

Abstract
Policy Points.
The US Food and Drug Administration (FDA) has in recent years allowed onto the market several drugs with limited evidence of safety and effectiveness, provided that manufacturers agree to carry out additional studies while the drugs are in clinical use.
Studies suggest that these postmarketing requirements (PMRs) frequently lack transparency, are subject to delays, and fail to answer the questions of greatest clinical importance. Yet, none of the literature speaks directly to the challenges that the FDA—as a regulatory institution—encounters in enforcing PMRs.
Through a series of interviews with FDA leadership, this article analyzes and situates those challenges in the midst of political threats to the FDA's public health mandate.
Context
Modern pharmaceutical regulation is premised on a rigorous examination of a drug's safety and effectiveness prior to its lawful sale. However, since the 1990s, the US Food and Drug Administration (FDA) has gradually shifted to a model of “lifecycle” regulation that increasingly relies on postmarketing requirements (PMRs) to encourage studies of drug safety and effectiveness following regulatory approval. This article examines the range of legal, institutional, and political challenges that FDA faces in the context of lifecycle regulation.
Methods
Document‐based legal and policy analysis was combined with a set of semistructured interviews of current and former FDA officials (n = 23) in order to explore the implications of the FDA's use of PMRs. The median interview time per official was 61 minutes, with a range of 24 to 227 minutes. All of the officials interviewed occupied positions of leadership and influence within the FDA, such as directors of an FDA center or office, key legal counsel on agency‐wide policy initiatives, and the commissioner of the FDA.
Findings
Insufficient resources and coordination within the FDA, inadequate legal authorities, and the political economy of withdrawing an approved indication in the face of opposition from companies and patients all contribute to the observed shortcomings in the FDA's use and enforcement of PMRs. Further, the FDA is fully aware of these challenges, yet is seemingly resigned to and resistant to criticism of its use of PMRs.
Conclusions
This study of the FDA's shift toward lifecycle regulation reveals not simply an agency in transition, but rather an agency on guard against a set of larger political threats to its mandate. This can be characterized as a state of institutional incumbency in which the agency is engaged in an effort to reproduce key features of the regulatory system—in concert with regulated industries and others—while simultaneously sanctioning significant changes to the regulatory standards the FDA has long applied, to the detriment of public health.
Keywords: Food and Drug Administration, pharmaceutical industry, regulation, prescription drugs
National pharmaceutical regulators are embattled agencies. The US Food and Drug Administration (FDA) in particular has endured repeated periods of intense political scrutiny over the course of its history. At times, the theme of a “drug lag” in the United States relative to Europe painted the bureaucracy as the enemy of pharmaceutical innovation.1, 2 During other periods, allegations of conflicts of interest among the agency's ranks or slow agency action in response to drug‐related adverse events have propelled a narrative of regulatory capture by industry.3, 4 As Carpenter2 explains, these lenses of regulatory laggard on the one hand and industry lackey on the other tend to obscure the accumulated weight of institutional practice within the agency—the many ways in which FDA officials exercise, subtlety but strategically, their discretion, reputation, and power in order to balance the agency's public health mission with appetites for pharmaceutical innovation.
One area where the agency has long struggled to balance these imperatives is often referred to as the postapproval or postmarket phase of a drug's lifecycle. Legally, the agency's powers in the premarket phase are “cosmic” by comparison to the postmarket phase.2 (p586) The power to say no to market entry conditions pharmaceutical companies to comply with FDA feedback. Upon approval, the dynamics change, in part, because the powers at the agency's disposal are considerably less once the drug is on the market. Institutionally, the FDA's organization and internal hierarchies appear to track this fundamental asymmetry between pre‐ and postapproval.2, 5, 6, 7 Agency officials with training in epidemiology, tasked with monitoring drugs on the market, have traditionally served as consultants to physician‐dominated drug reviewing divisions. Changes to a drug's labeling, or the prospect of product withdrawal, proposed by these postmarket monitors must be vetted and approved by the physician gatekeepers. All of which underscores a deep tension within the FDA, not only with respect to its differentiated legal powers pre‐ and postapproval, but also in terms of its institutional mandate for, as Carpenter describes it, “any success of postmarket epidemiology threatens to demonstrate a failure of premarket clinical trial design and review.”2 (p622)
Despite the longstanding divide between pre‐ and postapproval regulatory functions inside the agency, the FDA has in recent years embraced a lifecycle approach to drug regulation.6, 8 In particular, the FDA's leadership, including FDA commissioners and influential career officials, have published a number of articles that emphasize the value of what can be learned about a drug's and biologic's safety and effectiveness through the collection and analysis of real‐world evidence, that is, evidence generated during the postmarket use of a product.9, 10, 11, 12, 13 Although the power to say no to an indication remains absent after its approval, the agency gained substantial legal authorities for the purpose of postmarket surveillance with the passage of the Food and Drug Administration Amendments Act (FDAAA) of 2007,14 and it has made increasing use of these legal tools in recent years despite the resources challenges involved.15 Meanwhile, there is a growing empirical literature that calls into question the limited evidence behind many new drugs and biologics approved by the FDA on an expedited basis and/or underscores the many obstacles to gathering additional knowledge about their safety and effectiveness in the real world despite the agency's expanded scope of postmarket authority.16, 17, 18, 19, 20, 21
Using a mix of documentary and qualitative data, including interviews with 23 current and former influential FDA officials, this article assesses the state of lifecycle regulation at the FDA. In the first section of the article, I describe the ongoing shift to lifecycle regulation at the FDA, survey a series of review procedures, regulatory pathways, and statutory powers that underpin this shift, and situate the shift in terms of the larger political context. The second section of the article details the methodology I employ to investigate whether the observed shortfalls in the FDA's enforcement of postmarket studies is the result of insufficient resources and coordination within the agency, inadequate legal authorities, or a more fundamental political economy problem.22 In the third section, through an analysis of interview and documentary data, I find that all three—resources, legal authority, and the prevailing political economy—contribute to the status quo. The FDA is fully aware of these challenges and yet seemingly resigned to, and resistant to criticism of, the shift toward lifecycle regulation.
In the final section of the article I attempt to make sense of these findings by developing a concept of institutional incumbency. I abstract this concept from both the analysis of legal and policy materials related to lifecycle regulation alongside interviews with FDA officials, and my reading of a larger body of literature on government bureaucracies and the interactions between such bureaucracies, including the FDA, private sector firms, and other actors.23, 24, 25 With the term institutional I refer not just to the agency, but also how the FDA operates in dialogue with regulated industries and, increasingly, patient groups, to generate, shape, and delimit knowledge about drugs and biologics. And by incumbency I mean to evoke the strategies that established firms deploy to preserve positions of dominance26, 27 and extend this idea of defense‐by‐offense to the FDA. Through this conceptual lens of institutional incumbency I aim to reveal not simply an agency in transition, but an agency on guard, engaged in an effort to reproduce key features of the regulatory system—in concert with regulated industries and others—while simultaneously sanctioning significant changes to the regulatory standards the FDA has long applied. In the name of lifecycle regulation, the agency is preserving who produces information about the safety and effectiveness of drugs and biologics, while altering when, and under what circumstances, that information production occurs.
The Rise of Lifecyle Regulation
The concept of lifecycle regulation refers not only to regulating pharmaceutical products both prior to and following market approval, it also refers to an approach to regulation that factors the prospect of evidence generation postapproval into preapproval decision making. Since the 1930s the FDA has had a mandate to evaluate pharmaceuticals both pre‐ and postapproval.28 The blurring of pre‐ and postapproval phases of a drug's lifecycle is a more recent regulatory phenomenon.
The origins of lifecycle regulation are not located in a single piece of legislation. Rather, the move to lifecycle regulation is better observed through the steady accumulation of new review procedures, regulatory pathways, and statutory powers, that, in one way or another, blend the pre‐ and postapproval phases of the pharmaceutical lifecycle. These procedures, pathways, and powers have generally been codified in various laws, but were sometimes anticipated through administrative innovation by officials at the FDA.2 (p491),4(p45) Individually, the rationale, scope, and details behind these various procedures, pathways, and powers differ. Collectively, they have coalesced into a larger vision of lifecycle regulation that is now part and parcel of regulatory parlance at the FDA and beyond.
