

Abstract
CRISPR‐Cas9 has been shown to be a valuable tool in recent years, allowing researchers to precisely edit the genome using an RNA‐guided nuclease to initiate double‐strand breaks. Until recently, classical RAD51‐mediated homologous recombination has been a powerful tool for gene targeting in the moss Physcomitrella patens. However, CRISPR‐Cas9‐mediated genome editing in P. patens was shown to be more efficient than traditional homologous recombination (Plant Biotechnology Journal, 15, 2017, 122). CRISPR‐Cas9 provides the opportunity to efficiently edit the genome at multiple loci as well as integrate sequences at precise locations in the genome using a simple transient transformation. To fully take advantage of CRISPR‐Cas9 genome editing in P. patens, here we describe the generation and use of a flexible and modular CRISPR‐Cas9 vector system. Without the need for gene synthesis, this vector system enables editing of up to 12 loci simultaneously. Using this system, we generated multiple lines that had null alleles at four distant loci. We also found that targeting multiple sites within a single locus can produce larger deletions, but the success of this depends on individual protospacers. To take advantage of homology‐directed repair, we developed modular vectors to rapidly generate DNA donor plasmids to efficiently introduce DNA sequences encoding for fluorescent proteins at the 5′ and 3′ ends of gene coding regions. With regard to homology‐directed repair experiments, we found that if the protospacer sequence remains on the DNA donor plasmid, then Cas9 cleaves the plasmid target as well as the genomic target. This can reduce the efficiency of introducing sequences into the genome. Furthermore, to ensure the generation of a null allele near the Cas9 cleavage site, we generated a homology plasmid harboring a “stop codon cassette” with downstream near‐effortless genotyping.
Keywords: CRISPR, genome editing, homology‐directed repair, multiplexing, Physcomitrella patens, vector system
1. INTRODUCTION
Recent advances in genome editing are actively revolutionizing the fields of genetics, biotechnology, medicine, and agronomics. The creation of double‐strand breaks in DNA by site‐specific nucleases is an essential step in efficient genome editing. In the last few years, CRISPR has been employed in a wide array of organisms to create double‐strand breaks with great success, and the design is quite straightforward (Sander & Joung, 2014). Adopted from type II CRISPR systems in bacteria, introduction of the Cas9 enzyme and a programmable single‐guide RNA (sgRNA) into cells is among the most common to generate controlled DNA double‐strand breaks (Jinek et al., 2012; Makarova et al., 2011; Malzahn, Lowder, & Qi, 2017; Sander & Joung, 2014). The sgRNA contains ~20 bases homologous to a DNA target of interest at the 5’ end (known as the protospacer) and a region that binds the Cas9 nuclease (Jinek et al., 2012; Nishimasu et al., 2014). The Cas9:sgRNA complex binds and cleaves a target DNA sequence if the protospacer is directly 5′ to a protospacer adjacent motif (PAM) on the noncomplementary DNA strand (Jinek et al., 2012; Nishimasu et al., 2014).
Double‐strand breaks, which are a form of DNA damage, are repaired by one of two major pathways: nonhomologous end joining or homology‐directed repair (also referred to as homologous recombination) (Chang, Pannunzio, Adachi, & Lieber, 2017; Moynahan & Jasin, 2010; Sander & Joung, 2014). Double‐strand breaks repaired by nonhomologous end joining often result in inserted or deleted nucleotides (“indels”), especially when microhomology‐mediated end joining (also known as alternative end joining) is employed (Chang et al., 2017). The resulting indels often disrupt gene function by potentially altering the translational reading frame within a protein‐coding region. Alternatively, homology‐directed repair uses a DNA template that shares homology with both sides of the break to accurately repair the DNA (Moynahan & Jasin, 2010). By taking advantage of this pathway, it is possible to precisely alter a gene of interest by providing a DNA “donor” template containing desired modifications together with Cas9 and the sgRNA. However, the overall activity of homology‐directed repair is quite low in a variety of organisms, and thus, the majority of double‐strand breaks are repaired by nonhomologous end joining (Beucher et al., 2009; Puchta, 2005; Sargent, Brenneman, & Wilson, 1997).
In plants, CRISPR has been used to perform a variety of gene modifications, including the targeted mutagenesis of genes related to crop yield (Li et al., 2016) as well as host genes required for disease pathogenesis (Pyott, Sheehan, & Molnar, 2016; Wang et al., 2016). Successful editing events have been reported in rice (Shan et al., 2013), maize (Liang, Zhang, Chen, & Gao, 2014), and Arabidopsis thaliana (Feng et al., 2014; Li et al., 2013), as well as in polyploid crops deemed difficult for gene editing such as strawberry (Wilson, Harrison, Armitage, Simkin, & Harrison, 2019), wheat (Zhang et al., 2016), and cotton (Chen et al., 2017; Li, Unver, & Zhang, 2017), among many others (see Malzahn et al., 2017 for a review). The majority of these gene editing experiments resulted in gene knockout via nonhomologous end joining. Gene targeting using homology‐directed repair has been quite challenging in seed plants, with most attempts reporting low success rates (Butler, Baltes, Voytas, & Douches, 2016; Čermák, Baltes, Čegan, Zhang, & Voytas, 2015; Li et al., 2013; Schiml, Fauser, & Puchta, 2014; Shi et al., 2017; Svitashev, Schwartz, Lenderts, Young, & Cigan, 2016; Svitashev et al., 2015). The model moss Physcomitrella patens has been used over the last few decades to study various fundamental processes of plant biology. P. patens is known to be exceptionally amenable to genetic manipulation due to its ability to perform high rates of homologous recombination, especially when linear DNA is supplied (Kamisugi & Cuming, 2009; Prigge & Bezanilla, 2010; Schaefer & Zryd, 1997). Recently, both CRISPR‐mediated gene knockout (using nonhomologous end joining) (Lopez‐Obando et al., 2016) and gene knock‐in (using homology‐directed repair) (Collonnier et al., 2017) have high rates of success in P. patens, including the ability to target multiple genes in a single, transient transformation (i.e., “multiplexing”) (Lopez‐Obando et al., 2016).
Here, we describe an efficient and modular CRISPR vector system for use in Physcomitrella patens. In this system, protospacer sequences are synthesized as oligonucleotides and are efficiently ligated into entry vectors containing the sgRNA expression cassette, eliminating the need for gene synthesis. Using Multisite Gateway cloning (Invitrogen), multiple entry vectors recombine with a single destination vector containing Cas9 for efficient multiplexing. By co‐transforming three expression vectors with different antibiotic selection cassettes, it may be possible to target up to 12 genomic sites in a single, transient transformation. Here, we showcase the multiplexing capabilities of the system by simultaneously targeting six genes and successfully editing four in one transformation. We show that it is possible to create gene deletions using adjacent sgRNAs. In addition, we describe a simple, yet effective way to clone homology fragments flanking genes encoding fluorescent proteins to efficiently generate donor template DNA for use with homology‐directed repair. Likewise, we introduce a novel concept to use homology‐directed repair to knock‐in DNA encoding for multiple stop codons in each reading frame to allow for a controlled gene knockout with near‐effortless genotyping. Lastly, we explore the issues that arise when donor template DNA contains the protospacer sequence during homology‐directed repair experiments and describe ways to avoid these issues.
2. MATERIALS & METHODS
2.1. Protospacer sequence design and ligation
For each editing experiment, entry vectors were linearized with BsaI. The CRISPOR online software (crispor.tefor.net) (Haeussler et al., 2016) was used to design protospacers for each editing experiment using P. patens (Phytozome V11) and S. pyogenes (5′ NGG 3′) as the genome and PAM parameters, respectively. Protospacers were chosen based on high specificity scores and low off‐target frequency. The chosen protospacer for a given gene and its reverse complement were then constructed to have 4 nucleotides added to their 5′ ends such that, when annealed, they create sticky ends compatible with BsaI‐linearized entry vectors (Figure 2b). These were synthesized as oligonucleotides (Table S1) and annealed together using a PCR machine (500 pmol of each, 10 µl total volume with PCR machine setting: 98°C for 3 min, 0.1°C/s to oligo Tm, hold 10 min, 0.1°C/s to 25°C). The final product was ligated into BsaI‐linearized entry vector using Instant Sticky‐End Ligation Master Mix (New England Biolabs) following the manufacturer's recommendations.
Figure 2.
