC is expressed on the plasma membrane of TM cells. (2) PrPC maintains cell-ECM interactions by stabilizing β1-integrin and other proteins5,7. Pathological implications: (1) Downregulation of PrPC induces (2) aggregation of β1-integrin and (3) upregulation of fibronectin, collagen 1A, α-SMA, vimentin, and laminin, resulting in (4) increase in IOP and possibly POAG. AH: aqueous humor; PM: plasma membrane; Nu: nucleus.
Methods
Ethics statement
All animal procedures were in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Animal experiments were approved by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-approved Animal Resource Center (ARC) at Case Western Reserve University (CWRU) School of Medicine (SOM).
Antibodies and Chemicals
HRP-conjugated secondary antibodies (anti-mouse (NA931V), anti-rabbit (NA92V)) were from GE Healthcare (supplied by Sigma, USA). Alexa Fluor 546 (A11071, A11018) and Alexa Fluor 488 (A11017) tagged secondary antibodies were from Southern Biotech, USA and Molecular Probes (supplied by ThermoFisher, USA) respectively. A complete list of antibodies is provided in Table 1. PNGase F (P0704S) was from New England Biolabs (NEB), USA, Lipofectamine 3000 and Lipofectamine RNAiMax were from Invitrogen, USA. Dexamethasone (D1756) was from Sigma Aldrich, USA. siRNA against PrP (sc36318), and scrambled siRNA (sc37007) were from Santa Cruz Biotechnology, USA (sequences provided in Table 2).
Table 1.
List of antibodies.
| Antibody | Host species ** | Species reactivity ** | Company | Cat.No. | Dilution |
|---|---|---|---|---|---|
| PrP (3F4) IgG2a | m | h | Signet laboratories (Dedham, MA) | - |
WB-1:250 IHC-1:50 |
|
PrP (8H4) IgG2b |
m | m, h, b | Sigma Aldrich | P0110 |
WB-1:250 IHC-1:50 |
| PrP (8B4) IgG1 | m | m, h | Santa Cruz, USA | sc-47729 | WB-1:250 |
| PrP (SAF32) IgG2b | m | m, h, b | Cayman Chemical | 189720 |
WB-1:250 IHC-1:50 |
| PrP (G-12) IgG1 | m | m, h | Santa Cruz, USA | sc-398451 | WB-1:250 |
| PrP (2301) IgG | Rb | h | Gift by Dr. Shu Chen, PhD | - | WB-1:500 |
| Laminin receptor IgG | Rb | m, h | Abcam | ab137388 |
WB-1:1000 IHC-1:50 |
| Alpha smooth muscle actin IgG2a | m | m, Rb, h, b | Abcam | ab7817 |
WB-1:2000 IHC-1:100 |
| Laminin IgG | Rb | m, Rb, h, | Novus Biologicals | NB300-144 |
WB-1:1000 IHC-1:50 |
| Fibronectin IgG1 | m | h | Santa Cruz, USA | sc59826 |
WB-1:1000 IHC-1:50 |
| Vimentin IgG1 | m | h, b | Santa Cruz, USA | sc32322 |
WB-1:500 IHC-1:50 |
| Myocilin IgG2b | m | h | Santa Cruz, USA | sc137233 |
WB-1:500 IHC-1:50 |
| 9EG7 anti activated β1 integrin IgG2a | r | m, h | BD Biosciences | 553715 |
ICC- 1:50 WB-1:500 |
| Collagen 1A IgG | Rb | h, b | Rockland antibodies and assays | 600-401-103-0.1 |
WB-1:500 IHC-1:50 |
| β-actin IgG2bκ | m | All species with actin | Millipore, USA | MAB1501 | WB-1:5000 |
| Mouse IgG - Control | Abcam | ab37355 | IHC: 1:100 | ||
| Rabbit IgG, Control | Abcam | ab37415 | IHC: 1:100 |
**m: mouse, Rb: rabbit, b: bovine, h: human, r: rat.
Table 2.
