

Abstract
Diabetes mellitus elicits cellular, epigenetic, and post-translational changes that directly or indirectly affect the biology of the vasculature and other metabolic systems resulting in the apparition of cardiovascular disease. In this review, we provide a current perspective on the most recent discoveries in this field, with particular focus on hyperglycemia- induced pathology in the cardiovascular system. We also provide perspective on the clinical importance of molecular targeting of cardiovascular and diabetes mellitus therapies to treat hyperglycemia, inflammation, thrombosis, dyslipidemia, atherosclerosis, and hypertension.
Keywords: atherosclerosis, cardiovascular disease, cell signaling, cholesterol toxicity, diabetes mellitus, dyslipidemia, epigenetics, glucose toxicity, inflammation, pathophysiology
Introduction
Diabetes mellitus (DM) often coexists with cardiovascular disease (CVD) in clinical practice, but the pathophysiology of this comorbid condition could be rather confusing as the amount of scientific evidence is dispersed and has increased, especially in the last decade.
The strong link between these two diseases is evident. Patients with CVD share similar risk factors for DM onset such as unhealthy dietary and lifestyle habits, obesity, smoking, etc., or already have DM (http://www.who.int/mediacentre/factsheets/fs317/en/). Similarly, DM itself constitutes a risk factor for CVD and its complications, such as myocardial infarction (MI), stroke, amputation, etc. 1.
The mechanisms of the pathogenesis of CVD in diabetes are related to epigenetic, genetic, and cell-signaling defects in inter-related metabolic and inflammatory pathways. These metabolic defects (especially in the endothelium, liver, skeletal muscle, and β cells) can be triggered by various environmental factors such as high caloric intake, smoking, glycation end-products, glucose toxicity, and some medications 2. It could be stated that the expression of both type 2 diabetes mellitus (T2DM) and CVDs is an idiosyncratic response to the environment, guided by the biological capacity of cellular systems in patients 3.
These idiosyncrasies are expressed differently among patients and populations 3. Some patients may express clear or mixed phenotypes of hyperglycemia, dyslipidemia, hypertension, inflammation, or thrombosis, which also represent risk factors for CVD 4. Interestingly, all of these clinical manifestations share similar cellular mechanisms and molecular abnormalities. T2DM has multiple cell-signaling pathways in cell growth, survival, and proliferation such as pAkt, endothelial nitric oxide synthase pathway, and AMP-activated protein kinase pathways that could potentiate the development of CVD. In addition to this, glucose and oxidized lipids exert important effects in tissues at the epigenetic level 5–7. It is noteworthy that some of these epigenetic adaptations can even be passed down to several generations 8–10.
In this review, we provide a current perspective on the advances of such discoveries and for this purpose; we grouped them as CVD risk factors affecting the biology of patients with pre-existing DM. We also provide a brief overview on how cell-signaling pathology and post-translational changes in hypertension, dyslipidemia, inflammation, and hyperglycemia result in the early appearance of CVD phenotypes and the opportunity for new therapies. In an attempt to explain the major relevant pathophysiological pathways that are present in DM that are related to CVD and their therapeutic implications, we have listing them on the basis of their clinical phenotype.
Hyperglycemia
T2DM is considered a multifactorial disease that involves abnormalities in carbohydrate and lipid metabolism 11. Its most evident manifestation is chronic hyperglycemia. Recent evidence shows that T2DM patients have defects in epigenetic and post-translational modifications of the vascular architecture 2. The multiple endometabolic defects of diabetes impact behaviors such as overfeeding (leptin resistance or deficiency) as well as aspects of glucose and lipid metabolism such as cellular toxicity and insulin receptor (IR) effects of palmitic acid and other saturated fatty acids 12. Leptin is a protein secreted by white adipocytes and has the function of stimulating satiety (postprandial) and increase energy expenditure by binding its cognate receptor (leptin receptor B) 13. Mutations in either leptin protein (biologically inactive) or its receptor (defective activation) result in overfeeding behaviors, leading to profound obesity phenotypes with the association of peripheral insulin resistance and hyperglycemia 14.
