

Abstract
The genovariation of endothelin receptor type B (EDNRB) was identified in a Chinese family with Waardenburg syndrome type I (WS1) in the present study. WS1 was diagnosed in a 19-year-old young man, his older sister and aunt according to WS consortium criteria. After extracting genomic DNA from the peripheral blood samples, the coding exons and intronic regions of EDNRB were sequenced. A missense heterozygous mutation was found in the coding region of exon 2 in the EDNRB gene on chormosome 13q22.3 of the proband. The same mutation was detected in the proband's afflicted paternal aunt and first older sister. Subsequent polyphen analysis and three-dimensional modeling confirmed that the c.469A>G heterozygous mutation in EDNRB was possibly pathogenic. This is the first report of EDNRB mutation as a potential disease-causing mutation in Chinese patients with WS1.
Keywords: EDNRB, Chinese family, Waardenburg syndrome type I, gene mutation
INTRODUCTION
Waardenburg syndrome (WS) is a neurocristopathy characterized by pigmentation abnormality and sensorineural deafness[1]. WS cases are further divided into 4 subtypes based on additional signs. Dystopia canthorum is a typical feature of WS1, but it is absent in WS2. The two types can be distinguished according to the W index: W>1.95 is WS1 while W<1.95 is WS2. WS3 is similar to WS1 except for additional upper-limb abnormalities and WS4 resembles WS2 except for additional Hirschsprung disease (HD) or gastrointestinal atresia[2]. WS not only has heterogeneous clinical manifestations, but also presents genetic heterogeneity depending on the gene involved[3].
The endothelin pathway plays a key role in neural crest development[4]. Endothelin receptor type B (EDNRB) is integral to the survival, proliferation and differentiation of melanoblasts[5]. EDNRB mutations (mainly homozygous and very rare heterozygous) have been described in WS4 and its heterozygous mutations have been reported in isolated HD[6]. However, whether EDNRB mutations are associated with other subtypes of WS is still enigmatic. Here we report the EDNRB mutation in a Chinese family afflicted with WS1.
SUBJECTS AND METHODS
Ethical Approval
The study conformed to the requirements of the Declaration of Helsinki. Informed consent was obtained from all the subjects. This study was approved by the Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University.
Patient and Clinical Data
A 19 year-old young patient presented to our clinic with gradual exotropia for 2y. He had iris pigmentation abnormality in the left eye and deafness in the left ear while no signs of frontal white forelock or other hypo-pigmented areas were found. The paternal aunt and the first older sister of the proband presented with bilateral blue irises, unilateral deafness in the right ear and dystopia canthorum.
Routine clinical, audiological examination and ophthalmologic evaluation including anterior segment and fundus photography were performed on this patient. The W index was calculated using the following equation: W=X+Y+(A/B) {X=[2A-(0.2119C+3.909)]/C, Y=[2A-(0.249B+3.909)]/B}, wherein A referred to the inner canthus, B referred to pupillary distance and C referred to outer canthus.
Mutation Analysis
Peripheral blood samples of the proband, his mother, paternal aunt, two older sisters, aunt and cousins were collected at the Third Affiliated Hospital, Sun Yat-sen University. Genetic analyses were performed at Jinyu Laboratory Groups, Genetics Division, Guangzhou, China.
Protein Structure Prediction
Both the wild type and mutated EDNRB sequences were used to conduct protein structure prediction with an online tool I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) as previously described[7]. The pathogenicity of the mutation was evaluated using the PolyPhen (http://genetics.bwh.harvard.edu/pph) analytical tool.
RESULTS
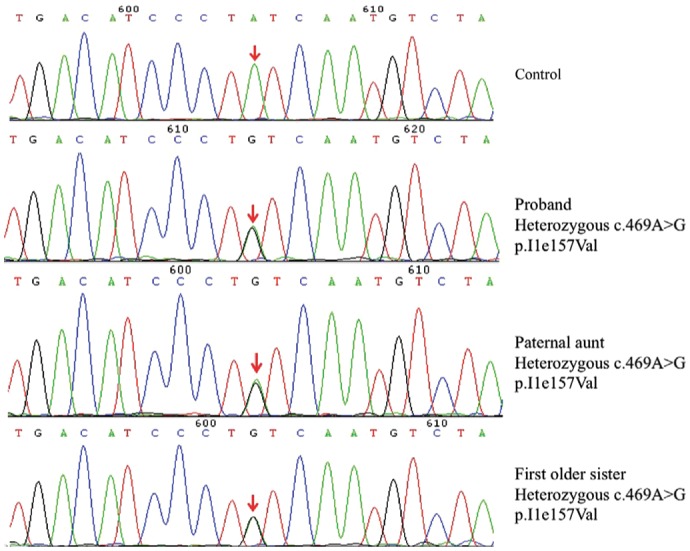
Dystopia canthorum and unilateral deafness were present in all affected patients, namely the proband (II3), his paternal aunt (I2) and first older sister (II1) (Figure 1A). A brilliant blue iris was present in the left eye of the proband (Figure 1B). On slit-lamp examination, complete heterochromia iridis in the left eye was found in the proband (Figure 1C and 1D). Fundoscopy revealed albinotic fundus in the left eye (Figure 1E). Auditory evoked potential objectified a unilateral sensorineural hearing loss requiring cochlear implant. The preoperative prismatic deviation was 37Δ at both near and distance. The extra circular muscle movements and ocular tension were normal. The proband underwent uniocular recession-resection surgery. Postoperatively, the calculated W index was 2.59. He was born of a non-consanguineous marriage. Both of his parents were normal on appearance. A heterozygous nucleotide substitution within exon 2 of EDNRB (OMIM 131244), c.469A>G; p.I1e157Val (NM_000115.3) was identified in the proband's genomic DNA as the sole putative causative mutation. The proband's afflicted paternal aunt and first older sister also carried the same mutation. This mutation was not found in other unaffected relatives and healthy controls (Figure 2).