List of biological samples and siRNA used in this study.
| PMI (h)/PCoD | Age (year) | Gender | Tissue region | |
|---|---|---|---|---|
| Human tissues used for primary TM cell culture and whole tissue lysates Source: Lions Gift of Sight | ||||
| 14 | 77 | F | Trabecular meshwork/Retina | |
| 4.5 | 70 | F | Trabecular meshwork/Retina/Optic nerve/Ciliary body | |
| 16/Dementia | 69 | M | Trabecular meshwork/Retina/Optic nerve/Ciliary body | |
| 5.5/ESLD | 56 | M | Trabecular meshwork/Retina/Optic nerve/Ciliary body | |
| 3/ESRD | 78 | F | Trabecular meshwork/Retina | |
| 12/Multiple system failure | 67 | M | Trabecular meshwork/Retina | |
| 7.5/Acute cardiac event | 65 | M | Trabecular meshwork/Retina | |
| TM cells from Dr. Rhee’s lab (from 8 different donors at passage 2). | ||||
| Mouse strain | ||||
| Wild-type and PrP knockout | 6 weeks/5M & 5F | C57BL/6 | Trabecular meshwork/Anterior Chamber | |
| Bovine samples | ||||
| Breed | Tissue region | |||
| Mixed | — | — | — | Trabecular meshwork/Anterior Chamber |
| siRNA | ||||
| Name | Company | Cat.No. | Sequence | |
| Scrambled siRNA | Santa Cruz | sc-37007 |
Sense: UUCUCCGAACGUGUCACGUtt Antisense: ACGUGACACGUUCGGAGAAtt |
|
| PrP siRNA (h) is a pool of 3 different siRNA duplexes | Santa Cruz | sc-36318 |
sc-36318A: Sense: GUGACUAUGAGGACCGUUAtt Antisense: UAACGGUCCUCAUAGUCACtt sc-36318B: Sense: GAGACCGACGUUAAGAUGAtt Antisense: UCAUCUUAACGUCGGUCUCtt sc-36318C: Sense: GUUGAGCAGAUGUGUAUCAtt Antisense: UGAUACACAUCUGCUCAACtt |
|
Culture and characterization of human TM cells
Primary cultures of human TM cells were obtained from the Rhee laboratory and established from eye globes using the standard protocol35,62, and characterized before by checking upregulation of myocilin in response to dexamethasone35 (Supplementary Fig. S1a-b). Primary human TM cell cultures were derived from several donors (age range: 56–78). After ciliary body (CB) was removed using scalpel, a cut in the rim of TM was made and the TM tissue was pulled out carefully using surgical grade forceps. Then, we made six sections of TM tissue using scalpel. Tissue sections were washed once in clean media and placed into the well of 6- wellplates. A coverslip was washed twice in clean media and placed gently over tissue sample keeping tissue sample toward center of well. Air bubbles were avoided during the whole process. The well was carefully handled in order not to scratch bottom of wells. Three milliliter of the media were added to each well dropping directly over the coverslip. The plate was incubated in 37 °C, 10% of CO2 incubator. The growth of TM was checked for 3 weeks. Once the cells grew, media was replaced twice a week until cells grew confluent. TM cells were maintained in the growth medium consisted of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20% fetal bovine serum (FBS) (Invitrogen-Gibco, Grand Island, NY), 1% L-glutamine (2 mM), and 0.5% or 0.1% gentamicin (50 or 10 µg/mL). For further experiment, TM cultures were seeded into 6-well plates, allowed to grow to confluence at 37 °C in a 10% CO2 atmosphere, and given an additional 2–3 days for differentiation. Confluent cultures of TM cells were used for all biochemical studies. For immunocytochemistry sub-confluent cultures were used to facilitate visualization of the plasma membrane. Both confluent and sub-confluent cultures responded to dexamethasone by upregulating myocilin (Supplementary Fig. S1c). To silence PrPC, the cells were transfected with PrP-specific or the corresponding scrambled siRNA using Lipofectamine RNAiMax as per manufacturer’s instructions. Desired downregulation of PrPC was confirmed by Western blotting.
