

Abstract
ATP‐citrate lyase (ACLY) catalyzes production of acetyl‐CoA and oxaloacetate from CoA and citrate using ATP. In humans, this cytoplasmic enzyme connects energy metabolism from carbohydrates to the production of lipids. In certain bacteria, ACLY is used to fix carbon in the reductive tricarboxylic acid cycle. The carboxy(C)‐terminal portion of ACLY shows sequence similarity to citrate synthase of the tricarboxylic acid cycle. To investigate the roles of residues of ACLY equivalent to active site residues of citrate synthase, these residues in ACLY from Chlorobium limicola were mutated, and the proteins were investigated using kinetics assays and biophysical techniques. To obtain the crystal structure of the C‐terminal portion of ACLY, full‐length C. limicola ACLY was cleaved, first non‐specifically with chymotrypsin and subsequently with Tobacco Etch Virus protease. Crystals of the C‐terminal portion diffracted to high resolution, providing structures that show the positions of active site residues and how ACLY tetramerizes.
Keywords: CoA‐binding, proteolysis, site‐directed mutagenesis
1. INTRODUCTION
In humans, ATP‐citrate lyase (ACLY, EC 2.3.3.8) is the cytoplasmic enzyme connecting energy metabolism from carbohydrates to the production of lipids. Within mitochondria, citrate synthase (CS) converts acetyl‐CoA and oxaloacetate to CoA and citrate as part of the tricarboxylic acid (TCA) cycle. While acetyl‐CoA cannot cross the mitochondrial membrane, citrate can be exported to the cytoplasm via transporters. In the cytoplasm, ACLY converts citrate back to acetyl‐CoA using CoA and ATP. This acetyl‐CoA can be used to synthesize cholesterol or fatty acids in the cytoplasm or to acetylate histones in the nucleus.1 Because ACLY produces cholesterol and fatty acids, the enzyme has been suggested as a target for drug design to treat cancer and obesity.2, 3
ACLY is a member of the acyl‐CoA synthetase (NDP‐forming) superfamily.4 The prototype for this superfamily is succinyl‐CoA synthetase (SCS) and the N‐terminal portion of human ACLY (hACLY) is similar to SCS (Figure 1). In the presence of Mg2+, ACLY catalyzes the reaction: citrate + CoA + ATP → ADP + Pi + acetyl‐CoA + oxaloacetate, while SCS catalyzes the reaction: succinate + CoA + ATP/GTP → ADP/GDP + Pi + succinyl‐CoA. Both SCS and ACLY have a catalytic histidine that transfers the phosphoryl group between nucleotide triphosphate and organic acid.5, 6 Structures of the N‐terminal portion of hACLY in complex with citrate7, 8 and Mg2+‐ADP9 have been determined using X‐ray crystallography. As in SCS, the distance between the two binding sites, site I and site II, is large, ~40 å. In both SCS and ACLY, the active site histidine is part of a loop thought to swing from one binding site to the other.8, 10 The phosphohistidine segment of the related acetyl‐CoA synthetase has been observed at each of the binding sites.11 Addition of the phosphoryl group to the organic acid makes the compound susceptible to attack by CoA to form the thioester, succinyl‐CoA in the case of SCS, citryl‐CoA in the case of ACLY, and acetyl‐CoA in the case of acetyl‐CoA synthetase.
Figure 1.
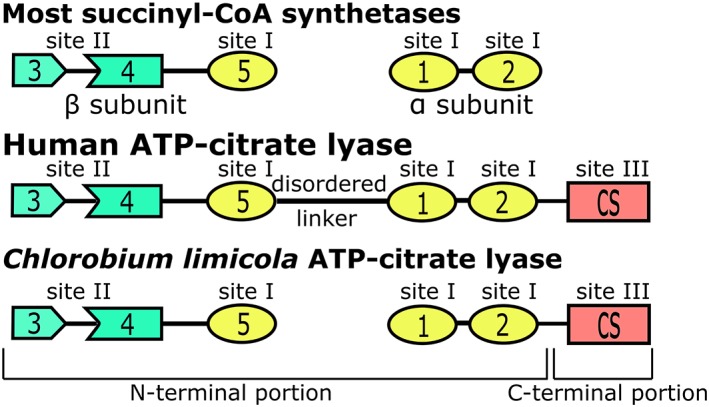
Domains of succinyl‐CoA synthetases and ATP‐citrate lyases. Members of the acyl‐CoA synthetase (NDP‐forming) superfamily have five domains in common. In most succinyl‐CoA synthetases, domains 1 and 2 form the α‐subunit, while domains 3, 4, and 5 form the β‐subunit. ATP‐citrate lyases also have a domain showing sequence similarity to citrate synthase (CS). Human ATP‐citrate lyase is a homotetramer where all domains are in a single polypeptide chain, but C. limicola ATP‐citrate lyase is a heterooctamer with two different polypeptide chains. Sites I, II, and III are the three catalytic sites. Site I is at the interface of domains 1, 2, and 5, site II is within the ATP‐grasp fold formed by domains 3 and 4, and site III is within the CS domain
The carboxy‐terminal (C‐terminal) portion of ACLY shows sequence similarity to CS of the TCA cycle (Figure 1). CS catalyzes first condensation of oxaloacetate and acetyl‐CoA to form citryl‐CoA, then hydrolysis of citryl‐CoA to citrate and CoA. The portion of ACLY showing similarity to CS would be expected to catalyze cleavage of citryl‐CoA to oxaloacetate and acetyl‐CoA (site III). CS is multimeric, existing as a dimer or hexamer, where the hexamer is a trimer of dimers.12 In either form, the active site is at the dimer interface.12, 13 Because hACLY exists as a homotetramer with 1,101 amino acid residues in each polypeptide, the C‐terminal portion would be expected to provide at least one of the sites of dimerization. Recent biophysical investigations support this hypothesis.14
To investigate the roles of residues of ACLY equivalent to the active site residues of CS (Figure S1), these residues were mutated, and the proteins investigated using kinetics assays and biophysical techniques. ACLY from the green sulfur bacterium Chlorobium limicola was used for these experiments. Chlorobium limicola uses ACLY in the reductive TCA cycle to fix carbon.15 Chlorobium limicola ACLY (ClACLY) is similar to hACLY, but is a heterooctamer with four copies of two different subunits, A and B.16 Subunit B and the N‐terminal portion of subunit A are similar to the N‐terminal portion of hACLY (Figure 1). The C‐terminal portion of subunit A is similar to the C‐terminal portion of hACLY and CS. To obtain the crystal structure of the C‐terminal portion of ACLY (prior to the recent publications17, 18), full‐length ClACLY was cleaved specifically with Tobacco Etch Virus (TEV) protease. Crystals diffracted to high resolution, providing structures that show the positions of active site residues and how ACLY tetramerizes.
