

The global tuberculosis (TB) epidemic has been exacerbated by the rise in drug-resistant TB cases worldwide. To tackle this crisis, it is necessary to identify new vulnerable drug targets in Mycobacterium tuberculosis, the causative agent of TB, and develop compounds that can inhibit the bacterium through novel mechanisms of action. The QcrB subunit of the electron transport chain enzyme cytochrome bc1 has recently been validated to be a potential drug target. In the current work, we report the discovery of a new class of QcrB inhibitors, 4-amino-thieno[2,3-d]pyrimidines, that potently inhibit M. tuberculosis growth in vitro. These compounds are chemically distinct from previously reported QcrB inhibitors, and therefore, 4-amino-thieno[2,3-d]pyrimidines represent a new scaffold that can be exploited to inhibit this drug target.
KEYWORDS: CydAB, Mycobacterium tuberculosis, QcrB, antibiotic, cytochrome, drug discovery, respiration
ABSTRACT
Antibiotic resistance is a global crisis that threatens our ability to treat bacterial infections, such as tuberculosis, caused by Mycobacterium tuberculosis. Of the 10 million cases of tuberculosis in 2017, approximately 19% of new cases and 43% of previously treated cases were caused by strains of M. tuberculosis resistant to at least one frontline antibiotic. There is a clear need for new therapies that target these genetically resistant strains. Here, we report the discovery of a new series of antimycobacterial compounds, 4-amino-thieno[2,3-d]pyrimidines, that potently inhibit the growth of M. tuberculosis. To elucidate the mechanism by which these compounds inhibit M. tuberculosis, we selected for mutants resistant to a representative 4-amino-thieno[2,3-d]pyrimidine and sequenced these strains to identify the mutations that confer resistance. We isolated a total of 12 resistant mutants, each of which harbored a nonsynonymous mutation in the gene qcrB, which encodes a subunit of the electron transport chain (ETC) enzyme cytochrome bc1 oxidoreductase, leading us to hypothesize that 4-amino-thieno[2,3-d]pyrimidines target this enzyme complex. We found that addition of 4-amino-thieno[2,3-d]pyrimidines to M. tuberculosis cultures resulted in a decrease in ATP levels, supporting our model that these compounds inhibit the M. tuberculosis ETC. Furthermore, 4-amino-thieno[2,3-d]pyrimidines had enhanced activity against a mutant of M. tuberculosis deficient in cytochrome bd oxidase, which is a hallmark of cytochrome bc1 inhibitors. Therefore, 4-amino-thieno[2,3-d]pyrimidines represent a novel series of QcrB inhibitors that build on the growing number of chemical scaffolds that are able to inhibit the mycobacterial cytochrome bc1 complex.
IMPORTANCE The global tuberculosis (TB) epidemic has been exacerbated by the rise in drug-resistant TB cases worldwide. To tackle this crisis, it is necessary to identify new vulnerable drug targets in Mycobacterium tuberculosis, the causative agent of TB, and develop compounds that can inhibit the bacterium through novel mechanisms of action. The QcrB subunit of the electron transport chain enzyme cytochrome bc1 has recently been validated to be a potential drug target. In the current work, we report the discovery of a new class of QcrB inhibitors, 4-amino-thieno[2,3-d]pyrimidines, that potently inhibit M. tuberculosis growth in vitro. These compounds are chemically distinct from previously reported QcrB inhibitors, and therefore, 4-amino-thieno[2,3-d]pyrimidines represent a new scaffold that can be exploited to inhibit this drug target.
INTRODUCTION
Infection with Mycobacterium tuberculosis resulted in over 9 million new cases of tuberculosis (TB) and 1.5 million deaths in 2017, making it the most deadly infectious agent in the world (1). This epidemic is exacerbated by the rise of multidrug-resistant (MDR) TB cases that are resistant to at least the two frontline antibiotics used to treat TB, isoniazid and rifampin. MDR-TB constituted 3.6% of new TB cases in 2017 and 17% of previously treated TB cases, with rates of MDR-TB being as high as 50% among previously treated TB cases in some countries (1). Furthermore, 8.5% of MDR-TB cases in 2017 were estimated to be extensively drug resistant (XDR), which are also resistant to a fluoroquinolone and a second-line injectable drug (1). This rise in drug resistance and the scarcity of drugs in the pipeline have made it clear that we are not equipped to successfully battle the ongoing TB epidemic.
In 2012, the diarylquinoline compound bedaquiline (Sirturo), which inhibits the mycobacterial ATP synthase (2), was approved to treat MDR-TB patients (3). The success of this new anti-TB antibiotic fueled interest in mycobacterial energy metabolism pathways as vulnerable targets for new antibiotic development. More recently, the imidazopyridine amide (IPA) Q203 (telacebec) was identified to be a potent antimycobacterial compound that targets QcrB, a subunit of the mycobacterial cytochrome bc1:aa3 oxidoreductase in the electron transport chain (ETC) (4). Q203 is currently in phase II clinical trials for the treatment of TB (5). Since the discovery of Q203 and additional imidazopyridine amides (4, 6–10), a number of compounds have been identified that are also reported to target QcrB, including pyrazolo[1,5-a]pyridine-3-carboxamides (11–13), imidazo[2,1-b]thiazole-5-carboxamides (14), pyrrolo[3,4-c]pyridine-1,3(2H)-diones (15), lansoprazole sulfide (16), 2-(quinolin-4-yloxy)acetamides (17, 18), arylvinylpiperazine amides (19), phenoxyalkylbenzimidazoles (20–22), and morpholino thiophenes (23).
Here, we present the discovery of 4-amino-thieno[2,3-d]pyrimidines as a new series of QcrB inhibitors that potently inhibit M. tuberculosis growth and that are chemically distinct from previously identified QcrB inhibitors. This work adds to the growing number of QcrB inhibitors that have recently been identified and contributes to our understanding of ways to exploit this target in the development of new chemotherapeutic strategies for TB treatment.
RESULTS
Identification of 4-amino-thieno[2,3-d]pyrimidines with growth-inhibitory activity in Mycobacterium smegmatis and M. tuberculosis.
In an effort to identify novel inhibitors of mycobacteria, we screened a selection of 78 small-molecule nucleotide mimetics purchased from ChemBridge Corporation for compounds that inhibit the growth of M. smegmatis in a high-throughput liquid culture assay. From these screens, we identified a 4-amino-thieno[2,3-d]pyrimidine (CB37) that inhibited the growth of M. smegmatis (Fig. 1A and B). We hypothesized that the charged carboxylate group on CB37 may greatly reduce penetration through the cell envelope of the mycobacteria and selected a set of 9 structurally related compounds that did not contain the carboxylate group but that contained the 2-ethyl-6-methylthieno[2,3-d]pyrimidine core (nearest neighbors) to purchase and assay for inhibition of M. smegmatis (Fig. 1; see also Fig. S1 and entries 2 to 10 in Table S1 in the supplemental material). Eight of the compounds showed either similar levels of growth inhibition against M. smegmatis as CB37 or no growth inhibition at all (Fig. S1). However, one of these compounds, CB81, showed improved growth inhibition in M. smegmatis (Fig. 1C and D). We resynthesized CB81 and henceforth designate it CWHM-728.
FIG 1.

