

Abstract
Translocation of virulence effector proteins through the type III secretion system (T3SS) is essential for the virulence of many medically relevant Gram‐negative bacteria. The T3SS ATPases are conserved components that specifically recognize chaperone–effector complexes and energize effector secretion through the system. It is thought that functional T3SS ATPases assemble into a cylindrical structure maintained by their N‐terminal domains. Using size‐exclusion chromatography coupled to multi‐angle light scattering and native mass spectrometry, we show that in the absence of the N‐terminal oligomerization domain the Salmonella T3SS ATPase InvC can form monomers and dimers in solution. We also present for the first time a 2.05 å resolution crystal structure of InvC lacking the oligomerization domain (InvCΔ79) and map the amino acids suggested for ATPase intersubunit interaction, binding to other T3SS proteins and chaperone–effector recognition. Furthermore, we validate the InvC ATP‐binding site by co‐crystallization of InvCΔ79 with ATPγS (2.65 å) and ADP (2.80 å). Upon ATP‐analogue recognition, these structures reveal remodeling of the ATP‐binding site and conformational changes of two loops located outside of the catalytic site. Both loops face the central pore of the predicted InvC cylinder and are essential for the function of the T3SS ATPase. Our results present a fine functional and structural correlation of InvC and provide further details of the homo‐oligomerization process and ATP‐dependent conformational changes underlying the T3SS ATPase activity.
Keywords: ATPase, bacterial pathogenesis, crystallography, multi‐angle light scattering, native mass spectrometry, Salmonella enterica, spectroscopy, type III secretion system (T3SS)
Short abstract
1. INTRODUCTION
The injectisome type III secretion system (T3SS) is a multi‐protein nanomachine essential for the virulence of many pathogenic Gram‐negative bacteria, including Salmonella, Shigella, Yersinia, enteropathogenic Escherichia coli, Chlamydia, and Pseudomonas aeruginosa that cause millions of deaths worldwide each year.1, 2, 3 The T3SS forms a syringe‐like structure extending from the bacterial cytosol across the bacterial membranes to the target cell to directly inject virulence effector proteins into its cytoplasm. Although the structural components of the T3SSs are highly conserved among bacterial species, the secreted effectors are pathogen‐specific.1, 4, 5, 6 Most of the effector proteins require the formation of complexes with their T3SS chaperones prior secretion. The chaperones maintain a region of the effectors partially unfolded to facilitate their subsequent secretion through the narrow aperture of the T3SS channel (20 å).7, 8, 9 The secretion mechanism of the T3SS critically depends on the hierarchical selection and delivery of effectors by the sorting platform complex.10 The Salmonella pathogenicity island 1 (SPI‐1) sorting platform is a dynamic complex that interacts with the cytosolic interface of the membrane‐embedded T3SS and forms cytosolic soluble intermediates.11, 12 This complex is constituted by a central ATPase oligomeric cylinder formed by InvC (SctN in the unified nomenclature), which is linked through its negative regulator OrgB (SctL) to the flagellar C‐ring orthologue SpaO (SctQ) and the accessory protein OrgA (SctK). Additionally, InvC interacts through its central pore to the stalk protein InvI (SctO).13, 14
InvC has been proposed to recognize and dissemble the chaperone–effector complexes and unfold the effectors in an ATP‐dependent manner.7 This function mirrors the AAA+ ATP‐driven translocase mechanism, which consists of hydrolyzing ATP to power conformational changes of its homo‐hexameric cylinder architecture and to trigger unfolding and translocation of substrates through the central pore of the cylinder.15 However, the T3SS ATPases share high sequence homology and three‐dimensional structural similarities with the β subunit of the F1FO ATPases.16, 17 Recently, the E. coli ATPase EscN has been shown to form a homo‐hexameric cylinder with six ATP‐binding sites located at the interface of adjacent dimer pairs, suggesting cooperativity between subunits.18 EscN binds to its central stalk protein and presents different functional states supporting a rotary catalytic mechanism homologous to the F1FO ATPases. in vivo and in vitro studies show that the T3SS ATPases form oligomers in solution and in association with the T3SS. The stoichiometry of the complexes in solution range from dimers to dodecamers depending on the bacterial species.16, 19, 20 The T3SS ATPases fold in three domains. The N‐terminal domain is considered to be important for stable assembly of higher oligomers; it binds to the ATPase negative regulator (OrgB in Salmonella) and presents lipid affinity.20, 21, 22, 23, 24 The predicted ATPase core is the central and most conserved domain. It contains the phosphate‐binding loop (P‐loop) with the Walker box A motif (GxGKT/S) characteristic for enzymes with ATP activity.25 The C‐terminal domain has moderate sequence similarity among different species and is the potential recognition site for chaperone–effector complexes.22
The atomic structures for the Shigella Spa47, E. coli EscN, Salmonella SPI‐2 SsaN, and Salmonella Flagellar FliI ATPases share high similarity in conformation and ATP‐analogues binding states.16, 17, 26, 27 Although the T3SS ATPases are conserved, little is known about the structure of the Salmonella Typhimurium SPI‐1 ATPase InvC. Here, we present the first structure of the Salmonella T3SS ATPase InvC lacking the first 79 residues (Δ79) in the apo‐state and in the presence of ATP‐analogues. We show that Salmonella InvCΔ79 dimerizes in solution in the absence of its N‐terminal domain. Our structural assignments allow mapping of amino acids and further interpretation of previous genetic and biochemical analysis fundamental for understanding the function of T3SS ATPases. Additionally, we characterize the structure of InvCΔ79 in presence of ADP or ATPγS. These structures reveal additional conformational changes of two loops outside of the ATP catalytic site that have been shown to be essential in the overall function of InvC. These structural evidences provide insights into the energizing mechanism of T3SS ATPases.
2. RESULTS
2.1. InvCΔ79 forms monomers and dimers in solution
The Salmonella InvC belongs to the conserved family of T3SS ATPases, sharing between 38 and 57% sequence identity with its orthologues (Figure S1). It was reported that the N‐terminal domain (amino acids 1–79) of the T3SS ATPases is involved in membrane anchoring and homo‐oligomer stabilization.21, 22 When we recombinantly expressed and purified the full length InvC, the N‐terminal domain suffered fast proteolysis (data not shown), suggesting that the N‐terminal domain or the linker to this domain are flexible regions. Hence, we removed the first 79 amino acids to generate a construct containing the predicted ATPase and C‐terminal domains followed by a Strep‐Tag that we termed InvCΔ79.
The E. coli T3SS ATPase forms a homo‐hexameric cylinder with ATP‐binding sites located at the interface of adjacent subunits,18 similar to the F1ATPases. We reasoned that interaction between ATPase subunits might occur also in the absence of its N‐terminal region and characterized the molecular size of InvCΔ79 in solution. Size‐exclusion chromatography coupled to multi‐angle light scattering (SEC‐MALS) analysis of InvCΔ79 resulted in two elution peaks, a major one assigned as Peak I and a minor one named Peak II. The weight‐averaged molecular masses of Peaks I and II corresponded well to a monomer (40 kDa) and a dimer (80 kDa) of InvCΔ79, respectively (Figure 1a, S2). Our results are in line with the E. coli ATPase EscNΔ7 lacking the first seven amino acids that forms dimers in solution.16 Further analysis of elution Peak I by native mass spectrometry (MS) confirmed the predominance of monomers in this fraction even though some dimers were also detected. In contrast, native MS of the Peak II presented mostly dimers and some monomers. The small amount of trimers and tetramers detected are likely unspecific clusters inherent to this method (Figure 1b, Table S1). Together, these results demonstrate that InvCΔ79 lacking the N‐terminal domain exists predominantly as monomer in solution and can self‐associate into dimers.
Figure 1.