Human and bovine eye samples
Human eye globes were acquired from Lions Gift of Sight eye bank (1000 Westgate Dr Ste 260 Saint Paul, MN 55114). The donors ranged in age from 42–78 years. Other available details of donors are provided in Table 2. Bovine eyes were collected from a local abattoir. The samples were either fixed in buffered formalin (1:10) for immunohistochemistry, or dissected to isolate the desired tissues.
Mouse strains
PrP-knock out (PrP−/−) mice were obtained from Jackson Laboratories (cat # 129-Prnptm2Edin/J Stock No: 012938) and crossed with C57BL/6 wild-type mice for 10 generations. F2 generation of wild type (PrP+/+) and corresponding PrP−/− (PrP−/−) were used for these studies. Control and experimental mice were ~6 weeks old, sex matched, and maintained under similar conditions.
Tissue preparation and Immunohistochemistry
Immunocytochemistry and immunohistochemistry were performed as described43. In short, thin sections of formalin-fixed TM tissue or primary human TM cells cultured on coverslips were processed for immunoreaction with the desired primary antibody followed by Alexa Fluor-conjugated secondary antibody. The nuclei were stained with Hoechst (#33342, Invitrogen, USA). Stained specimens were mounted and imaged with Leica inverted microscope (DMi8). Each experiment was repeated 3–4 times, and a representative image from 10 different fields is shown. Images of control sections reacted with isotype specific irrelevant primary antibody or buffer are shown in respective figures. Additional care was taken to identify the trabecular meshwork area for all sections in all the 3 species and the H&E staining for all sections analyzed were carried out and provided in the respective figures.
SDS-PAGE and Western blotting
Protein lysates and aqueous humor were fractionated by SDS-PAGE and analyzed by Western blotting as described30. For Collagen1A blotting, the samples were processed in a non-reducing and non-denaturing condition. Quantification of protein bands was performed by densitometry using UN-SCAN-IT gels (version6.1) software (Silk Scientific, USA) and ImageJ Software analyzed graphically using GraphPad Prism (Version 5.0) software (GraphPad Software Inc., USA) and Microsoft excel. Full-length blots are included in Supplementary Fig. S2.
IOP measurement
IOP was measured at the same time of day with TonoLab tonometer (Colonial Medical Supply; USA-Icare, Finland). Mice were anesthetized with ketamine/xylazine before the measurement, and six measurements were obtained for each eye per animal. Average of all values was used for analysis.
Statistical analysis
Quantification of protein bands was performed and presented as Mean ± SEM of the indicated n. Level of significance was calculated by Two-way ANOVA between the control and experimental groups.
Supplementary information
Acknowledgements
Funded by R01 NS 092145 to NS.
Author Contributions
A.A.: planned and performed all experiments, analyzed the data, wrote and modified the manuscript; N.S.: conceived the idea, overlooked experimental design, analyzed the data, wrote the manuscript; M.H.K.: harvested and cultured primary human T.M. cells, provided feedback; A.S.W.: performed de-glycosylation and Western blotting; W.M.J.: provided reagents and feedback; M.L.: cultured T.M. cells and performed immunohistochemistry; R.R.: assisted in Western blot analysis; D.R. and P.P.: provided feedback.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-49482-6.