CB37 and CB81 are 4-amino-thieno[2,3-d]pyrimidines that inhibit the growth of M. smegmatis. (A) Structure of CB37. (B) M. smegmatis (Msm) strain csm208 (Table 2) was incubated in LB medium in the presence of DMSO, 100 μM CB37, or 73 μM rifampin (Rif), and the optical density at 600 nm (ODλ600) was measured periodically over the course of 12 h in a plate reader (n = 2 replicates). (C) Structure of CB81, which was resynthesized and renamed CWHM-728. (D) M. smegmatis strain csm208 was incubated in LB medium in the presence of DMSO, 100 μM CB81, or 73 μM rifampin, and the ODλ600 was measured over the course of 12 h in a plate reader (n = 3 replicates).
Related 4-amino-thieno[2,3-d]pyrimidines do not inhibit the growth of M. smegmatis more than CB37 does. M. smegmatis strain csm208 was incubated in the presence of the indicated 4-amino-thieno[2,3-d]pyrimidine at 100 μM, and the optical density (OD) was measured periodically over the course of 12 h in a plate reader (n = 3 experiments). (A to F) DMSO and 73 μM rifampin were included as controls. The control data for panels A to F are the same in each panel because all the compounds were tested in the same assay plate. (G and H) DMSO and 1.1 mM streptomycin were included as controls. The control data for panels G and H are the same in both panels because these compounds were tested in the same assay plate. Download FIG S1, TIF file, 1.3 MB (1.3MB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Structure-activity relationship studies around CB37. aclogP, calculated log(partition coefficient, P). Values were calculated using the CDD Vault platform. bValues are from Fig. 2C. Download Table S1, DOCX file, 0.1 MB (141.6KB, docx) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To determine if CWHM-728 also had activity against M. tuberculosis, we performed zone-of-inhibition assays with the wild-type (WT) M. tuberculosis Erdman strain by spreading approximately 2.5 × 108 CFU of bacteria on an agar plate and spotting 5 μl of a 100 mM stock of CWHM-728 dissolved in dimethyl sulfoxide (DMSO) onto a disk in the center of the plate. After incubation at 37°C for 10 days, the bacteria formed a lawn and a zone absent of bacterial growth indicated growth inhibition by the compound. DMSO had no effect on M. tuberculosis growth in this assay and did not generate a zone of clearing on its own, whereas incubation of M. tuberculosis with CWHM-728 resulted in growth inhibition (Fig. 2A). To test if CWHM-728 has a bacteriostatic or a bactericidal effect on M. tuberculosis, we cultured M. tuberculosis in liquid medium in the presence of DMSO or 5 μM or 25 μM CWHM-728 and enumerated the viable CFU after 14 days of incubation (Fig. 2B). While exposure to 5 μM CWHM-728 for 14 days caused a slight but not statistically significant decrease in the number of CFU compared to that for the DMSO-treated control, exposure to 25 μM CWHM-728 caused a significant reduction in the number of CFU per milliliter compared to that for the DMSO-treated control. However, exposure to CWHM-728 did not decrease the number of viable CFU below the number of initial CFU on day 0, indicating that over 14 days, this concentration of CWHM-728 had a bacteriostatic effect on M. tuberculosis.
FIG 2.

CWHM-728 is a 4-amino-thieno[2,3-d]pyrimidine compound that exhibits anti-M. tuberculosis activity. (A) A zone-of-inhibition assay was performed by spreading 2.5 × 108 CFU of M. tuberculosis on a plate containing 7H11 agar medium, placing a sterile disk in the center, and pipetting 5 μl of 100% DMSO or 100 mM CWHM-728 on the disk. The plate was incubated at 37°C for 10 days. Representative images from at least 3 independent experiments are shown. (B) M. tuberculosis (Mtb) was incubated in 7H9 liquid medium in the presence of DMSO or CWHM-728 at the indicated concentrations. At the indicated time points, samples were collected and plated onto 7H11 agar medium containing no antibiotics to enumerate the CFU per milliliter (n = 3 replicates). Statistical comparisons for the number of CFU per milliliter on the final day of the treatment are depicted. P values were determined by two-way analysis of variance with Tukey’s posttest. *, P < 0.05; ns, not significant. (C) M. tuberculosis was incubated with increasing concentrations of CWHM-728, and bacterial respiration and metabolism were measured using the MABA (n = 3 replicates). The best-fit curve and IC50 values ± standard deviation were calculated using GraphPad Prism software.
To identify the half-maximal inhibitory concentration (IC50) of CWHM-728 against M. tuberculosis, we used a microplate alamarBlue assay (MABA), which is a high-throughput assay commonly used to evaluate the efficacy of antimycobacterial compounds (24). The MABA utilizes the redox-sensitive dye resazurin, which is blue in its oxidized form but becomes reduced to the pink fluorescent compound resorufin as a result of bacterial metabolism and respiration. Fluorescence can therefore be quantified as a readout for M. tuberculosis metabolism and respiration, which serves as a proxy for M. tuberculosis growth and/or survival. We incubated M. tuberculosis in the presence of increasing concentrations of CWHM-728 and found that CWHM-728 inhibited M. tuberculosis in the MABA with an IC50 of 2.7 ± 0.84 μM (Fig. 2C).
To explore chemical modifications that would improve upon the growth-inhibitory activity of CWHM-728 and develop structure-activity relationships (SAR), we used the MABA to test if the analogs that we had previously obtained in which the n-butyl side chain was replaced with other aliphatic groups had inhibitory activity against M. tuberculosis (see entries 1 to 10 in Table S1). Analogs that had side chains with ionizable groups (entries 1 and 3) and polar groups, such as hydroxyl (entry 4), had poor activity, with IC50 values being greater than 25 μM. Capping the hydroxyl group as methoxy (entries 5 and 6) restored some potency, and isopropoxy (entry 7) yielded a compound 8-fold more potent than the compound with a butyl side chain (entry 2). Cyclopropyl was tolerated (entry 8), with a submicromolar IC50, while slightly bulkier cyclopentyl and piperidine side chains resulted in very poor potency (entries 9 and 10).
To further extend the SAR, we synthesized analogs of CWHM-728 containing lipophilic side chains, and the resulting analogs were evaluated in the MABA (Table S2). Replacing the n-butyl side chain with t-butyl caused a nearly 10-fold decrease in potency (entry 2), but trifluoro-n-butyl and isopentyl variants (entries 3 and 4) were found to be greater than 10-fold more potent than the original n-butyl (entry 1). Capping the NH with a methyl group (entry 5) demonstrated little effect on potency. Finally, a series of phenyl and alkylphenyl side chains was prepared and showed remarkable SAR, with potency being strongly dependent on chain length (entries 6 to 9).
Structure-activity relationship studies around CWHM-728 and comparison to known M. tuberculosis inhibitors. aclogP, calculated log(partition coefficient, P). Values were calculated using the CDD Vault platform. bValues are from Fig. 3C. Download Table S2, DOCX file, 0.1 MB (130.1KB, docx) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The most potent compound from our SAR evaluation was CWHM-1023, which shared the 4-amino-thieno[2,3-d]pyrimidine core scaffold with CWHM-728 but which contained a 3-phenylpropyl side chain (Fig. 3A). We found that CWHM-1023 inhibited M. tuberculosis growth in the zone-of-inhibition assay (Fig. 3B) and inhibited M. tuberculosis in the MABA with an IC50 of 83 ± 5.4 nM (Fig. 3C). This is a 38-fold improvement in potency compared to that of CWHM-728, which had an IC50 of 3.2 ± 0.13 μM in this experiment (Fig. 3C). Thus, CWHM-728 and CWHM-1023 are 4-amino-thieno[2,3-d]pyrimidines that inhibit M. tuberculosis at low micromolar and submicromolar concentrations, respectively. Following this same trend in potency, we found that the IC50s of CWHM-728 and CWHM-1023 for M. smegmatis in the MABA were 52 ± 16 μM and 1.7 ± 0.49 μM, respectively (Fig. 3D).