2. RESULTS
2.1. Analysis of mutant proteins
Specific activities were measured for ClACLY and the mutant proteins, H491A, D543A, and the H491A&D543A double mutant. While ClACLY and the H491A mutant had specific activities of 1.4 ± 0.1 and 0.5 ± 0.1 U/mg, respectively, neither the D543A mutant nor the double mutant showed activity. Kinetics assays for ClACLY and the H491A mutant showed only small differences for ATP, CoA, and citrate (Table 1).
Table 1.
Kinetic parameters for ClACLY and the H491A mutant
| k cat (s−1) | K m app for ATP (mM) (R 2 value) | K m app for CoA (μM) (R 2 value) | K m app for citrate (mM) (R 2 value) | |
|---|---|---|---|---|
| Wild type | 0.022 ± 0.001 | 0.29 ± 0.04 (0.93) | 14.3 ± 1.6 (0.92) | 1.6 ± 0.2 (0.96) |
| H491A mutant | 0.008 ± 0.001 | 0.28 ± 0.04 (0.90) | 17.6 ± 2.4 (0.92) | 1.6 ± 0.4 (0.80) |
Thermal stabilities of ClACLY and the three mutants were tested using differential scanning fluorimetry (DSF). The melting temperature (T m) for ClACLY measured in 10 mM Tris HCl pH 7.0 was 48°C. None of the mutants showed significant changes from the T m of ClACLY under similar conditions.
Isothermal titration calorimetry proved that the double mutant still bound CoA. Figure S2 depicts the plots and Table 2 presents the fitted parameters. Both ClACLY and the double mutant bind CoA, although the conditions that best showed the binding were somewhat different.
Table 2.
Thermodynamic data for binding of CoA
| ClACLY | H491A&D543A double mutant | |
|---|---|---|
| Binding sites | 2.14 ± 0.21 | 2.00 ± 0.01 |
| Association constant (K b) | (2.03 ± 0.39) × 105 M−1 | (6.27 ± 0.71) × 105 M−1 |
| Dissociation constant (K d) | 4.93 ± 0.91 μM | 1.59 ± 0.18 μM |
| Binding enthalpy (ΔH) | −6.78 ± 0.09 kcal Mol−1 | −4.50 ± 0.08 kcal Mol−1 |
2.2. Structure determination of the C‐terminal portion of ClACLY
Initial crystals of chymotrypsin‐cleaved ClACLY grew from solutions containing acetyl‐CoA, suggesting that the C‐terminal portion crystallized. Mass spectrometry identified tryptic fragments that included residues 369–608 of subunit A. Residues 369–608 would have a mass of 26.3 kDa, but the polypeptide ran with a relative molecular weight of 28 kDa on sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE). PeptideCutter19 indicated two possible cleavage sites for chymotrypsin, one between 344 and 345 (29.1 kDa) and one between 354 and 355 (28.0 kDa). The shorter fragment was produced by truncating the gene and adding codons for a histidine‐tag at the N‐terminus. This fragment could be purified but did not behave well in crystallization experiments. Interestingly, the C‐terminal portion ran as a tetramer on gel filtration (data not shown).
Crystals of the C‐terminal portion of ClACLY cleaved by TEV protease (CterClACLY) contained residues 347–608 of subunit A. The best crystal diffracted to 1.9 å‐resolution (trigonal Table S2) and the second best to 2.05 å‐resolution (triclinic). Four copies of the 28.8 kDa polypeptide were predicted in the asymmetric unit of the trigonal crystal form and 12 in the triclinic. The initial model consisted of 705 alanine residues in space group P32. Autobuilding generated a tetramer with 255, 257, 253, and 248 residues in four chains, plus six additional residues. Refining and rebuilding resulted in nearly complete chains. In the triclinic crystal, two protomers display disorder. Because the models are similar, the P32 model is described (Table S2).
CterClACLY forms a tetramer with 222 symmetry (Figure 2). The N‐termini lie close to one 2‐fold axis and the C‐termini close to another. In one orientation, the tetramer appears elliptic, but the perpendicular view shows four domains extending from the central core. The P450 domains (CATH Classification 1.10.230.1022), which extend from 447 to 555, are swung away from the core of the structure. The tetramer has five domains, one forming the core and the other four extending from it.
Figure 2.
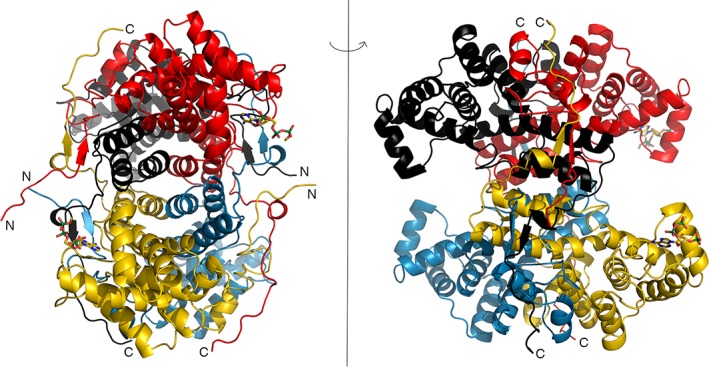
Crystal structure of the CterClACLY. The protein is a tetramer that possesses 222 symmetry. Both views are along twofold axes, with the model rotated 90° about the vertical axis between the views. The four polypeptide chains are colored differently in the ribbon diagrams, and the termini that are visible are labelled N or C. The two 3′‐phosphoadenosine portions of acetyl‐CoA are drawn as stick models. This figure and Figures 3 and 4 were drawn using PYMOL21
CterClACLY is predominantly α‐helical. However, residues 594–596 form a parallel β‐sheet with residues 355–357 of a different protomer (Figure 2). The polypeptide extends from residue 596 to the C‐terminus, wrapping around the other protomer. In this region and the three equivalent regions of the tetramer, residues from three different polypeptide chains interact. In the tetramer, each of the polypeptide chains interacts with all three of the other polypeptides.
Since acetyl‐CoA had been included in the crystallization experiment, it was possible that its binding site could be identified in the electron density maps. Adenine, ribose‐3‐phosphate, and diphosphate of the 3′‐phosphoadenosine end of acetyl‐CoA were fitted to two protomers (chains A and D). The diphosphate groups of the two acetyl‐CoA molecules show different conformations, influenced by crystal‐packing interactions. There was insufficient electron density to fit even the adenine base to chain B, the protomer that has no crystal‐packing interactions near the CoA‐binding site. There was no evidence of binding to chain C. Five phosphate ions and 16 molecules of glycerol were included in the model.
3. DISCUSSION
3.1. Identification of the active sites
The crystallographic model includes the 3′‐phosphoadenosine end of acetyl‐CoA at two active sites. Since acetyl‐CoA is a product of the reaction, it could bind if the concentration were high enough to lead to product inhibition; and the goal was to see the binding site. Acetyl‐CoA was used rather than CoA in case interactions of the acetyl group with residues of the active site could be seen. Isothermal titration calorimetry had shown that the N‐terminal portion of hACLY does not bind CoA, leading to the conclusion that the CoA‐binding site must be in the C‐terminal portion.7 However, a second possibility is that both the N‐ and C‐terminal portions are needed to form the CoA‐binding site. This is what differential scanning calorimetry indicated.14 In those experiments, CoA stabilized full‐length hACLY, but did not stabilize the individual N‐terminal portion nor the C‐terminal portion.