The FDA's expansive suite of expedited programs is emblematic of the agency's shift toward lifecycle regulation. The FDA has since the 1980s developed or overseen the implementation of six such expedited programs in the name of streamlining access to new drugs, beginning with the Orphan Drug Act of 1983,29 which created new market incentives and encouraged flexible clinical study designs in order to facilitate the development of therapies for rare diseases.30 As noted earlier, the HIV/AIDS epidemic motivated the creation of an expanded access program in 1987, which allowed patients with unmet medical needs to apply for and use unapproved, experimental therapies, as well as the fast track (1988) and accelerated approval (1992) pathways, which, respectively, encouraged faster agency reviews for therapies beyond phase 2 clinical trials and sanctioned reliance on surrogate markers, such as a reduction in tumor size, as opposed to clinically meaningful endpoints (eg, overall survival) in order to reduce the duration of premarket clinical studies.30 The FDA's priority review program, which requires reviews to be completed within six months (as opposed to the then‐typical twelve months) when the drug in question promises improvements over existing treatments in terms of safety, efficacy, or convenience, also began in 1992. Finally, in 2012, a breakthrough therapy designation was added to this array of procedures and pathways. Drugs that target a serious or life‐threatening illness for which preliminary evidence suggests may provide substantial improvement over currently available therapies are eligible for the breakthrough designation.30, 31
Although Congress was initially not persuaded to grant FDA authority to compel postmarket studies,4 (p52), 32 the agency began asking sponsors to carry out additional studies postapproval as it implemented its first expedited programs. The 1992 regulations that the FDA developed for the accelerated approval pathway, which were codified by Congress in 1996, operate differently. They vest the FDA with the authority to require a postmarket study to confirm the efficacy of the drug, backed by the threat of misbranding charges and/or civil monetary penalties if the sponsor delays the study without good cause. In the event efficacy is not subsequently shown, the FDA can withdraw its approval, effectively creating a disincentive for the manufacturer to conduct the study or at least an incentive to delay. Essentially the same power to compel—backed by either the threat of withdrawal or misbranding charges and monetary fines—a postmarket study was also incorporated into the animal efficacy rule, which gave the FDA authority to approve the use of drugs on the basis of animal studies alone in special circumstances and the Pediatric Research Equity Act (PREA),33 which encouraged the development of pediatric indications, in 2002 and 2003, respectively.34
Outside of the accelerated approval pathway, the majority of postmarket studies were voluntary and referred to as postmarketing commitments (PMCs) until 2007, when Congress passed the FDAAA. The thrust of the FDAAA was to bolster patient safety, in contrast to the three other types of postmarketing requirements (PMRs) embedded in administrative and/or legislative efforts to streamline drug review and approval, which were intended to address unmet medical needs (accelerated approval), respond to a public health emergency (animal efficacy), and encourage pediatric research (PREA). A series of high profile regulatory failures, including Merck & Co.’s rofecoxib (Vioxx) and GSK's paroxetine (Paxil) and rosiglitazone (Avandia), animated the legislation.5, 35 The PMR authority in the FDAAA thus gave the FDA new powers to compel postmarket safety studies either at the time of, or following, regulatory approval. (See Table 1 for a summary of the four types of PMRs and corresponding enforcement actions). In addition, the FDAAA also empowered the agency to require sponsors to develop and comply with a risk evaluation and mitigation strategy (REMS) once a drug was in use. A recent report by the US Department of Health and Human Services Office of Inspector General15 shows that the FDA's use of PMRs has, especially since the enactment of FDAAA, grown significantly. (See Figure 1 for a timeline of the expedited programs, key pieces of legislation, and rise of PMRs).
Table 1.
The FDA's Postmarket Requirement Authorities and Corresponding Enforcement Actions
| Available Enforcement Actions | ||||
|---|---|---|---|---|
| Statutory Authority | Key Parameters | Civil Monetary Penalties | Misbranding Charges | Withdrawal of Approval |
| Accelerated approval | • Must target a serious or life‐threatening illness and provide meaningful therapeutic benefit to patients over existing treatments | ✓ | ✓ | ✓ |
| • Approval based on a surrogate endpoint that is reasonably likely, based on epidemiologic, therapeutic, pathophysiologic, or other evidence, to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity | ||||
| Animal efficacy rule | • Must target serious or life‐threatening conditions caused by exposure to lethal or permanently disabling toxic biological, chemical, radiological, or nuclear substances | X | X | ✓ |
| • Applies only to products for which definitive human efficacy studies cannot be conducted because it would be unethical to deliberately expose healthy human volunteers to a lethal or permanently debilitating toxic biological, chemical, radiological, or nuclear substance, and field trials to study the product's effectiveness after an accidental or hostile exposure have not been feasible | ||||
| • Approval only when results of adequate and well‐controlled animal studies are reasonably likely to produce clinical benefit in humans | ||||
| Pediatric Research Equity Act (PREA) | • Applies when the FDA decides to defer the requirement of submitting safety and efficacy data in respect of a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration, until after approval of the drug product for use in adults | ✓ | ✓ | Xa |
| • Such a deferral may be granted if the drug is ready for approval in adults before studies in pediatric patients are complete or pediatric studies should be delayed until additional safety and effectiveness data have been collected | ||||
| Food and Drug Administration Amendments Act (FDAAA) | • May be required to assess a known serious risk related to the use of the drug or biologic, to assess signals of serious risk related to the use of the drug, or to identify an unexpected serious risk when available data indicates the potential for a serious risk | ✓ | ✓ | ? b |
| • Can be imposed at the time of, or following, approval | ||||
Withdrawal of approval is not applicable because in the case of a PMR issued under PREA, at the time of approval, no pediatric indication has been approved. Rather, the approval pertains to an adult indication, which carries a PMR to generate evidence as to whether one or more pediatric indications is also warranted. When the study or studies undertaken to fulfill a PMR under PREA is completed, the FDA evaluates whether the study ‘responds’ to the PMR, which, in turn, informs its decision to grant the pediatric indication(s) and as a result confer an additional period of six months market exclusivity to the sponsor for the approved pediatric indication(s).
In contrast to the accelerated approval pathway and the animal efficacy rule, the FDA lacks the legal authority to withdraw an approval that carries a PMR due to the sponsor's failure to fulfill the PMR. Rather, under FDAAA, s 505(o), the agency can only issue civil monetary penalties or pursue misbranding charges. If, however, a study undertaken pursuant to a PMR attached under FDAAA is completed, and its results yield new safety (or possibly efficacy) issues, then FDA may withdraw the approval under its general power to do so pursuant to s 505(e) of the FFDCA.
Figure 1.
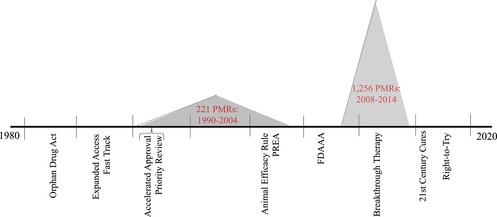
Timeline of Key Policy Changes, Adoption of Expedited Programs, and Increasing Use of Postmarket Requirementsa
aThe PMR data incorporated in this figure derive from two reports prepared by the US Department of Health and Human Services’ Office of Inspector General.5, 85 The two reports present PMR data differently; in particular, the 2006 report simply presents a lump sum total of all PMRs issued by FDA during the years 1990‐2004, as opposed to the number of PMRs issued per year, which the 2016 report documents. For consistency, a lump sum total of PMRs is therefore presented for the period covered by the 2016 report, ie, 2008‐2014. Importantly, the main contributor to the growth of PMRs in the latter period of time is the addition of the authority to issue PMRs under FDAAA, which was enacted in 2007 and came into force in 2008. As explained by OIG,85 (p8) the “number of PMRs that FDA issued increased by 111% from FY 2008 to FY 2009, and then remained fairly consistent through FY 2014.” [Color figure can be viewed at http://wileyonlinelibrary.com]
The question that has dominated the discussion around the FDA's embrace of lifecycle regulation is whether the addition of these expedited programs, coupled with the agency's increasing tendency to request (through PMCs) and/or demand (pursuant to a PMR) evidence generation post‐approval, has effectively diminished the regulator's governing standards for market approval. As Darrow and colleagues write, while the various expedited “programs did not formally change the legal standard for approval, which continues to require the demonstration of safety and ‘substantial evidence of effectiveness,’” they did establish “more flexible criteria for meeting this standard…and reduced the time available to the FDA to determine whether the standard has been met.”30 (p1444) Beyond this overarching concern, a growing body of literature raises questions about the FDA's record of enforcing the timely completion of PMCs and/or PMRs,16, 36, 37 and the quality of the studies carried out in respect to drugs approved through the expedited programs, in particular, studies relying on surrogate markers.17, 21, 38 In addition, notwithstanding the tenuous safety and effectiveness evidence behind these drugs at the time of FDA approval, many carry high prices, which the US Department of Health and Human Services has stipulated state Medicaid programs must cover.39, 40 Notably, these empirical studies have in turn motivated a variety of rebukes from FDA officials,10, 11, 41, 42 culminating in a new public report on the performance of PMRs and PMCs that paints a more optimistic picture about the timing and completion rates of postmarket studies.43
Amidst this evidentiary debate, the FDA has been under renewed pressure to expedite its approval processes, essentially deepening the lifecycle approach to regulation. In 2016, the US Congress passed the 21st Century Cures Act (Cures Act), containing a number of provisions that direct the FDA to approve products on the basis of “real world evidence” derived from “experience,” “observational studies,” “registries,” and in the case of medical devices, even “case reports” of individual patients as long as they are published in a journal.44 The legislation also encourages greater use of surrogate markers and to engage patients directly in research and development and regulatory processes, building on recent initiatives at the FDA, such as the Patient Engagement Advisory Committee and the Patient‐Focused Drug Development program.45, 46, 47
Whereas the Cures Act amounts to an endorsement by Congress of the direction that the FDA has long been headed with its expedited programs and more recent patient input initiatives, other legal developments—one from the courts, the other from the new administration—appear to sanction the agency for its alleged interference with commercial speech and/or patient choice. In a series of decisions beginning in the early 2000s, courts have chipped away at the FDA's efforts to curb the promotion of nonapproved, or off label, indications on the argument that the FDA was interfering with companies’ constitutionally protected free speech rights.2 (p620‐621),22(p2377‐2379) Most importantly, a federal appeals court ruled in 2012 in US v. Caronia 48 that manufacturers could lawfully engage in off‐label promotion provided the information used to promote the drug was “truthful and non‐misleading,” a standard that is both “far less rigorous than the FDA's current standards for determining a drug's efficacy and safety for a particular indication”49 and more challenging to enforce as FDA bears the burden of demonstrating an intention to mislead. In response to another company's assertion of its free speech rights, the FDA withdrew a warning letter from its website even though the company's promotion of a drug for unapproved uses was “extremely concerning from a public health perspective.”50 Perhaps fearing a more binding ruling, the FDA elected to settle or not to appeal in all of these cases and instead reexamined its approach,51 which recently resulted in a new guidance that appears to adopt the Caronia framework of permitting truthful and nonmisleading off‐label promotion.52 Even so, pressure to further relax the FDA's regulation of off‐label marketing is expected to increase with new bills introduced in state and federal legislatures.49
A libertarian think tank, the Goldwater Institute, which is behind these efforts to deregulate off‐label drug marketing is also a key driving force behind a spate of state and federal laws known as Right‐to‐Try.53, 54 The federal Right‐to‐Try Act, which was passed by Congress in May 2018,55 allows patients with life‐threatening diseases to access an experimental drug in development as long as it has completed phase 1 clinical trials (which typically assess safety in healthy volunteers or a small number of people with the disease in question). Unlike the FDA's longstanding expanded access program, which grants essentially all requests for access to experimental therapies, the Right‐to‐Try Act does not require the agency (or research ethics bodies) to assess the risks and benefits of the experimental treatment, to ensure that patients provide informed consent, or that sponsors share details of any observed adverse events. Moreover, even if such adverse events were reported to the agency, the FDA is precluded by the legislation, except in narrow circumstances, from utilizing that data in making its subsequent approval decisions. The law, in other words, allows patients to effectively bypass the FDA in the absence of a clear need to do so, impeding the agency's power to steer the production of information to inform patients about the actual safety and effectiveness of new drugs and biologics before they enter the market.22 Prior to its passage, then‐FDA Commissioner Scott Gottlieb had signaled a level of hesitation over Right‐to‐Try56; it remains to be seen whether its impact on the FDA can be moderated through careful implementation.57, 58
There are many ways to read these developments. From the outside, it appears the FDA is under significant attack. At the same time, the FDA has a long history of nimbly exercising its discretion, even in the face of much vaunted reforms to its governing laws, both in the service of public health and to blunt efforts to diminish the agency's reach and power.2 The postmarket setting is perhaps an exception to this pattern given the dominance of gatekeeping culture within the agency.2, 59 Armed with enhanced enforcement powers since 2007, it is plausible that the institutional culture is changing. The Government Accountability Office indeed found in 2009 that some progress had been made in terms of adding resources and improving communication between, and clarifying the roles of, premarket reviewers in the Office of New Drugs (OND) and postmarket monitors in the Office of Surveillance and Epidemiology (OSE). However, OND remained above the OSE in terms of the FDA's hierarchy, and the mechanism for resolving internal disputes among FDA officials lacked independence.7 In this study, then, I sought to uncover how officials inside the FDA view the project of lifecycle regulation, with particular attention to the challenges involved in enforcing requirements to carry out clinical studies after a drug or biologic has been approved for sale and use, while at the same time attending to the political economy of pharmaceutical regulation.