FIG 3.

CWHM-1023 is a 4-amino-thieno[2,3-d]pyrimidine with enhanced activity against mycobacteria. (A) Chemical structure of CWHM-1023. (B) Zone-of-inhibition assay with CHWM-1023. M. tuberculosis (2.5 × 108 CFU) was spread on a plate containing 7H11 agar medium, a sterile disk was placed in the center, and 5 μl of 100 mM CWHM-1023 was pipetted onto the disk. The plate was incubated at 37°C for 10 days. A representative image from at least 2 independent experiments is shown. (C and D) M. tuberculosis (Mtb) (C) or M. smegmatis (Msm) (D) was incubated in the presence of increasing concentrations of either CWHM-728 or CWHM-1023, before bacterial respiration and metabolism were measured using the MABA (n = 3 replicates). Best-fit curves and IC50 values were calculated using GraphPad Prism software. P values were determined by Student's t test. **, P < 0.01; ****, P < 0.0001.
Mutations in qcrB confer resistance to 4-amino-thieno[2,3-d]pyrimidines.
To identify the target of the 4-amino-thieno[2,3-d]pyrimidines, we selected for M. tuberculosis mutants that were resistant to CWHM-728 by plating approximately 2.5 × 108 CFU of WT M. tuberculosis Erdman on 7H11 agar medium containing 10 μM CWHM-728 and incubating the culture at 37°C for 12 weeks. We found that spontaneous CWHM-728-resistant mutant colonies emerged at an approximate frequency of 1 in 2.3 × 107. To identify the genetic basis for CWHM-728 resistance, we performed whole-genome sequencing on four CWHM-728-resistant strains and found that all four strains harbored missense mutations in the qcrB gene. Three of the strains, isolated from a single culture, had mutations resulting in an A178T amino acid change, and one strain, isolated from a second independent culture, had a mutation generating a V338G substitution (Table 1). We then sequenced the qcrB locus in 8 additional CWHM-728-resistant mutants isolated from a third independent culture and identified a missense mutation in qcrB in every isolate (Table 1). One strain harbored the A178T mutation, and 3 more had the V338G mutation that we had isolated previously. In addition, we found that the remaining isolates harbored a A178V, G175S, or G315S mutation in qcrB.
TABLE 1.
CWHM-728-resistant isolates harbor mutations in qcrBa
| Culture no. and resistant strain | Mutation in qcrB |
|---|---|
| Culture 1, CB81R | V338Gb |
| Culture 2 | |
| D3 | A178Tb |
| 81.2 | A178Tb |
| 81.5 | A178Tb |
| Culture 3 | |
| CB81R a | A178V |
| CB81R b | G175S |
| CB81R c | V338G |
| CB81R d | V338G |
| CB81R f | A178T |
| CB81R g | A178V |
| CB81R i | G315S |
| CB81R j | V338G |
The CWHM-728-resistant strains isolated in this study are listed with the mutation identified in qcrB.
The mutations were identified by whole-genome sequencing. All other mutations were identified by Sanger sequencing of the qcrB locus.
The qcrB gene is located within an operon containing ctaE, qcrC, and qcrA. The qcrCAB gene cassette encodes all 3 subunits of the cytochrome bc1 complex (25). This complex is localized to the plasma membrane and forms a supercomplex with the aa3-type cytochrome oxidase encoded by ctaBCDE (26, 27). The resulting cytochrome bc1:aa3 oxidase complex catalyzes the terminal electron transfer reaction in the mycobacterial ETC, transferring electrons from menaquinol, the lipid electron carrier in the membrane, to the terminal electron acceptor oxygen. We sought to confirm that mutations in qcrB were sufficient to confer resistance to 4-amino-thieno[2,3-d]pyrimidines to rule out the possibility that the CWHM-728-resistant mutants harbored mutations elsewhere in the genome that contributed to resistance. For these studies, we engineered M. tuberculosis strains that expressed either WT qcrCAB (qcrCABWT) or qcrCAB with the A178T substitution (qcrCABA178T) from the chromosomal attB site and deleted the endogenous qcrCAB locus. We then monitored the activity of CWHM-728 (Fig. 4A) and CWHM-1023 (Fig. 4B) against these strains using the MABA. We found that the strain expressing qcrCABA178T exhibited reduced sensitivity to both compounds compared to that of WT M. tuberculosis or the isogenic qcrCABWT control strain, confirming that this single amino acid change in qcrB confers resistance to 4-amino-thieno[2,3-d]pyrimidines. These data are consistent with a model in which the 4-amino-thieno[2,3-d]pyrimidines target cytochrome bc1.
FIG 4.

A mutation in qcrB confers resistance to 4-amino-thieno[2,3-d]pyrimidines. The M. tuberculosis (Mtb) WT or the M. tuberculosis ΔqcrCAB mutant complemented with either qcrCABWT or qcrCABA178T was incubated in the presence of increasing concentrations of CWHM-728 (A) or CWHM-1023 (B), and then bacterial respiration and metabolism were measured using the MABA (n = 3). Best-fit curves and IC50 values ± standard deviation (S.D.) were calculated using GraphPad Prism software. ns, not significant by Student's t test.
Based on our finding that mutations in the qcrB gene confer resistance to 4-amino-thieno[2,3-d]pyrimidines, we propose that this new class of antimycobacterial compounds targets QcrB. To better understand how mutations in qcrB confer resistance to 4-amino-thieno[2,3-d]pyrimidines, we used computational modeling to predict the structure of the M. tuberculosis QcrB protein based on a previously published structure for QcrB from Rhodobacter sphaeroides, which shares 16% amino acid identity with the M. tuberculosis QcrB (28) (Fig. 5A). We docked CWHM-728 onto the predicted structure of QcrB and found that it localized near the putative menaquinol binding site of QcrB. Based on our computational model, we speculate that the A178T, A178V, or G175S mutation in QcrB would disrupt the contacts required for 4-amino-thieno[2,3-d]pyrimidines to bind QcrB (Fig. 5B). The V338 and G315 residues are not located where they would directly interact with CWHM-728 but are positioned on neighboring helices within a very tightly packed region, such that changes in amino acid identity could possibly affect the position of those helices and disrupt the 4-amino-thieno[2,3-d]pyrimidine binding site.
FIG 5.

Molecular modeling of CWHM-728 in the predicted structure of M. tuberculosis QcrB. (A) QcrB homology model embedded in the lipid bilayer, with the lipid head group phosphorus being shown as magenta spheres. (B) QcrB binding site shown with CHWM-728 docked (green). The residues mutated in resistant mutants are highlighted in magenta.
The predicted binding site for the 4-amino-thieno[2,3-d]pyrimidines is similar to what has previously been reported from similar modeling studies for the QcrB inhibitor Q203 (29). We investigated whether the 4-amino-thieno[2,3-d]pyrimidine-resistant strain expressing qcrCABA178T was more resistant to Q203 than the isogenic qcrCABWT control strain and found that the qcrBA178T mutation did not affect Q203 sensitivity in the MABA (Fig. S2). These data suggest that while the qcrBA178T allele is sufficient to confer resistance to 4-amino-thieno[2,3-d]pyrimidines, it does not confer cross-resistance to Q203, possibly because the A187T mutation is not sufficient to disrupt the binding of Q203 to QcrB.