The 3′‐phosphoadenosine end of acetyl‐CoA interacts with the protein primarily through van der Waals interactions. L538 packs against adenine, while P485 packs near N6A. There is a weak hydrogen bond (3.2 å) donated by the amide of V486 to N1A. The only charged interaction supported by electron density is between K534 and the 3′‐phosphate. The diphosphate of CoA extends out of the active site cleft where electron density disappears, indicating that the rest of the molecule is disordered.
Phosphate ions are bound to all active sites. Phosphate is neither a substrate nor a product of the reaction catalyzed by CterClACLY. Its negative charge leads to binding to positively charged R568 and, in the P1 crystal form, a second phosphate ion is detected, bound to H415, H491, and R502 of nine of the protomers. Some residual electron density in the active site cleft was fitted with molecules of glycerol.
3.2. Comparison of ClACLY with CSs of the TCA cycle
ClACLY shows quaternary structure that has not been observed in structures of CSs. When one protomer of the CterClACLY is superposed on chicken CS (Protein Data Bank [PDB] rcsb.org 23 identifier (ID): 6CSC 20), ClACLY appears to be missing certain α‐helices of CS (Figure S3). In the ClACLY tetramer, α‐helices from a second protomer superpose with these α‐helices. A sequence alignment based on superposing the α‐helices in the two structures highlights the pseudosymmetry within CS (Figure S1). The C‐terminal copy has residues of both the CS domain and the smaller P450 domain, residues 271 to 388. The N‐terminal copy has only residues of the CS domain: it is missing the P450 domain.
In ClACLY, two pairs of protomers dimerize to form the tetramer in a similar way that the two protomers of CS dimerize. While the CS dimer has two active sites, the C‐terminal ClACLY tetramer has four. This is because the N‐terminal portion of CS lacks the P450 domain required to form an active site.
The hexameric form of CS found in Escherichia coli is allosterically inhibited by reduced nicotinamideadenine dinucleotide (NADH).24 The structure of NADH in complex with mutated E. coli CS shows the allosteric site at the interface of pairs of dimers, but NADH is predominantly bound to residues of the N‐terminal portion of CS.25 When a CoA analogue was soaked into the same crystal form, it bound at the same site, rather than at the active site.26 This allosteric site is located where the second P450 domain is missing from CS (Figure S3). CS could have lost this second P450 domain to allow allosteric regulation. However, dimeric forms of CS are not known to be regulated by allostery.
In the crystal structure of CterClACLY, the adenine base of CoA lies near the loop equivalent to where it binds in CS (Figure 3). The specific interactions that orient adenine in the CoA‐binding site of CS do not occur in the crystal structure of the C‐terminal portion of ClACLY. While the diphosphate of CoA extends out of the active site cleft, CoA bound to CS has a bent conformation extending into the active site.
Figure 3.
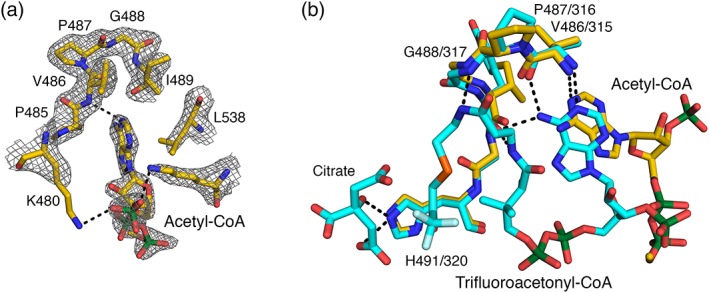
Binding of the 3′‐phosphoadenosine end of acetyl‐CoA to the C‐terminal portion of ClACLY. (a). The portion of acetyl‐CoA that could be modeled into electron density near chain A and the surrounding residues are drawn as stick models, colored according to atom type. Electron density from the 2Fo‐Fc map is displayed in grey, contoured at 1 rmsd. (b). The 3′‐phosphoadenosine end of acetyl‐CoA binds in a similar location to the acetyl‐CoA analogue, trifluoroacetonyl‐CoA, in chicken CS (PDB ID: 6CSC 20). The crystal structures were superposed based on main chain atoms of residues 485–495 of chain A of ClACLY and residues 314–324 of chain A of citrate synthase. Colors are similar to those in part A, but the carbon atoms in 6CSC are cyan. For reference, citrate is shown bound to CS. Only one of the two conformations of trifluoroacetonyl‐CoA is drawn. In both A and B, hydrogen bonds are represented by dashed lines. The adenine portion of acetyl‐CoA has only one hydrogen bond with the protein and interacts primarily via van der Waals interactions
CS adopts both an open and a closed conformation. When oxaloacetate and acetyl‐CoA are bound, CS closes to catalyze the condensation to form citryl‐CoA and the subsequent hydrolysis. In structures that show the closed conformation of CS, CoA analogues bind to the P450 domain, but also interact with residues of the second protomer in the dimer. The structure of CterClACLY is in the open conformation, despite the addition of acetyl‐CoA and the binding of phosphate ions in the active site. Residues that bind these phosphate ions, H415, H491, R502, and R568, are equivalent to residues that interact with citrate in the complex of chicken CS with trifluoroacetonyl‐CoA and citrate (PDB ID 6CSC 20): H238, H320, R329, and R401. A third arginine residue in chicken CS, R421', also interacts with citrate, the prime indicating that it is from the other protomer in the CS dimer. The equivalent arginine in CterClACLY, R588, adopts two conformations, one hydrogen bonding to D591 of the same chain and the second hydrogen bonding to D362 of a third protomer. It does not interact with either phosphate ion.
Recent publications also showed ACLY in an open conformation with CoA bound, and two structures in a closed conformation one with malate and acetyl‐CoA bound and one with citrate bound.17, 18 No structure had been determined with only the addition of acetyl‐CoA in the absence of an organic acid. The open conformation in the presence of acetyl‐CoA proves that binding of an appropriate organic acid is what induces a closed conformation, regardless of whether acetyl‐CoA is bound.