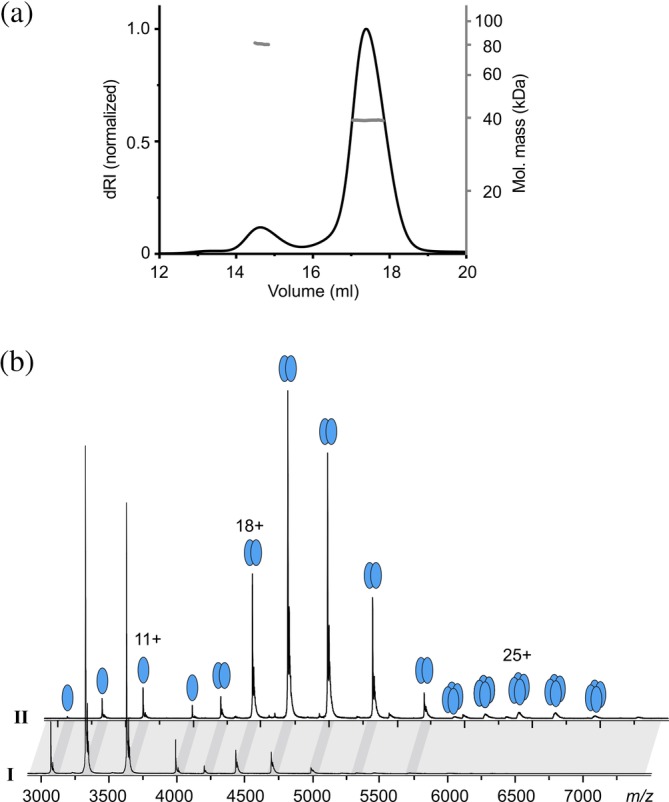
Stoichiometry of InvCΔ79. (a) SEC‐MALS analysis of InvCΔ79. The SEC profile (dRI, left axis) presents two elution peaks, I and II. The weight‐averaged molar masses (gray, right axis) across the elution peaks correspond to monomeric (40 kDa) and dimeric (80 kDa) states of InvCΔ79. (b) Representative native MS analysis of SEC‐peaks I and II demonstrating the monomeric (39,901.9 ± 0.6 Da) and dimeric state (79,808 ± 2 Da) of InvCΔ79, respectively. Dark gray bands highlight corresponding peaks from the two spectra. Masses are summarized in Table S1
2.2. Structure of InvCΔ79
To determine the structure of InvC, we performed crystallization trials using the monomeric size‐exclusion chromatography (SEC) fraction of InvCΔ79. We solved the X‐ray crystal structure of InvCΔ79 in the absence of ATP‐analogues at 2.05 å resolution (Figure 2, Table 1). The apo‐form folds in two structural domains, the ATPase core (amino acids R81 to T355) and the C‐terminal domain (T356 to N431) similar to its bacterial orthologues. The ATPase catalytic core is constituted by the α/β Rossmann fold25 with a parallel nine‐stranded twisted β‐sheet flanked by three helices at one side, and four helices at the other one. The phosphate‐binding loop motif (P‐loop) is constituted by the amino acid sequence GCGKT (162–166) and is located between α2 and β5 of the ATPase core. The smaller C‐terminal domain is composed of three helices and contains a helix–loop–helix motif that is proposed to interact with chaperone and effector proteins.27, 28
Figure 2.
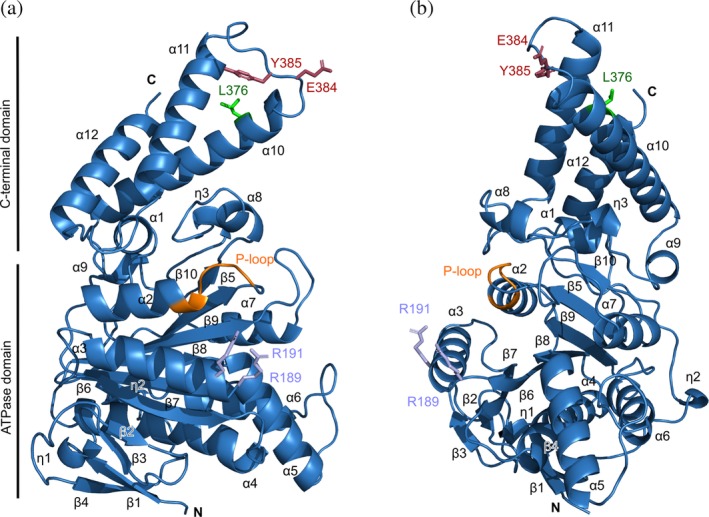
Overall architecture of InvCΔ79. (a, b) Two views of InvCΔ79 produced by 90° rotation showing secondary structure elements labeled as in Figure S1. The P‐loop region is highlighted in orange and previously studied amino acids are presented as sticks. R189 and R191 are related with intersubunit interaction (lilac).22 Y385 is essential for full type III secretion28 and the conserved E384 interacts with the stalk protein in EscN (raspberry).18 L376 interacts with chaperone–effector complexes (green)7
Table 1.
Data collection and refinement statistics
| InvCΔ79 | InvCΔ79‐ATPγS | InvCΔ79‐ADP | |
|---|---|---|---|
| Data collection | |||
| Wavelength (å) | 1.0332 | 1.0332 | 1.0332 |
| Space group | P 65 | P 65 | P 65 |
| Cell dimensions a, b, c (å) | 106.3, 106.3, 73.5 | 107.9, 107.9, 73.8 | 107.4, 107.4, 73.5 |
| Resolution (å) | 100–2.05 | 100–2.65 | 100–2.80 |
| (2.10–2.05) | (2.71–2.65) | (2.87–2.80) | |
| R merge | 0.102 (1.091) | 0.100 (1.596) | 0.086 (0.804) |
| R meas | 0.109 (1.167) | 0.106 (1.694) | 0.092 (0.864) |
| CC 1/2 | 99.6 (66.0) | 99.9 (54.1) | 99.9 (87.5) |
| I/σ(I) | 11.35 (1.98) | 16.58 (1.53) | 13.17 (1.52) |
| Total reflections | 234,745 (16,821) | 144,020 (9,272) | 80,465 (5,394) |
| Completeness (%) | 99.9 (99.2) | 99.8 (97.5) | 98.3 (95.5) |
| Multiplicity | 7.9 (7.8) | 10.0 (8.9) | 6.8 (6.5) |
| Refinement | |||
| Reflections used | 29,752 | 14,382 | 11,813 |
| R work /R free | 0.168/0.207 | 0.186/0.224 | 0.211/0.257 |
| No. atoms | |||
| Protein | 2,711 | 2,677 | 2,623 |
| Ligands | 8 | 57 | 86 |
| Water | 152 | 33 | 13 |
| B‐factors | |||
| Protein | 56.35 | 78.85 | 94.55 |
| Ligands | 75.96 | 105.98 | 136.29 |
| Water | 56.00 | 71.17 | 83.29 |
| R.M.S. deviations | |||
| Bond lengths (å) | 0.003 | 0.003 | 0.003 |
| Bond angles (°) | 0.615 | 0.663 | 0.644 |
| Ramachandran values | |||
| Favored (%) | 97.14 | 93.88 | 92.88 |
| Allowed (%) | 2.86 | 5.25 | 6.23 |
| Outliers (%) | 0.00 | 0.87 | 0.89 |
| Rotamer outliers (%) | 0.69 | 6.64 | 6.43 |
| Clashscore | 4.61 | 8.45 | 9.42 |
Statistics for the highest‐resolution shell are shown in parentheses.