A Situational Analysis of the FDA
In keeping with situational analysis methodology, this study involved a continuous process of mapping and triangulating multiple sources of data in order to formulate and test new hypotheses as the data were collected.60 Included in this research were a wide variety of documentary materials detailed further in the following section and extensive qualitative data collected through semistructured interviews with both former and current FDA officials occupying positions of leadership and influence within the agency. The focus on lifecycle regulation and in particular PMRs expanded as the research proceeded, which led to follow‐up interviews with some participants who did not engage that issue in depth during the first interview.
Document Analysis
Drawing on my legal training, I compiled all relevant legal and policy materials, including (1) court cases, legislation, regulations, and guidance documents issued by the FDA; (2) grey literature, including reports published by government bodies such as the US Department of Health and Human Services’ Office of Inspector General and the US Government Accountability Office, which have completed relevant studies regarding the FDA and postmarket studies; and (3) secondary materials (eg, scholarly literature, newspaper and media articles) relevant to the topic of lifecycle regulation. In addition, some participants shared materials such as their own unpublished writing and teaching slides that they developed based on their expertise and experiences at the FDA. These various documentary sources were used to identify key examples and issues to raise with interview participants.
FDA Official Interviews
The current research was reviewed and approved by a research ethics board (institutional review board) prior to recruiting participants. FDA officials were identified and approached through a snowball sampling technique. The principal funding organization for the research (The Commonwealth Fund) had a number of contacts within the FDA. During an initial visit to Washington, DC, I met with two former FDA officials to outline the nature of the research project; each official, in turn, suggested particular individuals (both previously and currently at the FDA) to approach for potential participation in the study and offered to contact those individuals on my behalf. When an individual agreed to participate in an interview, I asked for suggestions of other individuals to approach for potential inclusion in the study. A total of four invitations to participate in the study were declined (in one case the individual explicitly declined; in the other three, the email was forwarded to a general communications officer within the FDA, which indicated interviews were not generally possible). In all other cases, the individuals that were approached via a former or current FDA colleague agreed in principle to participate, and at the outset of the interview the informed consent process, including the participant's ability to withdraw at any stage, was discussed.
A total of 23 FDA officials ultimately participated in the semistructured interviews. Seven officials were currently employed at the FDA. The remaining 16 officials had previously departed or retired from the FDA; however, 8 of those 16 had left the FDA within the last five years. Fourteen of the officials participated in telephone interviews only; nine officials were interviewed in person. Follow‐up interviews were held with six officials in order to ensure ample opportunity to discuss issues and examples of particular interest as they emerged over the research process. The median interview time per official was 61 minutes, with a range of 24 to 227 minutes. All of the officials interviewed occupied positions of leadership and influence within the FDA, including at the level of the agency as a whole (FDA commissioner, deputy, or associate commissioner, n = 7), its Office of Chief Counsel (chief counsel, special counsel, other agency lawyers, n = 8), and the agency's centers, including the Center for Drug Evaluation and Research or “CDER” (center and office directors, n = 8). Two of the participating officials declined to be quoted in the analysis but agreed to participate by providing background for the research, and two officials chose to be identified by name.
Data Analysis
I analyzed the data using an open‐coding approach. I reviewed and reread interview transcripts in an iterative fashion throughout the data collection phase of the research, identifying themes and examples for closer investigation with participants. I grouped the data into broad descriptive themes, which surfaced in most, if not all, participant interviews. I then used those broad themes to reanalyze the data, identifying issues and examples of particular interest that arose in at least a sizeable minority (eg, five to seven) of interviews to corroborate or complicate the documentary materials I had compiled. Particular attention was given to discrepancies and similarities between officials’ comments in the published literature versus those given in the course of the research interviews. This iterative analytical process yielded unexpected points of interest that in turn informed follow‐up interviews with participants with whom the point in question may not have been discussed during the initial interview, as well as further analysis of the qualitative data alongside the relevant legal and policy materials and scholarly literature.
An Agency On Guard
I present the research findings in relation to three plausible explanations for why PMRs may be challenging to enforce; namely, resource issues, limitations in the agency's legal powers, and the political economy of postmarket drug regulation. I find that all three present challenges for enforcing PMR studies. However, the interview data—in particular officials’ intimate awareness of these challenges yet marked resistance to pausing lifecycle regulation—also reveal an agency strategizing for, and against, greater change. I characterize this as a moment of institutional incumbency in which, in contrast to the past, the agency deploys its considerable resources and procedural know‐how not to maintain or expand the regulatory standards it applies, but rather to stay changes that could thwart its role in the system of pharmaceutical governance. Indeed, I infer institutional incumbency in significant part from the tendency of officials to, on the one hand, claim that the system is in the main unchanged, while on the other hand, working strategically—in concert with regulated industries and, increasingly, patient representatives—to alter some and reproduce other key features of the system that the FDA has long curated. This state of affairs, if accurate, contrasts with leading accounts of the agency, which posit, in the extremes, that the agency deploys its reputation and power to advance its public health mission2 or falls prey to industry capture.4
A Question of Resources, Private Protocols, and Evolving Priorities
PMRs are often poorly defined at the time of approval by the FDA. They tend to be short descriptions of the question(s) that the FDA would like to have answered through one or more postmarket studies, but they often fail to specify what type of study design should be employed to answer the question(s) of interest. Of 110 clinical trials that were encompassed in a cross‐sectional analysis of all PMRs issued by the FDA during 2009‐2012, many, if not most, “did not report enough information to establish use of randomization, comparator type, allocation, outcome, and number of patients to be enrolled.”16
Resource challenges partly explain these ambiguities. Officials are candid about the agency's limitations in terms of specifying study designs leading up to marketing approval. “I don't think we have the time or the staffing…to come up with the protocol” for the sponsor, one director emphasized. According to another official, the aim is to capture the rationale for PMR studies in the review of the discipline that's recommending the PMR, but those “discussions between FDA and sponsors about the details of a PMR often occur pretty late in the review cycle.” The degree of coordination also appears to vary depending on the type of PMR. According to officials, PMRs issued pursuant to PREA tend to command greater coordination within the agency because there is a centralized internal committee focused on pediatric research. Thus, the study or studies to be conducted under a PREA PMR were more likely to be well defined at the time of approval. In the context of accelerated approval “the goal is to be recruiting by the time of approval” and protocols should already be in place, though officials are unsure how often that is the case. PMRs crafted under FDAAA, in contrast, tend to suffer from a lack of coordination according to some participants. The FDA has both a high‐level Drug Safety Advisory Board, and an advisory committee dedicated to safety issues; however, the advisory committee has, at times, met infrequently and the FDA has in the past struggled to fill committee vacancies,7 which may impede its ability to inform PMR decision making.
Beyond resource challenges, it is also apparent that the traditional dynamic between the agency and sponsor factor into the lack of specificity in PMRs. While the FDA wields the authority to apply the criteria under its expedited programs and attach PMRs as it sees fit, the agency sees the onus of designing postmarket studies as falling primarily to the sponsor. Although reviewers may be cognizant of important weaknesses in the premarket evidence base, the agency shifts the burden of postmarket study design to the sponsor in order to ensure user fee legislation timelines are met and allow reviewers to turn to other premarket review activities. It is not that the FDA's guidance on the particulars is unwelcome; rather, companies do not want guidance about PMRs within the four corners of the approval letter because, as one former director explains, “that letter is posted on the FDA's website.”