M. tuberculosis harboring the qcrBA178T allele is not cross-resistant to Q203. WT or M. tuberculosis (Mtb) ΔqcrCAB complemented with either qcrCABWT or qcrCABA178T was incubated in the presence of increasing concentrations of Q203, and then bacterial respiration and metabolism were measured using the MABA (n = 3 experiments). Best-fit curves and IC50 values ± standard deviation (S.D.) were calculated using GraphPad Prism software. P values were determined by one-way analysis of variance with Tukey’s posttest. *, P < 0.05; **, P < 0.01; ns, not significant. Download FIG S2, TIF file, 0.8 MB (820.3KB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
4-Amino-thieno[2,3-d]pyrimidines deplete ATP levels in M. smegmatis and M. tuberculosis.
Based on our model that 4-amino-thieno[2,3-d]pyrimidines target the QcrB subunit of cytochrome bc1, we hypothesized that CWHM-728 and CWHM-1023 inhibit the mycobacterial ETC. To test this hypothesis, we incubated M. tuberculosis in the presence of 1 μM CWHM-728, 1 μM CWHM-1023, or 400 nM Q203 for 24 h and measured the ATP levels in the bacteria using the BacTiter-Glo assay (Promega). We found that treatment with CWHM-728, CWHM-1023, and Q203 decreased the ATP levels in M. tuberculosis by 75%, 60%, and 58%, respectively (Fig. 6A). Similar results were observed in M. smegmatis, where 10 μM CWHM-1023 caused a 41% decrease in ATP levels in M. smegmatis (Fig. 6B). These data support the possibility that 4-amino-thieno[2,3-d]pyrimidines target a complex involved in energy generation, consistent with our hypothesis that these compounds inhibit QcrB in the mycobacterial ETC.
FIG 6.
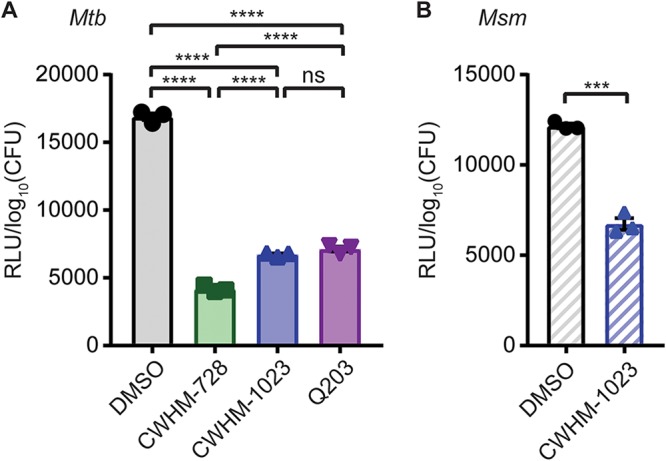
4-Amino-thieno[2,3-d]pyrimidines deplete mycobacterial ATP levels. (A) M. tuberculosis (Mtb) was incubated in the presence of 1 μM CWHM-728, 1 μM CWHM-1023, or 400 nM Q203 for 24 h, the samples were heat inactivated, and ATP was quantified using the BacTiter-Glo assay (n = 3). (B) M. smegmatis (Msm) was incubated in the presence of 10 μM CWHM-1023 for 12 h, and ATP was quantified using the BacTiter-Glo assay (n = 3). In both panels, ATP levels were normalized to the number of log10 CFU to account for differences in bacterial numbers due to differences in growth. P values were determined by one-way analysis of variance with Tukey’s posttest (A) or Student's t test (B). ***, P < 0.001; ****, P < 0.0001; ns, not significant.
M. smegmatis and M. tuberculosis lacking cytochrome bd are hypersensitive to 4-amino-thieno[2,3-d]pyrimidines.
Mycobacteria have a branched ETC that can terminate either in the cytochrome bc1:aa3 terminal oxidase or in the cytochrome bd terminal oxidase, encoded by cydAB (25, 30). Cytochrome bc1:aa3 oxidase and cytochrome bd oxidase have somewhat overlapping roles in the ETC to transfer electrons from menaquinol to oxygen, where the cytochrome bd oxidase can partially compensate for the loss of cytochrome bc1:aa3 oxidase activity, as evidenced by the observation that mutants of cytochrome bd exhibit increased sensitivity to cytochrome bc1 inhibitors (8, 31). Therefore, we hypothesized that 4-amino-thieno[2,3-d]pyrimidines would exhibit enhanced activity against mycobacteria lacking cytochrome bd compared with that of the WT strains. To test this hypothesis, we generated M. tuberculosis ΔcydA and M. smegmatis ΔcydA mutants, which lack cytochrome bd, and measured the sensitivity of these mutant strains to 4-amino-thieno[2,3-d]pyrimidines compared to that of the WT strains. We found that deletion of cydA increased the sensitivity of M. tuberculosis to CWHM-728 13.8-fold (Fig. 7A), CWHM-1023 22.6-fold (Fig. 7B), and Q203 7.7-fold (Fig. 7C), as measured by a decrease in the MABA IC50 compared to that for WT M. tuberculosis. Additionally, we found that CWHM-1023 had a 4.6-fold lower IC50 against M. smegmatis ΔcydA than against WT M. smegmatis, which was partially complemented by expressing cydAB in this strain (Fig. 7D). These data demonstrate that genetic deletion of cytochrome bd sensitizes mycobacteria to inhibition by 4-amino-thieno[2,3-d]pyrimidines, which is consistent with our hypothesis that these compounds target the QcrB subunit of cytochrome bc1.
FIG 7.

Mycobacteria lacking cytochrome bd have increased sensitivity to 4-amino-thieno[2,3-d]pyrimidines. (A to C) Either WT M. tuberculosis (Mtb) or M. tuberculosis ΔcydA was incubated in the presence of increasing concentrations of CWHM-728 (A), CWHM-1023 (B), or Q203 (C), and respiration and metabolism were measured using the MABA (n = 3 replicates). (D) WT M. smegmatis (Msm) or M. smegmatis ΔcydA complemented with the empty vector (EV) or with a plasmid harboring cydAB was incubated in the presence of increasing concentrations of CWHM-1023, and respiration and metabolism were measured using the MABA (n = 3 replicates). (A to D) Best-fit curves and IC50 values ± standard deviation (S.D.) were generated using GraphPad Prism software. P values were determined by Student's t test (A to C) or one-way analysis of variance with Tukey’s posttest (D). *, P < 0.05; ****, P < 0.0001; ns, not significant.
DISCUSSION
There is a dire need for new anti-TB drugs that shorten treatment regimens and that are effective against MDR and XDR strains of M. tuberculosis. To begin to address this need, two anti-TB agents have recently received accelerated regulatory approval from the U.S. Food and Drug Administration (FDA) (3). These two drugs, bedaquiline (Sirturo) and delamanid, are the first antibiotics to be approved for the treatment of TB in the last 50 years (32). The discovery and development of bedaquiline in particular have raised general interest in targeting M. tuberculosis ETC and respiration as a therapeutic strategy.
Simultaneously, numerous phenotypic screens have identified QcrB inhibitors that inhibit the growth of M. tuberculosis in axenic culture and/or in macrophages (4, 8, 15, 16, 19, 21, 23, 33), raising further interest in the ETC as a drug target. The most clinically advanced of these QcrB inhibitors is the imidazopyridine amide (IPA) Q203 (telacebec), which is currently in clinical trials for the treatment of TB (5). In this study, we report the discovery of a novel class of QcrB inhibitors, 4-amino-thieno[2,3-d]pyrimidines, that are chemically distinct from the previously identified QcrB inhibitors. Therefore, the new chemical scaffold described here adds to the growing chemical space that can be exploited to target the mycobacterial cytochrome bc1 complex. The most potent 4-amino-thieno[2,3-d]pyrimidine inhibitor of QcrB that we identified, CWHM-1023, had an IC50 of approximately 83 nM for M. tuberculosis in the MABA assay (see Table S2 in the supplemental material). This is in comparison with the IC50s for the known respiration inhibitors Q203, bedaquiline, and thioridazine, which are 1.5 nM, <78 nM, and 11.2 μM, respectively, against M. tuberculosis in the MABA (Table S2).