Analysis of the B‐factor values of InvCΔ79 after refinement shows that the ATPase core has an average value of 50.7. Most of this domain is rather rigid (blue) and contains two protruding loops with higher mobility (cyan to red in Figure 3a). The first loop region is located between α5 and α6 (amino acids A255 to L282) and the second is between β9 and α7 (L304 to S320). The average B‐factor of the C‐terminal domain is 75.3, showing a higher mobility in comparison with the ATPase core. This domain contains a third highly mobile loop region between α10 and α11 (K368 to R400). The β9‐α7 loop is highly conserved among the T3SS ATPases and the other two loops show a moderate sequence homology (Figure S1). The three loops are arranged in the same front of InvCΔ79 forming a mobile interface that could potentially be involved in conformational changes or interaction with other molecules during the ATPase activity.
Figure 3.
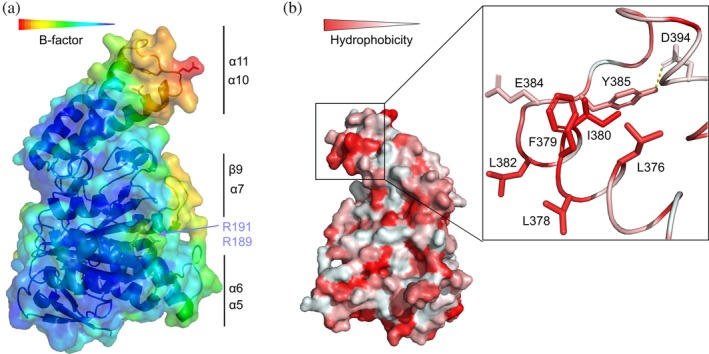
Structural analysis of InvCΔ79. (a) Structure colored by B‐factor values. Color‐coding bar shows lower (blue) to higher (red) B‐factor values for rigid to mobile regions, respectively. Flexible loops and amino acids for intersubunit interaction are labeled as in Figure 2. (b) Structure depicted by 180° rotation colored by higher (red) to lower (white) hydrophobicity of amino acids. The hydrophobic patch containing L376 is shown as inlet. Y385 buried (forming hydrogen bonds with D394) and E384 exposed in the loop are also depicted in the inlet
Taken together, InvCΔ79 shows high three‐dimensional structural similarity with other T3SS ATPases including Spa47 (RMSD of 1.05 å for 325 aligned Cα residues), EscN (1.51 å for 312 residues) (Figure S3), SsaN (1.63 å for 293 residues), and FliI (1.60 å for 317 residues). As predicted from the sequence alignment (Figure S1), the major differences between T3SS ATPase structures are located at the C‐terminal domains, probably due to its suggested function in recognizing specific effector‐bound chaperones.7, 27
2.3. Structural mapping of functional amino acids
Mutational analysis of InvC and its Shigella orthologue Spa47, identified residues R189 and R191 of the ATPase core (marked in lilac in Figure 2) as essential for ATPase activity, homo‐oligomerization, and type III secretion.22, 29 In our structure, these amino acids are located at the exposed surface of InvCΔ79 and their side chains present high mobility as denoted by their B‐factors (Figure 3a). It can be conceived that R189 and R191 play a role in dimerization of InvCΔ79 in the absence of the N‐terminal domain.
The C‐terminal domain of some T3SS ATPases contains conserved amino acids that are crucial for type III secretion. The InvC amino acid Y385 was shown to be essential for secretion of late effectors through the T3SS. The previously suggested mechanism included direct interaction of Y385 with effector–chaperone complexes.28 We show that Y385 forms a hydrogen bond with the side chain of D394, keeping most of its surface area buried within the structure (Figure 3b inlet). However, we cannot discard the possibility of a structural change around this amino acid upon ATP binding or chaperone–effector interaction. The amino acid E384 was reported to participate in the interaction with the stalk protein in Escherichia coli.18 E384 is surface exposed in our structure, and as Y385, is located in the α10‐α11 mobile loop (Figure 3a). Additionally, the InvC L376 was shown to play a role in recognition of chaperone‐bound to effectors.7, 27 Analysis of hydrophobicity distribution of InvCΔ79 shows that L376 is part of a hydrophobic patch together with the amino acids L378, F379, I380, and L382 (Figure 3b). These residues, except for L378, are conserved among T3SS ATPases and might be important candidates for chaperone–effector recognition by nonpolar interactions.
2.4. Conformational changes associated with ATP‐analogue binding to InvCΔ79 in solution
To understand the molecular mechanism of ATP recognition of InvC, we monitored the conformational changes of InvCΔ79 upon binding of different ATP‐analogues by using Fourier‐transform infrared (FTIR) and circular dichroism (CD) spectroscopy. ATPγS, AMP‐PNP, or ADP supplemented with equimolar concentrations of magnesium ions were used as ligands. FTIR difference spectroscopy of InvCΔ79 with ATP‐analogues showed a decrease of absorbance at 1655 cm−1, indicating a reduction of α‐helical content upon ligand binding (Figure 4a).30 The signal reduction is more pronounced for the ADP‐ and AMP‐PNP‐bound forms and moderate for the ATPγS‐bound form. CD analysis of InvCΔ79 indicates also a reduction of the α‐helical content (208 and 222 nm) in the presence of ADP or AMP‐PNP, while no major intensity change was detected for the ATPγS‐bound form (Figure 4b). Taken together, these results show that InvCΔ79 undergoes conformational changes upon binding to ADP and AMP‐PNP in solution, whereas the structural changes upon ATPγS interaction are barely detectable.
Figure 4.
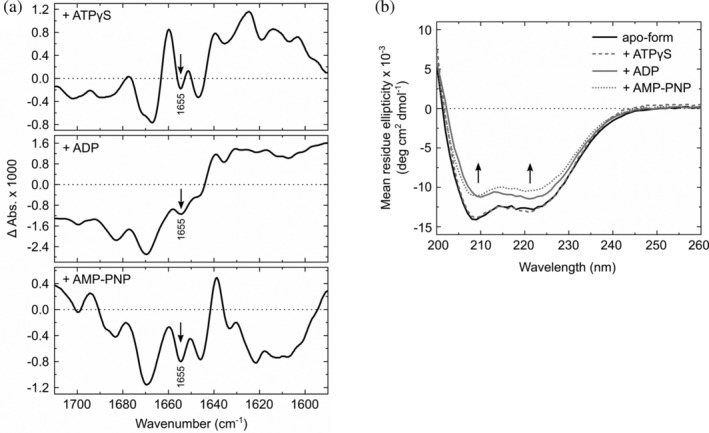
Conformational changes of InvCΔ79 upon ligand binding in solution. (a) FTIR difference spectra of InvCΔ79 bound to ATPγS, ADP, and AMP‐PNP in reference to its apo‐form. (b) Background‐corrected CD spectra of InvCΔ79 in the absence and presence of ATP‐analogues. Arrows indicate changes of α‐helical content upon nucleotide addition
2.5. Crystal structures of InvCΔ79 in the presence of ATP‐analogues
To further analyze the conformational changes of InvCΔ79 upon ligand binding, we performed co‐crystallization and soaking experiments of InvCΔ79 with ADP, ATPγS, or AMP‐PNP supplemented with magnesium ions. This allowed us to solve the crystal structures of InvCΔ79 co‐crystallized with ADP at 2.80 å resolution and InvCΔ79 bound to ATPγS by soaking experiments at 2.65 å resolution. However, we could not assign any ligand density for InvCΔ79 with AMP‐PNP and magnesium ions.