Similarly, saturated fatty acids such as palmitic acid have been proposed as insulin desensitizers not just in peripheral tissues but also in the hypothalamus. This dual effect leads to peripheral and central insulin resistance, resulting in hyperglycemia and dysregulation of energy balance in the whole organism 14,15. The chronicity of the insulin resistance along with the effect of saturated fatty acids, lipoproteins, leptin, and circulating proinflammatory cytokines translates into apoptosis of islet β cells 16. As glucose is taken up poorly by cells in the organism, this causes postprandial glucose levels to remain consistently high for prolonged periods of time, resulting in glucose-related tissue toxicity [production of receptor for advanced glycation end-products (RAGE), endothelial dysfunction, histone hyperacetylation, DNA methylation, etc.] 2. This toxicity affects microvessels and macro vessels (retinopathy, coronary arterial disease, etc.), nerves (peripheral neuropathy), and nephrons (decreased glomerular filtration and microalbuminuria), with deleterious clinical consequences. IR agonists such as chaetochromin derivatives and monoclonal antibodies with agonist activity on the IR have been reported to improve IR responsiveness and Akt activations, respectively, thus improving glucose metabolism at the cellular level in patients with peripheral insulin resistance 17,18.
Chronic inflammation and thrombosis
Glucose toxicity by aldose reductase activation initiates subsequent PKC-dependent nonosmotic nuclear factor (NF)-κB activation, resulting in the production and release of proinflammatory cytokines such as interleukin (IL)-6, IL-12, IL-10, tumor necrosis factor-α, etc. 19,20. Similarly, inflammation in adipose tissue leads to the release of adipocytokines such as adipsin, adiponectin, leptin, tumor necrosis factor-α, and plasminogen activator inhibitor I. The vascular redox state is affected by transduction signal signals originating from inflammatory, obesity, and insulin-related pathways. Importantly, adipose tissues can modify the secretory profile when sensing paracrine signals of cardiovascular (CV) oxidative stress or injury 21. Such inflammatory signals can transduce cellular signals in tissues such as fat, liver, muscle, heart, endothelium, etc., through toll-like receptor (TLR) signaling, which in turns activates inflammatory nuclear factors (NF-κB) feeding the chronic loop of persistent inflammation. In particular, TLR-2 and TLR-4 affect the frequency, plaque size, and lipid content of atherogenic plaque and the expression of inflammatory genes and cytokines (IL-12, monocyte chemoattractant protein-1, etc.) 22. In this respect, colchicine, an anti-inflammatory agent, has been shown to decrease IL-1b, MI, acute coronary syndrome, and noncardioembolic stroke in phase III studies 23,24. Canakinumab, a monoclonal antibody against IL-1b, is another perfect example of an anti-inflammatory drug that reduces recurrent CV events independent of lipid levels as shown in the CANTOS study 25.
For instance, circulating inflammatory factors can activate potentially life-threatening cell signaling such as thrombosis by platelet activation of both classical and alternative pathways 26. Platelets are easily activated and can aggregate quite fast in response to such circulating cytokines, especially in low-flow areas such as the coronaries, the lower extremities, the brain, etc. 26,27. The occlusion or sub occlusion of these vessels can result in infarctions or necrosis of important tissues such as the brain and the heart, increasing the risk for stroke and MIs 28–30.
In vessels that have an atherogenic lesion, in addition to the circulating inflammatory signals, local signals, along with plaque erosion, partial, or total rupture, can trigger thrombosis in the atherogenic suboccluded area or distal regions on that artery territory 29,30. Infiltration of immune cells can be found in plaques and although these cells repair and replace tissue, its presence and the release of inflammatory chemoattractive substances worsen the thrombotic state and increase the risk of further plaque core necrosis and plaque instability, with the subsequent release of debris into the distal portions of the artery lesion, a condition that worsens under low shear stress conditions 31–33.