3.3. Site‐directed mutagenesis of the catalytic residues
H491 and D543 were predicted to be catalytic residues based on sequence comparisons with CSs. These residues are equivalent to H320 and D375 of chicken CS (Figure S1), two residues important in binding substrates and catalyzing the condensation of oxaloacetate and acetyl‐CoA to form citryl‐CoA.13 H320 hydrogen bonds to the carbonyl of oxaloacetate (PDB ID: 1CSH 27) or the hydroxyl of citrate (PDB ID: 6CSC 20) and donates a proton to the carbonyl oxygen that becomes the hydroxyl of citrate.28 In the complex of CS with the acetyl‐CoA analogue amidocarboxymethyldethia coenzyme A (PDB ID: 1CSH 27), D375 accepts a hydrogen bond from the amide nitrogen. D375 acts as a base to remove the proton from the acetyl group in the enolization of acetyl‐CoA.28 H491 and D543 of ClACLY would be expected to serve similar roles to H320 and D375 in catalysis of the cleavage of citryl‐CoA to form acetyl‐CoA and oxaloacetate.
The specific activity measured for ClACLY, 1.4 ± 0.1 U/mg, is similar to values of 1.1 and 1.30 U/mg previously measured.29, 30 Mutation of H491 to alanine led to a significant loss of activity (Table 1). K M app for ATP and citrate remain the same, as expected because both binding sites are distant from the mutation.7, 9 K M app for CoA increased, but not significantly, showing that the protein folded correctly and loss of activity is due to loss of the side chain. Mutagenesis of the equivalent H in pig CS to G, R, N, or Q led to a decrease of greater than 99% in k cat.31 While CS uses H to bind oxaloacetate, this interaction would be less important in ClALCY, which binds citryl‐CoA and releases oxaloacetate. The interaction with the hydroxyl of citryl‐CoA could be replaced by interaction with water or a different residue of the H491A mutant protein.
Mutation of D543 to A led to no measurable activity, so the side chain of D543 is more critical than that of H491 for ClACLY. The result is consistent with neither of the D375N nor D375Q mutants of pig CS being active.32 Since N and Q can still hydrogen bond but neither has a dissociable proton, the acid–base property of D375 was concluded to be most important. This interpretation was supported by the higher activity of D375E mutant CS, at 0.2% that of wild type. Since the D375G mutant protein had 0.06% activity, it was thought that the lack of a side‐chain allowed a water molecule access to the active site to act as a general acid–base in catalysis. The total loss of activity when D543 of ClACLY is mutated to alanine indicates that neither water nor another residue of the mutant protein substitutes for the side chain of D543.
Since kinetic analyses could not be performed on either D543A ClACLY or the H491A&D543A double mutant, isothermal titration calorimetry was used to test the ability of the double mutant to bind CoA. Both ClACLY and the double mutant bound CoA (Figure S2), but the magnitude of the binding enthalpy was lower for the double mutant (Table 2). Surprisingly, only two binding sites were detected. Since ClACLY is a tetramer of heterodimers, it has four active sites and would be expected to bind four molecules of CoA, one at each active site. This stoichiometry was seen for hACLY, where 4.06 ± 0.04 binding sites and a binding enthalpy of −7.19 ± 0.09 kcal mol−1 were measured under similar conditions.7
Tests of thermal stability by DSF showed that the three mutants had the same T m as ClACLY, indicating that they were correctly folded. This contrasts with results obtained with pig CS by circular dichroism33 where mutation of D375 increased or decreased stability. However, pig CS has a higher T m, 55.7°C. The lower T m of 48°C for ClACLY could be due to unfolding of any domain, not specifically the CS domain.
3.4. Catalytic residues in the crystals
In the crystal structure, H491 and D543 adopt similar conformations to H320 and D375 of CS. ND1 of H491 accepts hydrogen bonds from the backbone nitrogen atoms of residues 493 and 494. If H491 is neutral, NE2 would be protonated. There is residual electron density in the active site, between H491 and R502. In the H491A mutant protein, either a water molecule or the amino group of K494 could interact with the hydroxyl of citryl‐CoA or the carbonyl of oxaloacetate. In CS, the residue equivalent to K494 is L323. This residue could not form a hydrogen bond through its side chain, so could not substitute for the imidazole side chain in the same way K494 could. This could be why the mutation of H to A has a less drastic effect on catalysis by ClACLY than by CS.31 In the crystal structure of CterClACLY, D543 interacts with a molecule of glycerol.
The catalytic H274 of CS does not align with a histidine residue in ClACLY, but with F450 (Figure S1). In CS, H274 interacts with the thioester and the enolate, donating a proton to the carbonyl of the acetyl group of acetyl‐CoA to stabilize the enol intermediate.28 F450 could not serve this role, but the crystal structure shows that the amide nitrogen of G451 could. The peptide bond between F450 and G451 is flipped relative to that in CS (Figure 4). The carbonyl accepts a hydrogen bond from G447, which adopts a different conformation than G271 of CS because of insertion of A270 (Figure S1). The amide nitrogen is directed to the substrate‐binding site. It interacts with a glycerol molecule in the active site of chain A and water molecules in the active sites of the other protomers in the P32 crystal form. It would interact with the carbonyl of the thioester in citryl‐CoA.
Figure 4.
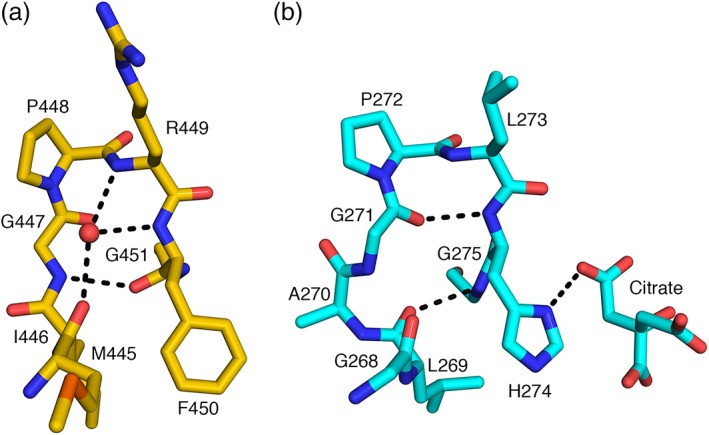
Substitution of histidine in CS with phenylalanine in ACLY. A conserved phenylalanine residue in ACLY (a) replaces the active site histidine residue in CS (b). In chicken CS, the imidazole donates a hydrogen bond to citrate. The phenylalanine has no hydrogen bond donor in its side chain, but the peptide bond between F450 and G451 is flipped relative to the peptide bond between H274 and G275. This allows the amide nitrogen to serve as the hydrogen bond donor. The crystal structures were superposed based on Cα atoms of residues 398–446 of chain A of ClACLY and residues 221–269 of chain A of chicken citrate synthase (PDB ID: 6CSC 20)
3.5. Delivery of citryl‐CoA from the N‐terminal portion of ACLY to the C‐terminal portion
Identification of the binding site for CoA and active site residues H491 and D543 in the C‐terminal portion leads to the question of how the two portions of ACLY transfer citryl‐CoA from site I to site III. We propose that the adenosine end of CoA binds to the C‐terminal portion of ACLY as seen in the crystal structure. CoA swings the free thiol into site I in the N‐terminal portion (Figure 1), where citrate and phosphohistidine also bind. The active site histidine residue would have been phosphorylated by ATP at site II, also in the N‐terminal portion. When the citryl‐CoA thioester bond has formed at site I, the citryl end swings into site III in the C‐terminal portion of ACLY. Once citryl‐CoA has bound, the active site closes to catalyze cleavage to oxaloacetate and acetyl‐CoA at site III. This proposal is consistent with the recently published structures of hACLY and Methanothrix soehngenii ACLY.17, 18 The location of the N‐terminal portion with respect to the C‐terminal portion shows that the citryl end of citryl‐CoA swings 35 å from site I to site III.