Mycobacteria have a branched ETC, whereby electrons can be shuttled either to cytochrome bc1 or to cytochrome bd to be transferred to the terminal electron acceptor, O2 (30). Cytochrome bc1 is proposed to be bioenergetically more efficient than cytochrome bd under standard aerobic conditions (25). However, cytochrome bd is induced under conditions of low oxygen and contributes to mycobacterial fitness under microaerobic conditions, suggesting that cytochrome bd can support mycobacterial survival under certain conditions (30). As such, while cytochrome bc1 is important for the growth of M. tuberculosis under laboratory conditions (25, 34–36), M. tuberculosis is able to survive during inhibition of cytochrome bc1 (37, 38), which is consistent with our data (Fig. 2B) and previously reported data indicating that QcrB inhibitors lack early bactericidal activity and are bacteriostatic for several weeks (19, 20, 31). Furthermore, it was reported that treatment of an M. tuberculosis mutant lacking cytochrome bd with QcrB inhibitors results in bacterial death (19, 31). Conversely, it was found that overexpression of cytochrome bd in M. tuberculosis can enable the bacteria to grow, despite cytochrome bc1 inhibition (8, 39). Together, these data indicate that cytochrome bd can compensate for cytochrome bc1 inhibition.
Expression of cytochrome bc1 and cytochrome bd has also been shown to change throughout the course of infection. In mice infected with M. tuberculosis, it was found that qcrC expression was the highest during acute infection (day 15) and that following 20 days of infection qcrC was transcriptionally downregulated 2- to 2.5-fold. In contrast, cydA was transcriptionally upregulated after 20 days, with the highest level of expression being detected at 30 days postinfection, when there was a 7-fold increase over the day 15 expression levels (40). These findings suggest that M. tuberculosis may rely more heavily on cytochrome bc1 during acute stages of infection, whereas cytochrome bd is activated at later stages of infection. These expression patterns may explain why it has been found that QcrB inhibitors work well to halt M. tuberculosis growth in the mouse when administered during the acute phase of infection (4, 12, 16, 19) but have a variable impact on the bacterial burden when treatment starts after the mice have been infected for more than 2 weeks, with most studies reporting modest decreases in the bacterial burden, if any decrease at all (4, 12, 19, 31). Some of the variability of QcrB inhibitor efficacy during the later stages of infection likely also depends on the timing of the treatment and the dosing of the compound.
Given the respiratory flexibilty in M. tuberculosis, interest has increased in using combinations of antibiotics that target different components of the ETC, a strategy that has been shown to lead to synergy in bactericidal effects (38, 41, 42). In particular, our studies with 4-amino-thieno[2,3-d]pyrimidines in the ΔcydA mutants imply that inhibition of QcrB would have an enhanced impact on M. tuberculosis in combination with inhibitors of cytochrome bd, as previously observed for Q203 (42). Together, these findings indicate that 4-amino-thieno[2,3-d]pyrimidines target QcrB and highlight the idea that the concurrent inhibition of both cytochrome bc1 and cytochrome bd is more effective than inhibition of cytochrome bc1 alone. In addition to potentiating other respiration inhibitors, a QcrB inhibitor has recently been shown to enhance pyrazinamide as well as rifampin monotherapy in mice, indicating that QcrB inhibitors may prove to be a useful addition to the current standard of care (12).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
M. tuberculosis Erdman was cultured in Middlebrook 7H9 liquid medium supplemented with 60 μl/liter oleic acid, 5 g/liter bovine serum albumin, 2 g/liter dextrose, and 0.003 g/liter catalase (OADC) plus 0.5% glycerol and 0.05% Tween 80. For solid medium, Middlebrook 7H10 or 7H11 agar medium supplemented with OADC and 0.5% glycerol was used. M. smegmatis mc2155 was cultured in LB medium supplemented with 0.5% glycerol, 0.5% dextrose, and 0.05% Tween 80. Genetic deletion mutant strains of M. tuberculosis and M. smegmatis (Table 2) were generated using specialized transduction with the conditionally replicating phage phAE87 as previously described (43, 44). When appropriate, mycobacterial strains were selected on 20 μg/ml kanamycin and/or 50 μg/ml hygromycin.
TABLE 2.
Strains and plasmids used in this study
| Strain or plasmid | Description |
|---|---|
| Strains | |
| M. tuberculosis | |
| WT | Erdman |
| qcrCABWT | Erdman ΔqcrCAB::Hygr attB::pMSG430-qcrCABMtbWT |
| qcrCABA178T | Erdman ΔqcrCAB::Hygr attB::pMSG430-qcrCABMtbA178T |
| ΔcydA | Erdman ΔcydA::Hygr attB::pMSG430 (EV) |
| M. smegmatis | |
| WT | mc2155 |
| csm208 | mc2155 ΔrelA attB::pMSG430-relAMtb53–394 |
| ΔqcrCAB | mc2155 ΔqcrCAB::Hygr |
| ΔcydA + EV | mc2155 ΔcydA::Hygr attB::pMSG430 (EV) |
| ΔcydA + cydAB | mc2155 ΔcydA::Hygr attB::pMSG430-cydABMsm |
| Plasmids | |
| pMSG430 | Shuttle vector that harbors the L5 recombinase and attP site and a constitutive Pmyc1-tetO promoter, Kanr |
| pMSG430-qcrCABMtbWT | pMSG430 with qcrCABMtbWT (Pmyc1-tetO promoter) |
| pMSG430-qcrCABMtbA178T | pMSG430 with qcrCABMtbA178T (Pmyc1-tetO promoter) |
| pMSG430-relAMtb53–394 | pMSG430 with relAMtb53–394 (Pmyc1-tetO promoter) |
| pMSG430-cydABMsm | pMSG430 with cydABMsm (Pmyc1-tetO promoter) |
| pMSG360 | Cloning vector that harbors regions of homology to the phAE87 phage genome and multiple cloning sites upstream and downstream of an Hygr marker for engineering specialized transducing phage, Hygr |
| pMSG360-qcrCABMtb | pMSG360 with homology to Erdman nucleotides 2448245–2448982 and 2452708–2453440 |
| pMSG360-cydAMtb | pMSG360 with homology to Erdman nucleotides 1809431–1810193 and 1807061–1808049 |
| pMSG360-qcrCABMsm | pMSG360 with homology to mc2155 nucleotides 4344291–4344965 and 4348476–4349188 |
| pMSG360-cydAMsm | pMSG360 with homology to mc2155 nucleotides 3317608–3318615 and 3315796–3316290 |
Initial screen.
Logarithmically growing M. smegmatis strain csm208 was inoculated into 96-well dishes containing 100 μM compounds in 200 μl LB at a starting optical density at 600 nm (ODλ600) of 0.2 and incubated with shaking at 37°C in a Tecan M200 Pro plate reader, with ODλ600 measurements being taken in each well every 15 s.
Microplate alamarBlue assays (MABAs).