This echoes the agency's longstanding practice of working out its decisions through repeated, confidential exchanges with sponsors. The agency's decision to refuse a new drug application (NDA) or biologics license application (BLA) are seldom made public, not because FDA lacks the authority to publish them, but rather because those applications are apt to return, whether in revised form or supported by new evidence, for reconsideration.61, 62, 63 When the FDA finally approves an NDA or BLA, substantial documentation becomes publicly available. However, that transparency can also mask the considerations that prove determinative in a given case, including in the context of PMRs. For instance, in February 2008, the FDA decided to approve bevacizumab (Avastin) for the treatment of metastatic breast cancer (subject to a PMR) only two months after the Oncologic Drug Advisory Committee voted to reject that indication. Apart from the company stating its willingness to conduct a postmarket study, it is not clear what shifted during the intervening two months.64 Under PREA the agency has on occasion held firm to its assessment that the studies carried out by the sponsor “fail to respond” to the PMR for a pediatric study.65 In other cases, though, senior officials have overridden the clinical reviewers’ assessment and granted the extension of six‐months exclusivity. Compare, for instance, the criticism contained in the medial review regarding the six‐month exclusivity for clopidogrel (Plavix) with the more senior officials’ positive assessment of the postmarket evidence.66 (p68), 67 Why senior management at the FDA chose to intervene in one case versus others is not obvious from the safety and effectiveness data alone.
Historically, officials charged with monitoring postmarket drug safety have been subordinate both to senior management as well as drug reviewing divisions because of the dominance of gatekeeping in FDA culture.2 (p608),5‐7 A current FDA director suggests that the work of the postmarket monitors is now more routinized and integrated into premarket reviews:
[W]hen I started in this…the way the premarket people worked with the postmarket people around the approval of a drug is about two to four weeks before the drug was approved, the premarket people would meet with the postmarket people and say, “Here's what we're about to approve. Here's what you might want to look for in your safety surveillance.” That's completely different now. We involve postmarket safety people as early as we need to…[they] are brought in at different times in this review on a structured and scheduled basis.
Despite this integration, however, the ultimate decision‐making power continues to rest with FDA gatekeepers according to the same official:
The Office of New Drugs, the premarket group, they retain regulatory control over that product throughout its lifecycle, so any requirement for a PMR, any postmarket label change, etc., is done through them and is signed by their office, not an independent postmarket safety office.
These patterns of shifting the burden to the sponsor, working out the details of a PMR in private, keeping disagreements within the agency outside of public purview, and preserving the institutional hierarchy of gatekeepers over monitors are indicative of the FDA's lasting priorities in the midst of the stated shift toward lifecycle regulation. After passage of the safety‐focused FDAAA, OND reviewers still stressed that “their primary focus is on completing premarket work within [user fee legislation] time frames, and issues related to postmarket safety receive lesser priority.”7 Nearly a decade later, officials continue to echo the need to prioritize premarket review activities, especially NDAs and BLAs that fall under one or more of the agency's expedited programs. “It's a bit of a fire drill at the end of the review process, ”explains Janet Woodcock, director of the FDA's Center for Drug Evaluation and Research (CDER). “FDA has worked diligently to put into place the procedures, personnel, and tracking systems in an effort to ensure postmarket studies receive adequate attention,” another official stresses, but “a breakthrough therapy [investigational new drug application] is likely to be given higher priority by officials than reviewing a postmarket study protocol.” The breakthrough therapy is “the squeaky wheel that gets the grease.”
Further, the prospect of deferring evidence generation until the postmarket phase influences premarket decision making. Officials are reticent to hold up approval because the specific details of a postmarket study are not yet settled. Indeed, officials intimate that the agency's power to attach PMRs likely smooths the way to approval. One former FDA director represents the influence of PMRs on approval decisions as follows:
The fact that the agency can require postmarketing commitments has probably allowed the agency to approve more drugs than they would have otherwise. In essence, one reviewer might question whether the drug should be approved before knowing X while others will stress that you do not really have to know X beforehand; a postmarket study can be used to answer X, Y, or Z after approval. In other words, the agency may look at the available evidence and decide that although the drug appears to be safe, they would be more comfortable if there was additional evidence in, for example, a subpopulation. In such a case, they can approve the drug, but require a postmarketing study in that subpopulation to generate additional safety data.
Law and Authority: Hesitant Efficacy, Inapparent Efficiency, and Empty Enforcement
Officials report a sense that prior to approval, sponsors facing the prospect of a PMR act cooperatively, yet the dynamic quickly shifts postapproval. Director Woodcock suggests that companies worry that “if they dispute [the PMR] or…enter into negotiation [over its terms], it will delay or prevent the approval. […] Then they have buyer's regret afterward because they don't necessarily agree.” However, the agency, too, exercises caution in what it expects sponsors to do under a PMR. In particular, outside of the accelerated approval pathway, which is specifically intended to postpone confirmation of effectiveness until the postmarket study phase, the agency seldom, if ever, crafts PMRs to encompass effectiveness outcomes. The wording of the law underpinning the FDAAA PMR power is indeed unclear, a point which several FDA officials expressed awareness of during interviews. According to Robert Temple, an official with more than 40 years of experience at the FDA, the agency is often very interested in such questions and in some cases postmarket studies designed to rule out a safety risk also yield efficacy findings. A 2008 guidance from the FDA, for example, used PMRs under FDAAA to mandate long‐term cardiovascular outcomes trials for all new type 2 diabetes drugs, at least four of which subsequently suggested a cardiovascular benefit in addition to generating further evidence about safety risks.68 Unless, however, “you can make a safety argument about it,” Temple believes the agency is on shaky legal footing.
Historically, the FDA frequently made that very argument in the name of expanding its jurisdiction. Before 1962, when the requirement to provide substantial evidence of effectiveness was explicitly incorporated into the FDA's governing legislation, agency “officials slowly and ambiguously nudged the concept [of safety] toward the inclusion of ‘therapeutic effect’ issues [through] public speech, in administrative practice, and in rulemaking.2 (p150) By the 1950s, FDA officials had crafted the requirements and format of the NDA so as to render safety and effectiveness “procedurally inseparable.”2 (p155ff) Despite the agency's interest in effectiveness in the context of postmarket studies, seemingly little to no administrative innovation is being deployed to require effectiveness end‐points under a PMR outside of the accelerated approval pathway. For instance, of 60 PMRs attached to approvals pursuant to the FDAAA that were examined by Wallach and colleagues, only seven had a clinical outcome as a primary endpoint.16 It is not that the FDA proposes an effectiveness outcome as part of an FDAAA PMR and companies resist on legal grounds; rather, Temple explains, “I don't think we even think we can insist” (emphasis added). Other than accelerated approval PMRs, then, the FDA appears to limit itself to requesting effectiveness data under a PMC as opposed to attempting to compel its production pursuant to a PMR.
Setting aside the question of scope, the FDA's legal authority to enforce PMRs is clear in law but questionable in practice. In principle, a sponsor's failure to conduct a postmarket study in accordance with the agreed‐upon timetable, whether attached under the accelerated approval, PREA, or FDAAA PMR authorities, can attract civil monetary penalties and/or misbranding charges provided the delays are not for “good cause” (Table 1).69 Failing to fulfill an accelerated approval PMR can also theoretically trigger proceedings to withdraw the approved indication. In practice, the FDA rarely threatens these actions,70 although the recently departed commissioner's decision to publicly admonish one company for failure to fulfill a PMR may signal a change in approach.71 A 2016 report by the US Department of Health and Human Services’ Office of Inspector General found that 90 PMRs issued between 2008 and 2014 had fallen one to five years behind schedule yet the FDA had issued only 32 letters of noncompliance, only one of which was a “warning letter” threatening enforcement action.15 The empirical literature about the status of PMRs thus continues to highlight room for improvement with respect to PMR enforcement.16, 36, 72 In the literature42 and in the context of my interviews, officials contend that the problem of PMR delays have been either misstated or overblown. According to one former FDA director, the publicly available PMR database, which outside researchers have relied on to carry out their studies of PMR enforcement, are inaccurate due to revisions made to a PMR's timetable following approval.
From another perspective, company compliance with PMRs appears remarkable, especially given the agency's internal thinking about the utility of enforcement tools at its disposal. The 90 PMRs issued between 2008 and 2014 that were delayed one to five years from the agreed‐upon schedule constitute only 7% of the total number of PMRs (1,256) issued by the FDA during that period.15 The FDA cannot, as Director Woodcock stresses, “just write something down and issue a fine. There's all this legal process we have to go through, [a] gigantic legal process.” Another official corroborates that thinking, noting that the agency has “to be ready to go to court as soon as they issue a warning letter or take any other action.” While that may not be technically accurate as the action must constitute final agency action to be ripe for judicial review, industry's general tendency to eventually satisfy PMRs in the face of the agency's hesitation to use the most immediate enforcement options at its disposal is telling. Sponsors have a shared interest in upholding an image of a well‐functioning system of lifecycle regulation.
The industry's shared interest does not extend to, or is at least more tenuous, when it comes to the prospect of withdrawing an accelerated approval, whether for failure to conduct a postmarket study or in the event that the results of such a study fail to confirm the product's effectiveness and/or safety. When the accelerated approval pathway was first designed in 1992, an accelerated withdrawal process was designed to go with it in order to make this enforcement option actionable in practice. The regulations expressly direct the FDA not to follow its most formal, labor‐intensive hearing procedure known as “separation of functions.” While accelerated approval is not technically a conditional approval, withdrawal should follow in the event that the product's benefits are not subsequently confirmed. However, according to interviewed officials, sponsors tend to view an FDA approval—notwithstanding the attached PMR(s)—as nothing short of a “vested right.”