It is noteworthy that another less characterized player, RAGE, is implicated in deleterious effects on energy expenditure, weight gain, adipose tissue inflammation, and insulin resistance together with a high-fat diet. RAGE protects against high-fat diet-induced systemic inflammation and weight gain 34. Also, elevated serum RAGE of more than 838.19 pg/ml can double the risk for CV events in patients with pre-existing CVD (a composite of MI, stroke, and CV death) 35.
Dyslipidemia and atherogenesis
Dysplipidemia and obesity are often present in patients with DM and can facilitate atherogenesis and atherosclerosis 36. Fat droplets in cells, especially adipocytes, are essentially ‘packed energy’ that our body can use as a fuel source in times of fasting or when there is a need for extraphysical activity 37. In contrast, carbohydrates are metabolized into energy using aerobic or anaerobic mitochondrial pathways. If all the elementary energy requirements of the cell are met, then lipids are synthetized from carbohydrates, a process called ‘de-novo lipogenesis’ 38. Lipids can be stored and converted back into burnable compounds (pyruvate) within the cell 37. Lipids can themselves be a ‘source of energy’ in times of fasting that, along with their high affinity to cell membranes, can access the cells with minimum effort (vectors-exosomes) 37,39. However, the distribution of lipids is aided by proteins as their physicochemical properties allow them to remain in the circulatory system, avoiding early absorption 40. The synthesis of those proteins is mainly orchestrated by the liver. Those proteins are categorized according to their molecular density into very low-density lipoprotein (LDL), LDL, and high-density lipoprotein. Along with triglycerides, which are clusters of lipids, lipoproteins travel along the circulatory system to distal organs and tissues 40.
Chronic high levels of atherogenic LDL cholesterol along with increased non-high-density lipoprotein C and ApoB values in patients have been related to the progression of atherogenesis 41. Oxidation of low-density lipoprotein (oxLDL) is an important condition that represents oxidative stress and increases the atherogenic and inflammatory properties of LDL 42. In addition, elevated serum levels of oxLDL are associated with the incidence of coronary disease 42,43. Therefore, a logical therapeutic target is the reduction of the LDL cholesterol by statins or by the novel proprotein convertase subtilisin/kexin type 9 inhibitors. Statins inhibit the production of cholesterol by inhibiting the transformation from hydroxymethylglutaryl-coenzyme A into mevalonic acid (primitive fatty acid) 44. In contrast, proprotein convertase subtilisin/kexin type 9 inhibitors increase LDL-receptor density on the cell surface, facilitating LDL intake by the cell and decreasing circulating LDL, thereby facilitating plaque regression as reported recently in the GLAGOV study 45.
Concurrently, hyperglycemia contributes toward the development of atherosclerosis and arterial stiffness 46. Chronic damage to the endothelium and the effects of inflammatory cytokines on endothelium play important roles in the genesis and stability of the plaque. The cellular mechanisms of media thickening and proliferation, presence of endothelium- adhesion molecules (vascular cell adhesion molecule 1 and intercellular adhesion molecule 1), and infiltration of macrophages in the subintima are regulated by epigenetic mechanisms and posttranslational modifications 47,48. Hyperglycemia induces hyperacetylation of histone H3K9/K14 in 88 genes codifying for diabetes, 52 genes for hypertension, and 84 genes for CV disorders among other diseases 49. It is particularly noteworthy that hyperacetylation of the histone H3K9/K14 in the endothelium results in the expression of important glucose metabolism and metalloproteinases regulating genes such as heme oxygenase 1 (HMOX1), IL-8 precursor, matrix metalloproteinase (MMP) protein-10, cysteine/glutamate transporter (SLC7A11), and MMP1 49. ILs and metalloproteinases are closely related as they are both regulated by proinflammatory signals and participate in vascular remodeling, particularly in plaque progression and plaque instability 50,51. MMP inhibitors have been used to stabilize plaques, but there is a need for more selective targeting of MMPs as broad-spectrum inhibitors exert dual effects on the plaque 51.