In the InvCΔ79 apo‐form, the putative ligand‐binding site is occupied by several water molecules (Figure 5). In both co‐crystal structures, most of the water molecules were replaced by the ligands. The modeling of ADP bound to InvCΔ79 was challenging because of its discontinuous density, likely due to partial occupancy of the site or flexibility of the ligand. We could model the ligand between two clear densities for the adenine and phosphate groups. However, no density for the ribose group was distinguished (Figure 5, S4). The phosphate groups of the ADP molecule form hydrogen bonds with the InvC P‐loop amino acids G164 and T166. The adenine group interacts with a water molecule bound to V411 by hydrogen bonds. In the ATPγS bound structure, a magnesium ion is coordinated by the β‐ and γ‐phosphates of the ligand, the side chains of D249 and T166, and one water molecule that forms hydrogen bonds with the α‐phosphate of ATPγS. The phosphate groups of the ligand interact with the P‐loop of InvCΔ79 forming hydrogen bonds with the amino acids G164 and T166, and a salt bridge with K165. The adenine group is stabilized by π‐π stacking with Y338. The interaction of ligands with InvCΔ79 resembles the ATP‐analogue binding observed for orthologue T3SS ATPases (Figure 6).16, 17, 22, 31 Additionally, single mutations of G164 and K165 in the InvC P‐loop result in loss of ATP‐hydrolysis function.22 Our structures validate the relevance of this loop for ATP recognition and allow us to further analyze the properties of the remaining InvC ligand‐binding site.
Figure 5.
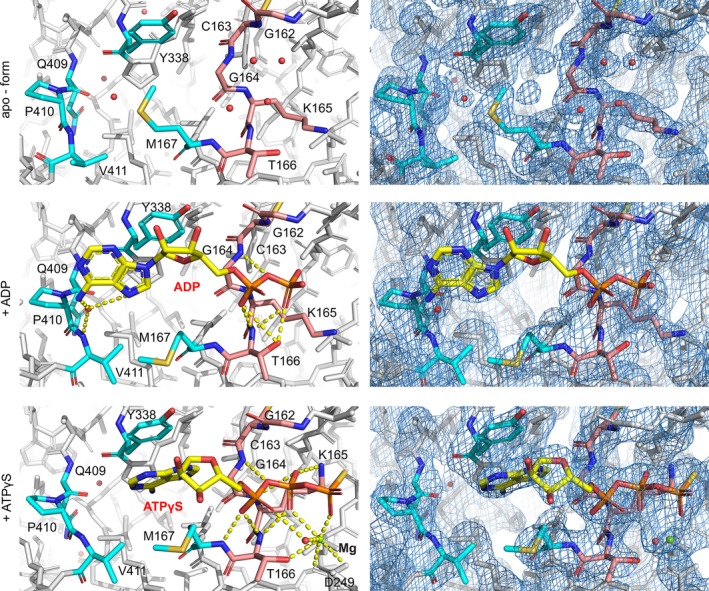
Structural changes of the InvCΔ79 ATP‐binding site in the presence of ATP‐analogues. Analysis of ligand interaction (left column) and electron density maps with 2Fo‐Fc contour at 0.8–1.5σ (right column). P‐loop (G162 to T166) interacting with the phosphate groups is colored in light orange and other key amino acids for ligand recognition are colored in cyan. Contacts involved in ligand stabilization are indicated by yellow dashed lines. ADP, ATPγS, and magnesium ion are labeled
Figure 6.
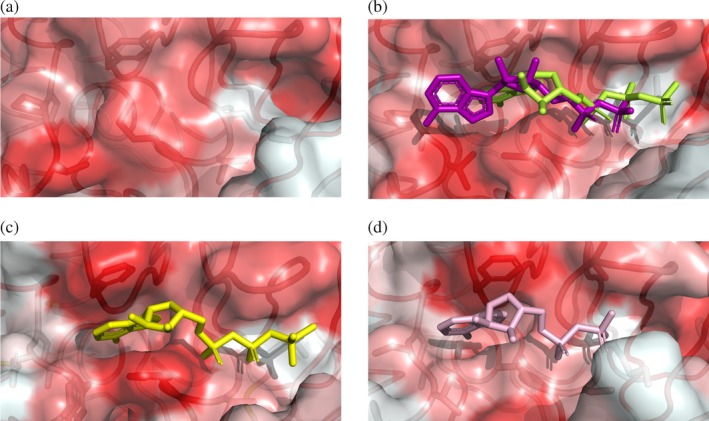
Opening of the hydrophobic pocket at the InvCΔ79 ATP‐binding site and comparison with T3SS ATPase orthologues. Surface and ribbon representations colored by higher (red) to lower (white) hydrophobicity of amino acids showing (a) the Salmonella InvCΔ79 apo‐form, (b) InvCΔ79 in presence of ADP (purple) and ATPγS (lime), (c) the Shigella Spa47Δ83 with ATPγS (PDB ID: 5ZT1) and (d) the E. coli EscNΔ102 with ADP (PDB ID: 2OBM). Ligands and side chains of amino acids stabilizing the ligands are depicted as sticks in the same orientation as in Figure 5
2.6. Remodeling of the ATP‐binding site upon ligand interaction
The phosphate groups of the ATP‐analogues interact with the InvCΔ79 P‐loop. Upon binding of ATPγS, K165 is displaced toward the β‐phosphate of the ligand interacting by a salt bridge. The adenine group of the ATP‐analogues binds to a hydrophobic pocket formed by Y338, P410, V411, and M167 in the InvC ligand‐binding site. This hydrophobic pocket is in closed conformation in the apo‐form and is opened upon interaction with ATP‐analogues (Figures 5 and 6a,b). The side chain of M167 is oriented toward V411 creating an open cavity to stabilize the adenine group. In addition, the loop including V411 (α11‐α12) is located closer to M167 surrounding and further defining the open hydrophobic pocket (Figure 5). A similar pocket is formed in the ligand‐binding site of other T3SS ATPases, although the amino acid composition in this region has little sequence similarity.16, 17, 31 A comparison with the nucleotide‐binding site of the Shigella Spa47 and E. coli EscN T3SS ATPases shows that the adenine double‐ring from the ADP‐InvCΔ79 structure is rather displaced from the inner region of the hydrophobic pocket but still captured on its surface (Figure 6).
2.7. Ligand induced conformational changes in two luminal loops
Analyzing the overall structures of the apo‐ and ligand bound states, the α‐helical content of InvCΔ79 decreased modestly upon recognition of ADP. The ADP‐InvCΔ79 structure presents loss of α‐helical arrangement mostly in the extremes of α10 and α11 facing the α10‐α11 loop. The ATPγS‐InvCΔ79 structure presents a scarce decrease of α‐helical content in α10 (Figures 7 and 8). The decrease of the α‐helical content in the ADP‐crystal structure and the partial maintenance of the secondary structure in the ATPγS‐InvCΔ79 structure follow the same tendency of the structural changes detected by FTIR and CD spectroscopy analysis.
Figure 7.
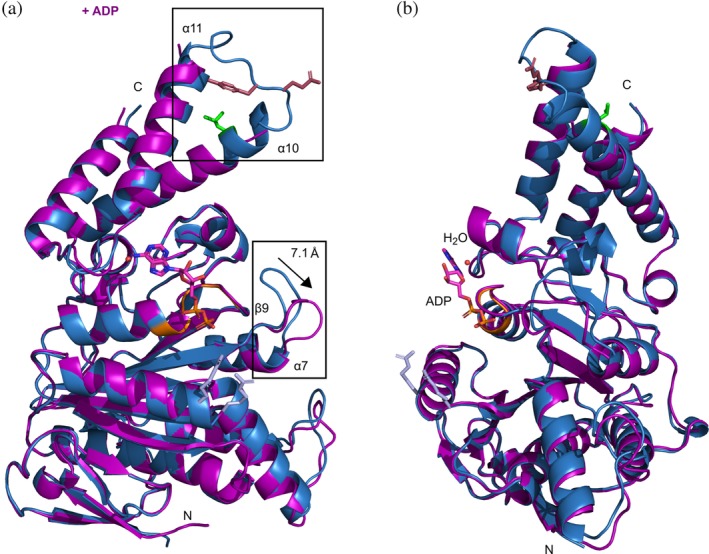
Overall structural changes of InvCΔ79 in the presence of ADP. (a) 3D structural alignment of the apo‐form (blue) with the ADP bound form (purple). Differences in loop conformations are highlighted in rectangles and key amino acids colored as in Figure 2a. The β9‐α7 loop moves a distance of approximately 7.1 å. (b) 90° rotation view showing ADP interaction in the ligand‐binding site
Figure 8.