Logarithmically growing M. tuberculosis was inoculated into 7H9 medium in 96-well plates with wells containing increasing concentrations of compound. M. tuberculosis was inoculated at an ODλ600 of 0.0008, corresponding to approximately 4 × 105 CFU/ml in 200 μl per well. The plates were incubated at 37°C in 5% CO2 for 1 week, at which point 32.5 μl of a mixture containing an 8:5 ratio of 0.6 mM resazurin (Sigma) dissolved in 1× phosphate-buffered saline to 20% Tween 80 was added, and the production of fluorescent resorufin was measured after incubation at 37°C in 5% CO2 overnight. For M. tuberculosis, samples were removed from the plate and mixed with formalin to kill the M. tuberculosis bacteria before measuring the fluorescence. For M. smegmatis, the assay plate was measured directly. Fluorescence was measured on a Tecan M200 Pro plate reader with an excitation λ of 530 nm and an emission λ of 590 nm. For each assay, medium alone served as a negative control, and untreated M. tuberculosis or M. smegmatis was included as a positive control. The percent inhibition was calculated as the {[(fluorescence of the positive control − fluorescence of the negative control) − (fluorescence of the sample − fluorescence of the negative control)]/(fluorescence of the positive control − fluorescence of the negative control)} × 100.
Selection and sequencing of resistant mutants.
Resistant mutants were selected on 7H11 agar medium containing 10 μM CWHM-728. Approximately 2.5 × 108 CFU of WT M. tuberculosis Erdman was spread per plate, and the plate was incubated at 37°C for 12 weeks. Genomic DNA was isolated using cetyltrimethylammonium bromide-lysozyme lysis, followed by chloroform-isoamyl alcohol extraction and isopropanol precipitation, as previously described (45). Whole-genome sequencing was performed by use of an Illumina HiSeq sequencer with 50-bp single-end reads. The identification of single nucleotide polymorphisms (SNPs) was done using SeqMan NGen software (DNASTAR). The genomes were assembled and compared to the genomic DNA from the WT parental control strain to identify SNPs that may be responsible for resistance to CWHM-728. The mutations identified in qcrB by whole-genome sequencing were subsequently confirmed by Sanger sequencing. For Sanger sequencing of the qcrB locus, the genomic region was PCR amplified using primers erdqcrBfwxbaI430 (GTCTAGAATGAGTCCGAAACTGAGTCCGCC) and RK27 (GAAGCTTTCGCCGGGCTAGTGCTCGCCGTC) and then sequenced using the same two primers (GeneWiz).
Computational modeling.
A homology model of M. tuberculosis QcrB was built using the crystal structure of Rhodobacter sphaeroides QcrB (PDB accession number 2QJP) (28) as the template structure, using the sequence alignment from Ko and Choi (29). The model, which was built in the Molecular Operating Environment (MOE 2016.08; Chemical Computing Group, Montreal, Quebec, Canada), was placed into a membrane environment (46), and energy was minimized. The geometry of the [2Fe-2S] center and the disulfide bond pattern adjacent to the inhibitor-binding site were assumed to match those of the template structure. CWHM-728 was docked into the homology model using the GOLD (v5.5) program (47).
ATP measurements.
M. tuberculosis or M. smegmatis was inoculated into 7H9 medium with or without the test compounds at an ODλ600 of 0.1 and incubated with shaking at 37°C for 24 h (M. tuberculosis) or 12 h (M. smegmatis). An aliquot of the culture was heat inactivated at 95°C for 20 min and diluted 1:100. Diluted samples were mixed with the BacTiter-Glo assay (Promega) reagent at a 1:1 ratio, and luminescence was quantified on a Tecan M200 Pro plate reader (integration = 1 s). Relative luminescence units (RLU) were normalized to the number of log10 CFU in the sample to account for differences in bacterial number.
Compounds.
CB37, CWHM-728 (CB81), CWHM-935, CWHM-936, CWHM-941, CWHM-950, CWHM-937, CWHM-946, CWHM-951, and CWHM-942 were purchased from ChemBridge Corporation. Synthesis of the compounds CWHM-1069, CWHM-1020, CWHM-1022, CWHM-1021, CWHM-1304, CWHM-1303, CWHM-1306, and CWHM-1023 is described in Text S1 in the supplemental material, and liquid chromatography-mass spectrometry (LC-MS), 1H nuclear magnetic resonance (NMR), and 13C NMR analyses were done on CWHM-1023 to confirm the purity and identity of the synthesized compound (Fig. S3 and S4). Q203 was acquired from Enamine (catalog number EN-300-218150), and both bedaquiline (catalog number 465749185) and thioridazine (catalog number 1662504) were purchased from Sigma-Aldrich; all three compounds were tested in the MABA for comparative purposes.
Supplemental information. Download Text S1, PDF file, 0.3 MB (346.6KB, pdf) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
LC-MS spectra for CWHM-1023. CWHM-1023 was subjected to LC-MS analysis as described in Text S1 in the supplemental material. Download FIG S3, TIF file, 0.6 MB (627.8KB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
NMR spectra for CWHM-1023. CWHM-1023 was subjected to 1H NMR (A) and 13C NMR (B) analysis. The inset in panel A depicts the chemical structure of CWHM-1023. Download FIG S4, TIF file, 0.4 MB (378KB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data availability.
The sequence data are publicly available in the NCBI Sequence Read Archive under accession no. PRJNA561987.
ACKNOWLEDGMENTS
This project was supported by NIH/National Center for Advancing Translational Sciences (NCATS) grant UL1 TR000448 as well as NIH grant R33AI111696 and NIH grant R01AI134847 to C.L.S. C.L.S. is also supported by a Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease award. G.A.H. is supported by National Science Foundation graduate research fellowship DGE-1745038 and NIGMS cell and molecular biology training grant GM007067. K.F. was supported by a pilot award from the Center for Women’s Infectious Disease Research at Washington University. R.L.K. is supported by a Potts Memorial Foundation postdoctoral fellowship, J.A. was supported by NIH grant R25HG006687, M.E.S. was supported through the Washington University BioMedRAP, and K.J. was supported through the Washington University Biology Summer Undergraduate Research Fellowship Program. The Genome Technology Access Center in the Department of Genetics at the Washington University School of Medicine is partially supported by NCI Cancer Center Support grant P30CA91842 to the Siteman Cancer Center and by ICTS/CTSA grant UL1TR000448 from the National Center for Research Resources (NCRR), a component of the NIH, and the NIH Roadmap for Medical Research.
We thank the Genome Technology Access Center in the Department of Genetics at the Washington University School of Medicine for help with genomic analysis.
This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. We have no competing financial interests to declare but acknowledge that C.L.S. has ownership in the company Fimbrion Therapeutics, which licenses 4-amino-thieno[2,3-d]pyrimidines as QcrB inhibitors in Mycobacterium tuberculosis and, therefore, may financially benefit if the company is successful in marketing its product. M.J.M. is also a consultant to Fimbrion Therapeutics.
REFERENCES
- 1.World Health Organization. 2018. Global tuberculosis report 2018. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Andries K, Verhasselt P, Guillemont J, Gohlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 3.Horsburgh CR, Barry CE, Lange C. 2015. Treatment of tuberculosis. N Engl J Med 373:2149–2160. doi: 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- 4.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim SA, Nam J, Kang H, Kwon H, Oh CT, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SPS, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han SJ, No Z, Lee J, Brodin P, Cho SN, Nam K, Kim J. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 5.National Library of Medicine. 2018. A phase 2 study to evaluate early bactericidal activity, safety, tolerability, and pharmacokinetics of multiple oral doses of telacebec (Q203). National Library of Medicine, Bethesda, MD. [Google Scholar]
- 6.Kang S, Kim RY, Seo MJ, Lee S, Kim YM, Seo M, Seo JJ, Ko Y, Choi I, Jang J, Nam J, Park S, Kang H, Kim HJ, Kim J, Ahn S, Pethe K, Nam K, No Z, Kim J. 2014. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J Med Chem 57:5293–5305. doi: 10.1021/jm5003606. [DOI] [PubMed] [Google Scholar]
- 7.Abrahams KA, Cox JAG, Spivey VL, Loman NJ, Pallen MJ, Constantinidou C, Fernandez R, Alemparte C, Remuinan MJ, Barros D, Ballell L, Besra GS. 2012. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS One 7:e52951. doi: 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arora K, Ochoa-Montaño B, Tsang PS, Blundell TL, Dawes SS, Mizrahi V, Bayliss T, Mackenzie CJ, Cleghorn LAT, Ray PC, Wyatt PG, Uh E, Lee J, Barry CE, Boshoff HI. 2014. Respiratory flexibility in response to inhibition of cytochrome c oxidase in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:6962–6965. doi: 10.1128/AAC.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moraski GC, Miller PA, Bailey MA, Ollinger J, Parish T, Boshoff HI, Cho S, Anderson JR, Mulugeta S, Franzblau SG, Miller MJ. 2015. Putting tuberculosis (TB) to rest: transformation of the sleep aid, Ambien, and “anagrams” generated potent antituberculosis agents. ACS Infect Dis 1:85–90. doi: 10.1021/id500008t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moraski GC, Markley LD, Cramer J, Hipskind PA, Boshoff H, Bailey MA, Alling T, Ollinger J, Parish T, Miller MJ. 2013. Advancement of imidazo[1,2-a]pyridines with improved pharmacokinetics and nM activity vs. Mycobacterium tuberculosis. ACS Med Chem Lett 4:675–679. doi: 10.1021/ml400088y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang J, Wang B, Wu T, Wan J, Tu Z, Njire M, Wan B, Franzblauc SG, Zhang T, Lu X, Ding K. 2015. Design, synthesis, and biological evaluation of pyrazolo[1,5-a]pyridine-3-carboxamides as novel antitubercular agents. ACS Med Chem Lett 6:814–818. doi: 10.1021/acsmedchemlett.5b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, Williams Z, Hards K, Tang J, Cheung C-Y, Aung HL, Wang B, Liu Z, Hu X, Lenaerts A, Woolhiser L, Hastings C, Zhang X, Wang Z, Rhee K, Ding K, Zhang T, Cook GM. 2019. Pyrazolo[1,5-a]pyridine inhibitor of the respiratory cytochrome bcc complex for the treatment of drug-resistant tuberculosis. ACS Infect Dis 5:239–249. doi: 10.1021/acsinfecdis.8b00225. [DOI] [PubMed] [Google Scholar]
- 13.Lu X, Tang J, Cui S, Wan B, Franzblauc SG, Zhang T, Zhang X, Ding K. 2017. Pyrazolo[1,5-a]pyridine-3-carboxamide hybrids: design, synthesis and evaluation of anti-tubercular activity. Eur J Med Chem 125:41–48. doi: 10.1016/j.ejmech.2016.09.030. [DOI] [PubMed] [Google Scholar]
- 14.Moraski GC, Seeger N, Miller PA, Oliver AG, Boshoff HI, Cho S, Mulugeta S, Anderson JR, Franzblau SG, Miller MJ. 2016. Arrival of imidazo[2,1- b] thiazole-5-carboxamides: potent anti-tuberculosis agents that target QcrB. ACS Infect Dis 2:393–398. doi: 10.1021/acsinfecdis.5b00154. [DOI] [PubMed] [Google Scholar]
- 15.van der Westhuyzen R, Winks S, Wilson CR, Boyle GA, Gessner RK, Soares de Melo C, Taylor D, de Kock C, Njoroge M, Brunschwig C, Lawrence N, Rao SPS, Sirgel F, van Helden P, Seldon R, Moosa A, Warner DF, Arista L, Manjunatha UH, Smith PW, Street LJ, Chibale K. 2015. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: a novel antimycobacterial class targeting mycobacterial respiration. J Med Chem 58:9371–9381. doi: 10.1021/acs.jmedchem.5b01542. [DOI] [PubMed] [Google Scholar]
- 16.Rybniker J, Vocat A, Sala C, Busso P, Pojer F, Benjak A, Cole ST. 2015. Lansoprazole is an antituberculous prodrug targeting cytochrome bc 1. Nat Commun 6:7659. doi: 10.1038/ncomms8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phummarin N, Boshoff HI, Tsang PS, Dalton J, Wiles S, Barry CE, Copp BR. 2016. SAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc1 inhibitors. Medchemcomm 7:2122–2127. doi: 10.1039/C6MD00236F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subtil FT, Villela AD, Abbadi BL, Rodrigues-Junior VS, Bizarro CV, Timmers L, de Souza ON, Pissinate K, Machado P, López-Gavín A, Tudó G, González-Martín J, Basso LA, Santos DS. 2018. Activity of 2-(quinolin-4-yloxy)acetamides in Mycobacterium tuberculosis clinical isolates and identification of their molecular target by whole-genome sequencing. Int J Antimicrob Agents 51:378–384. doi: 10.1016/j.ijantimicag.2017.08.023. [DOI] [PubMed] [Google Scholar]
- 19.Foo CS, Lupien A, Kienle M, Vocat A, Benjak A, Sommer R, Lamprecht DA, Steyn AJC, Pethe K, Piton J, Altmann K-H, Cole ST. 2018. Arylvinylpiperazine amides, a new class of potent inhibitors targeting QcrB of Mycobacterium tuberculosis. mBio 9:e01276-18. doi: 10.1128/mBio.01276-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandrasekera NS, Berube BJ, Shetye G, Chettiar S, O’Malley T, Manning A, Flint L, Awasthi D, Ioerger TR, Sacchettini J, Masquelin T, Hipskind PA, Odingo J, Parish T. 2017. Improved phenoxyalkylbenzimidazoles with activity against Mycobacterium tuberculosis appear to target QcrB. ACS Infect Dis 3:898–916. doi: 10.1021/acsinfecdis.7b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ananthan S, Faaleolea ER, Goldman RC, Hobrath JV, Kwong CD, Laughon BE, Maddry JA, Mehta A, Rasmussen L, Reynolds RC, Secrist JA, Shindo N, Showe DN, Sosa MI, Suling WJ, White EL. 2009. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb) 89:334–353. doi: 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandrasekera NS, Alling T, Bailey MA, Files M, Early JV, Ollinger J, Ovechkina Y, Masquelin T, Desai PV, Cramer JW, Hipskind PA, Odingo JO, Parish T. 2015. Identification of phenoxyalkylbenzimidazoles with antitubercular activity. J Med Chem 58:7273–7285. doi: 10.1021/acs.jmedchem.5b00546. [DOI] [PubMed] [Google Scholar]
- 23.Cleghorn LAT, Ray PC, Odingo J, Kumar A, Wescott H, Korkegian A, Masquelin T, Lopez Moure A, Wilson C, Davis S, Huggett M, Turner P, Smith A, Epemolu O, Zuccotto F, Riley J, Scullion P, Shishikura Y, Ferguson L, Rullas J, Guijarro L, Read KD, Green SR, Hipskind P, Parish T, Wyatt PG. 2018. Identification of morpholino thiophenes as novel Mycobacterium tuberculosis inhibitors, targeting QcrB. J Med Chem 61:6592–6608. doi: 10.1021/acs.jmedchem.8b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho S, Lee HS, Franzblau S. 2015. Microplate Alamar Blue assay (MABA) and low oxygen recovery assay (LORA) for Mycobacterium tuberculosis, p 281–292. In Parish T, Roberts DM (ed), Mycobacteria protocols. Springer New York, New York, NY. [DOI] [PubMed] [Google Scholar]