Hyperglycemia also induces DNA methylation of important genes for glucose metabolism, G-coupled protein receptors (GPRs), and insulin growth factor proteins such as ABCC11, ADAD1, ADAM8, BCL3, CCDC61, CEP120, CSF1R, CSTL1, CTTNBP2NL, EGLN3, ENOX1, ERAS, FAM107A, FASLG, GADD45B, GNG2, GPR39, GPR62, GRK7, HMGB2, HNRNPL, HYOU1, and IGFBPL1 49. Gene expression and suppression persist for up to 6 days in the endothelium after the hyperglycemic episode in vitro 2. Here is the importance of novel GPR agonists which currently are underway in an effort to improve GPR signaling in tissues and its metabolic benefits in patients with diabetes 52,53.
Other epigenetic mechanisms such as microRNAs (miR) can regulate gene expression post-transcriptionally, directly exert their effects in signal pathways, and reach other cells when included in extracellular vesicles called ‘exosomes’ 54. miR-941, miR-208b, miR-197, and miR-223 have been found to have diagnostic value in predicting CV events or CV death 55–57. miR-126-5p has been associated inversely with the complexity of CAD with low serum levels in multivessel disease and high SYNTAX scores in patients with stable angina 58. Some epigenetic therapies are underway as potential antithrombotics such as miR-19b for use in patients with unstable angina 59. Also, a bigger epigenetic factor, long noncoding RNAs in exosomes, such as exosomal long noncoding RNA-growth arrest-specific 5 (long noncoding RNA GAS5), can increase the apoptosis of macrophages and endothelial cells in atherosclerosis 60.
Hypertension
The renin–angiotensin–aldosterone system has been proposed as a feasible model to explain secondary hypertension as the cause of primary hypertension is unknown 61. Inflammatory cytokines have a major impact on the endothelium by affecting the capacity of energy metabolism (mitochondrial dysfunction) and the release endothelial nitric oxide synthase, which is an important vasodilator, thus affecting vascular relaxation and inducing arterial stiffness 62–64. These inflammatory cytokines chemoattract macrophages and lymphocytes, which can produce and release reactive oxygen species and angiotensin II (AngII) 62,63. Reactive oxygen species activates NF-κB signaling, amplifying the vicious cycle of local inflammatory response, and AngII increases the blood flow by inducing constriction of the media of arteries, thereby increasing blood pressure 65. As this inflammatory stage is chronic, AngII can consistently and continuously induce an increase in blood pressure. This high-flow system induces the development of media hypertrophy, reducing even more the arterial lumen, which in turn increases resting blood pressure values 66,67. Unchecked stages of this condition may result in the onset of secondary hypertension and the need for a medical intervention with lifestyle changes and antihypertensives 67.
The persistence of a high-flow system together with inflammation, dyslipidemia, and hyperglycemia increases the risk of atherogenic plaque erosion or rupture, hemorrhage (especially microcirculation), and thrombosis 68.
Sympathetic regulation of blood pressure by catecholamines also plays an important role in the presence and persistence of hypertension 61,69. Renal denervation was proposed to treat uncontrolled hypertension without relevant and consistent results in the SYMPLICITY HTN-3 trial, pointing to the utility of targeting renin–angiotensin–aldosterone system, as it may be more clinically relevant than the sympathetic pathway 70.
Discussion
The optimal balance between genes codifying for epigenetic modulators and associated proteins required for the transcription of these modulators can be affected by cellular toxic products such as glucose itself and glycation end-products leading to transcriptional stages of inflammation (oxidative stress, cytokine production, and release and apoptosis), endothelial dysfunction (decrease in nitric oxide production and release of AngII), and downregulation of GPR density. oxLDL plays a role in the pathogenesis of CVD by desensitizing the IR pathway and IR-dependent glucose uptake, thus reinforcing hyperglycemia and its toxic effects in cells. Another CVD risk factor described in this review is hypertension, triggered by inflammatory signals, together with the inability to control vascular relaxation by nitric oxide and angiotensin, both of which are endothelium-release-dependent factors. Taken together with atherovascular lesions, this could result in a mature hypertension phenotype and its associated increased risk for CV morbidity and/or CV death (Fig. 1).