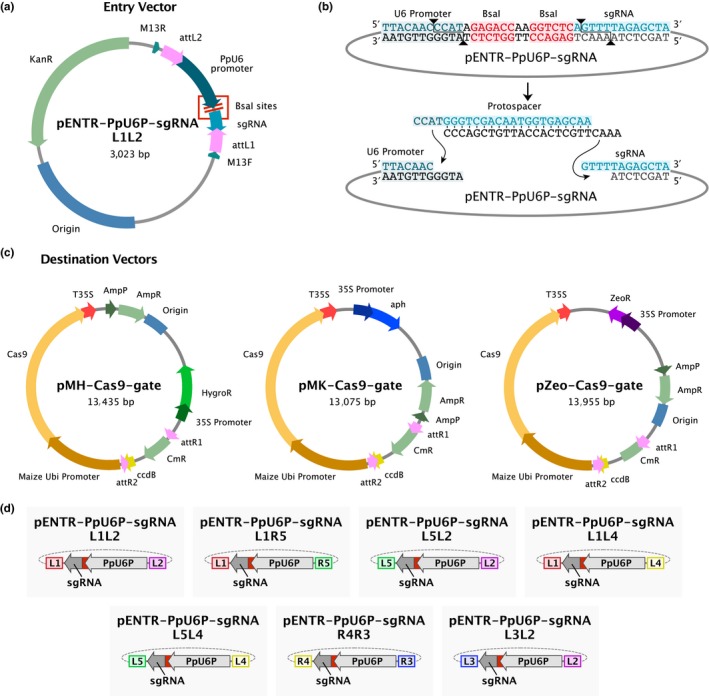
(a) A plasmid map of the entry vector containing the U6 promoter to drive expression of the sgRNA flanked by Gateway att sites. (b) The DNA sequence of the U6 promoter‐sgRNA junction within the entry clone separated by inverted BsaI sites. Upon digestion of the entry clone with BsaI, unique vector overhangs allow for directional ligation of custom oligonucleotides containing the protospacer sequence. (c) Plasmid maps of the destination vectors pMH‐Cas9‐gate, pMK‐Cas9‐gate, and pZeo‐Cas9‐gate for hygromycin, G418, and Zeocin selection in plants, respectively. (d) Entry vectors are shown that were generated based on the plasmid shown in (a) with modified att sites enabling compatibility with Multisite Gateway reactions
2.2. Polymerase III promoter assay
To build a CRISPR/Cas9 vector system for P. patens, we wanted to first assess RNA polymerase III promoter efficiency. The NLS‐4 moss line contains a transgene that codes for nuclear‐localized GFP fused to GUS (NLS‐GFP‐GUS) as described in Bezanilla, Pan, and Quatrano (2003). To target NLS‐GFP‐GUS using a rice U3 promoter, we ligated protospacer oligos into pENTR‐OsU3‐sgRNA (a gift from Devin O’Connor) to create pENTR‐OsU3‐sgRNA‐NGG. To target NLS‐GFP‐GUS using a P. patens U6 promoter, we removed the OsU3 promoter from pENTR‐OsU3‐sgRNA‐NGG using an AscI and SalI digest. We subsequently amplified the PpU6 promoter from wild‐type P. patens Gransden strain using primers with AscI and SalI sites (Table S1), digested with AscI and SalI, and ligated into linearized pENTR‐OsU3‐sgRNA‐NGG using Sticky‐End Ligation Master Mix (New England Biolabs) following the manufacturer's recommendations. These entry vectors were recombined with pH‐Ubi‐Cas9 (Miao et al., 2013) using an LR clonase reaction to create the final expression constructs, pH‐Ubi‐Cas9‐OsU3‐NGG and pH‐Ubi‐Cas9‐PpU6‐NGG.
Prior to imaging, we removed the labels from the plates such that the images were acquired by a blinded observer and drew a grid on the bottom of the plates to act as guides for counting. We counted 7‐day‐old plants and recorded the presence or absence of nuclear fluorescence for each plant using a fluorescence stereomicroscope (Leica MZ16FA), equipped with the following filter: excitation 480/40, dichroic 505 long pass, emission 510 long pass.
2.3. U6 promoter/sgRNA entry vector constructs
To generate pENTR‐PpU6‐sgRNA‐L1L2, the first Gateway entry vector for the P. patens vector system, we amplified the PpU6 and sgRNA fragments with two separate PCRs. For the PpU6 fragment, we used a forward primer containing an AscI site and a reverse primer containing two inverted BsaI sites at the 5′ ends (Table S1). Similarly, for the sgRNA fragment we used a forward primer containing two inverted BsaI sites and a reverse primer containing a SalI site (Table S1). The two fragments were then ligated using an overlap extension PCR and ligated into pGEM/T‐Easy (Promega). Positive clones were digested with AscI and SalI, and the dropout was subsequently ligated into an AscI‐ and SalI‐linearized pENTR‐PpU6‐sgRNA‐NGG plasmid.
To generate the six entry vectors compatible with Multisite Gateway (Invitrogen) for multiplexing experiments, we amplified the sgRNA expression cassette from pENTR‐PpU6‐sgRNA‐L1L2 using primers (Table S1) containing different Multisite Gateway attachment sites (attB) and subsequently recombined with the Multisite Gateway pDONR221 plasmid set (Invitrogen) using a BP clonase reaction following the manufacturer's recommendations.
2.4. Cas9/sgRNA destination and expression constructs
To generate the three destination vectors, we purified a fragment containing the Cas9 and Gateway cassette from pH‐Ubi‐Cas9 (Miao et al., 2013) digested with StuI and PmeI. This fragment was ligated into linearized pMH, pMK, and pZeo vectors by blunt‐end ligation to create pMH‐Cas9‐gate, pMK‐Cas9‐gate, and pZeo‐Cas9‐gate. Sequences are available on AddGene (https://www.addgene.org/kits/bezanilla-crispr-physcomitrella/). All of the Cas9/sgRNA expression vectors used in this study were generated using Gateway (for one sgRNA) or Multisite Gateway (for multiple sgRNAs) to recombine the entry vectors and destination vectors just described (Invitrogen).
2.5. Homology‐directed repair constructs
To generate pENTR‐R4R3‐stop (the “stop cassette”), we amplified 360 bp of the plasmid pBluescriptSK(+), including the multiple cloning site, with attB4r and attB3r Gateway primers (Table S1). The forward primer also contained three stop codons in each frame. We subsequently cloned the PCR fragment into pDONR221‐P4rP3r (Invitrogen) using a BP clonase reaction following the manufacturer's recommendations.
To generate the mEGFP and mRuby2 tagging entry vectors, we amplified mEGFP (Vidali et al., 2009) and mRuby2 (Lam et al., 2012) coding sequences using forward and reverse primers (Table S1) that contained attB4r and attB3r sites, respectively. The forward primers (Table S1) for both mEGFP and mRuby2 also contained a BamHI site. These PCR products were subsequently cloned into pDONR221‐P4rP3r (Invitrogen) using a BP clonase reaction to create pENTR‐R4R3‐mEGFP‐C and pENTR‐R4R3‐mRuby‐C. mEGFP‐pGEM, a vector described by Vidali et al. (2009), contains BamHI and BglII sites flanking the mEGFP coding sequence. We generated mRuby2‐pGEM, a vector constructed in the same way as mEGFP‐pGEM (Vidali et al., 2009). We digested these vectors with BamHI and BglII and the resulting fragments were ligated into BamHI‐digested pENTR‐R4R3‐mEGFP‐C and pENTR‐R4R3‐mRuby‐C to create pENTR‐R4R3‐2XmEGFP‐C and pENTR‐R4R3‐2XmRuby‐C, respectively. We linearized the resulting 2X constructs with BamHI and ligated the BamHI/BglII fragments from mEGFP‐pGEM and mRuby2‐pGEM to create the 3X constructs, pENTR‐R4R3‐3XmEGFP‐C and pENTR‐R4R3‐3XmRuby‐C, respectively. To create the mEGFP and mRuby2 N‐terminal constructs, the process above was repeated except the attB3r primers (Table S1) did not contain stop codons.
2.6. DNA donor templates
We used the three‐fragment Multisite Gateway cloning system (Invitrogen) to generate the final homology‐directed repair constructs. For Pp3c22_1100, we amplified 2 fragments of approximately 800 bp upstream and downstream of the Pp3c22_1100 stop codon. For Pp3c16_8300, we amplified 2 fragments of approximately 800 bp upstream and downstream of the expected start codon. For both genes, we cloned the upstream fragments into pDONR221‐P1P4 and the downstream fragments into pDONR221‐P3P2 using a BP clonase reaction. To create the final homology‐directed repair DNA donor plasmids, the resulting pENTR vectors from the BP reaction underwent an LR clonase reaction with the second‐position tagging vector (pENTR‐R4R3‐mEGFP‐C for Pp3c22_1100 and pENTR‐R4R3‐mRuby‐N for Pp3c16_8300) and the destination vector, pGEM‐gate (Vidali et al., 2009).
To restore efficient homology‐directed repair while tagging Pp3c16_8300, we performed site‐directed mutagenesis on the entry vector containing the 3′ homology fragment (pENTR‐L3L2‐3c16‐3Arm) to create pENTR‐L3L2‐3c16‐3Arm‐mut by altering the third nucleotide from the PAM (5’ NGG 3’) within the protospacer. We repeated the LR reaction using this third‐position entry vector to generate pGEM‐3c16‐mRuby‐HDR‐mut.