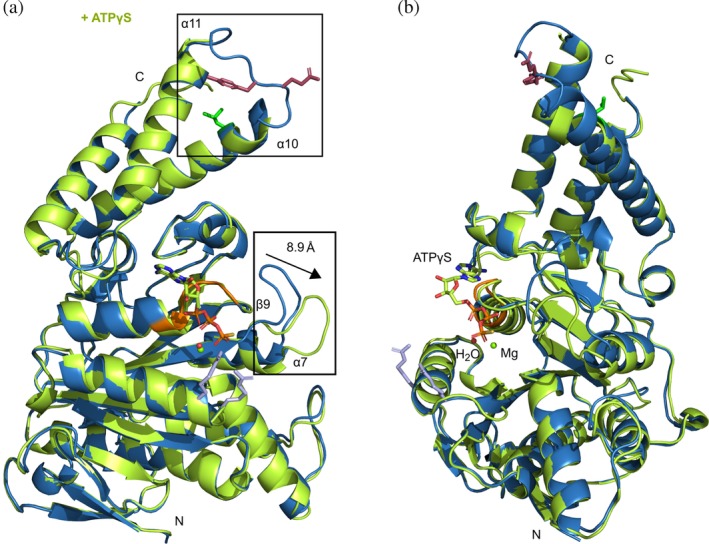
Overall structural changes of InvCΔ79 in the presence of ATPγS. (A) 3D structural alignment of the apo‐form (blue) with the ATPγS bound form (lime). Differences in loop conformations are highlighted in rectangles and key amino acids colored as in Figure 2a. The β9‐α7 loop moves a distance of approximately 8.9 å. (B) 90° rotation view showing ATPγS interaction in the ligand‐binding site
The apo‐, ADP‐ and ATPγS‐InvCΔ79 structures showed conformational differences for two of the most mobile loops (α10‐α11 and β9‐α7) (Figures 7 and 8). The α10‐α11 loop is located at the C‐terminal domain of InvC and is part of a helix–loop–helix motif. No density was observed for this loop in co‐crystals while the protein backbone was better defined in the apo‐form, indicating an increase of its flexibility upon ligand interaction (Figure S5). In our structures, α11 is closely followed by V411 of the ligand‐binding site. Upon ligand interaction V411 pulls α11 and gets closer to M167 (Figure 5), possibly affecting the α‐helical conformation of α11, α10, and the respective loop. Some amino acids relevant for T3SS ATPase function are located in the α10‐α11 loop. Indeed, point mutations in the InvC G383A, E384A, Y385A, and G388A were shown to cause a decrease of secretion of late effectors by the T3SS.28 Additionally, the amino acid L376 that interacts with chaperone–effector proteins7 is also part of this loop. The β9‐α7 loop is located next to the P‐loop and presents significant conformational changes in the presence of ATP‐analogues. Upon binding of ADP and ATPγS, the β9‐α7 loop swings away by approximately 7.1 and 8.9 å, respectively (Figures 7 and 8, S5). Point mutations in this region of InvC, including E306A, E308A, E309A, E310A, and D312A, were reported to affect secretion of late effectors by the T3SS.28 Similarly, the corresponding β9‐α7 loop of the Shigella Spa47Δ83 was shown to increase its flexibility upon interaction with ATP‐analogues.31 Interestingly, cryo‐EM analysis of the E. coli EscN homo‐hexameric cylinder showed differences in the α10‐α11 loop conformation for each ligand‐bound state and differences in the β9‐α7 loop upon ligand recognition.18
To analyze the location of the loops β9‐α7 and α10‐α11 in a putative hexameric complex, we built a model by superposing the InvCΔ79 apo‐form to the EscN asymmetric cylinder (PDB ID: 6NJP) using Pymol (Figure 9).18 In the model, β9‐α7 and α10‐α11 are located at the inner pore of the cylinder where the stalk protein (InvI in Salmonella) interacts with EscN. In addition, the hydrophobic patch containing the chaperone–effector interacting amino acid L3767 is mostly oriented to the inner pore but a prominent cleft observed between two subunits could probably allow the reported recognition site (Figure 9a). Moreover, the P‐loop, the amino acids participating in homo‐oligomerization of ATPase subunits (R198, R191),22, 29 and the amino acid for stabilization of ATP in orthologues (R349)16 were located at the interface of the homo‐hexameric model (Figure 9b). Our results show that InvCΔ79 contains two loops that undergo conformational changes upon interaction with ADP or ATPγS. The movement of these loops could be involved in the recognition of effector and chaperone proteins or in the interaction with other T3SS components including the stalk protein InvI during the dynamic process of type III secretion.
Figure 9.
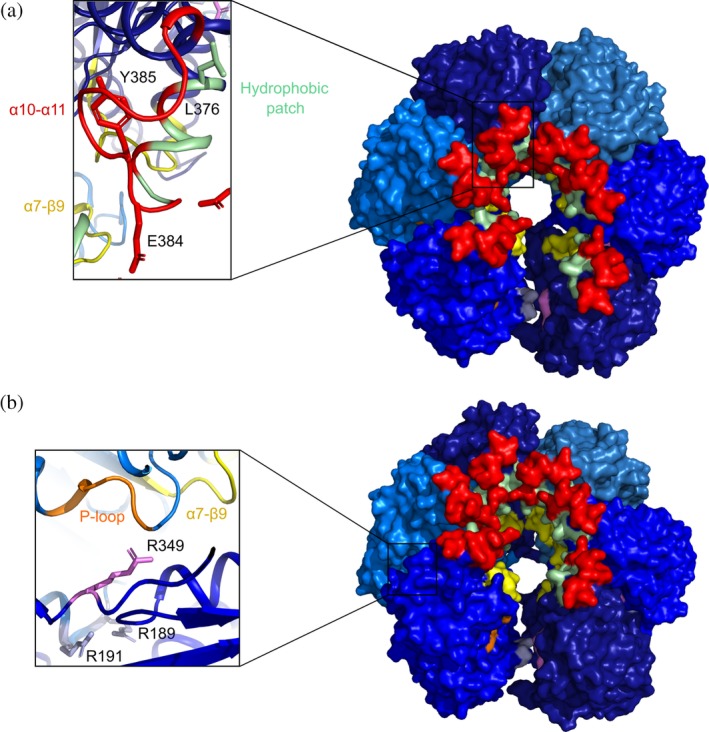
Homo‐hexameric model of InvCΔ79 apo‐form. (a) Top view of the InvCΔ79 subunits in blue tones showing the α10‐α11 loop (red), α7‐β9 loop (yellow), and hydrophobic patch (pale green). In the inlet, reported amino acids essential for chaperone–effector interaction (L376), stalk protein interaction in EscN (E384 in InvC), and type III secretion (Y385) are shown. (b) Angled view showing in the inlet the interface of two subunits with the P‐Loop (orange), the reported amino acid for ATP co‐stabilization in orthologues (R349), and the amino acids participating in intersubunit interactions (R189 and R191). The model was built by superposing the InvCΔ79 apo‐form to the E. coli EscN homo‐hexameric cylinder (PDB ID: 6NJP) using Pymol
3. DISCUSSION
The cytosolic interface of the membrane‐embedded T3SS seems to form a hetero‐complex with six or twelve ATPase subunits.11, 12, 13, 14, 32 However, in the soluble state, the isolated full‐length T3SS ATPases can form trimers, hexamers, and dodecamers.18, 19, 32 It was proposed that the N‐terminal domain of these ATPases (up to the first 79 to 104 amino acids depending on the bacterial species) is necessary for homo‐oligomerization. In this study, we show that InvCΔ79 can form dimer sets in solution in the absence of its N‐terminal domain (Figure 1), suggesting that additional domains contribute to the oligomerization state. The N‐terminal domain of orthologue T3SS ATPases can also bind to a negative regulator (OrgB in Salmonella) and display affinity for lipids.20, 21, 22, 23, 24 In our hands, the N‐terminal domain of the full length InvC suffered rapid proteolysis during and after purification (data not shown). It can be inferred that the linker to this domain is highly flexible in InvC and therefore more susceptible to proteolysis, or less stable in the absence of OrgB and lipids. Indeed, it is likely that further interaction of InvC with T3SS components including effector–chaperone complexes, the stalk protein InvI, or other sorting platform proteins (e.g., OrgB) is necessary for stable assembly of higher oligomers in the bacterial cells.
We present the first structure of InvCΔ79 alone (2.05 å resolution) and in complex with ADP (2.80 å) and ATPγS (2.65 å). Upon ligand interaction, the binding site of InvCΔ79 underwent conformational changes and opened its hydrophobic pocket for adenine stabilization (Figures 5 and 6a,b). The ligand density found in the ADP‐InvCΔ79 co‐crystal structure was not continuous, suggesting some flexibility or partial occupancy and posing a limit on the conclusions that can be drawn from this. Noteworthy, ligand densities found in E. coli ADP‐EscN, Salmonella Flagellar ADP‐FliI, and Shigella AMP‐PNP‐Spa47 crystal structures were also fragmented.16, 17, 31 Gao and coworkers have shown that ATP‐analogues can bind Spa47 with low affinity (K d ~ 21–322 μM).31 In addition, it has been reported for some T3SS ATPases that their monomers have significantly lower ATP hydrolyzing activity compared to the corresponding oligomers.19, 20, 21 Together, these results suggest that the monomeric InvCΔ79 can recognize ATPγS and ADP, although InvC oligomers might promote tighter ATP binding, probably by cooperative effects between subunits. Although the conformational changes observed in the monomeric structures might not represent the physiological state of the enzymatic active site, they provide important insights into the mechanisms of ATP binding.
Crystal structure analysis showed that the loops α5‐α6, β9‐α7, and α10‐α11 present high B‐factor values, indicating higher mobility in relation to the rest of the structure (Figure 3a). Upon interaction with ATP‐analogues, the loop β9‐α7 presents a significant conformational change and α10‐α11 is not resolved anymore, suggesting an increased protein flexibility upon ligand interaction. The changes in these loops at the ADP‐crystal structure affected partially the secondary structure of α10 and α11 (Figure 7), which is also detected by FTIR and CD spectroscopy in solution (Figure 4). Similarly, the α‐helical content of the ATPγS‐structure was scarcely affected (Figure 8), confirming the FTIR and CD spectroscopy results obtained in solution. Structural changes of the β9‐α7 and α10‐α11 loops upon ligand recognition were previously reported for the Shigella Spa47 and for the E. coli EscN ATPases.18, 31 Additionally, the integrity of the loops β9‐α7 and α10‐α11 has been shown to be essential for type III secretion.28 The conformational changes found in this study together with the functional relevance reported for β9‐α7 and α10‐α11 suggest that modifications in the structure of these loops upon ATP binding could be critical for InvC interaction with the stalk protein InvI or with chaperone and effector proteins during type III secretion.
4. MATERIALS AND METHODS
4.1. Plasmid design, gene expression, and protein purification
p‐invCΔ79 encodes the amino acids from 80 to 431 of the protein InvC from Salmonella typhimurium in fusion with a C‐terminal Strep‐tag. The construct was engineered by gene amplification from genomic DNA and ligated using BsaI restriction sites into the pASK‐IBA3+ vector (IBA GmbH, Göttingen, Germany). Escherichia coli BL21 (DE3) transformed with p‐invCΔ79 was grown to mid exponential phase at 37°C in LB medium (Luria/Miller) containing ampicillin and induced with 200 μg L−1 anhydrotetracycline (Sigma‐Aldrich, St. Louis, MO). Cells were harvested after 18 hr of expression at 20°C, resuspended in buffer B1 (100 mM Tris pH 7.5, 150 mM NaCl), lysed using the cell disruptor EmulsiFlex‐C3 (AVESTIN, Ottawa, ON, Canada), and centrifuged at 48,000g for 30 min. InvCΔ79 was pulled down by Strep‐Tactin affinity chromatography (IBA GmbH, Göttingen, Germany) and eluted with buffer B1 supplemented with 7.5 mM desthiobiotin. The protein product was further purified by SEC on a Superdex 200 10/300 GL column (GE Healthcare, Chicago, IL) equilibrated with buffer B2 (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA, and 1 mM DTT). Fractions containing InvCΔ79 were identified by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and dialyzed overnight at 4°C against buffer B3 (10 mM Tris pH 8.5, 10 mM NaCl, and 1 mM DTT).
4.2. Multi‐angle light scattering
Hundred microliters of affinity‐purified protein (9–10 mg/ml) was loaded onto a Superdex 200 10/300 GL SEC column equilibrated with buffer B2 and eluted at 0.4 ml min−1. The size‐exclusion column was coupled to a triple detector setting for UV absorption (λ = 280 nm), multi‐angle laser light scattering (MALS) with an in‐built quasi‐elastic light scattering module (Wyatt MiniDawn Treos) and a refractive index (RI) instrument (Wyatt Optilab T‐rEX). The RI increment of the protein was taken as 0.185 ml g−1 (25°C at 659 nm). Data analysis was done using ASTRA 7.0.1.24 (Wyatt Technologies, Santa Barbara, CA).
4.3. Native MS
Protein samples were buffer exchanged into 150 mM ammonium acetate, 1 mM DTT pH 8.0 with Micro Biospin 6 columns (Bio‐Rad, Hercules, CA) and analyzed at ~10 μM with a Q‐Tof 2 instrument (Waters, Manchester, UK and MS Vision, Almere, the Netherlands) modified for high mass.33
The nano‐electrospray ionization was accomplished with gold‐coated glass capillaries produced in‐house34 in positive ion mode. Source pressure was set to 10 mbar and voltages of 1,300 and 130 V were applied to the capillary and sample cone in given order. The argon filled collision cell was operated at a pressure between 1.7 and 1.8 × 10−2 mbar and an accelerating voltage of 50 V. Raw data were calibrated with cesium iodide clusters (25 mg/ml) and analyzed with Mass Lynx (Waters). Average masses, standard deviations, and full width at half maxima from multiple measurements were calculated (Table S1) and complex stoichiometry was confirmed by MS/MS (data not shown).
4.4. FTIR spectroscopy
FTIR spectra were recorded in transmission mode using a Bruker VERTEX 70 FTIR spectrometer (Bruker, Ettlingen, Germany) equipped with an AquaSpec cell (Bruker) and a liquid nitrogen cooled MCT detector. InvCΔ79 (0.2 mM) in buffer B3 was mixed with magnesium chloride and either ADP, AMP‐PNP (Sigma‐Aldrich), or ATPγS (Jena Bioscience, Jena, Germany) in a ratio of 1:10:10, respectively. All samples were degassed, equilibrated at room temperature, and measured against buffer complemented with the respective ligand and magnesium chloride at the same concentration. Spectra were recorded at 25°C and the sample temperature was controlled with a Huber Ministat 125 with Pilot ONE (Huber, Offenburg, Germany). The spectral resolution was set to 2 cm−1 and a total of 64 scans were averaged before Fourier transformation. Analysis of the spectra was performed using OPUS 7.8 (Bruker). Atmospheric compensation (H2O, CO2, and aqueous solution) was applied to the background‐corrected sample spectra. Vector normalization was performed between 1,710 and 1,500 cm−1 to correct for concentration differences. Difference spectra were obtained by subtraction of the InvCΔ79 apo‐form spectrum from the spectra of the ligand‐containing samples.
4.5. CD spectroscopy
The far‐UV CD spectra (200–260 nm) were recorded using an Aviv Model 425 Circular Dichroism Spectrometer (Aviv Biomedical, Lakewood, NJ). InvCΔ79 (6.6 μM) was mixed with magnesium acetate and either ADP, AMP‐PNP, or ATPγS in a ratio of 1:10:10 (buffer: 10 mM NaPO4 pH 8.5, 10 mM NaF). Spectra were recorded at 1 nm intervals using an averaging time of 9 s and a bandwidth of 1 nm. The measurements were performed at 25°C using a quartz cuvette with a path length of 1 mm. Spectra of the reference samples (buffer complemented with the respective ligand and magnesium acetate in the same concentration) were collected under the same conditions. Each sample spectrum was background‐corrected by subtracting the respective reference sample spectrum and expressed in units of mean residue ellipticity (deg cm2 dmol−1).
4.6. Crystallization and structure determination
InvCΔ79 was crystallized alone and in the presence of either 1 mM ATPγS, ADP or AMP‐PNP, and 1 mM magnesium chloride. Purified and dialyzed protein (8 mg/ml) was mixed with equal volumes of reservoir solution and crystals were grown at 19°C using sitting drop vapor diffusion. Crystal soaking experiments were conducted by transferring InvCΔ79 crystals to a drop of reservoir buffer containing up to 5 mM of ATP‐analogue and 5 mM magnesium chloride, incubating for 16 hr at 19°C. The best diffracting crystals were from the reservoir solutions containing 0.8 M succinic acid pH 7.0 for the apo‐form, 3 M sodium chloride pH 7.5 and 0.1 M Tris for the crystal soaked into the ATPγS containing mix, and 1.6 M magnesium sulfate and 0.1 M MES pH 6.5 for the co‐crystallization with the ADP containing mix. Crystals were cryo‐protected by soaking in buffer containing 30% (v/v) glycerol and flash frozen in liquid nitrogen. Crystal screening and data collection were performed at 100 K at the beam line P11 at the PETRA III storage ring (DESY, Hamburg, Germany).
Diffraction data were indexed, integrated, and scaled using XDS.35 Initial phases for the InvCΔ79 apo‐form were acquired by molecular replacement using Spa47Δ83 (PDB ID: 5SWJ)26 with Phaser.36 Further analysis of the ligand‐bound forms was performed by molecular replacement using the InvCΔ79 apo‐form. All crystals have one copy of the protein in the asymmetric unit with 59% solvent content. The models were manually adjusted and refined using Coot version 0.8.9.137 and PHENIX.refine version 1.14‐3260,38 including final TLS refinement. Cell parameters and statistics of data collection and refinement are provided in Table 1. Figures were prepared with Pymol version 2.2.3.39
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Table S1. Theoretical and measured masses of InvCΔ79
Figure S1. Multi‐sequence alignment of InvC and orthologues. Amino acid sequences of ATPases from the T3SS injectiosomes (Salmonella SPI‐1 InvC, Shigella Spa47, Yersinia YscN, E. coli EscN, Salmonella SPI‐2 SscN and Pseudomonas HrcN), the flagella apparatus (FliI) and the β subunit of the F1‐ATPase (F1‐b) were aligned. Identical residues are highlighted in red and conserved residues are shown in red letters. Secondary structure was assigned based on the InvCΔ79 crystal structure. Alignment was performed with Clustal Omega and ESPript 3.0.
Figure S2. Coomassie‐stained SDS‐PAGE gel of purified InvCΔ79. Elution products from Strep‐affinity purification and from SEC peaks I and II are shown. Molecular mass markers are included in lane M.
Figure S3. Structural comparison of ATPases. 3D structural alignment of Salmonella InvCΔ79 apo‐form (blue) and (A) Shigella Spa47Δ83 (yellow, PDB ID:5YBH) or (B) E. coli Δ102‐ V393P (light pink, PDB ID:2OBL).
Figure S4. Fo‐Fc electron density omit map of ligands. (A) Map contoured at 3.0σ for ATPγS, magnesium ion and water of the ligand binding site. (B) Map contoured at 1.8σ for ADP and water of the ligand binding site.
Figure S5. Conformational changes of the luminal loops upon ATP‐analogue interaction. Electron density maps with 2Fo‐Fc contour at 0.8 to 1.5σ.
ACKNOWLEDGEMENTS
We thank J. Börnicke for providing the p‐invCΔ1‐79 construct, C. Jeffries for the MALS analysis, G. Yang for experimental and analytical support and J. de Diego for her useful comments and critical reading of the manuscript. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III storage ring. The X‐ray diffraction data sets were collected at the beam line P11 and we thank O. Lorbeer and A. Burkhardt for the user assistance. The SEC‐MALS‐RI data were measured at the European Molecular Biology bioSAXS beam line P12 operated by EMBL Hamburg and we thank T. Graewert and M. A. Graewert for their assistance with the equipment. This work was funded by the European Research Council under the European Community's Seventh Frameweork Programme (FP7/2007–2013, grant no. 311374) and the Helmholtz Association funding agency IVF (Initiative and Networking Fund). The Heinrich Pette Institute, Leibniz Institute for Experimental Virology is supported by the Free and Hanseatic City of Hamburg and the German Federal Ministry of Health. C.U. acknowledges funding by the Leibniz‐Gemeinschaft through SAW‐2014‐HPI‐4 grant. I.B. and L.F. are funded by the "Promotion of young CSSB scientists" program by the Joachim Herz Stiftung.
Bernal I, Römermann J, Flacht L, Lunelli M, Uetrecht C, Kolbe M. Structural analysis of ligand‐bound states of the Salmonella type III secretion system ATPase InvC. Protein Science. 2019;28:1888–1901. 10.1002/pro.3704
Significance statement: Pathogenic Gram‐negative bacteria use a conserved ATPase to aid the delivery of virulence factors across the type III secretion system (T3SS) during the infection of human cells. Our high‐resolution structures of the Salmonella ATPase, named InvC, reveal how ATP‐analogues might induce allosteric regulation of its ATPase activity by moving two loops involved in the secretion of virulence factors. These observations shed novel structural and functional insights into the mechanism of activation of T3SS‐associated ATPases.
Data Availability Statement:The atomic coordinates and structure factors have been deposited in the Protein Data Bank (http://wwpdb.org/) with accession codes 6RAE (InvCΔ79 apo‐form), 6RAD (InvCΔ79‐ADP), and 6SDX (InvCΔ79‐ATPγS).
Funding information Free and Hanseatic City of Hamburg; German Federal Ministry of Health; Helmholtz Association funding agency IVF (Initiative and Networking Fund); "Promotion of young CSSB scientists" program by the Joachim Herz Stiftung; Leibniz‐Gemeinschaft, Grant/Award Number: SAW‐2014‐HPI‐4; European Research Council under the European Community's Seventh Frameweork Programme
DATA AVAILABILITY STATEMENT
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (http://wwpdb.org/) with accession codes 6RAE (InvCΔ79 apo‐form), 6RAD (InvCΔ79‐ADP), and 6SDX (InvCΔ79‐ATPγS).
REFERENCES
- 1. Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coburn B, Sekirov I, Finlay BB. Type III secretion systems and disease. Clin Microbiol Rev. 2007;20:535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marshall NC, Finlay BB. Targeting the type III secretion system to treat bacterial infections. Expert Opin Ther Targets. 2014;18:137–152. [DOI] [PubMed] [Google Scholar]
- 4. Ghosh P. Process of protein transport by the type III secretion system. Microbiol Mol Biol Rev. 2004;68:771–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luo Y, Bertero MG, Frey EA, et al. Structural and biochemical characterization of the type III secretion chaperones CesT and SigE. Nat Struct Biol. 2001;8:1031–1036. [DOI] [PubMed] [Google Scholar]
- 6. Thomas NA, Deng W, Puente JL, et al. CesT is a multi‐effector chaperone and recruitment factor required for the efficient type III secretion of both LEE‐ and non‐LEE‐encoded effectors of enteropathogenic Escherichia coli . Mol Microbiol. 2005;57:1762–1779. [DOI] [PubMed] [Google Scholar]
- 7. Akeda Y, Galán JE. Chaperone release and unfolding of substrates in type III secretion. Nature. 2005;437:911–915. [DOI] [PubMed] [Google Scholar]
- 8. Stebbins CE, Galan JE. Maintenance of an unfolded polypeptide by a cognate chaperone in bacterial type III secretion. Nature. 2001;414:77–81. [DOI] [PubMed] [Google Scholar]
- 9. Stebbins CE, Galán JE. Priming virulence factors for delivery into the host. Nat Rev Mol Cell Biol. 2003;4:738–744. [DOI] [PubMed] [Google Scholar]
- 10. Lara‐Tejero M, Kato J, Wagner S, Liu X, Galan JE. A sorting platform determines the order of protein secretion in bacterial type III systems. Science. 2011;331:1188–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Lara‐Tejero M, Bewersdorf J, Galan JE. Visualization and characterization of individual type III protein secretion machines in live bacteria. Proc Natl Acad Sci U S A. 2017;114:6098–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diepold A, Sezgin E, Huseyin M, Mortimer T, Eggeling C, Armitage JP. A dynamic and adaptive network of cytosolic interactions governs protein export by the T3SS injectisome. Nat Commun. 2017;8:15940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu B, Lara‐Tejero M, Kong Q, Galan JE, Liu J. In situ molecular architecture of the salmonella type III secretion machine. Cell. 2017;168:1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu B, Morado DR, Margolin W, et al. Visualization of the type III secretion sorting platform of Shigella flexneri. Proc Natl Acad Sci U S A. 2015;112:1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sauer RT, Bolon DN, Burton BM, et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell. 2004;119:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zarivach R, Vuckovic M, Deng W, Finlay BB, Strynadka NC. Structural analysis of a prototypical ATPase from the type III secretion system. Nat Struct Mol Biol. 2007;14:131–137. [DOI] [PubMed] [Google Scholar]
- 17. Imada K, Minamino T, Tahara A, Namba K. Structural similarity between the flagellar type III ATPase FliI and F1‐ATPase subunits. Proc Natl Acad Sci U S A. 2007;104:485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Majewski DD, Worrall LJ, Hong C, et al. Cryo‐EM structure of the homohexameric T3SS ATPase‐central stalk complex reveals rotary ATPase‐like asymmetry. Nat Commun. 2019;10:626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burgess JL, Jones HB, Kumar P, et al. Spa47 is an oligomerization‐activated type three secretion system (T3SS) ATPase from Shigella flexneri. Protein Sci. 2016;25:1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pozidis C, Chalkiadaki A, Gomez‐Serrano A, et al. Type III protein translocase: HrcN is a peripheral ATPase that is activated by oligomerization. J Biol Chem. 2003;278:25816–25824. [DOI] [PubMed] [Google Scholar]
- 21. Case HB, Dickenson NE. MxiN differentially regulates monomeric and oligomeric species of the Shigella type three secretion system ATPase Spa47. Biochemistry. 2018;57:2266–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akeda Y, Galán JE. Genetic analysis of the Salmonella enterica type III secretion‐associated ATPase InvC defines discrete functional domains. J Bacteriol. 2004;186:2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Auvray F, Ozin AJ, Claret L, Hughes C. Intrinsic membrane targeting of the flagellar export ATPase FliI: Interaction with acidic phospholipids and FliH. J Mol Biol. 2002;318:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bernal I, Bornicke J, Heidemann J, Svergun D, Horstmann JA, Erhardt M, Tuukkanen A, Uetrecht C, Kolbe M. Molecular Organization of Soluble Type III Secretion System Sorting Platform Complexes. J. Mol. Biol. 2019; 10.1016/j.jmb.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 25. Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha‐ and beta‐subunits of ATP synthase, myosin, kinases and other ATP‐requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burgess JL, Burgess RA, Morales Y, Bouvang JM, Johnson SJ, Dickenson NE. Structural and biochemical characterization of Spa47 provides mechanistic insight into type III secretion system ATPase activation and Shigella virulence regulation. J Biol Chem. 2016;291:25837–25852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Allison SE, Tuinema BR, Everson ES, et al. Identification of the docking site between a type III secretion system ATPase and a chaperone for effector cargo. J Biol Chem. 2014;289:23734–23744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kato J, Lefebre M, Galan JE. Structural features reminiscent of ATP‐driven protein translocases are essential for the function of a type III secretion‐associated ATPase. J Bacteriol. 2015;197:3007–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Demler HJ, Case HB, Morales Y, Bernard AR, Johnson SJ, Dickenson NE. Interfacial amino acids support Spa47 oligomerization and shigella type three secretion system activation. Proteins. 2019; 1–12. 10.1002/prot.25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barth A, Zscherp C. What vibrations tell us about proteins. Q Rev Biophys. 2002;35:369–430. [DOI] [PubMed] [Google Scholar]
- 31. Gao X, Mu Z, Yu X, et al. Structural insight into conformational changes induced by ATP binding in a type III secretion‐associated ATPase from Shigella flexneri. Front Microbiol. 2018;9:1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muller SA, Pozidis C, Stone R, et al. Double hexameric ring assembly of the type III protein translocase ATPase HrcN. Mol Microbiol. 2006;61:119–125. [DOI] [PubMed] [Google Scholar]
- 33. van den Heuvel RH, van Duijn E, Mazon H, et al. Improving the performance of a quadrupole time‐of‐flight instrument for macromolecular mass spectrometry. Anal Chem. 2006;78:7473–7483. [DOI] [PubMed] [Google Scholar]
- 34. Pogan R, Schneider C, Reimer R, Hansman G, Uetrecht C. Norovirus‐like VP1 particles exhibit isolate dependent stability profiles. J Phys Condens Matter. 2018;30:064006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 38. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. 2012;D68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeLano WL. The PyMOL molecular graphics system. San Carlos: Delano Scientific, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Theoretical and measured masses of InvCΔ79
Figure S1. Multi‐sequence alignment of InvC and orthologues. Amino acid sequences of ATPases from the T3SS injectiosomes (Salmonella SPI‐1 InvC, Shigella Spa47, Yersinia YscN, E. coli EscN, Salmonella SPI‐2 SscN and Pseudomonas HrcN), the flagella apparatus (FliI) and the β subunit of the F1‐ATPase (F1‐b) were aligned. Identical residues are highlighted in red and conserved residues are shown in red letters. Secondary structure was assigned based on the InvCΔ79 crystal structure. Alignment was performed with Clustal Omega and ESPript 3.0.
Figure S2. Coomassie‐stained SDS‐PAGE gel of purified InvCΔ79. Elution products from Strep‐affinity purification and from SEC peaks I and II are shown. Molecular mass markers are included in lane M.
Figure S3. Structural comparison of ATPases. 3D structural alignment of Salmonella InvCΔ79 apo‐form (blue) and (A) Shigella Spa47Δ83 (yellow, PDB ID:5YBH) or (B) E. coli Δ102‐ V393P (light pink, PDB ID:2OBL).
Figure S4. Fo‐Fc electron density omit map of ligands. (A) Map contoured at 3.0σ for ATPγS, magnesium ion and water of the ligand binding site. (B) Map contoured at 1.8σ for ADP and water of the ligand binding site.
Figure S5. Conformational changes of the luminal loops upon ATP‐analogue interaction. Electron density maps with 2Fo‐Fc contour at 0.8 to 1.5σ.
Data Availability Statement
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (http://wwpdb.org/) with accession codes 6RAE (InvCΔ79 apo‐form), 6RAD (InvCΔ79‐ADP), and 6SDX (InvCΔ79‐ATPγS).