References
- 1.Prusiner SB. A unifying role for prions in neurodegenerative diseases. Science. 2012;336:1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baskakov Ilia V. The many shades of prion strain adaptation. Prion. 2014;8(2):169–172. doi: 10.4161/pri.27836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma J, Wang F. Prion disease and the ‘protein-only hypothesis’. Essays in biochemistry. 2014;56:181–191. doi: 10.1042/bse0560181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh, N. The Role of Iron in Prion Disease and Other Neurodegenerative Diseases. Vol. 10 (2014). [DOI] [PMC free article] [PubMed]
- 5.Alleaume-Butaux A, et al. Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation. PLoS Pathog. 2015;11:e1005073. doi: 10.1371/journal.ppat.1005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong, Z. et al. Prion-like protein aggregates exploit the RHO GTPase to cofilin-1 signaling pathway to enter cells. EMBO J37, 10.15252/embj.201797822 (2018). [DOI] [PMC free article] [PubMed]
- 7.Kim HJ, et al. Regulation of RhoA activity by the cellular prion protein. Cell Death Dis. 2017;8:e2668. doi: 10.1038/cddis.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loubet D, et al. Neuritogenesis: the prion protein controls beta1 integrin signaling activity. FASEB J. 2012;26:678–690. doi: 10.1096/fj.11-185579. [DOI] [PubMed] [Google Scholar]
- 9.Martins VR, et al. Prion protein: orchestrating neurotrophic activities. Current issues in molecular biology. 2010;12:63. [PubMed] [Google Scholar]
- 10.Lima FR, et al. Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J Neurochem. 2007;103:2164–2176. doi: 10.1111/j.1471-4159.2007.04904.x. [DOI] [PubMed] [Google Scholar]
- 11.Mehrabian M, Ehsani S, Schmitt-Ulms G. An emerging role of the cellular prion protein as a modulator of a morphogenetic program underlying epithelial-to-mesenchymal transition. Front Cell Dev Biol. 2014;2:53. doi: 10.3389/fcell.2014.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abu-Hassan, D. W., Acott, T. S. & Kelley, M. J. The trabecular meshwork: a basic review of form and function. Journal of ocular biology2 (2014). [DOI] [PMC free article] [PubMed]
- 13.Rao PV, Pattabiraman PP, Kopczynski C. Role of the Rho GTPase/Rho kinase signaling pathway in pathogenesis and treatment of glaucoma: Bench to bedside research. Exp Eye Res. 2017;158:23–32. doi: 10.1016/j.exer.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pattabiraman PP, Maddala R, Rao PV. Regulation of plasticity and fibrogenic activity of trabecular meshwork cells by Rho GTPase signaling. J Cell Physiol. 2014;229:927–942. doi: 10.1002/jcp.24524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braunger BM, Fuchshofer R, Tamm ER. The aqueous humor outflow pathways in glaucoma: A unifying concept of disease mechanisms and causative treatment. Eur J Pharm Biopharm. 2015;95:173–181. doi: 10.1016/j.ejpb.2015.04.029. [DOI] [PubMed] [Google Scholar]
- 16.Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311:1901–1911. doi: 10.1001/jama.2014.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanna AP, Johnson M. Rho Kinase Inhibitors as a Novel Treatment for Glaucoma and Ocular Hypertension. Ophthalmology. 2018;125:1741–1756. doi: 10.1016/j.ophtha.2018.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torrejon KY, et al. TGFbeta2-induced outflow alterations in a bioengineered trabecular meshwork are offset by a rho-associated kinase inhibitor. Sci Rep. 2016;6:38319. doi: 10.1038/srep38319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garamszegi N, et al. Extracellular matrix-induced transforming growth factor-beta receptor signaling dynamics. Oncogene. 2010;29:2368–2380. doi: 10.1038/onc.2009.514. [DOI] [PubMed] [Google Scholar]
- 20.Gagen D, Faralli JA, Filla MS, Peters DM. The role of integrins in the trabecular meshwork. J Ocul Pharmacol Ther. 2014;30:110–120. doi: 10.1089/jop.2013.0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamuya FA, Duncan MK. aV integrins and TGF-beta-induced EMT: a circle of regulation. J Cell Mol Med. 2012;16:445–455. doi: 10.1111/j.1582-4934.2011.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montecchi-Palmer M, et al. TGFbeta2 Induces the Formation of Cross-Linked Actin Networks (CLANs) in Human Trabecular Meshwork Cells Through the Smad and Non-Smad Dependent Pathways. Invest Ophthalmol Vis Sci. 2017;58:1288–1295. doi: 10.1167/iovs.16-19672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honjo M, et al. Role of the Autotaxin-LPA Pathway in Dexamethasone-Induced Fibrotic Responses and Extracellular Matrix Production in Human Trabecular Meshwork Cells. Invest Ophthalmol Vis Sci. 2018;59:21–30. doi: 10.1167/iovs.17-22807. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi E, Inoue T, Fujimoto T, Kojima S, Tanihara H. Epithelial mesenchymal transition-like phenomenon in trabecular meshwork cells. Exp Eye Res. 2014;118:72–79. doi: 10.1016/j.exer.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Munger JS, Sheppard D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011;3:a005017. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghodrati F, et al. The prion protein is embedded in a molecular environment that modulates transforming growth factor beta and integrin signaling. Sci Rep. 2018;8:8654. doi: 10.1038/s41598-018-26685-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma S, et al. Proteomic Alterations in Aqueous Humor From Patients With Primary Open Angle Glaucoma. Invest Ophthalmol Vis Sci. 2018;59:2635–2643. doi: 10.1167/iovs.17-23434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gil M, et al. Cellular prion protein regulates invasion and migration of breast cancer cells through MMP-9 activity. Biochem Biophys Res Commun. 2016;470:213–219. doi: 10.1016/j.bbrc.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 29.Gauthier AC, Liu J. Epigenetics and Signaling Pathways in Glaucoma. Biomed Res Int. 2017;2017:5712341. doi: 10.1155/2017/5712341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ashok, A. et al. Prion protein modulates iron transport in the anterior segment: Implications for ocular iron homeostasis and prion transmission. Experimental eye research (2018). [DOI] [PMC free article] [PubMed]
- 31.Gu Y, et al. Mutant prion protein‐mediated aggregation of normal prion protein in the endoplasmic reticulum: implications for prion propagation and neurotoxicity. Journal of neurochemistry. 2003;84:10–22. doi: 10.1046/j.1471-4159.2003.01255.x. [DOI] [PubMed] [Google Scholar]
- 32.Yang C-YC, Huynh T, Johnson M, Gong H. Endothelial glycocalyx layer in the aqueous outflow pathway of bovine and human eyes. Experimental eye research. 2014;128:27–33. doi: 10.1016/j.exer.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altmeppen HC, et al. Proteolytic processing of the prion protein in health and disease. Am J Neurodegener Dis. 2012;1:15–31. [PMC free article] [PubMed] [Google Scholar]
- 34.Fujimoto T, et al. Involvement of RhoA/Rho-associated kinase signal transduction pathway in dexamethasone-induced alterations in aqueous outflow. Invest Ophthalmol Vis Sci. 2012;53:7097–7108. doi: 10.1167/iovs.12-9989. [DOI] [PubMed] [Google Scholar]
- 35.Keller KE, et al. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp Eye Res. 2018;171:164–173. doi: 10.1016/j.exer.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nuvolone M, Sorce S, Paolucci M, Aguzzi A. Extended characterization of the novel co-isogenic C57BL/6J Prnp(−/−) mouse line. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2017;24:36–37. doi: 10.1080/13506129.2017.1289913. [DOI] [PubMed] [Google Scholar]
- 37.Nuvolone M, et al. Strictly co-isogenic C57BL/6J-Prnp−/− mice: A rigorous resource for prion science. The Journal of experimental medicine. 2016;213:313–327. doi: 10.1084/jem.20151610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork. Experimental eye research. 2008;86:543–561. doi: 10.1016/j.exer.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen SG, et al. Truncated forms of the human prion protein in normal brain and in prion diseases. Journal of Biological Chemistry. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- 40.Watt NT, et al. Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellular response to oxidative stress. J Biol Chem. 2005;280:35914–35921. doi: 10.1074/jbc.M507327200. [DOI] [PubMed] [Google Scholar]
- 41.Altmeppen HC, et al. Roles of endoproteolytic alpha-cleavage and shedding of the prion protein in neurodegeneration. FEBS J. 2013;280:4338–4347. doi: 10.1111/febs.12196. [DOI] [PubMed] [Google Scholar]
- 42.Lewis V, et al. Prion protein “gamma-cleavage”: characterizing a novel endoproteolytic processing event. Cell Mol Life Sci. 2016;73:667–683. doi: 10.1007/s00018-015-2022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashok, A. & Singh, N. Prion protein modulates glucose homeostasis by altering intracellular iron. Scientific reports8 (2018). [DOI] [PMC free article] [PubMed]