Fig. 1.
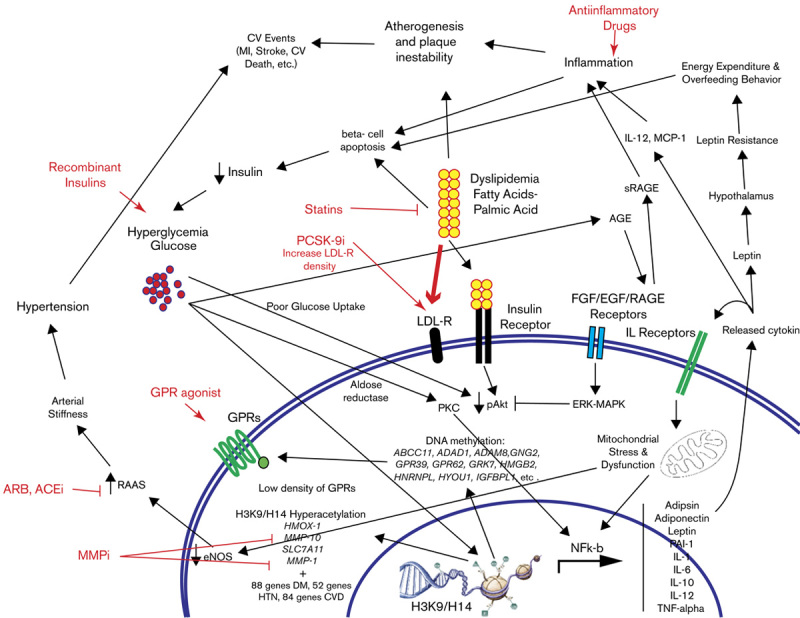
Cellular and clinical implications in DM that precipitate CVD and their importance for therapeutics.
There are still a few gaps in the understanding of these signals. For instance, soluble RAGE characterization at the epigenetic level and its inflammatory and Akt signal competition properties should be investigated further. Exosome-mediated long or short RNA information transfer and signal transductions have not been fully characterized and standardized for any ethnical or environmental variations. However, as the field advances, it is even more evident that some or most of the signal pathways are inter-related following a pattern that starts with the cellular response to high concentrations of glucose and cholesterols.
Conclusion
Diabetes is characterized by the presence of risk factors and common important epigenetic, genetic, and cellular signaling mechanisms that lead to or accelerate the development of CV disease and progression.
A better understanding of such cellular mechanisms can translate into a more selective and personalized therapy for the primary and secondary prevention of CV events in patients with diabetes.
Acknowledgements
Conflicts of interest
There are no conflicts of interest.