To date, the option has been exercised on only one occasion by the FDA in the face of company opposition and it appears this enforcement action is unlikely to be deployed again by the agency, at least in the absence of safety concerns. The lone case to date involved the drug bevacizumab (Avastin), which has a number of approved indications. In 2008, under the accelerated approval authority, the FDA added metastatic breast cancer to bevacizumab's list of approved indications. The sponsor, Genentech, complied with the terms of the PMR and carried out the postmarket study with relative efficiency; however, the postmarket trial did not—in the FDA's eyes—confirm the drug's effectiveness against metastatic breast cancer. The company and scores of patient groups contested the agency's assessment of the postmarket evidence in the context of a public hearing. The FDA was at pains to accommodate patient perspectives, adopting, contrary to the wording of the regulations, its elaborate separation of functions procedure to ensure a degree of impartiality to the hearing, which ultimately resulted in an exhaustively argued 70‐page decision by then‐FDA Commissioner Margaret Hamburg holding firm to the agency's decision to withdraw the indication.73 One former director who was closely involved in the case describes the episode as “Armageddon” while another FDA counsel shares that it “was so hard and so time consuming that if you suggest now that somebody take something off the market, they just roll their eyes at you.”
The Political Economy of Patient Engagement and Real‐World Evidence
The challenge in the bevacizumab case was in part owing to the sponsor's differing interpretation of the postmarket effectiveness data. More fundamentally, the case highlights the dilemma involved in saying no and withdrawing an indication—let alone a drug or biologic—after having previously said yes to its lawful use. In a period of six weeks, from when the FDA announced a public hearing would be held and the date of the hearing itself, some 450 submissions were sent to the FDA, “mostly by consumers asking the FDA to maintain the indication as their perception was the drug had benefit.”64 (p3) Others went further, suggesting the agency had no place to act by questioning the FDA's right “to make a decision that should be left to a woman and her doctor.”64 (p5) The FDA has difficulty enforcing the timely completion of PMRs, but acting upon PMR data once in hand is more vexed:
[E]ven if you get the study, and you often do, sometimes they don't confirm the efficacy of the product. […] What is FDA supposed to do with that? There's now a huge and vocal constituency for the product. Whether or not the study showed it worked, there are people out there who think it worked, and lots of people with a financial stake in it. It becomes a political nightmare to try to take a product off the market that's already developed that constituency by being approved for a period of time. […] [I]n some ways [it's] a bigger problem than whether you get the data. It's whether you can do anything with it when you have it.
Director Woodcock offers a similar perspective: “The idea that we could withdraw the drug, which we could, is usually not very palatable to anybody. We want the information, but we don't want to rip the drug out of the hands of patients and clinicians.”
Meanwhile, officials dismiss the suggestion that the agency should stay its embrace of lifecycle regulation in light of these enforcement and decision‐making challenges. That is “where the academic community is really off base,” contends Director Woodcock. “We know far more now about a drug when it is approved than in the past… [PMRs] are things that would be good to know, but wouldn't be necessary for approval.” Another FDA director maintains that the standard, that is, the legal requirement that a drug's safety and substantial evidence of effectiveness be shown prior to approval, is—and remains—the governing standard. These are remarkable claims to make in light of the growing body of literature showing that therapies approved on the basis of surrogate markers are seldom studied for their impact on clinically meaningful endpoints; of the few that are, only a small minority (< 10%) show clinical evidence of effectiveness, backed by a published, peer‐reviewed postmarket study.18, 19, 20, 38
During interviews officials pointed to the future to highlight encouraging developments outside the FDA, in particular, new insights from genomics and epigenomics being incorporated into drug development under the rubric of precision medicine and our increasingly digitized, information environment. They linked those developments back to the agency's efforts to modernize its review processes and deeply integrate patients in drug development and regulation. The Cures Act articulated these same linkages, earmarking considerable resources, which the agency has since prioritized for spending on patient‐focused research and development ($25.8 M), advancing new therapies ($95.3 M), modern trial design ($57.8 M), patient access to therapies ($185.2 M), and medical device innovations ($109 M) between 2017 and 2025 (FDA 2017). However, as Robert Califf, FDA commissioner at the time the Cures Act was passed, pointed out, the agency was in many respects already working toward those same goals.13, 74 Those critical of the FDA and its ongoing project of lifecycle regulation are, according to commissioner Califf, “overly cautious,”10 a point echoed by other forward‐looking officials.
Officials confirm that real‐world evidence is the “direction that everything is going,” while some register concerns with the significant methodological challenges involved. One current official explains:
You just let patients take them, and somehow all of these massive insurance databases will cough up an answer at the end of a year or two about how well the products work and how safe they are. We're not really anywhere near that degree of sophistication in our ability to analyze those big datasets, but there's a huge amount of pressure to push off lots of data collection there without a lot of methodology to do it.
However, other developments, especially Congress’ enactment of Right‐To‐Try legislation, are seen as qualitatively different than the challenges posed by lifecycle regulation. The latter pose technical, resource, and legal challenges; the former threaten the agency's very role in the system—across the lifecycle—insofar as it purports to allow, outside the ambit of FDA oversight, patients to access experimental therapies promoted as such by companies, to the detriment of the agency's ability to spur the generation of safety and effectiveness information.22 Officials did not explicitly connect the advent of Right‐to‐Try with the agency's deepening embrace of PMRs and lifecycle regulation more generally. Yet many were quick to stress that every patient organization that registered a public position on the legislation came out against it, manifesting the increasingly close nexus between patients and the FDA. There are “huge offices at FDA working on [patient engagement],” an official underscores. Ten years ago FDA reviewers would have dismissed patients as uninformed. Today, in contrast, “all of this effort [is] being expended on both hearing from patients in an anecdotal way, but also trying to figure out how to craft patient‐reported outcomes that can be evaluated in a way that gives you real evidence at the end.”
Institutional Incumbency and Pharmaceutical Knowledge Production
The FDA's stated commitment to lifecycle regulation implies a distribution of resources and decision‐making authority across the premarket and postmarket phases as new evidence about a drug's safety and effectiveness emerges. Despite the rhetorical turn toward the postmarket phase of a drug's lifecycle, this redistribution of resources and decision making does not appear to be occurring. The FDA's resources remain concentrated on the premarket side of the equation. After the FDAAA expanded the agency's ability to apply user fees to postmarket surveillance activities, the FDA planned to essentially double its postmarket‐related budget to over $100 million by 2012.7 (p13) By 2012 the FDA exceeded that goal; however, the budget for postmarket oversight ($187,275,000) remains less than half of what is allocated for new drug review ($440,970,000).75 All of the new resources secured by FDA under the Cures Act are, moreover, earmarked for expediting and modernizing premarket review processes rather than postmarket monitoring activities.76
Internal coordination of pre‐ and postmarket expertise appears to have improved over the last decade, with one director claiming that the FDA's epidemiology experts are now integrated as part of premarket reviewer teams and involved in the decision making as “early as they need to be” (though other officials suggest coordination is generally lacking outside of PREA PMRs, given the high importance accorded to pediatric interventions within the FDA). Beyond capturing the questions of greatest interest to the FDA, however, the task of defining postmarket study protocols is seen as the sponsor's responsibility, replicating the model of premarket agency/industry interaction in the postmarket phase.
Most importantly, decision‐making power within the agency remains concentrated among those vested with the authority to approve NDAs and BLAs for market entry. Signing off on the terms of a PMR, revisiting an approval based on PMR data, making changes to the drug's labeling, or contemplating withdrawal, remains rooted within the reviewing divisions of the OND, not the OSE. Those who monitor postmarket data may, in other words, be more integrated during the premarket phase yet hold little actual decision‐making power as a drug's evidentiary profile evolves. In some cases, it appears that the OND has precluded OSE officials from participating in advisory committee meetings, ostensibly, because of their public nature. (Carpenter2 [p631] cites examples in 2003 and 2004 involving the drugs leflunomide (Arava) and paroxetine (Paxil), respectively.) Other key FDA safety critics, including David Graham, celebrated for his efforts to uncover the increased risks of myocardial infarction associated with rofecoxib (Vioxx), and Thomas Marciniak have been cast as lone wolves and marginalized within the agency.77 Graham, for example, has in recent years been noticeably absent from high‐profile public meetings such as the 2013 hearings on the diabetes drug rosiglitazone (Avandia),78 and he no longer appears on CDER's list of key officials.79 Marciniak is no longer employed by the FDA. Thus, while more resources may help, the observed shortfalls in the timeliness, quality, and decision making around postmarket studies to date may be less about the agency's available resources, and more a reflection of who continues to wield them. In 2009, the Government Accountability Office called upon the FDA to “develop a comprehensive plan for transferring additional regulatory authorities from OND to OSE that includes time frames for the transfer and steps to ensure resources are properly aligned to allow OSE to assume those responsibilities.”7 (p40) The current research suggests little has changed; the reviewing divisions—as gatekeepers to the market—remain the locus of power within the FDA.
The discursive strategies employed by agency leadership has changed significantly. In the immediate aftermath of Vioxx and other drug safety scandals of the early 2000s, key officials, including then‐Deputy Commissioner Scott Gottlieb, Robert Temple, and others, took a tack of “defending randomized controlled trials and…disparaging pharmacoepidemiology” in order to avoid ceding “more control of the pharmaceutical market to David Graham and his colleagues” at OSE in the wake of the rofecoxib (Vioxx) disaster.2 (p610) In contrast, those who have risen with the FDA's ranks in recent years tend to hail from the reviewing divisions and rehearse the benefits of lifecycle regulation, real world evidence, and patient engagement throughout the research and regulatory process in response to concerns over the timeliness and quality of postmarket studies expressed in the published literature. For example, CDER Director Woodcock11 offered numerous “factual corrections” and rebuttals to the concerns raised by Wallach and colleagues80regarding the FDA's expedited programs. Former Commissioner Robert Califf went further, positing that the “convergence of ubiquitous digital data and the recognition of the importance of patient involvement” (since the HIV/AIDS crisis), “combined with enlightened leadership from FDA, have made it possible to introduce considerable flexibility in the drug development paradigm.”10 (p236),12 These in‐house proponents are candid about some of the challenges involved in lifecycle regulation, such as ensuring judgments of a drug's safety and effectiveness are informed by robustly generated real‐world evidence rather than clouded by noisy real‐world data.10, 12 But they are noticeably quiet about others; namely, the structural conflict of interest posed by embedding the ultimate authority over postmarket surveillance within the agency's drug reviewing divisions,2 (p630) and the task of making decisions in the absence of the agency's most powerful regulatory tool—the power to say no to market entry—in the face of opposition from, as one official puts it, a “vocal constituency” that favors a drug staying on the market whether or not it is safe and effective. Instead, FDA leaders aim to actively build that vocal constituency under the rubric of enhanced patient engagement.
It is in this selective presentation of the challenges involved that the FDA's project of lifecycle regulation takes on a political character. Facing concerted threats to its public health mandate from the president, congress, state legislatures, the courts, and other quarters, ranging from Right‐to‐Try legislation and constitutional challenges to its jurisdiction to a memorandum from the attorney general undermining the agency's ability to enforce its own guidances,81 the FDA's embrace of real world evidence and deeper patient engagement appears designed to insulate the agency against these larger political threats. The agency is aligning itself with patient groups, highlighting their public opposition to the Right‐to‐Try Act, emphasizing that patients are prepared to “accept more uncertainty”82 all the while knowing, in the wake of the bevacizumab (Avastin) saga, the complications that such constituencies will create if and when the drug's effectiveness is called into question. Agency action may be more likely when a safety issue is identified in the postmarket setting but the FDA's decision making has been uneven even in some of those recent instances. Compare, for instance, the agency's response to postmarketing studies carried out in respect of the weight loss drug sibutramine (Meridia) versus the type 2 diabetes drug saxagliptin (Onglyza). In the case of sibutramine, the drug was withdrawn (with the sponsor's cooperation) based on a 16% increased risk for a composite cardiovascular outcome and a finding of decreased effectiveness relative to the premarket trial evidence.83 In the case of saxagliptin, the FDA attached increased safety warnings to the product label based on a 21% increased risk for heart failure hospitalizations (a secondary cardiovascular outcome) and all‐cause mortality based on the FDA's own sensitivity analysis.61
Finally, the agency's leadership is downplaying, in a variety of ways, concerns about the relaxation of its governing standards, from characterizing postmarket studies as things that would simply be good to know but not necessary for approval and publishing analyses suggesting that the FDA seldom makes mistakes under the accelerated approval pathway,41 to noting in the course of interviews for the present study that data derived from “historical controls” has always qualified as “adequate and well‐controlled” in the FDA's eyes. Rather than agitating for greater resources or reallocating them and decision making authority across the lifecycle to enhance the coordination, quality, and timely enforcement of PMRs, much less publicizing the limitations of its legal powers in the postmarket space, agency leadership appear to be working to preserve its institutional hierarchies and preferred processes while simultaneously overseeing deep alterations to when, and on what basis, drugs are approved. It is this kind of dualism—of expressing confidence in the shift toward lifecycle regulation without pursuing concomitant institutional changes in support of that very shift—that suggests FDA is more focused on shoring up its place in pharmaceutical governance than using its discretion, reputation, and power to implement laws in a manner consistent with its public health mission.
This state of institutional incumbency envelops not only the FDA but also regulated pharmaceutical industries inasmuch as they have a shared interest in lifecycle regulation. Deferring studies until the postmarket phase is obviously attractive to industry if it expedites market entry. Abiding by the terms of PMRs, albeit slowly, ensures that companies—in concert with the FDA—maintain tight control over what studies are done versus which ones are deemed infeasible, how they are designed, and ultimately disseminated. If companies were instead to resist PMRs en masse it might underscore concerns about the quality of the evidence pre‐ and postapproval and, more fundamentally still, about who to rely on to generate the evidence behind pharmaceutical interventions. Industry's surprising level of compliance with PMRs in the absence of any “good options” at the agency's disposal for enforcing their timely completion can be understood as a way of staving off more sweeping reform, that is, that the industry's grip on pharmaceutical knowledge production must be loosened.84, 85, 86 It may also reflect the relational nature of firm‐FDA interactions: companies likely abide by the terms of a PMR not because they fear FDA enforcement, but rather because the approved indication carrying the PMR is one of several indications in a sequence that they plan to submit to the agency. And with industry support, the FDA can work to reproduce select features of the system it has long governed (eg, preserving private spaces for dialogue with industry and concentrating decision‐making authority over PMRs within its reviewing divisions) while modernizing others (eg, patient engagement in the research and regulatory process) despite an acute awareness of that those very features are likely to impede its ability to revise its regulatory decisions as a drug's safety and effectiveness profile evolves.
Further empirical inquiry is needed to corroborate this concept of institutional incumbency. Officials were asked in a variety of ways during interviews about whether there was a connection between extrinsic political developments such as the Right‐to‐Try legislation, and the agency's increasing shift toward lifecycle regulation. Most refuted such a connection or carefully measured their assessment of the influence of the political sphere upon agency decision making. The concept instead emerged through an analysis of developments outside the FDA that threaten its core mandate, the selective presentation of the challenges involved in relying on PMRs by agency leadership, the apparent absence of changes within the institution that distribute resources and decision‐making authority across the pharmaceutical lifecycle, and industry's tacit support of the agency's overall policy direction.
Importantly, the FDA's storied history of innovative administrative practice in the service of public health may prove an effective counter to the present state of affairs. That is, the agency's reputation may, with time, ensure that the incorporation of real‐world evidence into its process does not compromise the FDA's commitment to scientific rigor. The agency's inculcation of effectiveness into the premarket assessment of safety before the 1962 amendments2 is perhaps the most important example of FDA's proactive ability to push regulatory standards in a progressive direction. When needed, the agency has also deployed similar savvy in order to blunt or at least forestall policy proposals that run up against its public health and scientific commitments. Here, the FDA Oncologic Drug Advisory Committee's repeated cautions against relying on tumor response rate as a valid surrogate endpoint during the 1980s and 1990s, even after the accelerated approval regulations were codified, is especially notable.4 (p55ff) More recently, the agency's decision to withdraw approval of bevacizumab's breast cancer indication following a postmarket study that failed to confirm the biologic's effectiveness is a testament to the agency's continuing commitment to public health and, in turn, its own institutional credibility.87 The problem is that, in contrast to these historical and contemporary instances of administrative action in the name of public health, the agency now reads its scope of authority more narrowly, sets the bar for approval more flexibly, and hesitates to revise its decisions, in the context of lifecycle regulation. As communicated by officials during interviews, the FDA does not even entertain the idea that it can compel sponsors to examine effectiveness outside of the accelerated approval pathway, using tumor response rate has been normalized as an acceptable basis for approval despite continuing concern in the literature,17, 19, 20 and the withdrawal of bevacizumab's breast cancer indication from the market is understood internally to be both the first and last time agency will undertake that type of regulatory action. These are markers of a more timid agency, seemingly set on defending its place with industry, and increasingly patients, as coproducers of pharmaceutical knowledge at the expense of the evidentiary standards that FDA has long developed and applied.
Funding/Support
This research was supported by the Commonwealth Fund and Canadian Institutes of Health Research (CIHR PJT 156256).
Conflict of Interest Disclosures: The author completed the ICMJE Form for Disclosure of Potential Conflicts of Interest. No conflicts were reported.
Acknowledgments: This research benefited greatly from the author's visits to, and engagement with, the members of the Yale Collaboration for Research Integrity and Transparency (CRIT). In particular, Amy Kapczynski, Joseph Ross, Margaret McCarthy, Joshua Wallach, and Jeanie Kim offered critical insights and support during the research process. Finally, this analysis also benefited significantly from feedback from my colleague Janice Graham and timely research assistance from Gregory Rinkamp, a JD student at the University of Toronto.
References
- 1. Merrill RA. The architecture of government regulation of medical products. VA Law Rev. 1996;82(8):1753‐1866. [Google Scholar]
- 2. Carpenter DP. Reputation and Power: Organizational Image and Pharmaceutical Regulation at the FDA. Princeton, NJ: Princeton University Press; 2010. [Google Scholar]
- 3. Richert L. Conservatism, Consumer Choice, and the Food and Drug Administration during the Reagan Era: A Prescription for Scandal. Plymouth, United Kingdom: Lexington Books; 2014. [Google Scholar]
- 4. Davis C, Abraham J. Unhealthy Pharmaceutical Regulation: Innovation, Politics and Promissory Science. London, UK: Palgrave Macmillan; 2013. [Google Scholar]
- 5. US Government Accountability Office , Report to Congressional Requesters Drug Safety: Improvement Needed in FDA's Postmarket Decision‐making and Oversight Process; GAO 06–402. Washington, DC: US Government Accountability Office; 2006. https://www.gao.gov/new.items/d06402.pdf. Published March 2006. Accessed November 30, 2018. [Google Scholar]
- 6. Baciu A, Stratton K, Burke SP, eds; Committee on the Assessment of the US Drug Safety System The Future of Drug Safety: Promoting and Protecting the Health of the Public. Washington, DC: Institute of Medicine of the National Academies; 2007. https://www.nap.edu/catalog/11750/the-future-of-drug-safety-promoting-and-protecting-the-health. Accessed November 28, 2018. [Google Scholar]
- 7. US Government Accountability Office , Report to the Ranking Member, Committee on Finance, US Senate Drug Safety: FDA Has Begun Efforts to Enhance Postmarket Safety, but Additional Actions Are Needed; GOA 10–68. Washington, DC: US Government Accountability Office; 2009. https://www.gao.gov/assets/300/298135.pdf. Published November 2009. Accessed November 30, 2018. [Google Scholar]
- 8. Psaty BM, Meslin EM, Breckenridge A. A lifecycle approach to the evaluation of FDA approval methods and regulatory actions: opportunities provided by a new IOM report. JAMA. 2012;307(23):2491‐2492. [DOI] [PubMed] [Google Scholar]
- 9. US Department of Health and Human Services, Food and Drug Administration . Framework for FDA's Real‐World Evidence Program. Silver Spring, MD: US Food and Drug Administration; 2018. https://www.fda.gov/media/120060/download. Published December 2018. Accessed June 28, 2019. [Google Scholar]
- 10. Califf RM. Expedited and facilitated drug evaluations and evidence of benefit and risk: the cup is half‐full. Clinical Trials. 2018;15(3):235‐239. [DOI] [PubMed] [Google Scholar]
- 11. Woodcock J. Expediting drug development for serious illness: trade‐offs between patient access and certainty. Clinical Trials. 2018;15(3):230‐234. [DOI] [PubMed] [Google Scholar]
- 12. Sherman RE, Davies KM, Robb MA, Hunter NL, Califf RM. Accelerating development of scientific evidence for medical products within the existing US regulatory framework. Nat Rev Drug Discov. 2017;16:297‐298. [DOI] [PubMed] [Google Scholar]
- 13. Sherman RE, Anderson SA, Dal Pan GJ, et al. Real‐world evidence—What is it and what can it tell us ? N Engl J Med. 2016;375(23):2293‐2297. [DOI] [PubMed] [Google Scholar]
- 14. Food and Drug Administration Amendments Act of 2007 , Pub L No. 110–85, 121 Stat 823 (2007).
- 15. US Department of Health and Human Services, Office of Inspector General . FDA Is Issuing More Postmarketing Requirements, But Challenges with Oversight Persist; OEI‐01‐14‐00390. Washington, DC: Office of the Inspector General; 2016. https://oig.hhs.gov/oei/reports/oei-01-14-00390.pdf. Published July 2016. Accessed November 30, 2018. (OIG 2016) [Google Scholar]
- 16. Wallach JD, Egilman AC, Dhruva SS, et al. Postmarket studies required by the US Food and Drug Administration for new drugs and biologics approved between 2009 and 2012: cross‐sectional analysis. BMJ. 2018;361:k2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wallach JD, Ciani O, Pease AM, et al. Comparison of treatment effect sizes from pivotal and postapproval trials of novel therapeutics approved by the FDA based on surrogate markers of disease: a meta‐epidemiological study. BMC Medicine. 2018;16:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pease AM, Krumholz, HM , Downing NS, Shah ND, Ross JS. Postapproval studies of drugs initially approved by the FDA on the basis of limited evidence: Systematic Review. BMJ. 2017;357:j1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bikdeli B, Punnanithinont N, Akram Y, Lee I, Desai NR, Ross JS, Krumholz HM. Two decades of cardiovascular trials with primary surrogate endpoints: 1990‐2011. J Am Heart Assoc. 2017;6(3): e005285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim C, Prasad V. Cancer drugs approved on the basis of a surrogate endpoint and subsequent overall survival: an analysis of five years of US Food and Drug Administration approvals. JAMA Internal Med. 2015;175(12):1992‐1994. [DOI] [PubMed] [Google Scholar]
- 21. Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005‐2012. JAMA. 2014;311(4):368‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kapczynski A. Dangerous times: The FDA's role in information production, past and future. Minn. L. Rev. 2018; 102:2357‐2382. [Google Scholar]
- 23. Moe TM. The politics of structural choice: toward a theory of public bureaucracy In: Williamson OE, ed. Organization Theory: From Chester Barnard to the Present and Beyond. New York, NY: Oxford University Press; 1995. [Google Scholar]
- 24. Carpenter DP. Protection without capture: product approval by a politically responsive, learning regulator. Am Polit Sci Rev. 2004;98(4):613‐631. [Google Scholar]
- 25. Moffitt SL. Promoting agency reputation through public advice: advisory committee use in the FDA. Journal of Politics. 2010;72(3):880‐893. [Google Scholar]
- 26. Zucker LG, Darby MR. Present at the biotechnological revolution: transformation of technological identity for a large incumbent pharmaceutical firm. Research Policy. 1997;26:429‐446. [Google Scholar]
- 27. Rothaermel FT, Thursby M. The nanotech versus the biotech revolution: sources of productivity in incumbent firm research. Research Policy. 2007;36:832‐849. [Google Scholar]
- 28. Food, Drug, and Cosmetic Act , Pub L No. 75–717, 52 Stat 1040 (1938).
- 29. Orphan Drug Act of 1983 . Pub L. No. 97–414, 96 Stat. 2049 (1983).
- 30. Darrow JJ, Avorn J, Kesselheim AS. The FDA breakthrough‐drug designation—four years of experience. N Engl Jl Med. 2018;378(15):1444‐1453. [DOI] [PubMed] [Google Scholar]
- 31. Food and Drug Administration Safety and Innovation Act , Pub L No. 112–144, 126 Stat 993 (2012).
- 32. Temple RJ. The effect of the Drug Regulation Reform Act of 1978 on clinical research, drug availability, and the public health. Annals of NY Acad Sci. 1981;368:175‐185. [DOI] [PubMed] [Google Scholar]
- 33. Pediatric Research Equity Act , Pub. L. No. 108–155, 117 Stat. 1936 (2003).
- 34. US Department of Health and Human Services, Food and Drug Administration . New drug and biological drug products; evidence needed to demonstrate effectiveness of new drugs when human efficacy studies are not ethical or feasible. Fed Regist.2002;67:37988‐37998. To be codified at 21 CFR 314. https://www.federalregister.gov/documents/2002/05/31/02-13583/new-drug-and-biological-drug-products-evidence-needed-to-demonstrate-effectiveness-of-new-drugs-when. Accessed December 3, 2018. [PubMed] [Google Scholar]
- 35. Topol EJ. Failing the public health—Rofecoxib, Merck, and the FDA. N Engl J Med. 2004;351:1707‐1709. https://www.nejm.org/doi/full/10.1056/NEJMp048286. Accessed November 30, 2018. [DOI] [PubMed] [Google Scholar]
- 36. Woloshin S, Schwartz LM, White B, Moore TM. The fate of FDA postapproval studies. N Engl J Mede. 2017;377:114‐1117. [DOI] [PubMed] [Google Scholar]
- 37. Hwang TJ, Orenstein L, Kesselheim AS, Bourgeois FT. Completion rate and reporting of mandatory pediatric postmarketing studies under the US Pediatric Research Equity Act. JAMA Pediatr. 2019;173(1):68‐74. 10.1001/jamapediatrics.2018.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Naci H, Smalley KR, Kesselheim AS. Characteristics of preapproval and postapproval studies for drugs granted accelerated approval by the US Food and Drug Administration. JAMA. 2017;318(7):626‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brennan Z. CMS to states: Medicaid must cover drugs approved under AA pathway. Regulatory Focus. June 28, 2018. https://www.raps.org/news-and-articles/news-articles/2018/6/cms-to-states-medicaid-must-cover-drugs-approved. Accessed November 28, 2018. (2018b). [Google Scholar]
- 40. Gellad WF, Kesselheim AS. Accelerated approval and expensive drugs—a challenging combination. N Engl J Med. 2017;376:2001‐2004. [DOI] [PubMed] [Google Scholar]
- 41. Beaver JA, Howie LJ, Pelosof L, et al. A 25‐year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics: a review. JAMA Oncol. 2018;4(6):849‐856. [DOI] [PubMed] [Google Scholar]
- 42. Kashoki M, Lee C, Jenkins J. Drug postmarketing studies. JAMA. 2013;310(22):2459. [DOI] [PubMed] [Google Scholar]
- 43. US Department of Health and Human Services, Food and Drug Administration . Report on the Performance of Drugs and Biologics Firm in Conducting Postmarketing Requirements and Commitments, Fiscal Year 2017. Silver Spring, MD: US Food and Drug Administration; 2018. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Post-marketingPhaseIVCommitments/UCM623499.pdf. Published November 19, 2018. Accessed November 30, 2018. (FDA 2018c) [Google Scholar]
- 44. 21 Century Cures Act , Pub L No. 114–255, 130 Stat 1033 (2016).
- 45. Hunter NL, O'Callaghan KM, Califf, RM . Engaging patients across the spectrum of medical product development: view from the US Food and Drug Administration. JAMA. 2015;314(23):2499‐2500. [DOI] [PubMed] [Google Scholar]
- 46. Paradise J. 21st century citizen pharma: the FDA and patient‐focused product development. Am J Law Med. 2018;44:309‐327. [DOI] [PubMed] [Google Scholar]
- 47. US Department of Health and Human Services, Food and Drug Administration . Developing and Submitting Proposed Draft Guidance Relating to Patient Experience Data: Guidance for Industry and Other Stakeholders. Silver Spring, MD: US Food and Drug Administration; 2018. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM628903.pdf. Published December 2018. Accessed April 8, 2019. [Google Scholar]
- 48. United States v. Caronia , 703 F.3d 149 (2d Cir. 2012).
- 49. Sinha MS, Kesselheim AS. The next forum for unraveling FDA off‐label marketing rules: state and federal legislatures. PLoS Med. 2018;15(5):e1002564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Silverman E. FDA settles lawsuit, rescinds warning letter. STAT News December 15, 2015. https://www.statnews.com/pharmalot/2015/12/15/fda-lawsuit-warning-letter/. Accessed November 30, 2018.
- 51. Kim J, Kapczynski A. Promotion of drugs for off‐label uses: the US Food and Drug Administration at a crossroads. JAMA Int Med. 2017;177(2):157‐158. [DOI] [PubMed] [Google Scholar]
- 52. US Department of Health and Human Services, Food and Drug Administration . Medical Product Communications That Are Consistent With the FDA‐Required Labeling‐Questions and Answers: Guidance for Industry. Silver Spring, MD: US Food and Drug Administration; 2018. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm537130.pdf. Published June 2018. Accessed November 30, 2018. [Google Scholar]
- 53. Brennan Z. Updated: off‐label promotion: are FDA's rules about to unravel? Regulatory Focus. May 9 , 2018. https://www.raps.org/news-and-articles/news-articles/2018/5/off-label-promotion-are-fdas-rules-about-to-unra. Accessed November 28, 2018. [Google Scholar]
- 54. Bateman‐House A, Folers KM, Caplan A. ‘Right To Try’ won't give patients access to experimental drugs. Here's what will. Health Affairs Blog. May 3, 2017. https://www.healthaffairs.org/do/10.1377/hblog20170503.059926/full/. Accessed November 28, 2018. [Google Scholar]
- 55. Right to Try Act of 2017 , Pub L No. 115–176 (2017).
- 56. Becker MD. Right To Try Act poses big challenge for FDA. Shots: Health news from NPR. May 24, 2018. https://www.npr.org/sections/health-shots/2018/05/24/614105544/right-to-try-act-poses-big-challenge-for-fda. Accessed: November 28, 2018. [Google Scholar]
- 57. Lynch HF, Zettler PJ, Sarpatwari A. Promoting patient interests in implementing the federal Right to Try Act. JAMA. 2018;320(9):869‐870. [DOI] [PubMed] [Google Scholar]
- 58. Brennan Z. Diminishing the FDA's power was my intent: Right‐to‐Try author scolds Scott Gottlieb as agency implements new law. Endpoints News. June 1, 2018. https://endpts.com/diminishing-the-fdas-power-was-my-intent-right-to-try-author-scolds-scott-gottlieb-as-fda-implements-new-law/. Accessed November 28, 2018. [Google Scholar]
- 59. Avorn J. Powerful Medicines: The Benefits, Risks, and Costs of Prescription Drugs. New York, NY: Vintage Books; 2004. [Google Scholar]
- 60. Clarke A. Situational Analysis: Grounded Theory After The Postmodern Turn. London, UK: Sage; 2005. [Google Scholar]
- 61. US Department of Health and Human Services, Food and Drug Administration . FDA Drug Safety Communication: FDA adds warnings about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. https://www.fda.gov/Drugs/DrugSafety/ucm486096.htm. Published April 5, 2016. Accessed June 28, 2019.
- 62. Herder M. Toward a jurisprudence of drug regulation. J Law Med Ethics. 2014;July:244‐262. [DOI] [PubMed] [Google Scholar]
- 63. Herder M. Reviving the FDA's authority to publicly explain why new drug applications are approved or rejected. JAMA. 2018;178(8):1013‐1014. [DOI] [PubMed] [Google Scholar]
- 64. Vitry A, Nguyen T, Entwistle V, Roughhead E. Regulatory withdrawal of medicines marketed with uncertain benefits: the bevacizumab case study. JPharm Policy Prac. 2015;8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim J, Ross JS, Kapczynski A. Pediatric exclusivity and regulatory authority: implications of Amgen v HHS. JAMA. 2017;319(1):21‐22. [DOI] [PubMed] [Google Scholar]
- 66. US Department of Health and Human Services, Food and Drug Administration , Center for Drug Evaluation and Research, Application Number 20–839/S‐051. Medical Reviews. 2010. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/020839Orig1s051MedR.pdf Accessed December 3, 2018.
- 67. US Department of Health and Human Services, Food and Drug Administration , Center for Drug Evaluation and Research; Application Number 20–839/S‐051; Summary Review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/020839Orig1s051SumR.pdf Published May 5, 2011. Accessed December 3, 2018.
- 68. Cefalu WT, Kaul S, Gerstein HC, et al. Cardiovascular outcomes trials in type 2 diabetes: where do we go from here? Reflections from a Diabetes Care editors’ expert forum. Diabetes Care. 2018;41(1):14‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. US Department of Health and Human Services, Food and Drug Administration . Guidance for Industry: Postmarketing Studies and Clinical Trials – Implementation of Section 505(o)(3) of the Federal Food, Drug, and Cosmetic Act. Silver Spring, MD: US Food and Drug Administration; 2011. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM172001.pdf. Published April 2011. Accessed November 30, 2018. [Google Scholar]
- 70. Yale Collaboration for Research Integrity and Transparency . What's in your medicine cabinet? Ensuring the safety and efficacy of prescription drugs, biologics and medical devices in the United States. https://law.yale.edu/system/files/area/center/crit/document/crit_policy_paper_february_2018_3rd_edition.pdf. Published February 2018. Accessed December 1, 2018.
- 71. Mezher M. FDA cites Perrigo for missing postmarketing requirement deadline. Regulatory Focus. June 29, 2018. https://www.raps.org/news-and-articles/news-articles/2018/6/fda-cites-perrigo-for-missing-postmarketing-requir?feed=Regulatory-Focus. Accessed November 30, 2018. [Google Scholar]
- 72. Fain K, Daubresse M, Alexander GC. The Food and Drug Administration Amendments Act and postmarketing commitments. JAMA. 2014;310(2):202‐204. [DOI] [PubMed] [Google Scholar]
- 73. US Department of Health and Human Services, Food and Drug Administration . Proposal to Withdraw Approval for the Breast Cancer Indication for AVASTIN (Bevacizumab). Decision of the Commissioner; Docket. No. FDA‐2010‐N‐0621. Silver Spring, MD: US Food and Drug Administration; 2011. https://www.fda.gov/downloads/NewsEvents/Newsroom/UCM280546.pdf. Published November 18, 2011. Accessed November 30, 2018. [Google Scholar]
- 74. US Department of Health and Human Services, Food and Drug Administration . 21st Century Cures Act: making progress on shared goals for patients. https://blogs.fda.gov/fdavoice/index.php/2016/12/21st-century-cures-act-making-progress-on-shared-goals-for-patients/. Published December 13, 2016. Accessed November 30, 2018.
- 75. US Department of Health and Human Services, Food and Drug Administration . Fiscal Year 2013, Justification of Estimates for Appropriations Committees. Silver Spring, MD: US Food and Drug Administration; http://wayback.archive-it.org/7993/20180125110935/https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/BudgetReports/UCM291555.pdf. Accessed November 30, 2018. [Google Scholar]
- 76. US Department of Health and Human Services, Food and Drug Administration . Submission to Congress: Food & Drug Administration Work Plan and Proposed Funding Allocations of FDA Innovation Account. Silver Spring, MD: US Food and Drug Administration; 2017. https://www.fda.gov/downloads/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/21stCenturyCuresAct/UCM562852.pdf. Published June 6, 2017. Accessed November 30, 2018. [Google Scholar]
- 77. Cohen D. FDA official: “clinical trial system is broken.” BMJ. 2013;347:f7980. [DOI] [PubMed] [Google Scholar]
- 78. Husten L. Spinning RECORD: battle over Rosiglitazone heats up two weeks before crucial FDA meeting. Forbes. May 24, 2013. https://www.forbes.com/sites/larryhusten/2013/05/24/spinning-record-battle-over-rosiglitazone-heats-up-two-weeks-before-crucial-fda-meeting/#736b249a4eeb. Accessed November 30, 2018. [Google Scholar]
- 79. US Department of Health and Human Services, Food and Drug Administration . Center for Drug Evaluation and Research Key Officials. https://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ContactCDER/UCM070722.pdf. Published August 1, 2018. Accessed November 30, 2018.
- 80. Wallach JD, Ross JS, Naci H. FDA's expedited approval programs: evidentiary standards, regulatory trade‐offs, and potential improvements. Clinical Trials. 2018;15(3):243‐246. [DOI] [PubMed] [Google Scholar]
- 81. Associate attorney general announces end to use of civil enforcement authority to enforce agency guidance documents [news release] . Washington, DC: US Department of Justice Office of Public Affairs; January 25, 2018. https://www.justice.gov/opa/pr/associate-attorney-general-brand-announces-end-use-civil-enforcement-authority-enforce-agency. Accessed November 30, 2018.
- 82. Fagan A, Kaufman M. More and more, new drugs clear the FDA with ‘accelerated approval.’ UNDARK: Truth, Beauty, Science. September 10, 2018. https://undark.org/article/fda-drugs-accelerated-approval/. Accessed November 30, 2018. [Google Scholar]
- 83. US Department of Health and Human Services, Food and Drug Administration . FDA Drug Safety Communication: FDA recommends against the continued use of Meridia (sibutramine). https://www.fda.gov/Drugs/DrugSafety/ucm228746.htm. Published October 8, 2010. Accessed June 28, 2019.
- 84. Sismondo S. Key opinion leaders and the corruption of medical knowledge: what the Sunshine Act will and won't cast light on. J Law Med Ethics. 2013; 41(3):635‐643. [DOI] [PubMed] [Google Scholar]
- 85. Baker D. The benefits and savings from publicly funded clinical trials of prescription drugs. International Journal of Health Services. 2008;38(4):731‐750. [DOI] [PubMed] [Google Scholar]
- 86. Jayadev A, Stiglitz J. Two ideas to increase innovation and reduce pharmaceutical costs and prices. Health Aff(Millwood). 2009;28(1):w165‐w168. [DOI] [PubMed] [Google Scholar]
- 87. Carpenter D, Kesselheim AS, Joffe S. Reputation and precedent in the bevacizumab decision. N Engl J Med. 2011;365:e3. [DOI] [PubMed] [Google Scholar]
- 88.US Department of Health and Human Services, Office of Inspector General. FDA's Monitoring of Postmarketing Study Commitments. Washington, DC: Office of the Inspector General; 2006. https://oig.hhs.gov/oei/reports/oei-01-04-00390.pdf. Published June 2006. Accessed November 30, 2018. [Google Scholar]