2.7. Moss tissue culture and transformation
We propagated moss tissue weekly by light homogenization and subsequently plated on 10‐cm petri dishes to maintain the protonemal growth stage. Dishes contained 25 ml PpNH4 growth medium (103 mM MgSO4, 1.86 mM KH2PO4, 3.3 mM Ca(NO3)2, 2.72 mM (NH4)2‐tartrate, 45 µM FeSO4, 9.93 µM H3BO3, 220 nM CuSO4, 1.966 µM MnCl2, 231 nM CoCl2, 191 nM ZnSO4, 169 nM KI, and 103 nM Na2MoO4) with 0.7% agar covered with cellophane disks. Plants were grown in daily cycles of 16‐hr light/8‐hr dark with 85 µmol photons m−2 s−1. For transformation, protoplasts were transformed with 30 µg of each DNA construct using PEG‐mediated transformation protocol (as described in Augustine, Pattavina, Tuzel, Vidali, and Bezanilla (2011)). Plants were allowed to regenerate on plant regeneration media (PRMB) for 4 days (described in (Wu & Bezanilla, 2014)) atop of cellophane disks. Depending upon the selection cassette present on the expression vector, we subsequently moved plants to PpNH4 growth media containing either hygromycin (15 µg/ml), G418 (20 µg/ml), or Zeocin (50 µg/ml). Plants were not selected for homology‐directed repair DNA donor vectors. After 7 days on selection, we moved plants to PpNH4 media without antibiotics for maximal growth, except for plants that were transformed to compare the efficiency of the rice U3 and moss U6 promoters—those plants remained on selection and were imaged after 2 weeks. All other plants were allowed to grow for 2–3 weeks until tufts were 0.5–1 cm in diameter for DNA extraction.
2.8. DNA extraction and genotyping
We extracted DNA from plants that were 3–4 weeks old (0.5–1 cm in diameter) using the protocol as described in Augustine et al. (2011). For editing experiments, we used PCR primers (Table S1) surrounding the expected Cas9 cleavage site (~300–400 bp on each side). For homology‐directed repair experiments, we used PCR primers outside of the homology region to avoid amplification of residual DNA donor template. To perform PCR, we used Q5 polymerase (New England Biolabs) using the manufacturer's recommendations.
2.9. T7 endonuclease assay
To detect CRISPR edits, we amplified a 0.5 to 1 kb genomic region flanking the potential CRISPR editing site by PCR. The PCR product from each candidate plant was mixed with a roughly equivalent amount of wild‐type PCR product of the same locus. The mixture was denatured and annealed in a PCR machine and subsequently digested with 1 µl of T7 endonuclease (New England Biolabs) following the manufacturer's recommendations. We examined the digest on a 1% agarose gel.
2.10. Laser scanning confocal microscopy
For confocal imaging, moss protonemal tissue was grown in microfluidic imaging device as described in Bascom, Wu, Nelson, Oakey, and Bezanilla (2016). The imaging device is filled with half‐strength Hoagland's medium (4 mM KNO3, 2 mM KH2PO4, 1 mM Ca(NO3)2, 89 μM Fe citrate, 300 μM MgSO4, 9.93 μM H3BO3, 220 nM CuSO4, 1.966 μM MnCl2, 231 nM CoCl2, 191 nM ZnSO4, 169 nM KI, 103 nM Na2MoO4) and kept at room temperature on the benchtop with overhead fluorescent lights. Images were acquired on a Nikon A1R confocal microscope system with a 1.49 NA 60x oil immersion objective (Nikon Apo TIRF 60x Oil DIC N2) at room temperature. 488 nm laser illumination was used for mEGFP excitation. Emission filter was 525/50 nm for mEGFP. Image acquisition was controlled by NIS‐Elements software (Nikon). Images were processed using NIS‐Elements software (Nikon): advanced denoising with regression and other parameters set to default.
3. RESULTS
3.1. Optimal expression of the sgRNA results in high‐efficiency editing
The P. patens U6 promoter has recently been shown to be successful in driving guide RNA expression in P. patens (Collonnier et al., 2017; Lopez‐Obando et al., 2016). To test whether RNA polymerase III promoters from other organisms could be used in moss, we compared the efficiency of genome editing using either a rice U3 or the P. patens U6 promoter to drive expression of the guide RNA. As a rapid visual test for genome editing, we designed a protospacer that targets the 5′ end of the coding sequence of green fluorescent protein (GFP) (Figure 1a). The final Cas9 expression plasmids harboring either the PpU6 or the OsU3 promoter were then transformed separately into NLS‐4, a moss line that stably expresses nuclear‐localized GFP fused to β‐glucuronidase (GUS) (Bezanilla et al., 2003). Thus, plants lacking nuclear green fluorescence after transformation represent genome editing events leading to a loss‐of‐function mutation in the GFP:GUS fusion protein.
Figure 1.
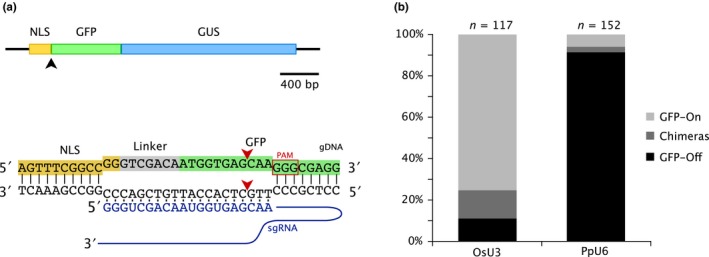
(a) A gene model of the NLS‐GFP‐GUS reporter. The black arrowhead indicates the expected Cas9 cleavage site. Below the gene model, the DNA sequence at the junction of the NLS and GFP gene fragments is shown with the sgRNA binding site. Red arrowheads indicate the expected cleavage site by Cas9 at the 5′ end of the GFP coding region. The PAM sequence is indicated with a red box. (b) A stacked bar graph representing the efficiency of editing the NLS‐GFP‐GUS gene using either OsU3 or the PpU6 promoters to drive expression of the sgRNA. Plants expressing the reporter (GFP‐on) are most likely not edited. Plants lacking NLS‐GFP‐GUS expression (GFP‐off) are edited. Chimeras indicate plants with GFP‐GUS expression in only a portion of the plant
After two weeks on selection, we imaged transformed plants using fluorescence microscopy to visualize the presence (or absence) of nuclear fluorescence. We found that plants transformed with the OsU3::sgRNA resulted in 11.1% of plants lacking GFP signal. In comparison, 91.4% of plants transformed with the PpU6::sgRNA lacked GFP signal (Figure 1b). We verified that NLS‐GFP‐GUS was edited in a subset of plants lacking GFP signal via Sanger sequencing (n = 10, OsU3::sgRNA transformants, Figure S1). Additionally, we observed that 13.7% of plants transformed with OsU3::sgRNA contained nuclear fluorescence in a portion of the plant, giving rise to a population of chimeric plants. Interestingly, of the plants transformed with PpU6::sgRNA, we only observed chimeras in 2.6% of the plants (Figure 1b). We reasoned that chimeras arise as a result of the Cas9 nuclease cleaving the NLS‐GFP‐GUS reporter after the initial cell division of the transformed protoplast. Given that the OsU3 promoter resulted in fewer plants lacking GFP and a larger percentage of chimeric plants as compared to the PpU6 promoter, our results suggest that the PpU6 promoter is more efficient at expressing the sgRNA in moss protoplasts.
3.2. Flexible vector system enables simultaneous targeting of multiple genomic sites
Here, we present the development of a vector system that rapidly and flexibly allows for simultaneous targeting of multiple genomic sites. Our system builds on vectors developed for rice (Miao et al., 2013) with modifications and enhancements to increase expression and transformation efficiency in P. patens. In the moss vector system, the sgRNA expression cassette resides in a Gateway entry vector (Invitrogen) and consists of the PpU6 promoter followed by DNA encoding the sgRNA (Figure 2a). The protospacer sequence is easily modified to target a specific genomic locus. Two reverse‐complementary oligonucleotides containing a custom protospacer sequence are annealed together and directionally ligated into an entry vector that has been linearized by two BsaI sites (Figure 2b). Using site‐specific recombination, the ligated entry vector recombines with a destination vector containing the Cas9 expression cassette in which Cas9 expression is driven by the maize ubiquitin promoter to create a final expression vector containing both components. We chose to use the maize ubiquitin promoter as this is a well‐documented promoter for high‐level expression in moss (Bezanilla et al., 2003; Saidi et al., 2005).
For maximum flexibility, we generated three destination vectors comprising different antibiotic resistance genes for selection in plants (Figure 2c). To target multiple genomic sites in one transformation, we took advantage of Multisite Gateway (Invitrogen), which enables directional stitching of up to four DNA fragments. We generated Multisite Gateway entry vectors enabling construction of a single expression vector that expresses Cas9 and up to four unique sgRNAs simultaneously (Figure 2d). Taking advantage of co‐transformation and simultaneous selection with hygromycin, G418, and Zeocin this vector system could target up to 12 different genomic sites.
3.3. Targeting multiple, distant genomic sites
To test successful targeting of multiple genomic sites in one transformation, we designed protospacer oligos to target six genomic sites (site 1: Pp3c8_18830V3.1; site 2: Pp3c18_4770V3; site 3: Pp3c22_15110V3; site 4: Pp3c4_16430V3; site 5: Pp3c8_18850V3; site 6: Pp3c23_15670V3). Of these sites, two of them (site #1 and site #5) have targeting sites 8,727 bp apart on the same chromosome and the remaining four have targeting sites on different chromosomes. We generated two expression vectors, each harboring three sgRNA expression cassettes (Figure 3a). Protoplasts were subsequently co‐transformed and selected for both expression constructs. We subsequently genotyped the remaining plants using T7 endonuclease, an enzyme that recognizes and cleaves regions of mismatching bases present in dsDNA. Thus, indel mutations can be easily detected when the DNA to be tested is annealed to wild‐type DNA. To start, we screened 24 plants at site #1 and site #4. In these 24 plants, we were unable to obtain T7 cleavage at site #1. However, 15 plants resulted in T7 cleavage at site #4. We sequenced these plants and confirmed the presence of indel mutations in all 15 plants, 6 plants of which contained frameshift mutations (Figure 3b). We subsequently screened the remaining sites (sites #2, #3, #5, and #6) in these 6 plants and found edits at sites #3, #5, and #6, but not at site #2 (Figure 3b; Figure S2). Taken together, these results provide evidence that sgRNA expression from two separate vectors enables targeting of multiple genes in a single transformation event.