References
- 1.[No authors listed]. Diabetes mellitus: a major risk factor for cardiovascular disease. A joint editorial statement by the American Diabetes Association; The National Heart, Lung, and Blood Institute; The Juvenile Diabetes Foundation International; The National Institute of Diabetes and Digestive and Kidney Diseases; and The American Heart Association. Circulation 1999; 100:1132–1133. [DOI] [PubMed] [Google Scholar]
- 2.Keating ST, Plutzky J, El-Osta A. Epigenetic changes in diabetes and cardiovascular risk. Circ Res 2016; 118:1706–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gluckman PD, Low FM, Buklijas T, Hanson MA, Beedle AS. How evolutionary principles improve the understanding of human health and disease. Evol Appl 2011; 4:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matheus AS, Tannus LR, Cobas RA, Palma CC, Negrato CA, Gomes MB. Impact of diabetes on cardiovascular disease: an update. Int J Hypertens 2013; 2013:653789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turdi S, Li Q, Lopez FL, Ren J. Catalase alleviates cardiomyocyte dysfunction in diabetes: role of Akt, Forkhead transcriptional factor and silent information regulator 2. Life Sci 2007; 81:895–905. [DOI] [PubMed] [Google Scholar]
- 6.Ren J, Duan J, Thomas DP, Yang X, Sreejayan N, Sowers JR, et al. IGF-I alleviates diabetes-induced RhoA activation, eNOS uncoupling, and myocardial dysfunction. Am J Physiol Regul Integr Comp Physiol 2008; 294:R793–R802. [DOI] [PubMed] [Google Scholar]
- 7.Kusmic C, L’Abbate A, Sambuceti G, Drummond G, Barsanti C, Matteucci M, et al. Improved myocardial perfusion in chronic diabetic mice by the up-regulation of pLKB1 and AMPK signaling. J Cell Biochem 2010; 109:1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun X, Wong D. Long non-coding RNA-mediated regulation of glucose homeostasis and diabetes. Am J Cardiovasc Dis 2016; 6:17–25. [PMC free article] [PubMed] [Google Scholar]
- 9.Sales VM, Ferguson-Smith AC, Patti ME. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab 2017; 25:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li P, Ruan X, Yang L, Kiesewetter K, Zhao Y, Luo H, et al. A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell Metab 2015; 21:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers 2015; 1:15019. [DOI] [PubMed] [Google Scholar]
- 12.Harris WS, Luo J, Pottala JV, Margolis KL, Espeland MA, Robinson JG. Red blood cell fatty acids and incident diabetes mellitus in the Women’s Health Initiative Memory Study. PLoS One 2016; 11:e0147894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab 2010; 21:643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes 2001; 50:2786–2791. [DOI] [PubMed] [Google Scholar]
- 15.Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J Clin Invest 2009; 119:2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, Reinecke M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005; 54 (Suppl 2):S108–S113. [DOI] [PubMed] [Google Scholar]
- 17.Qiang G, Xue S, Yang JJ, Du G, Pang X, Li X, et al. Identification of a small molecular insulin receptor agonist with potent antidiabetes activity. Diabetes 2014; 63:1394–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhaskar V, Goldfine ID, Bedinger DH, Lau A, Kuan HF, Gross LM, et al. A fully human, allosteric monoclonal antibody that activates the insulin receptor and improves glycemic control. Diabetes 2012; 61:1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yerneni KK, Bai W, Khan BV, Medford RM, Natarajan R. Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes 1999; 48:855–864. [DOI] [PubMed] [Google Scholar]
- 20.Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes 2004; 53:2910–2920. [DOI] [PubMed] [Google Scholar]
- 21.Oikonomou EK, Antoniades C. Immunometabolic regulation of vascular redox state: the role of adipose tissue. Antioxid Redox Signal 2017. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 22.Curtiss LK, Tobias PS. Emerging role of toll-like receptors in atherosclerosis. J Lipid Res 2009; 50 (Suppl):S340–S345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu M, Tsai SF, Kuo YM. The therapeutic potential of anti-inflammatory exerkines in the treatment of atherosclerosis. Int J Mol Sci 2017; 18:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol 2013; 61:404–410. [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017; 377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 26.Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol 2010; 47:2170–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bassiouny HS, Song RH, Kocharyan H, Kins E, Glagov S. Low flow enhances platelet activation after acute experimental arterial injury. J Vasc Surg 1998; 27:910–918. [DOI] [PubMed] [Google Scholar]