Figure 1. Clinical features of the proband and pedigree chart of this family.
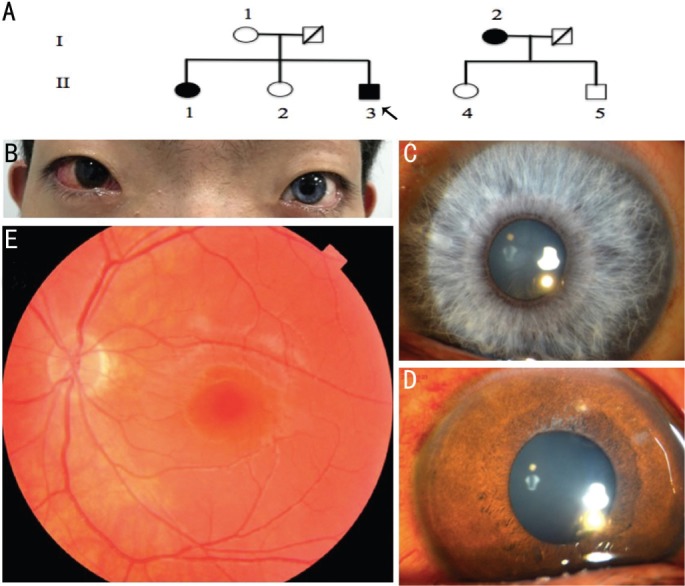
Figure 2. Sequence chromatography.
As detected by PolyPhen, the substitution of isoleucine with valine results in a “possibly damaging” effect. The mutation involves a highly conserved residue. As shown in the red square, the isoleucine at position 157 is highly conserved for EDNRB, which is demonstrated by analysis of 6 orthologs from different vertebrate species (Figure 3). The wild type EDNRB protein consists of ten helices (Figure 4A), whereas the EDNRB missense mutation results in a protein consisting of nine helices (Figure 4B).
Figure 3. Analysis of the conservation of the EDNRB mutation [c.469A>G (p. I1e157Val)].

Figure 4. Protein structure prediction of wild-type EDNRB protein (A) and mutated EDNRB (B).

DISCUSSION
At least 15% of WS cases cannot be explained at the molecular level, which suggests the potential involvement of other uncovered gene mutations[8]. The EDNRB gene spanning 24 kb and comprising 7 exons is located in 13q22.3[9]. To date, 32 EDNRB mutations have been shown to be related to WS (http://grenada.lumc.nl/LOVD2/WS), whereas the mutation in exon 2, c.469A>G has not been reported.
EDN3/EDNRB interaction is indispensable to the development of melanocytes and enteric neurons that originate from neural crest[10]. Mutation in the EDNRB gene was previously thought to be causative for WS4. WS4 featured the common manifestations of WS and the additional presence of HD[11]. However, apart from heterochromia iridis, sensorineural deafness and dystopia canthorum, the affected patients suffered no congenital aganglionic megacolon or gastrointestinal atresia.
In most WS cases, one or both of the proband's parents are affected[12]. In a smaller proportion of WS1 cases, there isn't any affected parent and the proband is presumed to develop WS1 as a result of a sporadic de novo pathogenic variant. In this family, the classical symptoms of WS1 in the proband included heterochromia iridum, sensorineural hearing disorder and dystopia canthorum. WS1 with exotropia is uncommon and only one case of WS with exotropia and anisocoria was reported previously[13]. In contrast, WS patients usually have esoiropia. The diverse clinical manifestations of WS patients with heterozygous mutations might be a result of interaction between environment, multigenic inheritance and the impact of modifier genes on cell fate in early embryogenesis[14].
Taken together, a missense heterozygous mutation [c.469A>G (p. I1e157Val)] of EDNRB was detected in the present study, which might be disease-causing for WS1.