Figure 3.
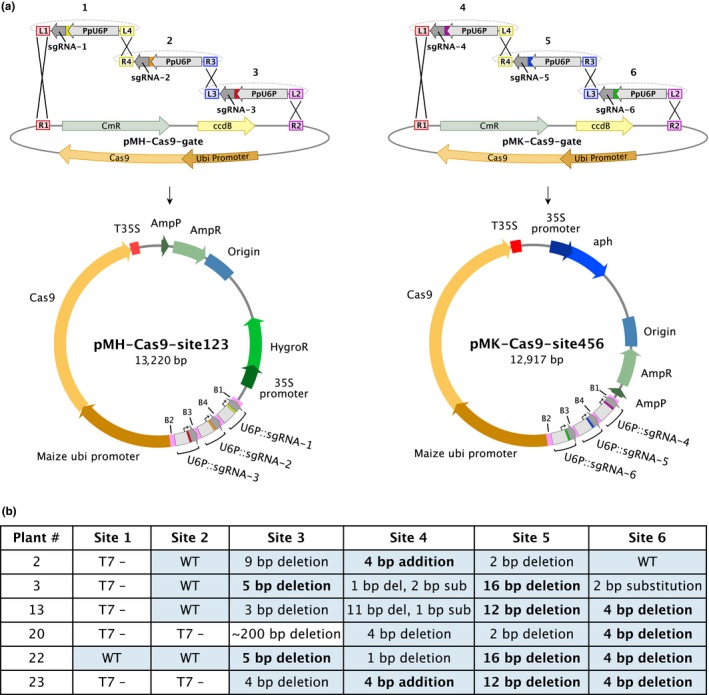
(a) A schematic showing the directional Multisite Gateway LR recombination reactions between entry clones containing the sgRNA expression cassette and each destination vector to create two final expression constructs (final plasmid maps are drawn to scale). (b) A table showing the genotyping results of 6 plants regenerated from protoplasts that were transformed with both plasmids simultaneously. Plants were initially screened by a T7 endonuclease assay. “T7‐” indicates that T7 endonuclease was unable to recognize a mismatch when transformant gDNA was paired with WT gDNA. Blue‐shaded cells indicate sequencing was performed on that site. Bolded sequencing results with identical values in the same column indicate identical edits at that site. For sequencing data, see Figure S2. The site #3 amplicon in plant 20 is ~200 bp smaller than the wild‐type amplicon
3.4. Targeting adjacent genomic sites can result in large deletions
The ability to target a region with multiple sgRNAs has been shown to be beneficial in creating knockout mutations and large deletions in other plant systems, including soybean (Cai et al., 2018), rice (Srivastava, Underwood, & Zhao, 2017; Wang et al., 2017; Zhou, Liu, Weeks, Spalding, & Yang, 2014), tomato (Brooks, Nekrasov, Lippman, & Eck, 2014), Arabidopsis thaliana (Gao, Chen, Dai, Zhang, & Zhao, 2016; Ordon et al., 2017), and Nicotiana benthamiana (Ordon et al., 2017), as well as in human cells (He et al., 2015), zebrafish (Xiao et al., 2013), and yeast (Hao et al., 2016). Protospacer sequences have variable probabilities of producing out‐of‐frame mutations, depending on the surrounding microhomology in the genomic DNA available for microhomology‐mediated end‐joining repair (Bae, Kweon, Kim, & Kim, 2014). Additionally, an indel that would cause a frameshift mutation in a protein‐coding gene may not disrupt the function of noncoding DNA. To increase the chances of making knockout mutations and easily visualizing them on a gel, it may be beneficial to excise a region of the gene using two sgRNAs. To test this, we designed two protospacer oligos (sgRNA‐7 and sgRNA‐8) 196 bp apart to simultaneously target a small region in the gene Pp3c16_8300 (Figure 4a). Genotyping revealed that two plants contained apparent deletions larger than 100 bp (n = 31) (Figure 4a, gel). To investigate further, we sequenced these plants (plants #4 and #9) as well as three other plants exhibiting a similar band size to wild type (plants #5, #6, and #7) (Figure 4a). Interestingly, of the plants with visible deletions, plant #4 was solely targeted by sgRNA‐8 and resulted in a 101 bp deletion (Figure 4a, gene models). The other deletion mutant, plant #9, was targeted by both sgRNA‐7 and sgRNA‐8 and resulted in two separate deletions of 152 bp and 10 bp, respectively. In this case, the cleavage of both target sites did not result in complete excision. Similarly, plant #5 was also targeted by both sgRNAs, though it only resulted in a total deletion of 8 bp. Plant #6 was only targeted by sgRNA‐8, and plant #7 was not edited (Figure 4a, gene models).
Figure 4.
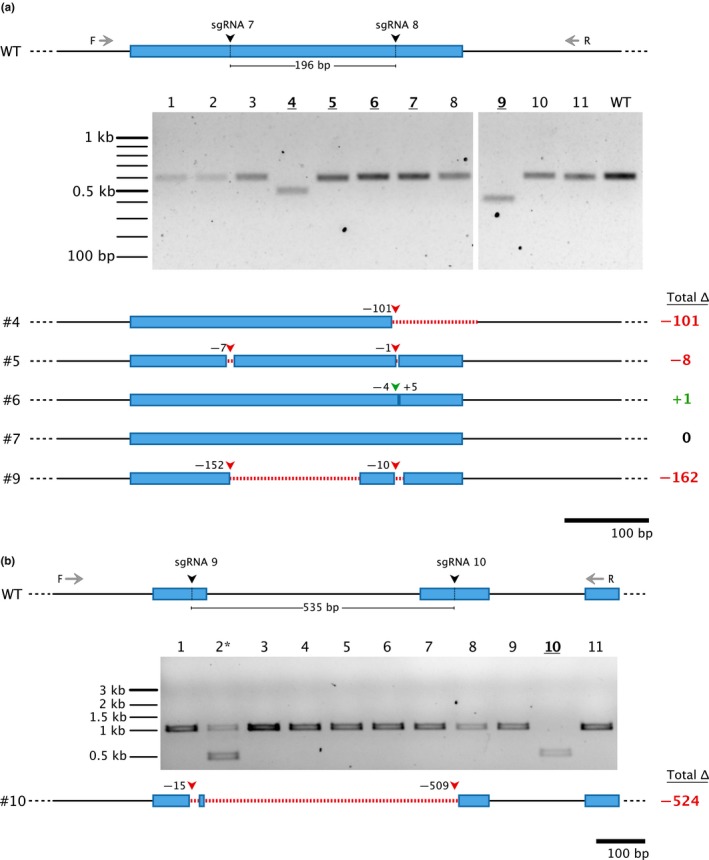
Testing genome editing efficiency in moss using two sgRNAs at adjacent sites within the same locus. The gene model of wild‐type (WT) and the expected sgRNA cleavage sites (196 bp apart for (a) and 535 bp apart for (b)) is displayed. Forward (F) and reverse (R) primers for genotyping are shown, with the genotyping PCR results displayed on the gel. Bolded/underlined numbers represent plants that were sequenced at the target locus. Gene models of the sequencing results are displayed below the gel with red dashed lines representing deleted regions. Red arrowheads indicate net deletion, and green arrowheads represent net addition of bases at that particular sgRNA site, with the corresponding number of bases deleted or added next to each arrowhead. The total net change in base pair length for each plant is indicated to the right. The asterisk in (b) represents a chimeric plant in which the editing is suspected to have occurred after the first cell division
We performed a similar experiment on a different gene (Pp2c9_8040) in which we designed two protospacer oligos with expected cleavage sites 535 bp apart (Figure 4b). Genotyping revealed two deletion mutants (plants #2 and #10), one of which is likely a chimeric plant due to the presence of an additional, wild‐type‐sized fragment (plant #2) (Figure 4b, gel). Sequencing of plant #10 revealed cleavage at both sgRNA sites: a 15 bp deletion at sgRNA‐9 and a 509 bp deletion at sgRNA‐10. Interestingly, there remained 15 bp of wild‐type sequence between the two deletions, indicating that the two sgRNAs failed to excise the fragment entirely and that the targeted sites repaired separately (Figure 4b, gene model). Thus, uncovering large deletions may not necessarily occur at a high frequency when simultaneously targeting adjacent sites.