- 28.DeFilippis AP, Chernyavskiy I, Amraotkar AR, Trainor PJ, Kothari S, Ismail I, et al. Circulating levels of plasminogen and oxidized phospholipids bound to plasminogen distinguish between atherothrombotic and non-atherothrombotic myocardial infarction. J Thromb Thrombolysis 2016; 42:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed GL, Houng AK, Wang D. Microvascular thrombosis, fibrinolysis, ischemic injury, and death after cerebral thromboembolism are affected by levels of circulating alpha2-antiplasmin. Arterioscler Thromb Vasc Biol 2014; 34:2586–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz RS, Burke A, Farb A, Kaye D, Lesser JR, Henry TD, Virmani R. Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death: relation to epicardial plaque histopathology. J Am Coll Cardiol 2009; 54:2167–2173. [DOI] [PubMed] [Google Scholar]
- 31.Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation 1994; 90:775–778. [DOI] [PubMed] [Google Scholar]
- 32.Xia J, Yin A, Li Z, Liu X, Peng X, Xie N. Quantitative Analysis of Lipid-Rich Necrotic Core in Carotid Atherosclerotic Plaques by In Vivo Magnetic Resonance Imaging and Clinical Outcomes. Med Sci Monit 2017; 23:2745–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seneviratne A, Hulsmans M, Holvoet P, Monaco C. Biomechanical factors and macrophages in plaque stability. Cardiovasc Res 2013; 99:284–293. [DOI] [PubMed] [Google Scholar]
- 34.Song F, Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, Xu X, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes 2014; 63:1948–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reichert S, Triebert U, Santos AN, Hofmann B, Schaller HG, Schlitt A, Schulz S. Soluble form of receptor for advanced glycation end products and incidence of new cardiovascular events among patients with cardiovascular disease. Atherosclerosis 2017; 266:234–239. [DOI] [PubMed] [Google Scholar]
- 36.Bamba V, Rader DJ. Obesity and atherogenic dyslipidemia. Gastroenterology 2007; 132:2181–2190. [DOI] [PubMed] [Google Scholar]
- 37.Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 2012; 81:687–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parks EJ. Dietary carbohydrate’s effects on lipogenesis and the relationship of lipogenesis to blood insulin and glucose concentrations. Br J Nutr 2002; 87 (Suppl 2):S247–S253. [DOI] [PubMed] [Google Scholar]
- 39.Skotland T, Sandvig K, Llorente A. Lipids in exosomes: current knowledge and the way forward. Prog Lipid Res 2017; 66:30–41. [DOI] [PubMed] [Google Scholar]
- 40.Ramasamy I. Recent advances in physiological lipoprotein metabolism. Clin Chem Lab Med 2014; 52:1695–1727. [DOI] [PubMed] [Google Scholar]
- 41.Puri R, Nissen SE, Shao M, Elshazly MB, Kataoka Y, Kapadia SR, et al. Non-HDL cholesterol and triglycerides: implications for coronary atheroma progression and clinical events. Arterioscler Thromb Vasc Biol 2016; 36:2220–2228. [DOI] [PubMed] [Google Scholar]
- 42.Trpkovic A, Resanovic I, Stanimirovic J, Radak D, Mousa SA, Cenic-Milosevic D, et al. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit Rev Clin Lab Sci 2015; 52:70–85. [DOI] [PubMed] [Google Scholar]
- 43.Koenig W, Karakas M, Zierer A, Herder C, Baumert J, Meisinger C, Thorand B. Oxidized LDL and the risk of coronary heart disease: results from the MONICA/KORA Augsburg Study. Clin Chem 2011; 57:1196–1200. [DOI] [PubMed] [Google Scholar]
- 44.Warita K, Warita T, Beckwitt CH, Schurdak ME, Vazquez A, Wells A, Oltvai ZN. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep 2014; 4:7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholls SJ, Puri R. Implications of GLAGOV study. Curr Opin Lipidol 2017; 28:465–469. [DOI] [PubMed] [Google Scholar]