- 25.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. 2005. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J Bacteriol 187:6300–6308. doi: 10.1128/JB.187.18.6300-6308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim M-S, Jang J, Ab Rahman NB, Pethe K, Berry EA, Huang L-S. 2015. Isolation and characterization of a hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis cytochrome bcc and Mycobacterium smegmatis cytochrome aa3. J Biol Chem 290:14350–14360. doi: 10.1074/jbc.M114.624312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Megehee JA, Hosler JP, Lundrigan MD. 2006. Evidence for a cytochrome bcc-aa3 interaction in the respiratory chain of Mycobacterium smegmatis. Microbiology 152:823–829. doi: 10.1099/mic.0.28723-0. [DOI] [PubMed] [Google Scholar]
- 28.Esser L, Elberry M, Zhou F, Yu C-A, Yu L, Xia D. 2008. Inhibitor-complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides. J Biol Chem 283:2846–2857. doi: 10.1074/jbc.M708608200. [DOI] [PubMed] [Google Scholar]
- 29.Ko Y, Choi I. 2016. Putative 3D structure of QcrB from Mycobacterium tuberculosis cytochrome bc1 complex, a novel drug-target for new series of antituberculosis agent Q203. Bull Korean Chem Soc 37:725–731. doi: 10.1002/bkcs.10765. [DOI] [Google Scholar]
- 30.Kana BD, Weinstein EA, Avarbock D, Dawes SS, Rubin H, Mizrahi V. 2001. Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis. J Bacteriol 183:7076–7086. doi: 10.1128/JB.183.24.7076-7086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalia NP, Hasenoehrl EJ, Ab Rahman NB, Koh VH, Ang MLT, Sajorda DR, Hards K, Gruber G, Alonso S, Cook GM, Berney M, Pethe K. 2017. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc Natl Acad Sci U S A 114:7426–7431. doi: 10.1073/pnas.1706139114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gualano G, Capone S, Matteelli A, Palmieri F. 2016. New antituberculosis drugs: from clinical trial to programmatic use. Infect Dis Rep 8:6569. doi: 10.4081/idr.2016.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballell L, Bates RH, Young RJ, Alvarez-Gomez D, Alvarez-Ruiz E, Barroso V, Blanco D, Crespo B, Escribano J, González R, Lozano S, Huss S, Santos-Villarejo A, Martín-Plaza JJ, Mendoza A, Rebollo-Lopez MJ, Remuiñan-Blanco M, Lavandera JL, Pérez-Herran E, Gamo-Benito FJ, García-Bustos JF, Barros D, Castro JP, Cammack N. 2013. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 8:313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, Ioerger TR. 2017. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis mBio 8:e02133-16. doi: 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Malley T, Alling T, Early JV, Wescott HA, Kumar A, Moraski GC, Miller MJ, Masquelin T, Hipskind PA, Parish T. 2018. Imidazopyridine compounds inhibit mycobacterial growth by depleting ATP levels. Antimicrob Agents Chemother 62:e02439-17. doi: 10.1128/AAC.02439-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamprecht DA, Finin PM, Rahman MA, Cumming BM, Russell SL, Jonnala SR, Adamson JH, Steyn A. 2016. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat Commun 7:12393. doi: 10.1038/ncomms12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Small JL, Park SW, Kana BD, Ioerger TR, Sacchettini JC, Ehrt S. 2013. Perturbation of cytochrome c maturation reveals adaptability of the respiratory chain in Mycobacterium tuberculosis. mBio 4:e00475-13. doi: 10.1128/mBio.00475-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. 2005. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc Natl Acad Sci U S A 102:15629–15634. doi: 10.1073/pnas.0507850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berube BJ, Parish T. 2017. Combinations of respiratory chain inhibitors have enhanced bactericidal activity against Mycobacterium tuberculosis. Antimicrob Agents Chemother 62:e01677-17. doi: 10.1128/AAC.01677-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu P, Asseri AH, Kremer M, Maaskant J, Ummels R, Lill H, Bald D. 2018. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci Rep 8:2625. doi: 10.1038/s41598-018-20989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bardarov S, Bardarov S, Pavelka MS, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG, and M. smegmatis. Microbiology 148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 44.Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, Glickman MS. 2009. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138:146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs William RJ. 2007. Genetic manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol Chapter 10:Unit10A.2. doi: 10.1002/9780471729259.mc10a02s6. [DOI] [PubMed] [Google Scholar]
- 46.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. 2012. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones G, Willett P, Glen RC, Leach AR, Taylor R. 1997. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related 4-amino-thieno[2,3-d]pyrimidines do not inhibit the growth of M. smegmatis more than CB37 does. M. smegmatis strain csm208 was incubated in the presence of the indicated 4-amino-thieno[2,3-d]pyrimidine at 100 μM, and the optical density (OD) was measured periodically over the course of 12 h in a plate reader (n = 3 experiments). (A to F) DMSO and 73 μM rifampin were included as controls. The control data for panels A to F are the same in each panel because all the compounds were tested in the same assay plate. (G and H) DMSO and 1.1 mM streptomycin were included as controls. The control data for panels G and H are the same in both panels because these compounds were tested in the same assay plate. Download FIG S1, TIF file, 1.3 MB (1.3MB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Structure-activity relationship studies around CB37. aclogP, calculated log(partition coefficient, P). Values were calculated using the CDD Vault platform. bValues are from Fig. 2C. Download Table S1, DOCX file, 0.1 MB (141.6KB, docx) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Structure-activity relationship studies around CWHM-728 and comparison to known M. tuberculosis inhibitors. aclogP, calculated log(partition coefficient, P). Values were calculated using the CDD Vault platform. bValues are from Fig. 3C. Download Table S2, DOCX file, 0.1 MB (130.1KB, docx) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
M. tuberculosis harboring the qcrBA178T allele is not cross-resistant to Q203. WT or M. tuberculosis (Mtb) ΔqcrCAB complemented with either qcrCABWT or qcrCABA178T was incubated in the presence of increasing concentrations of Q203, and then bacterial respiration and metabolism were measured using the MABA (n = 3 experiments). Best-fit curves and IC50 values ± standard deviation (S.D.) were calculated using GraphPad Prism software. P values were determined by one-way analysis of variance with Tukey’s posttest. *, P < 0.05; **, P < 0.01; ns, not significant. Download FIG S2, TIF file, 0.8 MB (820.3KB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental information. Download Text S1, PDF file, 0.3 MB (346.6KB, pdf) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
LC-MS spectra for CWHM-1023. CWHM-1023 was subjected to LC-MS analysis as described in Text S1 in the supplemental material. Download FIG S3, TIF file, 0.6 MB (627.8KB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
NMR spectra for CWHM-1023. CWHM-1023 was subjected to 1H NMR (A) and 13C NMR (B) analysis. The inset in panel A depicts the chemical structure of CWHM-1023. Download FIG S4, TIF file, 0.4 MB (378KB, tif) .
Copyright © 2019 Harrison et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The sequence data are publicly available in the NCBI Sequence Read Archive under accession no. PRJNA561987.