3.5. Generating vectors for homology‐directed repair
Homology‐directed repair is an endogenous pathway that repairs DNA double‐strand breaks using an available DNA template that shares regions of homology on both sides of the break site (Moynahan & Jasin, 2010). Using CRISPR/Cas9, homology‐directed repair can be exploited by supplying a DNA donor template together with the Cas9 enzyme and the sgRNA to repair the targeted region with extreme accuracy. To test for Cas9‐induced homology‐directed repair in moss, we designed a strategy to insert sequences encoding for mEGFP (Vidali et al., 2009) at the 3′ end of Pp3c22_1100 (Figure 5a). We identified a protospacer sequence that spanned the junction between the coding region and the 3′ UTR of Pp3c22_1100, which was an ideal site to target cleavage by Cas9. We generated entry clones containing homology fragments upstream and downstream of the desired insertion site (Figure 5b) and subsequently recombined them to generate the DNA donor vector containing the 5′ homology, mEGFP, and 3′ homology fragments (Figure 5c). We co‐transformed the Cas9/sgRNA co‐expression vector and the DNA donor vector into protoplasts. Genotyping revealed that 6 plants (28.6%) contained fragments of similar size expected for mEGFP insertion (n = 21, Figure 5d). Sequencing of one plant revealed seamless insertion of an in‐frame mEGFP flanked by the expected attB4 and attB3 sites. Imaging revealed accumulation of GFP fluorescence along the plasma membrane (Figure S3). Due to the high rate of successful insertions, we constructed several second‐fragment entry vectors for use with three‐fragment Multisite Gateway recombination for fluorescent protein gene tagging experiments (Figure 5e). These include genes encoding mEGFP (1X, 2X, and 3X) and mRuby2 (1X, 2X, and 3X) with or without DNA encoding for stop codons for C‐ and N‐terminal protein tagging, respectively.
Figure 5.
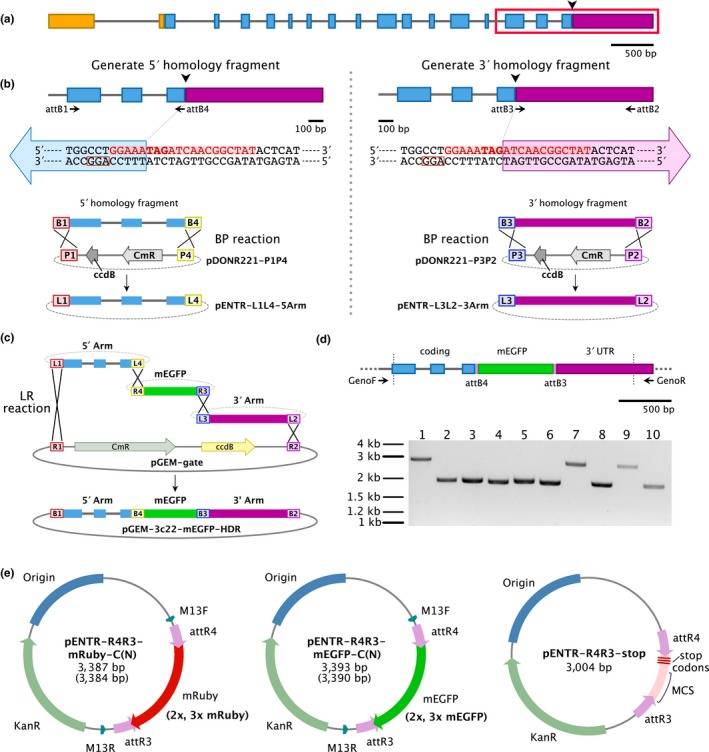
Cloning strategy for insertion of sequences encoding for mEGFP at the 3′ end of the Pp3c22_1100 gene. (a) The gene model representing the Pp3c22_1100 gene. The arrowhead represents the desired mEGFP insertion site. Exons are represented with colored boxes, and introns are represented as gray lines. The 5′ UTR, the coding region, and the 3′ UTR are represented by orange, blue, and purple, respectively. The red outlines the section of the gene model shown in (b). (b) Generation of the 5′ and 3′ homology fragments using PCR and primers with attB overhangs and subsequent BP reaction into first‐ and third‐element pDONR vectors, respectively. The DNA sequence of the junction between the coding region and the 3′ UTR is shown, with bolded “TAG” representing the stop codon, red sequence representing the protospacer binding site, and the red box representing the PAM. The large blue and purple arrows represent the 5′ and 3′ homology fragments, respectively. (c) A schematic showing the entry vectors created in (b) undergoing a Gateway Multisite LR reaction (Invitrogen) with a second‐element mEGFP entry vector and a destination vector to make the final DNA homology donor plasmid. (d) A gene model representing the genotyping strategy with forward and reverse primers. The region between the dashed, vertical lines represents the region present in the homology donor vector. The genotyping results are displayed on the gel. Larger bands show insertion of mEGFP into the Pp3c22_1100 locus. (e) Second‐element entry vectors constructed to facilitate insertion of sequences encoding fluorescent proteins and the stop cassette using homology‐directed repair
We reasoned that homology‐directed repair would also provide an ideal method to generate reliable and clean knockout alleles by, for example, inserting a fragment with an in‐frame stop codon. To do this, we constructed a plasmid encoding three stop codons in each reading frame fused to a 200 bp multiple cloning site (pENTR‐R4R3‐stop, Figure 5e). Insertion of this “stop cassette” allows for easy identification of knockout mutants. Additionally, the presence of a multiple cloning site following the stop codons allows the use of restriction enzymes during genotyping to further validate the insertion of the stop cassette.
3.6. Cas9 cleaves genomic DNA in the presence of competing plasmid DNA in moss cells
To perform precise genome editing using CRISPR‐induced homology‐directed repair, it may not always be possible to identify a protospacer sequence with high specificity near the desired editing site. Preserving the genomic sequence between the protospacer target site and the desired editing site necessitates the presence of the protospacer sequence within the DNA donor template, which is problematic. First, we reasoned that saturating amounts of DNA donor template containing the protospacer sequence might titrate away Cas9 from cleaving the genomic site. And second, the Cas9 enzyme could potentially cleave the plasmid DNA, rendering the DNA donor template inoperable during repair.
To test whether cleavage of a genomic site is possible in the presence of a DNA donor template that contains the protospacer sequence, we designed the following experiment. We generated a vector (pMK‐Cas9‐Hyg) expressing Cas9 and a sgRNA designed to target the hygromycin resistance gene. Transformation of pMK‐Cas9‐Hyg alone into a moss line containing a single copy of the hygromycin resistance gene should result in genome editing that renders the plant hygromycin sensitive. However, if a donor DNA template harboring the hygromycin resistance gene is co‐transformed with pMK‐Cas9‐Hyg, then we would expect this template to protect the genomic site from being edited, resulting in a larger percentage of hygromycin‐resistant plants. Surprisingly, we did not observe significant differences between the number of plants sensitive to hygromycin with (58.5%) or without (52.4%) donor template co‐transformation (n = 82 plants, Figure 6). These data indicate that the presence of a plasmid containing a protospacer binding site does not inhibit Cas9 from cleaving genomic DNA in moss cells.
Figure 6.
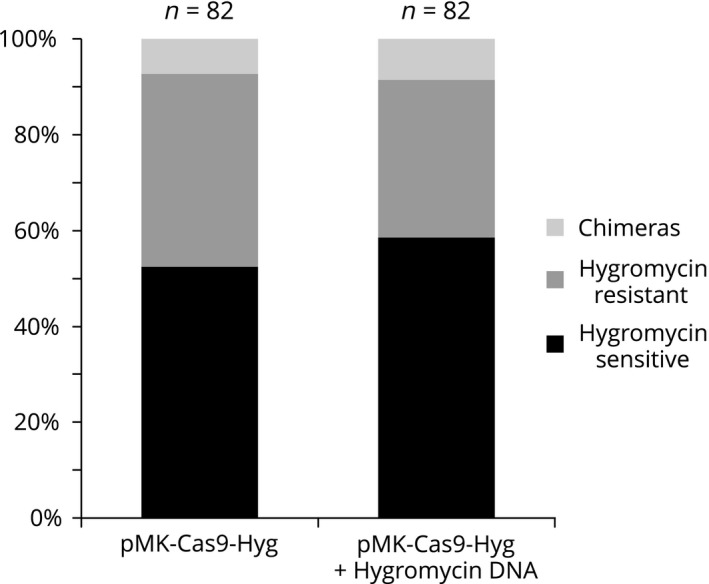
A stacked bar graph representing the amount of plants exhibiting different sensitivity to hygromycin after targeting the stable hygromycin cassette with Cas9 and with (right) or without (left) co‐transformation of a plasmid containing the hygromycin sgRNA targeting sequence
3.7. Cas9 cleaves plasmid DNA in moss cells
Given that genomic sites were still accessible to Cas9 cleavage even in the presence of donor template DNA harboring the same target site, we wondered if Cas9 could cleave plasmid DNA in moss cells. To test this, we compared the viability of plants co‐transformed with a hygromycin‐resistant plasmid (pGL2 as described in Bilang, Iida, Peterhans, Potrykus, and Paszkowski (1991) and either a Cas9 plasmid targeting the hygromycin resistance gene (pMK‐Cas9‐Hyg) or a Cas9 plasmid targeting a nonexistent site (pMK‐Cas9‐NGG). As expected, selection for the Cas9 plasmid resulted in similar numbers of surviving plants (733, hygromycin target; 833, nonexistent site). However, selection for the hygromycin‐resistant plasmid yielded strikingly different numbers (9.5, hygromycin target; 157, nonexistent site). These results show that the majority of the hygromycin‐resistant plasmid is cleaved by Cas9, indicating that Cas9 is able to cleave plasmid DNA that contains a protospacer sequence.