- 46.Rubin J, Nambi V, Chambless LE, Steffes MW, Juraschek SP, Coresh J, et al. Hyperglycemia and arterial stiffness: the Atherosclerosis Risk in the Communities study. Atherosclerosis 2012; 225:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baccarelli A, Tarantini L, Wright RO, Bollati V, Litonjua AA, Zanobetti A, et al. Repetitive element DNA methylation and circulating endothelial and inflammation markers in the VA normative aging study. Epigenetics 2010; 5:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khyzha N, Alizada A, Wilson MD, Fish JE. Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med 2017; 23:332–347. [DOI] [PubMed] [Google Scholar]
- 49.Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 2011; 21:1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu P, Sun M, Sader S. Matrix metalloproteinases in cardiovascular disease. Can J Cardiol 2006; 22 (Suppl B):25B–30B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med 2007; 17:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mancini AD, Poitout V. GPR40 agonists for the treatment of type 2 diabetes: life after ‘TAKing’ a hit. Diabetes Obes Metab 2015; 17:622–629. [DOI] [PubMed] [Google Scholar]
- 53.Reimann F, Gribble FM. G protein-coupled receptors as new therapeutic targets for type 2 diabetes. Diabetologia 2016; 59:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao J, Pan Y, Li XH, Yang XY, Feng YL, Tan HH, et al. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis 2016; 7:e2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bai R, Yang Q, Xi R, Li L, Shi D, Chen K. miR-941 as a promising biomarker for acute coronary syndrome. BMC Cardiovasc Disord 2017; 17:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, Yuan L, Chen F, Zhang L, Chen X, Yang C, Han Z. Circulating miR-208b: a potentially sensitive and reliable biomarker for the diagnosis and prognosis of acute myocardial infarction. Clin Lab 2017; 63:101–109. [DOI] [PubMed] [Google Scholar]
- 57.Schulte C, Molz S, Appelbaum S, Karakas M, Ojeda F, Lau DM, et al. miRNA-197 and miRNA-223 predict cardiovascular death in a cohort of patients with symptomatic coronary artery disease. PLoS One 2015; 10:e0145930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li HY, Zhao X, Liu YZ, Meng Z, Wang D, Yang F, Shi QW. Plasma microRNA-126-5p is associated with the complexity and severity of coronary artery disease in patients with stable angina pectoris. Cell Physiol Biochem 2016; 39:837–846. [DOI] [PubMed] [Google Scholar]
- 59.Li S, Ren J, Xu N, Zhang J, Geng Q, Cao C, et al. MicroRNA-19b functions as potential anti-thrombotic protector in patients with unstable angina by targeting tissue factor. J Mol Cell Cardiol 2014; 75:49–57. [DOI] [PubMed] [Google Scholar]
- 60.Chen L, Yang W, Guo Y, Chen W, Zheng P, Zeng J, Wusong T. Exosomal lncRNA GAS5 regulates the apoptosis of macrophages and vascular endothelial cells in atherosclerosis. PLoS One 2017; 12:e0185406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carretero OA, Scicli AG. Local hormonal factors (intracrine, autocrine, and paracrine) in hypertension. Hypertension 1991; 18 (Suppl):I58–I69. [DOI] [PubMed] [Google Scholar]
- 62.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 2009; 78:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J 2010; 4:302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anderson TJ. Arterial stiffness or endothelial dysfunction as a surrogate marker of vascular risk. Can J Cardiol 2006; 22 (Suppl B):72B–80B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 2011; 21:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosendorff C. The renin-angiotensin system and vascular hypertrophy. J Am Coll Cardiol 1996; 28:803–812. [DOI] [PubMed] [Google Scholar]
- 67.Stumpe KO, Agabiti-Rosei E, Zielinski T, Schremmer D, Scholze J, Laeis P, et al. Carotid intima-media thickness and plaque volume changes following 2-year angiotensin II-receptor blockade. The Multicentre Olmesartan atherosclerosis Regression Evaluation (MORE) study. Ther Adv Cardiovasc Dis 2007; 1:97–106. [DOI] [PubMed] [Google Scholar]
- 68.Alshehri AM. Metabolic syndrome and cardiovascular risk. J Family Community Med 2010; 17:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dominiak P, Grobecker H. Elevated plasma catecholamines in young hypertensive and hyperkinetic patients: effect of pindolol. Br J Clin Pharmacol 1982; 13 (Suppl 2):381S–390S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bhatt DL, Kandzari DE, O’Neill WW, D’Agostino R, Flack JM, Katzen BT, et al. A controlled trial of renal denervation for resistant hypertension. N Engl J Med 2014; 370:1393–1401. [DOI] [PubMed] [Google Scholar]