- 44.Asthana A, et al. Prion protein facilitates retinal iron uptake and is cleaved at the β-site: Implications for retinal iron homeostasis in prion disorders. Scientific reports. 2017;7:9600. doi: 10.1038/s41598-017-08821-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linsenmeier L, et al. Diverse functions of the prion protein - Does proteolytic processing hold the key? Biochim Biophys Acta Mol Cell Res. 2017;1864:2128–2137. doi: 10.1016/j.bbamcr.2017.06.022. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Castineiras S. Iron, the retina and the lens: a focused review. Experimental eye research. 2010;90:664–678. doi: 10.1016/j.exer.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mays CE, et al. Endoproteolytic processing of the mammalian prion glycoprotein family. The FEBS journal. 2014;281:862–876. doi: 10.1111/febs.12654. [DOI] [PubMed] [Google Scholar]
- 48.McDonald AJ, Millhauser GL. PrP overdrive: Does inhibition of α-cleavage contribute to PrPC toxicity and prion disease? Prion. 2014;8:183–191. doi: 10.4161/pri.28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haigh CL, Collins SJ. Endoproteolytic cleavage as a molecular switch regulating and diversifying prion protein function. Neural regeneration research. 2016;11:238. doi: 10.4103/1673-5374.177726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonald AJ, Dibble JP, Evans EG, Millhauser GL. A new paradigm for enzymatic control of α-cleavage and β-cleavage of the prion protein. Journal of Biological Chemistry. 2014;289:803–813. doi: 10.1074/jbc.M113.502351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jarosz-Griffiths HH, et al. Proteolytic shedding of the prion protein via activation of metallopeptidase ADAM10 reduces cellular binding and toxicity of amyloid-β oligomers. Journal of Biological Chemistry. 2019;294:7085–7097. doi: 10.1074/jbc.RA118.005364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altmeppen HC, et al. The sheddase ADAM10 is a potent modulator of prion disease. Elife. 2015;4:e04260. doi: 10.7554/eLife.04260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor DR, et al. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. Journal of Biological Chemistry. 2009;284:22590–22600. doi: 10.1074/jbc.M109.032599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Linsenmeier L, et al. Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein. Mol Neurodegener. 2018;13:18. doi: 10.1186/s13024-018-0248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noy PJ, et al. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: EVIDENCE FOR DISTINCT BINDING MECHANISMS FOR DIFFERENT TspanC8 PROTEINS. J Biol Chem. 2016;291:3145–3157. doi: 10.1074/jbc.M115.703058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Groef L, Van Hove I, Dekeyster E, Stalmans I, Moons L. MMPs in the trabecular meshwork: promising targets for future glaucoma therapies? Investigative ophthalmology & visual science. 2013;54:7756–7763. doi: 10.1167/iovs.13-13088. [DOI] [PubMed] [Google Scholar]
- 57.Haigh CL, et al. Dominant roles of the polybasic proline motif and copper in the PrP23-89-mediated stress protection response. Journal of cell science. 2009;122:1518–1528. doi: 10.1242/jcs.043604. [DOI] [PubMed] [Google Scholar]
- 58.Pattabiraman, P. P. & Rao, P. V. Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. American Journal of Physiology-Cell Physiology (2009). [DOI] [PMC free article] [PubMed]
- 59.Richardson DD, et al. The prion protein inhibits monocytic cell migration by stimulating beta1 integrin adhesion and uropod formation. J Cell Sci. 2015;128:3018–3029. doi: 10.1242/jcs.165365. [DOI] [PubMed] [Google Scholar]
- 60.Ezpeleta J, et al. Protective role of cellular prion protein against TNFα-mediated inflammation through TACE α-secretase. Scientific reports. 2017;7:7671. doi: 10.1038/s41598-017-08110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alleaume-Butaux A, et al. Cellular prion protein is required for neuritogenesis: fine-tuning of multiple singaling pathways involved in focal adhesions and actin cytoskeleton dynamics. Cell Health and Cytoskeleton. 2013;5:1–12. [Google Scholar]
- 62.Oh D-J, et al. Overexpression of SPARC in Human Trabecular Meshwork Increases Intraocular Pressure and Alters Extracellular MatrixSPARC Overexpression Increases IOP and TM ECM. Investigative ophthalmology & visual science. 2013;54:3309–3319. doi: 10.1167/iovs.12-11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.