3.8. Mutagenesis of the DNA donor template containing a protospacer sequence increases the efficiency of Cas9‐induced homology‐directed repair
CRISPR‐mediated homology‐directed insertion of the mEGFP gene at the 3’ end of the Pp3c22_1100 coding region was highly successful (Figure 5). In this experiment, the protospacer sequence was at an ideal position as it extended across the end of the coding sequence to the beginning of the 3′ untranslated region and included the DNA encoding the stop codon (Figure 5b). In this instance, the protospacer sequence was not present on the DNA donor template. Rather, portions of the sequence were shared among the two homology fragments separated by the mEGFP coding sequence. In cases where protospacer design is suboptimal and the protospacer sequence resides on the donor DNA template, we reasoned that mutagenesis of either the protospacer sequence or the PAM in the DNA donor template should help to achieve accurate homology‐directed repair. To test this, we designed a strategy to insert a gene encoding mRuby2 between the 5′ untranslated region and the beginning of the coding region of Pp3c16_8300. In this case, the protospacer targeting sequence resides within the coding region and the DNA donor plasmid, therefore, contains the protospacer sequence within the 3′ homology fragment (Figure 7a). We altered the DNA donor template to create a silent mutation within the protospacer sequence 3 bp from the PAM (5′ NGG 3′) (Figure 7b). We co‐transformed protoplasts with the Cas9/sgRNA co‐expression plasmid and either the mutagenized or unmutagenized DNA donor template. As expected, we were unable to detect any homology‐directed repair events in plants transformed with the DNA donor template containing the protospacer sequence (n = 23 plants) (Figure 7a, gel). Conversely, homology‐directed repair events were readily detected in plants transformed with mutagenized DNA donor templates: 6 out of 17 plants contained insertions with the expected size (Figure 7b). We sequenced PCR products from two of these plants and verified successful homology‐directed insertions. These data demonstrate that this DNA donor template containing the protospacer sequence resulted in inefficient homology‐directed repair.
Figure 7.
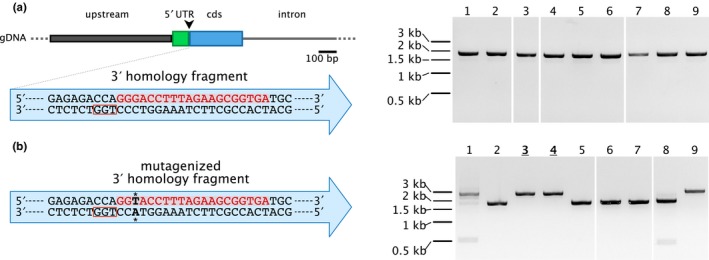
Homology‐directed repair with a DNA donor homology plasmid that contains a protospacer targeting sequence. (a) A partial gene model representing the 5′ region of Pp3c16_8300. The black arrowhead represents the desired mRuby2 insertion site. The DNA sequence of the 3′ homology fragment is shown within the blue arrow, with the protospacer binding sequence shown in red and the PAM shown in a red box. Plants co‐transformed with the DNA donor template and the Cas9 plasmid were unable to integrate the mRuby2 sequence into the locus (right). (b) The DNA sequence of the 3′ homology fragment after introduction of a silent mutation within the protospacer binding sequence (bold, asterisks). Mutagenesis allows mRuby2 integration upon co‐transformation with the Cas9 plasmid (right). Sequenced plants are indicated with bold and underlined numbers
4. DISCUSSION
The ability to precisely edit the genome has been shown to be extremely reliable and rapid using CRISPR/Cas9 in a variety of organisms (Malzahn et al., 2017; Sander & Joung, 2014). Based on modifications of the vectors described by Miao et al. (2013), we have described a CRISPR/Cas9 vector system to enhance CRISPR editing in Physcomitrella patens with methods that can be easily translated for use in other species. Rapid and efficient ligation of a pre‐assembled, double‐stranded oligo containing a custom protospacer sequence into an entry vector eliminates the need for gene synthesis. The final expression vector contains the sgRNA and the Cas9 expression cassettes. This results in equal stoichiometric amounts of DNA sequences coding for each component within each protoplast, which may be beneficial when targeting multiple genomic sites. We have also demonstrated that the native P. patens U6 promoter is highly efficient at expressing the sgRNA in comparison with the rice U3 promoter and therefore have included it in our vector system. Additionally, we have described an approach to rapidly generate DNA donor templates for homology‐directed repair using a three‐fragment Multisite Gateway reaction (Invitrogen).
As expansion of gene families is common in Physcomitrella patens (Lang et al., 2018; Zimmer et al., 2013), multiple family members can be targeted in a single transformation, eliminating repetitive and prolonged procedures. We have demonstrated the ability of our vector system to perform multiplex editing by targeting six different genomic sites in a single transformation using two expression vectors. In this test case, four of the six genomic sites were successfully edited. The absence of edits in two of the sites could be explained by differences in protospacer efficiency.
Multiplexing is not solely limited to different genes, as multiple sgRNAs can be used to target a single region. This has been shown to be successful in creating gene deletions as well as large‐scale chromosomal deletions (Cai et al., 2018; Hao et al., 2016; He et al., 2015; Mali et al., 2013; Xiao et al., 2013). In our case, we tested the ability to create easy‐to‐detect knockout mutations using two sgRNAs separated by ~200 and ~500 base pairs in protein‐coding genes. Indeed, visible deletions were detected in both cases, but at relatively low frequencies. Additionally, we were unable to obtain complete removal of the intervening region which may result from differences in protospacer efficiency. It is possible that one Cas9:sgRNA complex cleaved one site more than the other site, and/or that one site repaired before the other site was cleaved.
Induction of homology‐directed repair by Cas9‐mediated double‐strand breaks has been shown to be highly successful in P. patens (Collonnier et al., 2017). Traditionally, homology‐directed repair has been used in P. patens for decades in which a linear template containing regions of homology flanking a selectable marker is transformed into protoplasts and is integrated into the genome (Kamisugi & Cuming, 2009; Prigge & Bezanilla, 2010). In this case, stable integration of a selectable marker is used to isolate recombinant plants. However, integration of a selectable marker can limit genetic manipulation: N‐terminal protein tagging at an endogenous gene locus is challenging due to the placement of the selection cassette, and in other instances, loxP “scar” sequences remain after successful removal of the selection cassette upon expression of the Cre recombinase (Sander & Joung, 2014). In the latter case, it should be noted that removal of the selection cassette requires transient expression of Cre and therefore takes substantially longer to obtain the desired modifications. CRISPR‐induced homology‐directed repair is a way to combat these issues in which the selection cassette is transiently expressed from the Cas9‐sgRNA expression vector. Thus, there is no need to remove it from the genome, opening the ability make extremely precise alterations to the genome.
CRISPR‐induced homology‐directed repair in plants has been successful using ssDNA oligos (Shan et al., 2013; Svitashev et al., 2016, 2015), linear dsDNA (Schiml et al., 2014; Sun et al., 2016), and circular (Butler et al., 2016; Čermák et al., 2015; Gil‐Humanes et al., 2017; Li et al., 2013; Svitashev et al., 2015) DNA donor templates. Here, we demonstrated the ability to perform highly efficient gene tagging of Pp3c22_1100 using a circular donor template constructed with Multisite Gateway (Invitrogen). Due to the high rate of success, we constructed a variety of second‐fragment entry vectors compatible with three‐fragment Multisite Gateway recombination (Invitrogen) for rapid cloning of fluorescent protein‐tagging constructs (Figure 5e). These flexible, modular vectors provide a streamlined system to rapidly generate DNA donor plasmids that will enable tagging the same gene with single, double, triple, red, or green fluorescent proteins.
Homology‐directed repair experiments are not solely limited to gene knock‐in, however. Because moss plants can be propagated asexually, it is possible to use homology‐directed repair to rapidly generate the same allele in different genetic backgrounds without the need to perform genetic crosses. Additionally, homology‐directed repair can be used to create clean and easy‐to‐detect knockout mutants: we constructed a “stop cassette” entry vector containing a multiple cloning site with stop codons in each reading frame. In this way, knockout mutants can be readily detected by a simple shift in band size. Generating knockout mutants solely with Cas9 and sgRNA is feasible, but may be less efficient due to the possibility that the surrounding microhomology could favor in‐frame repair (Bae et al., 2014; Chang et al., 2017). Nevertheless, this latter approach may yield more variability in the different mutations created which could be beneficial in determining gene function. Thus, having all of these options available when performing CRISPR‐mediated knockout or knock‐in experiments gives us the ability to have tight control over the desired results.
A careful choice of protospacer is essential for successful homology‐directed repair. Insertion of the mEGFP coding sequence between the Pp3c22_1100 coding and 3′ UTR regions did not contain the protospacer sequence on the DNA donor template. In this scenario, the protospacer was divided into two portions on the DNA donor template separated by the mEGFP coding sequence. In cases where this particular design is not possible, we performed a series of experiments to test whether Cas9 cleaves in cases where the protospacer sequence remains on the donor template. We validated that Cas9 cleaves plasmid DNA that contains a protospacer sequence in moss cells, rendering the DNA donor template useless during repair. We also demonstrated that, while large amounts of DNA that contain the protospacer sequence are present within the cell, Cas9 retains the ability to cleave genomic DNA in moss cells. We have shown that these complications can be avoided by introducing a point mutation within the protospacer sequence present on the DNA donor template. In this case, a silent mutation 3 bases from the PAM restored successful homology‐directed repair events (Figure 7b).