Acknowledgments
The authors thank the patients and their family members for their collaboration in this study. The authors also thank Dr. Yi-Zhao Luan for his technical assistance.
Foundations: Supported by Medical Scientific Research Foundation of Guangdong Province (No.A2016271); Guangdong Natural Science Foundation (No.2016A030313208).
Conflicts of Interest: Cheng HH, None; Ling SQ, None; Zhao PZ, None; Li WL, None; Deng J, None.
REFERENCES
- 1.Yang T, Li X, Huang Q, Li L, Chai Y, Sun L, Wang X, Zhu Y, Wang Z, Huang Z, Li Y, Wu H. Double heterozygous mutations of MITF and PAX3 result in Waardenburg syndrome with increased penetrance in pigmentary defects. Clin Genet. 2013;83(1):78–82. doi: 10.1111/j.1399-0004.2012.01853.x. [DOI] [PubMed] [Google Scholar]
- 2.Ma J, Zhang TS, Lin K, Sun H, Jiang HC, Yang YL, Low F, Gao YQ, Ruan B. Waardenburg syndrome type II in a Chinese patient caused by a novel nonsense mutation in the SOX10 gene. Int J Pediatr Otorhinolaryngol. 2016;85:56–61. doi: 10.1016/j.ijporl.2016.03.043. [DOI] [PubMed] [Google Scholar]
- 3.Haddad NM, Ente D, Chouery E, Jalkh N, Mehawej C, Khoueir Z, Pingault V, Mégarbané A. Molecular study of three lebanese and Syrian patients with waardenburg syndrome and report of novel mutations in the EDNRB and MITF genes. Mol Syndromol. 2011;1(4):169–175. doi: 10.1159/000322891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pla P, Larue L. Involvement of endothelin receptors in normal and pathological development of neural crest cells. Int J Dev Biol. 2003;47(5):315–325. [PubMed] [Google Scholar]
- 5.Saldana-Caboverde A, Kos L. Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010;23(2):160–170. doi: 10.1111/j.1755-148X.2010.00678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puffenberger EG, Hosoda K, Washington SS, Nakao K, deWit D, Yanagisawa M, Chakravart A. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell. 1994;79(7):1257–1266. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 7.Hazan F, Ozturk AT, Adibelli H, Unal N, Tukun A. A novel missense mutation of paired box 3 gene in a Turkish family with Waardenburg syndrome type 1. Mol Vis. 2013;19:196–202. [PMC free article] [PubMed] [Google Scholar]
- 8.Lin YC, Lai HS, Hsu WM, Lee PI, Chen HL, Chang MH. Mutation analysis of endothelin-B receptor gene in patients with Hirschsprung disease in Taiwan. J Pediatr Gastroenterol Nutr. 2008;46(1):36–40. doi: 10.1097/01.mpg.0000304451.54057.df. [DOI] [PubMed] [Google Scholar]
- 9.Yoshihara M, Sato T, Saito D, Ohara O, Kuramoto T, Suyama M. A deletion in the intergenic region upstream of Ednrb causes head spot in the rat strain KFRS4/Kyo. BMC Genet. 2017;18(1):29. doi: 10.1186/s12863-017-0497-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee HO, Levorse JM, Shin MK. The endothelin receptor-B is required for the migration of neural crest-derived melanocyte and enteric neuron precursors. Dev Biol. 2003;259(1):162–175. doi: 10.1016/s0012-1606(03)00160-x. [DOI] [PubMed] [Google Scholar]
- 11.Núñez-Ramos R, Fernández RM, González-Velasco M, Ruiz-Contreras J, Galán-Gómez E, Núñez-Núñez R, Borrego S. A scoring system to predict the severity of hirschsprung disease at diagnosis and its correlation with molecular genetics. Pediatr Dev Pathol. 2017;20(1):28–37. doi: 10.1177/1093526616683883. [DOI] [PubMed] [Google Scholar]
- 12.Yang SZ, Dai P, Liu X, Kang DY, Zhang X, Yang WY, Zhou CY, Yang SM, Yuan HJ. Genetic and phenotypic heterogeneity in Chinese patients with waardenburg syndrome type II. PLoS One. 2013;8(10):e77149. doi: 10.1371/journal.pone.0077149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dastur YK, Dudhani A, Chitale A, Dasgupta S. Waardenburg syndrome with anisocoria and exotropia. J Postgrad Med. 1995;41(4):111–112. [PubMed] [Google Scholar]
- 14.Pingault V, Bondurand N, Lemort N, Sancandi M, Ceccherini I, Hugot JP, Jouk PS, Goossens M. A heterozygous endothelin 3 mutation in Waardenburg-Hirschsprung disease: is there a dosage effect of EDN3/EDNRB gene mutations on neurocristopathy phenotypes? J Med Genet. 2001;38(3):205–209. doi: 10.1136/jmg.38.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]