The locations of the four N‐termini of CterClACLY indicate that site III is not located in the same polypeptide chain as sites I and II. Figure 2 shows that the N‐terminus of one polypeptide is closer to a different polypeptide's active site III. The tetramer needs to be formed before the enzyme is active.
4. MATERIALS AND METHODS
4.1. Expression and purification of ClACLY
Genes for subunits B and A of Chlorobium limicola ATP‐citrate lyase (ClACLY) and the intergenic region were synthesized by Genscript. Codons were chosen for optimal production in E. coli and a His8‐tag was added to the C‐terminus of subunit B. NdeI and AvrII sites were included at the 5′‐ and 3′‐ends for insertion in the pET‐42b(+)plasmid (Novagen). After sequencing (University Core DNA Services), the expression plasmid was transformed into E. coli BL21(DE3), and 100 ml Luria‐Bertani broth containing 30 μg/ml kanamycin was inoculated with a single colony. After overnight culture at 37°C with shaking at 225 rpm, 50 ml was used to inoculate 1 L Terrific Broth (12 g tryptone, 24 g yeast extract, 4 ml glycerol, 2.31 g KH2PO4, and 12.54 g K2HPO4 per liter) (TB). This culture was grown until the optical density at 600 nm reached 1.6. Expression was induced with 0.1 mM isopropyl‐β‐d‐thiogalactoside and the temperature was dropped to 21°C. After ~16 hr, cells were harvested by centrifugation (20 min, 4,000g) at 4°C and stored at −80°C.
The protein was purified by affinity, gel filtration, and anion exchange chromatography. Cells were thawed in lysis buffer (50 mM sodium phosphate, 300 mM NaCl, 15% [vol/vol] glycerol, 10 mM imidazole, and pH 8.0) and lysed by sonication. The lysate was separated by centrifugation (30 min, 11,000g) and loaded on a Ni‐NTA Agarose (Qiagen) column. Bound protein was washed with buffer containing an additional 10 mM imidazole and eluted with buffer containing 100 mM imidazole. Protein was precipitated in 80% saturated ammonium sulfate and stored at 4°C. Prior to gel filtration on a 120‐ml Superdex 200 PG (GE Healthcare) column, the precipitate was collected by centrifugation (60 min, 7,800g) and dissolved in a minimal volume of running buffer [150 mM NaCl, 50 mM tris(hydroxymethyl)aminomethane hydrochloride (TrisHCl) pH 8.0, 15% glycerol, 10 mM 2‐mercaptoethanol (2‐ME)]. Fractions containing protein of the expected size were pooled and the protein was precipitated and stored at 4°C. The precipitate was collected, dissolved in low salt buffer (20 mM TrisHCl, 15% glycerol, 10 mM 2‐ME, and pH 8.0), then injected onto a 60‐ml Sephadex G‐25 (GE Healthcare) column connected to a 1‐ml HiTrap Q HP column (GE Healthcare). Protein was eluted with a salt gradient (20 mM Tris HCL, 15% glycerol, 10 mM 2‐ME, 1 M KCl, and pH 8.0). The protein was concentrated using an Amicon Ultra‐30 centrifuge filter, exchanging the buffer to 10 mM Tris HCl pH 8.0, 10 mM 2‐ME. The sample was flash frozen in 20 μl aliquots and stored at −80°C. Typical yield from 1 L TB was 7 mg ClACLY, calculated using its predicted absorbance 0.1% at 280 nm, that is, 0.913.
4.2. Site‐directed mutagenesis of ClACLY
Two residues, H491 and D543 of subunit A, were mutated to A. Mutagenesis followed the Quikchange Lightning protocol (Agilent) with the modification from Wang and Malcolm.34 Primers are listed in Table S1.
Specific activities of wild‐type ClACLY and the mutants were measured using a coupled‐enzyme assay with malate dehydrogenase (MDH).35 MDH catalyzes reduction of oxaloacetate by NADH. The assay solution contained 3.3 U MDH, 0.2 mM NADH, 2 mM sodium citrate, 50 μM CoA, 1 mM ATP, 10 mM MgCl2, 10 mM dithiothreitol (DTT), 100 mM Tris HCl, and pH 8.4. For the kinetics assays, the concentration of citrate, CoA, or ATP was varied, with the other substrates and MDH maintained at 2 mM sodium citrate, 50 μM CoA, and 1 mM ATP. Experiments were performed in triplicate and analyzed using GraphPad Prism.
DSF was performed to test whether the mutants displayed similar thermal stability to wild‐type ClACLY. The conditions mimicked those used during purification: 10 mM either Tris HCl, pH 7.0, 7.5, 8.0, 8.5, or 9.0 or HEPES, pH 6.8, 7.3, 7.8, or 8.3; 0, 100, or 200 mM NaCl; 0, 10, or 20% glycerol; 2 μl 100× SYPRO Orange Protein Dye (Life Technologies) and 1 μM protein in 40 μl total volume. Temperature was increased every 5 s in 0.2°C increments from 25 to 90°C with measurements every 5 s at 470 and 555 nm (Qiagen Rotor‐Gene Q). Three replicates were analyzed using GraphPad Prism to calculate an average T m with an analysis of variance two‐factor statistical analysis.
Isothermal titration calorimetry was used to test whether the double mutant bound CoA. Wild‐type ClACLY and the double mutant were purified by affinity chromatography and dialyzed overnight at 4°C against 10 mM MgCl2, 10 mM sodium citrate, 4 mM 2‐ME and 10 mM potassium phosphate, pH 7.5. Ten micromolar CoA was prepared using the dialysis solution. Both solutions were centrifuged to remove aggregates (10 min, 14,000g) prior to loading protein in the cell and CoA in the syringe of a MicroCal VP‐ITC isothermal titration calorimeter (Malvern). A titration consisted of a single 1‐μl injection of CoA followed by 59 4‐μl injections at 5‐min intervals at 30°C. Once appropriate concentrations had been determined, three replicates were done and the thermodynamic parameters for binding CoA to ClACLY and to the double mutant were determined using the MicroCal VP‐ITC Origin‐7™ software.