In conclusion, the CRISPR/Cas9 vector system presented here represents a powerful toolkit for rapid and efficient genome editing in the model moss Physcomitrella patens. Through nonhomologous end joining, CRISPR/Cas9 permits the study of essential genes by creating potential hypomorphs as well as of nonessential genes by creating allelic variants. Through homology‐directed repair, sequences can be precisely inserted or removed with co‐transformation of a DNA donor template constructed in vitro. The simple and modular design of our vector system allows fast vector construction. Additionally, editing of large gene families in a single, transient transformation is achievable in a short timeframe. Furthermore, due to the modular design, our CRISPR/Cas9 vector system could be employed in other organisms keeping in mind that rates of homology‐directed repair could be lower and species‐specific modifications to promoter sequences would likely be required.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
D.R.M., M.C., X.C., and M.B. designed the research. D.R.M., M.C., and X.C. performed the research. D.R.M. and M.B. wrote the article.
Supporting information
ACKNOWLEDGMENTS
We thank Fabien Nogué for the Physcomitrella patens U6 promoter sequence and Shu‐Zon Wu for imaging the mEGFP tagged line. M.C. received support from the Plant Biology Graduate Program at the University of Massachusetts, Amherst, and X.C. received support from the Molecular and Cellular Biology Graduate Program at Dartmouth College. Additionally, this work was supported by NSF grants MCB‐1330171 and MCB‐1715785 to M.B.
Mallett DR, Chang M, Cheng X, Bezanilla M. Efficient and modular CRISPR‐Cas9 vector system for Physcomitrella patens . Plant Direct. 2019;3:1–15. 10.1002/pld3.168
Mallett and Chang contributed equally.
This manuscript was previously deposited as a preprint at https://www.biorxiv.org/content/10.1101/674481v1
REFERENCES
- Augustine, R. C. , Pattavina, K. A. , Tuzel, E. , Vidali, L. , & Bezanilla, M. (2011). Actin interacting protein1 and actin depolymerizing factor drive rapid actin dynamics in Physcomitrella patens . The Plant Cell, 23(10), 3696–3710. 10.1105/tpc.111.090753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae, S. , Kweon, J. , Kim, H. S. , & Kim, J.‐S. (2014). Microhomology‐based choice of cas9 nuclease target sites. Nature Methods, 11(7), 705–706. 10.1038/nmeth.3015 [DOI] [PubMed] [Google Scholar]
- Bascom, C. S. , Wu, S.‐Z. , Nelson, K. , Oakey, J. , & Bezanilla, M. (2016). Long‐term growth of moss in microfluidic devices enables subcellular studies in development. Plant Physiology, 172, 28–37. 10.1104/pp.16.00879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beucher, A. , Birraux, J. , Tchouandong, L. , Barton, O. , Shibata, A. , Conrad, S. , … Löbrich, M. (2009). ATM and artemis promote homologous recombination of radiation‐induced DNA double‐strand breaks in G2. The EMBO Journal, 28(21), 3413–3427. 10.1038/emboj.2009.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla, M. , Pan, A. , & Quatrano, R. S. (2003). RNA interference in the moss Physcomitrella patens . Plant Physiology, 133(2), 470–474. 10.1104/pp.103.024901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilang, R. , Iida, S. , Peterhans, A. , Potrykus, I. , & Paszkowski, J. (1991). The 3′‐terminal region of the hygromycin‐B‐resistance gene is important for its activity in Escherichia coli and Nicotiana tabacum . Gene, 1119(91), 247–250. 10.1016/0378-1119(91)90375-L [DOI] [PubMed] [Google Scholar]
- Brooks, C. , Nekrasov, V. , Lippman, Z. B. , & Van Eck, J. (2014). Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR‐associated9 system. Plant Physiology, 166(3), 1292–1297. 10.1104/pp.114.247577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, N. M. , Baltes, N. J. , Voytas, D. F. , & Douches, D. S. (2016). Geminivirus‐mediated genome editing in potato (Solanum tuberosum L.) using sequence‐specific nucleases. Frontiers in Plant Science, 7, 1045 10.3389/fpls.2016.01045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y. , Chen, L. , Sun, S. , Wu, C. , Yao, W. , Jiang, B. , … Hou, W. (2018). CRISPR/Cas9‐mediated deletion of large genomic fragments in soybean. International Journal of Molecular Sciences, 19(12), 3835 10.3390/ijms19123835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čermák, T. , Baltes, N. J. , Čegan, R. , Zhang, Y. , & Voytas, D. F. (2015). High‐frequency, precise modification of the tomato genome. Genome Biology, 16(1), 232 10.1186/s13059-015-0796-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H. H. Y. , Pannunzio, N. R. , Adachi, N. , & Lieber, M. R. (2017). Non‐Homologous DNA end joining and alternative pathways to double‐strand break repair. Nature Reviews Molecular Cell Biology, 18(8), 495–506. 10.1038/nrm.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Lu, X. , Shu, N. , Wang, S. , Wang, J. , Wang, D. , … Ye, W. (2017). Targeted mutagenesis in cotton (Gossypium hirsutum L.) using the CRISPR/Cas9 system. Scientific Reports, 7(1), 44304 10.1038/srep44304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collonnier, C. , Epert, A. , Mara, K. , Maclot, F. , Guyon‐Debast, A. , Charlot, F. , … Nogué, F. (2017). CRISPR‐Cas9‐mediated efficient directed mutagenesis and RAD51‐dependent and RAD51‐independent gene targeting in the moss Physcomitrella patens . Plant Biotechnology Journal, 15(1), 122–131. 10.1111/pbi.12596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z. , Mao, Y. , Xu, N. , Zhang, B. , Wei, P. , Yang, D.‐L. , … Zhu, J.‐K. (2014). Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas‐induced gene modifications in Arabidopsis . Proceedings of the National Academy of Sciences, 111(12), 4632–4637. 10.1073/pnas.1400822111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X. , Chen, J. , Dai, X. , Zhang, D. , & Zhao, Y. (2016). An effective strategy for reliably isolating heritable and Cas9‐free Arabidopsis mutants generated by CRISPR/Cas9‐mediated genome editing. Plant Physiology, 171(3), 1794–1800. 10.1104/pp.16.00663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil‐Humanes, J. , Wang, Y. , Liang, Z. , Shan, Q. , Ozuna, C. V. , Sánchez‐León, S. , … Voytas, D. F. (2017). High‐efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. The Plant Journal, 89(6), 1251–1262. 10.1111/tpj.13446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler, M. , Schönig, K. , Eckert, H. , Eschstruth, A. , Mianné, J. , Renaud, J.‐B. , … Concordet, J.‐P. (2016). Evaluation of off‐target and on‐target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology, 17(1), 148 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, H. , Wang, X. , Jia, H. , Yu, M. , Zhang, X. , Tang, H. , & Zhang, L. (2016). Large Fragment deletion using a CRISPR/Cas9 system in Saccharomyces cerevisiae . Analytical Biochemistry, 509, 118–123. 10.1016/j.ab.2016.07.008 [DOI] [PubMed] [Google Scholar]
- He, Z. , Proudfoot, C. , Mileham, A. J. , McLaren, D. G. , Bruce, C. , Whitelaw, A. , & Lillico, S. G. (2015). Highly efficient targeted chromosome deletions using CRISPR/Cas9. Biotechnology and Bioengineering, 112(5), 1060–1064. 10.1002/bit.25490 [DOI] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J. A. , & Charpentier, E. (2012). A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisugi, Y. , & Cuming, A. C. (2009). Gene targeting In Knight C. D., Perroud P.‐F., & Cove D. J. (Eds.), The moss Physcomitrella Patens (pp. 76–112). Oxford, UK: Wiley‐Blackwell. [Google Scholar]
- Lam, A. J. , St‐Pierre, F. , Gong, Y. , Marshall, J. D. , Cranfill, P. J. , Baird, M. A. , … Lin, M. Z. (2012). Improving FRET dynamic range with bright green and red fluorescent proteins. Nature Methods, 9(10), 1005–1012. 10.1038/nmeth.2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, D. , Ullrich, K. K. , Murat, F. , Fuchs, J. , Jenkins, J. , Haas, F. B. , … Rensing, S. A. (2018). The Physcomitrella patens chromosome‐scale assembly reveals moss genome structure and evolution. The Plant Journal, 93(3), 515–533. 10.1111/tpj.13801 [DOI] [PubMed] [Google Scholar]
- Li, C. , Unver, T. , & Zhang, B. (2017). A high‐efficiency CRISPR/Cas9 system for targeted mutagenesis in cotton (Gossypium hirsutum L.). Scientific Reports, 7(1), 43902 10.1038/srep43902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J.‐F. , Norville, J. E. , Aach, J. , McCormack, M. , Zhang, D. , Bush, J. , … Sheen, J. (2013). Multiplex and homologous recombination‐mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nature Biotechnology, 31(8), 688–691. 10.1038/nbt.2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Li, X. , & Zhou, Z. , Wu, P. , Fang, M. , Pan, X. , … Li, H. (2016). Reassessment of the four yield‐related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Frontiers in Plant Science, 7, 377 10.3389/fpls.2016.00377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Z. , Zhang, K. , Chen, K. , & Gao, C. (2014). Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system. Journal of Genetics and Genomics, 41(2), 63–68. 10.1016/j.jgg.2013.12.001 [DOI] [PubMed] [Google Scholar]