4.3. Crystallization of the C‐terminal portion of ClACLY using chymotrypsin
The first crystals were grown by cleaving full‐length ClACLY with chymotrypsin. Chymotrypsin was added to the crystallization experiment and crystals grew in hanging drops using 17.5–20% PEG3350, 100 mM Tris HCl pH 7.0–8.5, 125–180 mM lithium citrate, or 150–175 mM sodium potassium tartrate. The protein solution contained 10 mM Tris HCl pH 8.0, 5 mM acetyl‐CoA, and 5 mg/ml ClACLY and was mixed in a 1:1 ratio with the precipitant solution. To identify what portion crystallized, crystals were dissolved in 20 μl loading buffer for SDS‐PAGE. After SDS‐PAGE, the band was excised and tryptic peptides were analyzed by mass spectrometry (Southern Alberta Mass Spectrometry Facility).
4.4. Introduction of the TEV protease cleavage site and purification of the CterClACLY
Two regions in subunit A of ClACLY were chosen to introduce the TEV protease cleavage site. The site that worked best was introduced with site‐directed mutagenesis, changing residue 345 from K to F and inserting Y before it, to match the consensus sequence EXXYXQ^S/G36 with EVMYFQ^G, where ^ represents the cleavage site. Primers are given in Table S1. Sequences were confirmed and the plasmid was used to transform E. coli strain BL21 STAR™ (DE3). ClACLY with the TEV protease cleavage site was produced and purified by affinity chromatography and stored as an ammonium sulfate precipitate. The precipitate was collected by centrifugation and dissolved in cleavage buffer (50 mM Tris HCl pH 8.00, 0.5 mM EDTA, 1 mM DTT). TEV protease was added in a 1:100 wt/wt ratio and the sample was left for 24 hr at 4°C. After digestion, the sample was diluted with cation exchange buffer (20 mM MES pH 6.3, 15% glycerol, and 10 mM 2‐ME) and loaded onto a HiTrap SP HP column (GE Healthcare). The protein eluted with a salt gradient (20 mM MES, 15% glycerol, 10 mM 2‐ME, 1 M KCl, and pH 6.3). Eluted protein was diluted with anion exchange buffer (20 mM Tris HCl, 15% glycerol, 10 mM 2‐ME, and pH 8.0), loaded onto a HiTrap Q HP column and eluted with a salt gradient (20 mM Tris, 15% glycerol, 10 mM 2‐ME, 1 M KCl, and pH 8.0). Eluted protein was concentrated and exchanged to 20 mM potassium phosphate pH 7.0, 10 mM 2‐ME before storage at −80°C. The protein concentration was measured spectrophotometrically using its predicted absorbance 0.1% at 280 nm, that is, 1.141.
4.5. Crystal structure of the C‐terminal portion of ClACLY cleaved by TEV protease
Screening used the PEG/Ion Screen (Hampton Research) and hanging drops with 0.5 μl protein solution (5 mg/ml protein, 5 mM acetyl‐CoA and 20 mM potassium phosphate pH 7.0) and 0.5 μl well solution. Optimized well solution contained 20% wt/vol PEG3350, 200 mM sodium acetate, 100 mM MES pH 6.3. Seeds for streak seeding were grown with a well solution containing 17% wt/vol PEG3350 and 200 mM sodium acetate pH 5.2. A hair was touched to these crystals, then streaked across a drop containing 10% wt/vol PEG3350, 100 mM sodium acetate, 50 mM MES pH 6.2, 2.5 mg/ml protein, 2.5 mM acetyl‐CoA, and 10 mM potassium phosphate equilibrated for 1 hr over well solutions containing 20% wt/vol PEG3350, 200 mM sodium acetate, 100 mM MES pH 6.2. Crystals grew to 25 × 25 × 100 μM3 were cryo‐protected using 20% vol/vol glycerol, 20% wt/vol PEG 3350, 200 mM sodium acetate, 100 mM MES pH 6.3, vitrified in nitrogen at 100 K, stored under liquid nitrogen, and shipped to the synchrotron.
Two high‐resolution data sets were collected, one at the CMCF‐ID beamline at the Canadian Light Source on a Pilatus3 S 6M detector and the second at IMCA‐CAT beamline 17‐ID at the Advanced Photon Source on a Pilatus 6M detector. Data were processed using XDS or DIALS via XIA2.37, 38, 39, 40, 41, 42 ARCIMBOLDO Shredder43 was run on a HTCondor Grid of 130 nodes44 to determine initial phases using a truncated model of Acetobacter aceti CS (PDB ID: 2H12 45). It performed eLLG‐guided location of template‐derived fragments with PHASER.46 Additional degrees of freedom modified the fragments during molecular replacement through gyre refinement against the rotation function and gimble refinement after placement.47 Best scored phase sets were subject to density modification and autotracing with SHELXE48 leading to a main chain trace comprising 672 residues and characterized by a CC of 31%. The model and electron density maps were visualized using COOT49 and the model was refined using phenix.refine.50, 51 This first model was used to solve the second structure by molecular replacement using PHASER.52 TLS refinement in PHENIX improved both models.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Supplementary Figure S1 Structural alignment of the sequence of the C‐terminal portion of ClACLY with the N‐ and C‐terminal portions of chicken citrate synthase. The sequences were aligned by identifying which residues had similar positions in the □‐helices. The □‐helices of the C‐terminal portion of ClACLY are highlighted by red coils above the sequences. When the polypeptide in one structure took a different path from the others, the residues were aligned with gaps in the other sequences. Residues are colored according to the default of Clustal X.53 The residues mutated in this work are highlighted by rectangles. This figure was drawn using Chimera.54
Supplementary Figure S2 Binding of CoA to ClACLY and to the H491A&D543A double mutant. The top panels depict raw data from isothermal titration calorimetry, that is, heat generated as a function of time. The bottom panels show the integrated area of each peak plotted against the molar ratio, as well as the curve fitted to the data by nonlinear regression. A. 0.7 mM CoA was titrated into 5 μM ClACLY. B. 0.3 mM CoA was titrated into 15 μM H491A&D543A double mutant.
Supplementary Figure S3 Superposition of the C‐terminal portion of ClACLY on CS and the CS allosteric site. A. Chicken CS (PDB ID: 6CSC22) is represented by the semitransparent gray and pink ribbon diagram. B. One protomer of the C‐terminal portion of ClACLY (black) is superposed on the C‐terminal portion of a protomer of chicken CS. C. Two protomers of the C‐terminal portion of ClACLY (black and red) are superposed on a protomer of chicken CS. E and F. Three and four protomers of the C‐terminal portion of ClACLY (yellow, black, red and blue) are superposed. D. NADH is shown as a red stick model bound to the allosteric site of E. coli CS (PDB ID: 1NXG24). The allosteric site is located where the second P450 domain is missing from CS. This figure was drawn using PYMOL.52
Supplementary Table 1 Oligonucleotides with mismatches underlined.
Supplementary Table 2 Statistics for X‐ray diffraction data and model.
ACKNOWLEDGMENTS
Research was supported by a Discovery Grant from NSERC (MEF), Canada Graduate Scholarship—Master's Program (NS), Queen Elizabeth II Scholarship (VHN). I.U. was supported by Grants BIO2015‐64216‐P and MDM2014‐0435, A.M. is supported by a fellowship BES‐2017‐080368 (Spanish Ministry of Science and Innovations). Beamline 08ID‐1 at the Canadian Light Source is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada (NSERC), University of Saskatchewan, Government of Saskatchewan, Western Economic Diversification Canada, National Research Council Canada, and Canadian Institutes of Health Research. Use of resources at the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA‐CAT) beamline 17‐ID was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman‐Woodward Medical Research Institute. The Advanced Photon Source is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357.
Nguyen VH, Singh N, Medina A, Usón I, Fraser ME. Identification of the active site residues in ATP‐citrate lyase's carboxy‐terminal portion. Protein Science. 2019;28:1840–1849. 10.1002/pro.3708
Significance statement: ATP‐citrate lyase (ACLY) is an important enzyme to make a key molecule used in lipid production. Understanding how ACLY functions can aid in drug development to limit lipid production as a potential treatment for obesity and cancer. Key catalytic residues were identified, and their importance was determined. The structure of a portion of ACLY was determined that provides atomic resolution view of how this part of ACLY facilitates catalysis.
Funding information Natural Sciences and Engineering Research Council of Canada, Grant/Award Number: Discovery Grant; Spanish Ministry of Science and Innovations, Grant/Award Numbers: BES‐2017‐080368, BIO2015‐64216‐P, MDM2014‐0435; Argonne National Laboratory, Grant/Award Number: DE‐AC02‐06CH11357; Canadian Institutes of Health Research; National Research Council Canada; Western Economic Diversification Canada; Government of Saskatchewan; University of Saskatchewan; Natural Sciences and Engineering Research Council of Canada (NSERC); Canada Foundation for Innovation; Queen Elizabeth II Scholarship; Canada Graduate Scholarship—Master's Program; NSERC
Contributor Information
Isabel Usón, Email: uson@ibmb.csic.es.
Marie E. Fraser, Email: frasm@ucalgary.ca.
REFERENCES
- 1. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP‐citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jain KS, Kulkarni RR, Jain DP. Current drug targets for antihyperlipidemic therapy. Mini Rev Med Chem. 2010;10:232–262. [DOI] [PubMed] [Google Scholar]
- 3. Zu X‐Y, Zhang Q‐H, Liu J‐H, et al. ATP citrate lyase inhibitors as novel cancer therapeutic agents. Recent Pat Anticancer Drug Discov. 2012;7:154–167. [DOI] [PubMed] [Google Scholar]
- 4. Sanchez LB, Galperin MY, Muller M. Acetyl‐CoA synthetase from the amitochondriate eukaryote Giardia lamblia belongs to the newly recognized superfamily of acyl‐CoA synthetases (NDP‐forming). J Biol Chem. 2000;275:5794–5803. [DOI] [PubMed] [Google Scholar]
- 5. Ramaley RF, Bridger WA, Moyer RW, Boyer PD. The preparation, properties, and reactions of succinyl coenzyme A synthetase and its phosphorylated form. J Biol Chem. 1967;242:4287–4298. [PubMed] [Google Scholar]
- 6. Cottam GL, Srere PA. Nature of the phosphorylated residue in citrate cleavage enzyme. Biochem Biophys Res Commun. 1969;35:895–900. [DOI] [PubMed] [Google Scholar]
- 7. Sun T, Hayakawa K, Bateman KS, Fraser ME. Identification of the citrate‐binding site of human ATP‐citrate lyase using X‐ray crystallography. J Biol Chem. 2010;285:27418–27428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu J, Komakula A, Fraser ME. Binding of hydroxycitrate to human ATP‐citrate lyase. Acta Crystallogr. 2017;D73:660–671. [DOI] [PubMed] [Google Scholar]
- 9. Sun T, Hayakawa K, Fraser ME. ADP‐Mg2+ bound to the ATP‐grasp domain of ATP‐citrate lyase. Acta Crystallogr. 2011;F67:1168–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fraser ME, James MNG, Bridger WA, Wolodko WT. A detailed structural description of Escherichia coli succinyl‐CoA synthetase. J Mol Biol. 1999;285:1633–1653. [DOI] [PubMed] [Google Scholar]
- 11. Weiße RH‐J, Faust A, Schmidt M, Schönheit P, Scheidig AJ. Structure of NDP‐forming acetyl‐CoA synthetase ACD1 reveals a large rearrangement for phosphoryl transfer. Proc Natl Acad Sci U S A. 2016;113:E519–E528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen NT, Maurus R, Stokell DJ, Ayed A, Duckworth HW, Brayer GD. Comparative analysis of folding and substrate binding sites between regulated hexameric type II citrate synthases and unregulated dimeric type I enzymes. Biochemistry. 2001;40:13177–13187. [DOI] [PubMed] [Google Scholar]
- 13. Remington S, Wiegand G, Huber R. Crystallographic refinement and atomic models of two different forms of citrate synthase at 2.7 and 1.7 A resolution. J Mol Biol. 1982;158:111–152. [DOI] [PubMed] [Google Scholar]
- 14. Bazilevsky GA, Affronti HC, Wei X, Campbell SL, Wellen KE, Marmorstein R. ATP‐citrate lyase multimerization is required for coenzyme‐A substrate binding and catalysis. J Biol Chem. 2019;294(18):7259–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ivanovsky R, Sintsov N, Kondratieva E. ATP‐linked citrate lyase activity in the green sulfur bacterium Chlorobium limicola Forma thiosuifatophilumgy. Arch Microbiol. 1980;128:239–241. [Google Scholar]
- 16. Kanao T, Fukui T, Atomi H, Imanaka T. ATP‐citrate lyase from the green sulfur bacterium Chlorobium limicola is a heteromeric enzyme composed of two distinct gene products. Eur J Biochem. 2001;268:1670–1678. [PubMed] [Google Scholar]
- 17. Wei J, Leit S, Kuai J, et al. An allosteric mechanism for potent inhibition of human ATP‐citrate lyase. Nature. 2019;568:566–570. [DOI] [PubMed] [Google Scholar]
- 18. Verschueren KHG, Blanchet C, Felix J, et al. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature. 2019;568:571–575. [DOI] [PubMed] [Google Scholar]
- 19. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server The proteomics protocols handbook. Totowa, NJ: Humana Press, 2005; p. 571–607. [Google Scholar]
- 20. Usher K, Remington S. PDB ID: 6CSC Chicken citrate synthase complex with trifluoroacetonyl‐CoA and citrate. 1997.
- 21. DeLano WL. The PyMOL molecular graphics system. San Carlos: Delano Scientific, 2002. [Google Scholar]
- 22. Dawson NL, Lewis TE, Das S, et al. CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017;45:D289–D295. https://academic.oup.com/nar/article/45/D1/D289/2605733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weitzman PDJ. Regulation of citrate synthase activity in Escherichia coli . Biochim Biophys Acta. 1966;128:213–215. [DOI] [PubMed] [Google Scholar]
- 25. Maurus R, Nguyen NT, Stokell DJ, et al. Insights into the evolution of allosteric properties. The NADH binding site of hexameric type II citrate synthases. Biochemistry. 2003;42:5555–5565. [DOI] [PubMed] [Google Scholar]
- 26. Duckworth HW, Nguyen NT, Gao Y, et al. Enzyme‐substrate complexes of allosteric citrate synthase: Evidence for a novel intermediate in substrate binding. Biochim Biophys Acta Prot Proteom. 2013;1834:2546–2553. [DOI] [PubMed] [Google Scholar]
- 27. Usher KC, Remington SJ, Martin DP, Drueckhammer DG. A very short hydrogen bond provides only moderate stabilization of an enzyme‐inhibitor complex of citrate synthase. Biochemistry. 1994;33:7753–7759. [DOI] [PubMed] [Google Scholar]
- 28. Karpusas M, Branchaud B, Remington SJ. Proposed mechanism for the condensation reaction of citrate synthase: 1.9‐å structure of the ternary complex with oxaloacetate and carboxymethyl coenzyme A. Biochemistry. 1990;29:2213–2219. [PubMed] [Google Scholar]
- 29. Antranikian G, Herzberg C, Gottschalk G. Characterization of ATP citrate lyase from Chlorobium limicola. J Bacteriol. 1982;152:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanao T, Fukui T, Atomi H, Imanaka T. Kinetic and biochemical analyses on the reaction mechanism of a bacterial ATP‐citrate lyase. Eur J Biochem. 2002;269:3409–3416. [DOI] [PubMed] [Google Scholar]
- 31. Evans CT, Kurz LC, James Remington S, Srere PA. Active site mutants of pig citrate synthase: Effects of mutations on the enzyme catalytic and structural properties. Biochemistry. 1996;35:10661–10672. [DOI] [PubMed] [Google Scholar]
- 32. Alter GM, Casazza JP, Zhi W, Nemeth P, Srere PA, Evans CT. Mutation of essential catalytic residues in pig citrate synthase. Biochemistry. 1990;29:7557–7563. [DOI] [PubMed] [Google Scholar]
- 33. Zhi W, Srere PA, Evans CT. Conformational stability of pig citrate synthase and some active‐site mutants. Biochemistry. 1991;30:9281–9286. [DOI] [PubMed] [Google Scholar]
- 34. Wang W, Malcolm BA. Two‐stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange site‐directed mutagenesis. Biotechniques. 1999;26:680–682. [DOI] [PubMed] [Google Scholar]
- 35. Srere PA. The citrate cleavage enzyme I. Distribution and purification. J Biol Chem. 1959;234:2544–2547. [PubMed] [Google Scholar]
- 36. Carrington JC, Dougherty WG. A viral cleavage site cassette: Identification of amino acid sequences required for tobacco etch virus polyprotein processing (potyvirus proteinase/celi‐free expression/cleavage site requirements/protein engineering). Biochemistry. 1988;85:3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. 2011;D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Winter G. Xia2: An expert system for macromolecular crystallography data reduction. J Appl Crystallogr. 2010;43:186–190. [Google Scholar]
- 39. Evans P. Scaling and assessment of data quality. Acta Crystallogr. 2006;D62:72–82. [DOI] [PubMed] [Google Scholar]
- 40. Kabsch W. Integration, scaling, space‐group assignment and post‐refinement. Acta Crystallogr. 2010;D66:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Winter G, Waterman DG, Parkhurst JM, et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. 2018;D74:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr. 2013;D69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Millán C, Sammito MD, McCoy AJ, et al. Exploiting distant homologues for phasing through the generation of compact fragments, local fold refinement and partial solution combination. Acta Crystallogr. 2018;D74:290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sammito M, Millán C, Frieske D, Rodríguez‐Freire E, Borges RJ, Usón I. ARCIMBOLDO_LITE: Single‐workstation implementation and use. Acta Crystallogr. 2015;D71:1921–1930. [DOI] [PubMed] [Google Scholar]
- 45. Francois JA, Starks CM, Sivanuntakorn S, et al. Structure of a NADH‐insensitive hexameric citrate synthase that resists acid inactivation. Biochemistry. 2006;45:13487–13499. [DOI] [PubMed] [Google Scholar]
- 46. Oeffner RD, Afonine PV, Millán C, et al. On the application of the expected log‐likelihood gain to decision making in molecular replacement. Acta Crystallogr. 2018;D74:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCoy AJ, Oeffner RD, Millán C, Sammito M, Usón I, Read RJ. Gyre and gimble: A maximum‐likelihood replacement for Patterson correlation refinement. Acta Crystallogr. 2018;D74:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Usón I, Sheldrick GM. An introduction to experimental phasing of macromolecules illustrated by SHELX; new autotracing features. Acta Crystallogr. 2018;D74:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr. 2010;D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: A comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. 2012;D68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. 2007;D63:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 Structural alignment of the sequence of the C‐terminal portion of ClACLY with the N‐ and C‐terminal portions of chicken citrate synthase. The sequences were aligned by identifying which residues had similar positions in the □‐helices. The □‐helices of the C‐terminal portion of ClACLY are highlighted by red coils above the sequences. When the polypeptide in one structure took a different path from the others, the residues were aligned with gaps in the other sequences. Residues are colored according to the default of Clustal X.53 The residues mutated in this work are highlighted by rectangles. This figure was drawn using Chimera.54
Supplementary Figure S2 Binding of CoA to ClACLY and to the H491A&D543A double mutant. The top panels depict raw data from isothermal titration calorimetry, that is, heat generated as a function of time. The bottom panels show the integrated area of each peak plotted against the molar ratio, as well as the curve fitted to the data by nonlinear regression. A. 0.7 mM CoA was titrated into 5 μM ClACLY. B. 0.3 mM CoA was titrated into 15 μM H491A&D543A double mutant.
Supplementary Figure S3 Superposition of the C‐terminal portion of ClACLY on CS and the CS allosteric site. A. Chicken CS (PDB ID: 6CSC22) is represented by the semitransparent gray and pink ribbon diagram. B. One protomer of the C‐terminal portion of ClACLY (black) is superposed on the C‐terminal portion of a protomer of chicken CS. C. Two protomers of the C‐terminal portion of ClACLY (black and red) are superposed on a protomer of chicken CS. E and F. Three and four protomers of the C‐terminal portion of ClACLY (yellow, black, red and blue) are superposed. D. NADH is shown as a red stick model bound to the allosteric site of E. coli CS (PDB ID: 1NXG24). The allosteric site is located where the second P450 domain is missing from CS. This figure was drawn using PYMOL.52
Supplementary Table 1 Oligonucleotides with mismatches underlined.
Supplementary Table 2 Statistics for X‐ray diffraction data and model.