- Lopez‐Obando, M. , Hoffmann, B. , Géry, C. , Guyon‐Debast, A. , Téoulé, E. , Rameau, C. , … Nogué, F. (2016). Simple and efficient targeting of multiple genes through CRISPR‐Cas9 in Physcomitrella patens . G3: Genes|genomes|genetics, 6(11), 3647–3653. 10.1534/g3.116.033266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova, K. S. , Haft, D. H. , Barrangou, R. , Brouns, S. J. J. , Charpentier, E. , Horvath, P. , … Koonin, E. V. (2011). Evolution and classification of the CRISPR–Cas systems. Nature Reviews Microbiology, 9(6), 467–477. 10.1038/nrmicro2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P. , Yang, L. , Esvelt, K. M. , Aach, J. , Guell, M. , DiCarlo, J. E. , … Church, G. M. (2013). RNA‐guided human genome engineering via Cas9. Science, 339(6121), 823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malzahn, A. , Lowder, L. , & Qi, Y. (2017). Plant genome editing with TALEN and CRISPR. Cell & Bioscience, 7(1), 21 10.1186/s13578-017-0148-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, J. , Guo, D. , Zhang, J. , Huang, Q. , Qin, G. , Zhang, X. , … Qu, L.‐J. (2013). Targeted mutagenesis in rice using CRISPR‐Cas system. Cell Research, 23(10), 1233–1236. 10.1038/cr.2013.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan, M. E. , & Jasin, M. (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature Reviews Molecular Cell Biology, 11(3), 196–207. 10.1038/nrm2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu, H. , Ann Ran, F. , Hsu, P. D. , Konermann, S. , Shehata, S. I. , Dohmae, N. , … Nureki, O. (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell, 156(5), 935–949. 10.1016/j.cell.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordon, J. , Gantner, J. , Kemna, J. , Schwalgun, L. , Reschke, M. , Streubel, J. , … Stuttmann, J. (2017). Generation of chromosomal deletions in dicotyledonous plants employing a user‐friendly genome editing toolkit. The Plant Journal, 89(1), 155–168. 10.1111/tpj.13319 [DOI] [PubMed] [Google Scholar]
- Prigge, M. J. , & Bezanilla, M. (2010). Evolutionary crossroads in developmental biology: Physcomitrella patens . Development, 137(21), 3535–3543. 10.1242/dev.049023 [DOI] [PubMed] [Google Scholar]
- Puchta, H. (2005). The repair of double‐strand breaks in plants: mechanisms and consequences for genome evolution. Journal of Experimental Botany, 56(409), 1–14. 10.1093/jxb/eri025 [DOI] [PubMed] [Google Scholar]
- Pyott, D. E. , Sheehan, E. , & Molnar, A. (2016). Engineering of CRISPR/Cas9‐mediated potyvirus resistance in transgene‐free Arabidopsis plants. Molecular Plant Pathology, 17(8), 1276–1288. 10.1111/mpp.12417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidi, Y. , Finka, A. , Chakhporanian, M. , Zrÿd, J.‐P. , Schaefer, D. G. , & Goloubinoff, P. (2005). Controlled expression of recombinant proteins in Physcomitrella patens by a conditional heat‐shock promoter: A tool for plant research and biotechnology. Plant Molecular Biology, 59(5), 697–711. 10.1007/s11103-005-0889-z [DOI] [PubMed] [Google Scholar]
- Sander, J. D. , & Joung, J. K. (2014). CRISPR‐Cas systems for editing, regulating and targeting genomes. Nature Biotechnology, 32(4), 347–355. 10.1038/nbt.2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent, R. G. , Brenneman, M. A. , & Wilson, J. H. (1997). Repair of site‐specific double‐strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Molecular and Cellular Biology, 17(1), 267–277. 10.1128/MCB.17.1.267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, D. G. , & Zryd, J.‐P. (1997). Efficient gene targeting in the moss Physcomitrella patens . The Plant Journal, 11(6), 1195–1206. 10.1046/j.1365-313X.1997.11061195.x [DOI] [PubMed] [Google Scholar]
- Schiml, S. , Fauser, F. , & Puchta, H. (2014). The CRISPR/Cas system can be used as nuclease for in Planta gene targeting and as paired nickases for directed mutagenesis in Arabidopsis resulting in heritable progeny. The Plant Journal, 80(6), 1139–1150. 10.1111/tpj.12704 [DOI] [PubMed] [Google Scholar]
- Shan, Q. , Wang, Y. , Li, J. , Zhang, Y. , Chen, K. , Liang, Z. , … Gao, C. (2013). Targeted genome modification of crop plants using a CRISPR‐Cas system. Nature Biotechnology, 31(8), 686–688. 10.1038/nbt.2650 [DOI] [PubMed] [Google Scholar]
- Shi, J. , Gao, H. , Wang, H. , Lafitte, H. R. , Archibald, R. L. , Yang, M. , … Habben, J. E. (2017). ARGOS8 variants generated by CRISPR‐Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnology Journal, 15(2), 207–216. 10.1111/pbi.12603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, V. , Underwood, J. L. , & Zhao, S. (2017). Dual‐targeting by CRISPR/Cas9 for precise excision of transgenes from rice genome. Plant Cell, Tissue and Organ Culture (PCTOC), 129(1), 153–160. 10.1007/s11240-016-1166-3 [DOI] [Google Scholar]
- Sun, Y. , Zhang, X. , Wu, C. , He, Y. , Ma, Y. , Hou, H. , … Xia, L. (2016). Engineering herbicide‐resistant rice plants through CRISPR/Cas9‐mediated homologous recombination of acetolactate synthase. Molecular Plant, 9(4), 628–631. 10.1016/j.molp.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Svitashev, S. , Schwartz, C. , Lenderts, B. , Young, J. K. , & Cigan, A. M. (2016). Genome editing in maize directed by CRISPR–Cas9 ribonucleoprotein complexes. Nature Communications, 7(1), 13274 10.1038/ncomms13274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitashev, S. , Young, J. K. , Schwartz, C. , Huirong Gao, S. , Falco, C. , & Cigan, A. M. (2015). Targeted mutagenesis, precise gene editing, and site‐specific gene insertion in maize using Cas9 and guide RNA. Plant Physiology, 169(2), 931–945. 10.1104/pp.15.00793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidali, L. , van Gisbergen, P. A. C. , Guerin, C. , Franco, P. , Li, M. , Burkart, G. M. , … Bezanilla, M. (2009). Rapid formin‐mediated actin‐filament elongation is essential for polarized plant cell growth. Proceedings of the National Academy of Sciences, 106(32), 13341–13346. 10.1073/pnas.0901170106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F. , Wang, C. , Liu, P. , Lei, C. , Hao, W. , Gao, Y. , … Zhao, K. (2016). Enhanced rice blast resistance by CRISPR/Cas9‐targeted mutagenesis of the ERF transcription factor gene OsERF922 . PLoS ONE, 11(4), e0154027 10.1371/journal.pone.0154027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Geng, L. , Yuan, M. , Wei, J. , Jin, C. , Li, M. , … Li, X. (2017). Deletion of a target gene in indica rice via CRISPR/Cas9. Plant Cell Reports, 36(8), 1333–1343. 10.1007/s00299-017-2158-4 [DOI] [PubMed] [Google Scholar]
- Wilson, F. M. , Harrison, K. , Armitage, A. D. , Simkin, A. J. , & Harrison, R. J. (2019). CRISPR/Cas9‐mediated mutagenesis of phytoene desaturase in diploid and octoploid strawberry. Plant Methods, 15(1), 45 10.1186/s13007-019-0428-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S.‐Z. , & Bezanilla, M. (2014). Myosin VIII associates with microtubule ends and together with actin plays a role in guiding plant cell division. Elife, 3, e03498 10.7554/eLife.03498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, A. , Wang, Z. , Hu, Y. , Wu, Y. , Luo, Z. , Yang, Z. , … Zhang, B. (2013). Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Research, 41(14), e141 10.1093/nar/gkt464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Liang, Z. , Zong, Y. , Wang, Y. , Liu, J. , Chen, K. , … Gao, C. (2016). Efficient and transgene‐free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nature Communications, 7(1), 12617 10.1038/ncomms12617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Liu, B. , Weeks, D. P. , Spalding, M. H. , & Yang, B. (2014). Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Research, 42(17), 10903–10914. 10.1093/nar/gku806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, A. D. , Lang, D. , Buchta, K. , Rombauts, S. , Nishiyama, T. , Hasebe, M. , … Reski, R. (2013). Reannotation and extended community resources for the genome of the non‐seed plant Physcomitrella patens provide insights into the evolution of plant gene structures and functions. BMC Genomics, 14(1), 498 10.1186/1471-2164-14-498 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials