

Abstract
Twenty crystal structures of the complexes of l‐asparaginase with l‐Asn, l‐Asp, and succinic acid that are currently available in the Protein Data Bank, as well as 11 additional structures determined in the course of this project, were analyzed in order to establish the level of conservation of the geometric parameters describing interactions between the substrates and the active site of the enzymes. We found that such stereochemical relationships are highly conserved, regardless of the organism from which the enzyme was isolated, specific crystallization conditions, or the nature of the ligands. Analysis of the geometry of the interactions, including Bürgi–Dunitz and Flippin–Lodge angles, indicated that Thr12 (Escherichia coli asparaginase II numbering) is optimally placed to be the primary nucleophile in the most likely scenario utilizing a double‐displacement mechanism, whereas catalysis through a single‐displacement mechanism appears to be the least likely.
Keywords: Bürgi–Dunitz angles, double‐displacement, enzyme mechanism, Flippin–Lodge angles, single‐displacement
Abbreviations
- ASNAses II
type II l‐asparaginases
- ASNAses
l‐asparaginases
- EcAII
type II Escherichia coli l‐asparaginase
- ErA
Dickeya dadantii l‐asparaginase
1. INTRODUCTION
l‐asparaginase (EC 3.5.1.1) catalyzes the deamidation reaction leading to conversion of asparagine to aspartate. The existence of such a catalytic activity has been known for almost a century,1 but the medically important biological activity of this enzyme was first noticed by Kidd,2 who reported anticancer properties of the guinea pig serum. The basis of such an activity was later shown to be starvation of some types of cancer cells of an amino acid that was essential for them, but not for normal cells.3 This observation ultimately led to the use of l‐asparaginase in clinical practice in the treatment of acute lymphoblastic childhood leukemia (ALL), with the approval by the FDA of the Escherichia coli asparaginase (Elspar©) in 1978. Although the nature of the activity of the enzyme isolated from E. coli (EcAII) and from other bacterial and mammalian sources was unambiguous, the identity of the catalytic pocket become clear only once crystal structures of l‐asparaginases became available 15 years later.
The first accurate structure of a bacterial l‐asparaginase4 in complex with aspartic acid, the product of catalytic reaction, led to a postulate of the existence of a catalytic triad involving Thr89, Lys162, and Asp90, reminiscent of a triad present in serine proteases.5 However, another threonine (Thr12) is also located near the ligand‐binding site. These observations, supported by additional structures of the orthologous enzymes from other organisms, initiated a still‐unsettled debate on the nature of the catalytic mechanism of this enzyme. The initial question involved identification of the residue acting as the primary nucleophile in the enzymatic reaction, if the mechanism follows double displacement (Figure 1a). Two main reasons fueled this debate. While positions of both threonines relative to the substrate are similar, Thr12 seems structurally to be more favorable to play that role. Although Thr89 is a member of a putative catalytic triad, Thr12 is assisted only by the hydroxyl group of Tyr25. However, recently a third option was introduced and discussed,6 with the authors suggesting a possibility of a single displacement mechanism of catalysis (Figure 1b), in which neither of the threonine residues would chemically engage the substrate.
Figure 1.
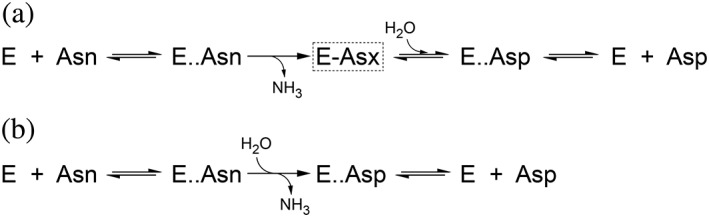
Schematic representation of the putative (a) double displacement mechanism of l‐asparaginase, and (b) single displacement mechanism. In the former case, the boxed structure indicates a covalent intermediate
In the first step of a double‐displacement model of the mechanism of catalysis by l‐asparaginases, either Thr12 or Thr89 would form a covalent acyl‐enzyme intermediate with the substrate through nucleophilic attack, with release of the first product (NH3 in the case of l‐Asn being the substrate) upon completion of this step. Subsequently the covalent intermediate would be hydrolyzed in the second step by one of the water molecules (or a hydroxyl anion) present in the active site. Such nucleophilic substitution would result in formation of the second product (l‐Asp) and its subsequent release from the active site pocket. Both nucleophilic substitutions would proceed through a tetrahedral transition state. A general scheme of the double displacement mechanism, as putatively utilized by l‐asparaginases, is shown in Figure 1a. According to an alternative single‐displacement scenario, a substrate molecule complexed with the enzyme assumes the position and conformation such that it could be subjected to direct nucleophilic attack by a water molecule (or OH−). The reaction would proceed through a tetrahedral transition state, but with no formation of a covalent intermediate, and both products would be released simultaneously. A general scheme for this mechanism is shown in Figure 1b.
Since the release of the first structure of type II l‐asparaginase from E. coli (EcAII), a total of over 70 structures of closely related l‐asparaginases have been deposited in the Protein Data Bank (PDB). They represent enzymes from at least 12 different organisms, and many of these structures describe complexes with products or substrates. In this report, we present an analysis of the geometric relationships and the distributions of the atomic displacement parameters (B‐factors) of selected atoms located within the active site pockets of complexes of Type I and Type II l‐asparaginases from five different sources. The principal aim of this work was twofold. First, we investigated the extent of conservation of these relationships for all the available structures representing complexes of l‐asparaginases, as well as for their specific subsets (i.e., Type I, II, E. coli, etc.). Second, we used the results of this analysis as the basis of discriminating between three possible scenarios of the catalytic mechanism (distinguishing between single‐displacement and the two possibilities of double‐displacement, the latter utilizing different threonine residues as primary nucleophiles), to check for possible preferences or contradictions. To add more experimental data we determined 11 new high‐resolution structures of complexes formed between the wild‐type or mutated l‐asparaginases with l‐Asn or l‐Asp. Combined, our analysis included 120 crystallographically independent active sites of L‐asparaginases from five different organisms, for which structures were determined in a relatively wide range of conditions.
2. RESULTS
2.1. Active site of l‐asparaginase
In the canonical reaction catalyzed by l‐asparaginases the carboxyamide of L‐Asn is converted into carboxylate via hydrolysis, resulting in release of ammonia and L‐Asp. It was also previously shown that the β‐carboxylate of L‐Asp bound to the active site of EcAII undergoes oxygen exchange catalyzed by the enzyme,7 a process that is equivalent to hydrolysis of this “substrate.” Such an oxygen exchange process may take place in the crystalline state perpetually, as the substrate is never depleted. Kinetic studies of mutated L‐asparaginases identified five residues, almost entirely conserved in all Types I and II L‐asparaginases that are directly involved in catalysis.8, 9 In EcAII, this quintet comprises Thr12, Tyr25, Thr89, Asp90, and Lys162, with the first two residues contributed by a flexible N‐terminal hairpin. Thr12 and Tyr25 are adjacent to each other during the reaction, whereas the structurally conserved triad consists of Thr89, Asp90, and Lys162. Because of its therapeutic significance,10 EcAII has been studied most extensively, therefore this enzyme usually provides a reference for the whole family. Consequently, when referring to one of the five active site residues mentioned above, we will use the sequence numbers of EcAII regardless of the organism from which the enzyme was isolated. For further clarification of the equivalences between these five residues for l‐asparaginases used in this analysis, see Table S1. With the exception of Tyr25 all five residues are contributed by a single monomer, but in l‐asparaginases from guinea pig and from Pyrococcus furiosus the active site Tyr is supplied by a different monomer. However, even in these cases, location of the tyrosine hydroxyl is conserved, suggesting its equivalent role in catalysis; therefore, we will also refer to this residue as Tyr25. Similarly, while the catalytic efficiency of Type I and Type II l‐asparaginases (i.e., EcAI and EcAII) is quite different, striking similarity of their active sites suggests a common catalytic mechanism.
2.2. l‐asparaginase structures included in the analysis
In the analysis presented here, we investigated stereochemical relationships of the carboxyamide (or the equivalent carboxylate) groups with several adjacent side chains within the active site. Therefore, only complexes of l‐asparaginase with l‐Asn, l‐Asp, or succinic acid were considered. We excluded from analysis all complexes with ligands larger than l‐Asn, such as l‐Glu or l‐Gln, as well as any covalent complexes. Furthermore, all structures included here retain at least one of the two threonine residues in the active site (Thr12 or Thr89). A search of the PDB revealed 20 entries satisfying these conditions (Table 1). Additionally, we included in the analysis 11 structures of the wild‐type and mutated EcAII, or Dickeya dadantii l‐asparaginase (for historical reasons referred to as ErA), determined in our laboratory but not previously reported (Table 2).
Table 1.
A list of structures previously deposited in the PDB and included in the analysis of the active site geometry and B‐factor distribution
| PDB ID | Enzyme source/type | Ligand/pH | Resolution (å)a |
|---|---|---|---|
| 3eca | EcAII (wt) | l‐Asp/5.0 | 2.4 |
| 5mq5 | EcAII (N24S) | l‐Asp/7.5 | 1.6 |
| 1nns | EcAII (wt) | l‐Asp/6.0 | 1.95 |
| 1ho3 | EcAII (Y25F) | l‐Asp/4.8 | 2.5 |
| 2jk0 | D. dadantii (wt) | l‐Asp/7.0 | 2.5 |
| 2gvn | D. dadantii (wt) | l‐Asp/5.5 | 1.9 |
| 5f52 | Dickeya dadantii (wt) | l‐Asp/7.5 | 1.63 |
| 5i3z | D. dadantii (E63Q) | l‐Asp/7.5 | 2.05 |
| 5i48 | D. dadantii (A31I,E63Q) | l‐Asp/7.5 | 1.5 |
| 5i4b | D. dadantii (E63Q,S254N) | l‐Asp/7.5 | 1.6 |
| 1hg0 | D. dadantii (wt) | Succinate/5.4 | 1.9 |
| 5k3o | Wolinella succinogenes (P121) | l‐Asp/7.5 | 1.696 |
| 5k4g | W. succinogenes (S121) | l‐Asp/7.5 | 1.6 |
| 4nje | Pyrococcus furiosus (wt) | l‐Asp/6.0 | 2.5 |
| 5b5u | P. furiosus (wt) | l‐Asp/8.5 | 2.61 |
| 2wlt | Helicobacter pylori (wt) | l‐Asp/7.0 | 1.4 |
| 4r8l | Guinea pig (wt) | l‐Asp/7.0 | 2.41 |
| 5dnc | Guinea pig (T19A) | l‐Asp/7.0 | 2.01 |
| 5dnd | Guinea pig (T116A) | l‐Asp/7.0 | 2.29 |
| 5dne | Guinea pig (K188 M) | l‐Asp/7.0 | 2.39 |
As reported in the original depositions.
Table 2.
A list of newly determined structures included in the analysis of the active site geometry and B‐factor distributions
| ID | Enzyme source/type | Ligand/pH | Resolution (å) | Active sites in a.u. |
|---|---|---|---|---|
| 1 | EcAIIwt | l‐Asp/7.0 | 1.73 | 4 |
| 2 | EcAIIwt | l‐Asp/5.6 | 1.60 | 4 |
| 3 | EcAIIT12V | l‐Asp/5.5 | 1.85 | 4 |
| 4 | EcAIIT12V | l‐Asn/7.0 | 1.88 | 2 |
| 5 | EcAIIT89V/K162T | l‐Asn/7.0 | 1.65 | 4 |
| 6 | EcAIIT89V/K162T | l‐Asn/7.0 | 1.90 | 4 |
| 7 | EcAIIT89V/K162T | l‐Asp/7.0 | 1.85 | 4 |
| 8 | EcAIIT89V/K162T | l‐Asn/8.3 | 2.12 | 4 |
| 9 | EcAIIT89V/K162T | l‐Asn/8.3 | 2.00 | 8 |
| 10 | EcAIIK162M | l‐Asp/5.6 | 1.90 | 4 |
| 11 | ErA | l‐Asp/5.6 | 1.59 | 4 |
The structures included in this analysis originated from crystals grown under widely different conditions and characterized by different crystal packing. They were determined at different resolution and were refined with different software utilizing different restraints. Finally, the quality of structural characterization also varied. We did not attempt, however, to correct for such discrepancies and used the coordinates directly as retrieved from the PDB. An exception was made for only one entry, 5mq5, representing the structure of the EcAII(N24S) mutant in complex with l‐Asp.11 In that case, the original authors deposited the structure in which the occupancies for all atoms in l‐Asp were set to 0.0 (despite evident presence of this ligand in the active sites), resulting in significantly incorrect contacts between the enzyme and the ligand. We reset the occupancies of l‐Asp to 1.0 and performed 10 cycles of refinement with the program Refmac512 against structure factors deposited in the PDB. The resulting coordinates were used in analysis.
2.3. New structures included in analysis
The newly determined structures of the native and mutated forms of EcAII describe complexes with l‐Asp or l‐Asn, as well as ErA in complex with l‐Asp. Structures were carefully refined with care taken to minimize potential bias introduced by use of the molecular replacement method for their determination (see Table 3 and Section 4). Although not particularly novel, these structures still provide some important new information. The structure of the EcAIIT12V variant has shown that mutation Thr12Val does not change the mode of binding of the substrate. Structures of the complexes of inactive variants (i.e., EcAIIT12V and EcAIIT89V/K162T) with l‐Asn indicated that the placement of l‐Asp and l‐Asn in the active site of l‐asparaginase is virtually indistinguishable. The new structures represent four different variants of EcAII (wild type and three‐mutated forms), crystallized in three different space groups under different conditions (pH between 5.5 and 8.3), adding high‐quality description of 46 independent active sites of Type II l‐asparaginase in complex with substrates.
Table 3.
Statistics of data collection and refinement. Individual structures are identified by the numbers introduced in Table 2
| Data set | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Resolutiona | 40–1.73 (1.775–1.73) | 40–1.6 (1.63–1.6) | 40–1.85 (1.9–1.85) | 40–1.88 (1.93–1.88) | 40–1.65 (1.71–1.65) | 40–1.90 (1.95–1.93) |
| Space group | C2 | C2 | C2 | P21212 | C2 | C2 |
| Unit cell axes (å) | 151.31, 62.37, 140.93 | 151.18, 62.40, 140.66 | 151.84, 62.47, 143.37 | 61.07, 68.91, 130.97 | 152.85, 62.99, 141.29 | 151.79, 62.746, 140.97 |
| Unit cell angles (°) | 90, 117.6, 90 | 90, 117.6, 90 | 90, 118.1, 90 | 90, 90, 90 | 90, 117.8, 90 | 90, 117.7, 90 |
| Completeness (%)a | 94.9 (92.7) | 88.0 (54.9) | 91.0 (55.1) | 94.0 (67.1) | 99.7 (97.0) | 99.4 (98.8) |
| Redundancya | 2.7 (2.6) | 2.5 (1.4) | 3.0 (1.9) | 5.6 (3.8) | 3.7 (3.1) | 3.0 (2.0) |
| <I>/σ<I>a | 23.4 (1.9) | 36.1 (3.1) | 19.2 (1.9) | 33.3 (2.05) | 20.7 (2.3) | 14.6 (2.0) |
| Rsym (%)a | 3.7 (46.9) | 1.9 (26.9) | 5.3 (39.4) | 4.4 (61.4) | 7.7 (43.1) | 4.4 (42.5) |
| Structure refinement | ||||||
| Number of refl. Refined/free | 112,878/2548 | 131,536/3432 | 86,417/4403 | 40,674/2081 | 139,105/2723 | 83,883/4394 |
| Resolution (å) | 40–1.73 | 40–1.6 | 40–1.85 | 40–1.88 | 40–1.6 | 40–1.90 |
| Completeness (%) | 98.6 | 88.0 | 89.5 | 93.5 | 99.5 | 95.1 |
| R/Rfree | 13.7/17.8 | 13.0/17.2 | 16.9/22.9 | 15.7/20.9 | 14.6/18.1 | 14.6/19.4 |
| Bond rmsd (å) | 0.019 | 0.020 | 0.020 | 0.020 | 0.021 | 0.019 |
| Angle rmsd (°) | 1.96 | 1.94 | 1.89 | 1.91 | 1.96 | 1.85 |
| PDB ID | 6PAB | 6PAC | 6PAA | 6PA9 | 6PA3 | 6PA8 |
| Data set | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|
| Data collection | |||||
| Resolutiona | 40–1.85 (1.775–1.73) | 40–2.12 (1.63–1.6) | 40–2.0 (2.03–2.0) | 40–1.9 (1.95–1.9) | 30–1.6 (1.66–1.6) |
| Space group | C2 | C2 | P21 | P21 | C2 |
| Unit cell axes (å) | 151.40, 62.41, 142.40 | 151.22, 62.40, 140.875 | 140.53, 62.33, 151.05 | 62.18, 124.47, 81.74 | 106.16, 90.40, 127.36 |
| Unit cell angles (°) | 90, 118.0, 90 | 90, 117.7, 90 | 90, 117.7, 90 | 90, 112.4,90 | 90, 91.9, 90 |
| Completeness (%)a | 95.2 (63.4) | 98.4 (93.7) | 95.8 (87.3) | 99.4 (98.8) | 85.8 (81.6) |
| Redundancya | 3.0 (1.9) | 3.1 (3.1) | 2.9 (2.5) | 5.5 (5.1) | 2.9 (2.0) |
| <I>/σ < I>a | 21.8 (1.65) | 13.2 (1.9) | 14.3 (1.9) | 14.8 (1.95) | 18.2 (3.4) |
| Rsym (%)a | 4.3 (50.8) | 7.1 (42.5) | 6.6 (49.0) | 7.3 (54.6) | 5.5 (33.5) |
| Structure refinement | |||||
| Number of refl. Refined/free | 92,741/2798 | 63,323/1977 | 146,167/4012 | 80,759/2462 | 133,097/3368 |
| Resolution (å) | 40–1.85 | 40–2.12 | 26.2–2.0 | 26.1–1.90 | 29.6–1.59 |
| Completeness (%) | 94.5 | 98.4 | 95.5 | 91.8 | 85.4 |
| R/Rfree | 15.6/20.0 | 16.3/23.1 | 17.0/21.3 | 13.6/18.9 | 13.8/17.8 |
| Bond rmsd (å) | 0.019 | 0.020 | 0.020 | 0.020 | 0.019 |
| Angle rmsd (°) | 1.88 | 1.93 | 1.97 | 2.02 | 2.19 |
| PDB ID | 6PA4 | 6PA6 | 6PA5 | 6PA2 | 6PAE |
Highest‐resolution shell in parentheses.
2.4. Stereochemistry of nucleophilic substitution
High efficiency of nucleophilic substitution requires the fulfillment of several conditions. From a chemical viewpoint, the atom initiating the reaction needs to display nucleophilic properties (be a Lewis base). In each of the three scenarios of the catalytic reaction considered here, the primary nucleophile is an oxygen atom contributed by either one of the two threonine residues, or alternatively by a water molecule (or OH−). Whereas negatively charged OH− is a strong nucleophile, neither water nor a hydroxyl group of threonine mirror this property. In the two latter cases, a potential weak nucleophile needs to be accompanied by a proton acceptor. An accompanying residue, however, does not need to be a strong base that enhances nucleophilicity of the reactive oxygen. A group may assist in proton transfer if it forms part of a well‐defined H‐bonded network linking the nucleophile with a proton “sink” (either a basic residue or an arrangement of proton‐deficient groups).
In terms of stereochemical requirements, the nucleophile must be located along or near the optimal trajectory. In the case of nucleophilic substitution on the carbonyl carbon (i.e., ketones or aldehydes), Bürgi and Dunitz first outlined the optimal geometric parameters,13, 14 the most widely cited of which is the angle of approach by a nucleophile relative to the C=O bond, commonly called the Bürgi–Dunitz angle (αBD). Definition of αBD is graphically explained in Figure 2a. Based on limited crystallographic data for small molecules available at the time and on quantum chemistry calculations, Bürgi and Dunitz predicted the optimal value for αBD to be 105 ± 5°. From the time of its introduction, αBD was adapted to describe reactions with other carbonyl‐containing electrophiles, such as esters or amides. Since αBD does not unambiguously characterize the trajectory of a nucleophile, the second angle, αFL, was introduced by Heathcock, Flippin, and Lodge,15, 16 derived from somewhat similar principles as those used by Bürgi and Dunitz. The optimal αFL angle was proposed to be close to 0°, deviating slightly (±7°) if two substituents (other than the carbonyl oxygen) of electrophilic carbon are different. More recent re‐evaluation of αBD and αFL values for serine proteases17 or for small molecules18 suggested ~90° as the optimal value of αBD. In the case of αFL, however, available experimental information aligns quite well with original predictions and its values departing by more than 10–15° from the optimal are rarely observed.17 Both αBD and αFL depend somewhat on the specific chemical/structural properties of both the nucleophile and the assembly containing the electrophilic reaction center. Whereas the theoretical basis of Bürgi and Dunitz predictions is quite strong, it is possible that the predicted properties of a nucleophile trajectory are manifested at somewhat shorter distances than those observed in a majority of crystallographic reports. This problem, however, is beyond the scope of our investigation. Instead, we aim to utilize the best experimentally obtained values of the αBD and αFL angles as benchmarks for grading a potential of a specific atom to be the nucleophile in the process catalyzed by l‐asparaginases. Therefore, in our analysis we evaluated both angles for each of the two potential nucleophiles, Thr12 and Thr89. We consider that a more likely nucleophile will be the one for which the observed values of αBD will be ~90° and of αFL ~0°, allowing for a spread of both values by several degrees.
Figure 2.
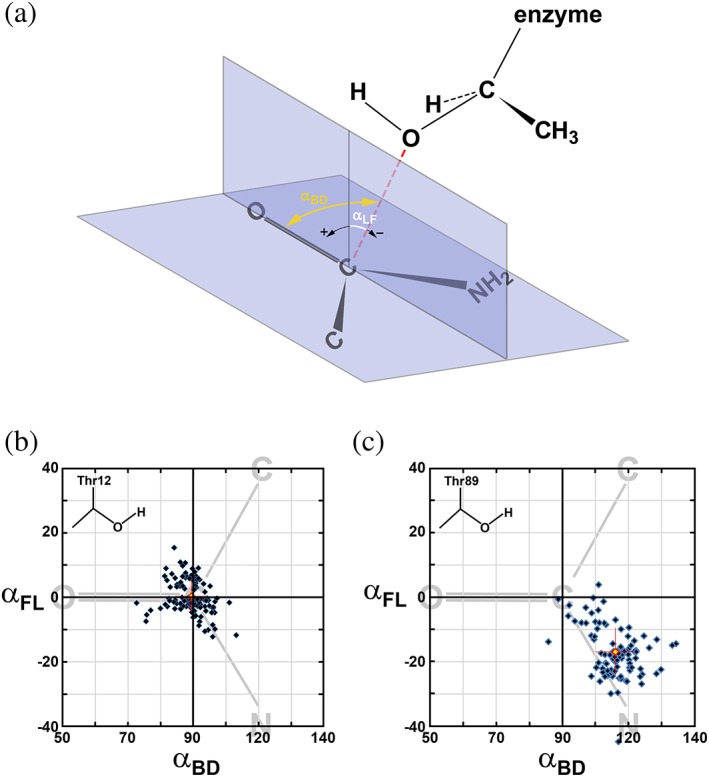
Distribution of putative Bürgi–Dunitz (αBD) and Flippin–Lodge (αFL) angles for the Thr12(Oγ) and Thr89(Oγ) atoms in l‐asparaginases. (a) Angle between the direction of nucleophilic attack (red dashed line) and the carbonyl bond corresponds to αBD. Angle between the direction of nucleophilic attack and its projection on the plane perpendicular the carboxyamide group is defined as αFL. (b) Distribution αBD and αFL for 111 independent active sites of l‐asparaginases complexes with a substrate and calculated based on assumption that Thr12(Oγ) atom is a nucleophile. Each blue diamond represents a single active site and the red diamond represents the average (αBD, αFL). The latter is accompanied by red crosshairs representing values of standard deviations for both angles. The area of distribution is limited to the ranges of 50–140° for αBD and −40 to 40° for αFL which encompasses all data points. This area is coplanar with the carboxyamide group that is schematically represented by simplified formula (shown in gray) and oriented appropriately relative to αBD and αFL axes. (c) The same distribution as described in Panel B, but representing αBD and αFL for 95 independent active sites of l‐asparaginases complexes with a substrate and calculated based on an assumption that Thr89(Oγ) is the nucleophile
Whereas positions of the hydroxyl groups in both threonine residues are known and can be evaluated in terms of stereochemical criteria, no water molecule located in a position near the optimal trajectory for nucleophilic substitution was ever observed in the structures of l‐asparaginase. Therefore, to consider a possibility of direct displacement as a viable mechanism of catalysis by these enzymes, one needs to analyze the requirements necessary to achieve proper presentation of a water molecule (or OH−) in relation to the electrophilic Cγ atom of a substrate.
It is generally agreed that crystal structures do not provide direct information about molecular dynamics. However, if carefully assessed, estimates of such properties of proteins can be obtained with confidence, especially if they are consistent across the structures of homologous molecules, independently determined from crystals grown under different conditions. The atomic displacement parameters (Bf's) are larger for the more flexible parts of the structures, while the most flexible areas of the enzymes may not be modeled at all due to the lack of electron density. Conversely, rigid regions of protein molecules or side chains engaged in stabilizing interactions are characterized by the low Bf values. The absolute values of atomic Bf's from different structures are usually not directly comparable due to differences in resolution, software, protocols, and restraints applied during refinement. However, for each individual structure, Bf's can be converted into unitless (or relative) parameters, rBf = Bfatom/<Bfstructure>. Values of rBf's from different, but related structures are suitable for comparisons.
2.5. Analysis of the geometric and thermal parameters of the active sites
Twelve atoms, labeled in Figure 3, were selected for analysis of the stereochemical parameters that relate an active site bound substrate (product) molecule to the adjacent residues of the enzyme. In addition to the previously mentioned angles αBD and αFL, we defined 11 additional parameters, calculated (whenever possible) for each active site. These parameters, consisting of five distances (d's) and six angles (α's), are defined in Figure 3. Parameters d1 and d2 describe the distances between the potential nucleophiles, n1 (Oγ1 of Thr12) or n2 (Oγ1 of Thr89) and the reaction center RC (Cγ‐carbon of a substrate), respectively. Parameter d3 is the distance between n2 and the leaving group l (NH2 in l‐Asn or OH in l‐Asp/succinate). Distances d1 and d2 complement the angles αBD and αFL in an analysis of the trajectory of potential nucleophilic substitution by Thr12 or Thr89, respectively, whereas d3 characterizes an interaction of the latter residue (n2 atom) with the leaving group L. Presentation of n2 toward L is further described by the angles α1 and αC2n2L (see Figure 3). These three parameters allow to assess the potential for forming H‐bond interactions between n2 and L, especially for the tetrahedral intermediate formed during catalysis. The remaining six parameters illuminate geometric relations of the two potential nucleophiles, n1 and n2, with structurally adjacent residues, Tyr25 and Lys162, respectively. An interaction between n1 and the OH atom of Tyr25 is described by the distance dx and the two angles, αCTOHn1 and αC1n1OH. Similarly, an interaction between n2 and the NZ atom of Lys162 is characterized by the distance dy and two angles, αCLNZn2 and αC2n2NZ.
Figure 3.
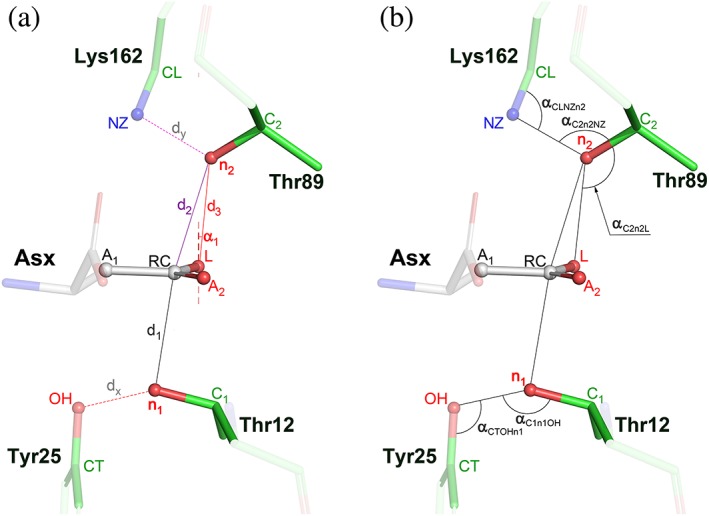
Geometrical relations in an asparaginase active site, occupied by a substrate. Substrate molecule is represented by l‐aspartate (labeled Asx). (a) Side chains of two threonine residues, 12 and 89 (EcAII numbering), sandwiching the reaction center, RC (the Cγ atom of Asx), and adjacent to them Tyr25 and Lys162, are accentuated with brighter colors. Two, potentially nucleophilic, Oγ1 atoms are labeled as n1 (in Thr12) and n2 (in Thr89). Adjacent Cβ atoms in both threonine residues are labeled C1 and C2. Label L denotes the leaving group of the substrate (NH2 in l‐Asn or OH in l‐Asp or succinate). Six additional atoms labeled in this figure are A1 (Cβ) and A2 (Oδ1) in a substrate molecule, OH and CZ (Cζ) from Tyr25, or NZ and CL (Cε) NZ Lys162, respectively. Definitions of distances (d1, d2, d3, dx, and dy) are self‐explanatory. Angle α1 is between the vector normal to the best plane defined by A1, A2, RC, and L, and the vector connecting n2 with L. (b) Five additional angles calculated in this analysis are defined in this panel
The average values, their standard deviations, and the number of independent active sites used for averaging are presented in Table 4. Additionally, for angles αBD and αFL, we prepared distribution diagrams (see Figure 2b for Thr12 and Figure 2c for Thr89) that allow visual assessment of the spread of these parameters across all crystal structures of l‐asparaginases considered here. An analysis of the parameters is presented below in the context of specific scenarios for nucleophilic substitution. However, several observations are quite significant regardless of the mechanism of catalysis. An overview of data shown in Table 4 indicates that most parameters are relatively well conserved among all asparaginases. For instance, the average values of αBD1 vary within a range of 5.5°, with the extreme values of 85.0° and 90.5°. Similarly, the average values of αC2n2L are clustered between 111.9° and 124.9°. An even higher degree of conservation is observed for the three parameters describing the interaction between Thr89 and Lys162, namely dy, αCLNZn2, and αC2nzNZ. The spread of values for other parameters is only slightly higher than in the examples above. Such high conservation of the interactions between the active sites and substrate molecules is quite remarkable, since the enzymes originate from biologically very divergent organisms (i.e., E. coli, guinea pig, or Helicobacter pylori). While the sequence of EcAII is 60% identical to the sequence of ErA, it is only 21% identical to the sequence of guinea pig l‐asparaginase. Furthermore, the active sites of enzymes from guinea pig and P. furiosus include contribution from different structural motifs than those in Type II l‐asparaginases. Additionally, the structures included in this study describe crystals grown under very different conditions. Therefore, an assumption that the catalytic mechanism is common to all l‐asparaginases seems to be justified.
Table 4.
Geometrical characterization of the ASNase active site, occupied by a substrate molecule. The reported values represent averages calculated over all relevant entries available in the PDB (Table 1) and 11 additional structures determined during this study (Table 2)
| Parametera | All entriesb | ASNase II | Escherichia coli | Dickeya | Guinea pig | Extremophiles |
|---|---|---|---|---|---|---|
| αBD1 (°) | 89.0 ± 4.9 (111) | 89.3 ± 4.7 (96) | 90.5 ± 5.2 (53) | 87.9 ± 3.7 (39) | 87.2 ± 6.4 (12) | 85.0 ± 2.1 (3) |
| αFL1 (°) | 0.4 ± 5.1 (111) | 0.5 ± 5.1 (96) | −0.4 ± 5.2 (53) | 2.5 ± 5.0 (39) | −2.7 ± 3.5 (12) | 1.0 ± 2.1 (3) |
| d1 (å) | 2.83 ± 0.18 (111) | 2.84 ± 0.19 (96) | 2.86 ± 0.20 (53) | 2.86 ± 0.17 (39) | 2.70 ± 0.07 (12) | 2.95 ± 0.22 (3) |
| αBD2 (°) | 106.5 ± 5.8 (95) | 106.3 ± 5.6 (80) | 104.5 ± 6.8 (33) | 108.0 ± 4.5 (39) | 107.3 ± 8.8 (11) | 109.9 ± 6.2 (3) |
| αFL2 (°) | −17.5 ± 6.9 (95) | −18.9 ± 6.0 (80) | −19.1 ± 8.4 (33) | −18.4 ± 3.7 (39) | −7.9 ± 5.3 (11) | −18.5 ± 6.3 (3) |
| d2 (å) | 2.98 ± 0.11 (95) | 2.97 ± 0.11 (80) | 3.01 ± 0.13 (33) | 2.95 ± 0.08 (39) | 3.04 ± 0.08 (11) | 3.05 ± 0.18 (3) |
| d3 (å) | 2.73 ± 0.19 (95) | 2.69 ± 0.18 (80) | 2.75 ± 0.25 (33) | 2.67 ± 0.11 (39) | 2.96 ± 0.16 (11) | 2.75 ± 0.20 (3) |
| α1 (°) | 9.1 ± 6.3 (95) | 7.8 ± 5.5 (80) | 10.3 ± 6.4 (33) | 6.6 ± 4.4 (39) | 18.0 ± 4.8 (11) | 9.6 ± 6.3 (3) |
| αC2n2L (°) | 120.7 ± 5.2 (84) | 121.6 ± 4.7 (73) | 120.5 ± 6.0 (26) | 122.2 ± 6.0 (39) | 111.9 ± 5.4 (7) | 124.9 ± 6.9 (3) |
| dx (å) | 2.81 ± 0.24 (62) | 2.82 ± 0.26 (47) | 2.68 ± 0.11 (24) | 3.03 ± 0.26 (19) | 2.81 ± 0.06 (12) | 2.68 ± 0.22 (3) |
| αC1n1OH (°) | 108.0 ± 15.7 (62) | 112.0 ± 13.8 (47) | 101.4 ± 4.2 (24) | 127.8 ± 4.3 (19) | 94.7 ± 3.0 (12) | 98.0 ± 23.3 (3) |
| αCTOHn1 (°) | 106.2 ± 11.0 (62) | 107.0 ± 10.9 (47) | 100.0 ± 4.7 (24) | 117.6 ± 4.5 (19) | 102.6 ± 3.5 (12) | 105.2 ± 10.0 (3) |
| dy (å) | 2.73 ± 0.06 (84) | 2.73 ± 0.07 (73) | 2.72 ± 0.09 (26) | 2.73 ± 0.06 (39) | 2.71 ± 0.04 (7) | 2.75 ± 0.07 (3) |
| αCLNZn2 (°) | 118.7 ± 5.9 (84) | 119.0 ± 5.6 (73) | 120.3 ± 5.8 (26) | 118.0 ± 5.8 (39) | 120.4 ± 0.7 (7) | 106.6 ± 14.8 (3) |
| αC2n2NZ (°) | 119.1 ± 4.3 (95) | 119.3 ± 4.4 (80) | 120.0 ± 5.2 (33) | 118.0 ± 5.0 (39) | 118.2 ± 3.0 (11) | 116.0 ± 7.8 (3) |
Data shown in Table 4 also indicate that the spatial relationship between Thr89 and Lys162 (see parameters dy, αCLNZn2, and αC2n2NZ) is particularly invariant, as illustrated by a very small spread of the average values of these parameters calculated for different subsets of enzymes, as well as by their relatively small standard deviations. Also, a spatial relationship of the n2 atom (from Thr89) with RC and L atoms of the substrate is common to all subsets of enzymes within the uncertainties of structure determination. While some deviations can be seen for ErA, this subset (7–11 active sites) is relatively small, and the observed differences have no qualitative significance. Structural invariance of this motif (Thr89‐Lys162‐substrate) is paralleled by its apparent positional rigidity (see Table 5). The values of rBf's for both Thr89 and Lys162 are among the lowest for the whole enzyme and those calculated for the substrate correspond to the average values of all non‐H enzyme atoms. The central role of both Thr89 and Lys162 in catalysis has already been well established through studies of mutated forms19, 20; however, results of this analysis indicate that both residues retain near‐fixed positions throughout the catalytic cycle.
Table 5.
Relative atomic displacement parameters (rBf's) for selected atoms in the active sites of l‐asparaginase
| Escherichia coli residue | Atoma | rBfAvr ± rBfStdDev (count)b | E. coli residue | Atoma | rBfAvr ± rBfStdDev (count)b |
|---|---|---|---|---|---|
| Substrate | A1 (CB) | 0.990 ± 0.280 (120) | Thr89 | C2 (CB) | 0.658 ± 0.168 (95) |
| A2 (OD1) | 0.913 ± 0.267 (120) | n2 (OG1) | 0.685 ± 0.169 (95) | ||
| RC (CG) | 1.013 ± 0.302 (120) | Lys162 | CL (CE) | 0.700 ± 0.182 (88) | |
| L (NH2,OH) | 1.009 ± 0.286 (120) | NZ (NZ) | 0.721 ± 0.190 (88) | ||
| Thr12 | C1 (CB) | 1.024 ± 0.334 (111) | Tyr25 | CT (CZ) | 1.539 ± 0.633 (66) |
| n1 (OG1) | 1.036 ± 0.361 (111) | OH (OH) | 1.503 ± 0.717 (66) |
The names of the atoms used in this analysis defined in Figure 2 are shown in bold and are followed by conventional names of these atoms (in parentheses).
For each included atom i the value of rBfAvr = rBf = Bfatom/<Bfstructure>, represents average over all structures of l‐asparaginases included here, followed by associated value of a standard deviation (± rBfStdDev) and number of independent active sites used in calculations (in parenthesis).
Another significant (although indirect) observation may be derived from data presented here. Although structures included in this study represent complexes with l‐Asp, l‐Asn, and even succinic acid, positioning of the reactive carboxyamide (l‐Asn) or carboxylate (l‐Asp and succinic acid) groups against the residues of l‐asparaginases implicated in the catalytic process is virtually invariant. This observation is strongly supported by the values of parameters presented in Table 4. No different mode of substrate binding has been reported for l‐asparaginases. Therefore, it is conceivable that a substrate molecule is tightly restrained by interactions with the active site groups and is not subjected to extreme conformational (or positional) rearrangements throughout the catalytic reaction. Such conclusion needs to be accounted for in consideration of any plausible mechanism of catalysis.
2.6. Double‐displacement mechanism with Thr12 as the primary nucleophile
The first clue supporting the possibility of n1 (Oγ1 atom of Thr12) being the primary nucleophile is its average distance of 2.83 å (d1 in Table 4) from RC (Cγ atom of substrate), calculated for 111 available active sites. In protein structures carbon and oxygen atoms rarely reach separation shorter than the sum of their van der Waals radii (3.25 å). In l‐asparaginases, the n1‐RC distance is not enforced by external restraints, since Oγ1 of Thr12 is contributed by a highly mobile structural element. In contrast, it appears that both atoms display mutual attraction. Estimates of partial charges of atoms in the carboxyamide group of l‐Asn, derived from both experimental data21 and from theoretical predictions22 indicate that RC carries a partial positive charge. This would suggest that one of the two lone electron pairs of n1 is attracted to RC via electrostatic interactions, a prerequisite for nucleophilic attack. The distance d1, observed across all individual active sites, varies quite significantly (between 2.5 å and 3.4 å), with a majority being close to the calculated average value. This variability is significantly larger than expected from the estimated coordinate errors. Many structures included in this analysis were determined for active enzymes in complex with of l‐Asp, assuring a perpetual catalytic reaction (see above). The rate of catalysis in such crystals certainly depends on pH, buffer composition, enzyme modifications, substrate concentration, and other factors. However, the resulting structure represents an average of states representing all individual active sites. Both the short distances between n1 and RC and a wide range of these distances among different structures can be rationalized assuming that Thr12 participates directly in the catalytic reaction. A short value of d1, however, does not immediately support the role of n1 as the primary nucleophile. Additional clues emerge through analysis of αBD1 and αFL1 angles (Table 4), which are quite consistent across different subsets of l‐asparaginases. Furthermore, these average values, as well as distributions representing structures of all available active sites (see Figure 2b) are in good agreement with data published earlier for serine/cysteine proteases17 and for small molecules.18
In the structures of complexes of enzymatically active l‐asparaginases, n1 is always assisted by OH (primarily the OH atom of Tyr25). The average separations of both atoms, dx (2.81 å for 62 active sites), as well as the relevant angles αC1n1OH (107.4° for 62 active sites), and αCTOHn1 (105.7 for 62 active sites) indicate that the OH groups of Tyr25 and Thr12 are in near‐optimal positions to form strong H‐bonds. This does not necessarily mean that Tyr25 enhances the nucleophilic properties of n1, as a tyrosine residue does not have properties necessary for such a function. Instead, the OH group of Tyr25 participates in a network of H‐bonds connecting n1 with a proton‐deficient (basic) group or arrangement, thus it plays a role of a proton conveyor. It has been previously suggested that the basic residue enhancing nucleophilic properties of Thr12 may be Glu283, which in EcAII forms a pseudo‐triad, together with Tyr25 and Thr12.23 However, this is quite unlikely to be a general phenomenon since many l‐asparaginases lack the equivalent of Glu28324, 25 and mutants of Glu283 in EcAII are catalytically active.19 Therefore, if n1 is the primary nucleophile, its activation is provided by more distant structural elements, networked through hydrogen bonds with this atom through additional components, most likely water molecules observed in the active site region in all high‐resolution crystal structures of l‐asparaginases (Figure 4). However, detailed analysis of the role of other residues that might assist catalysis is beyond the scope of this report.
Figure 4.
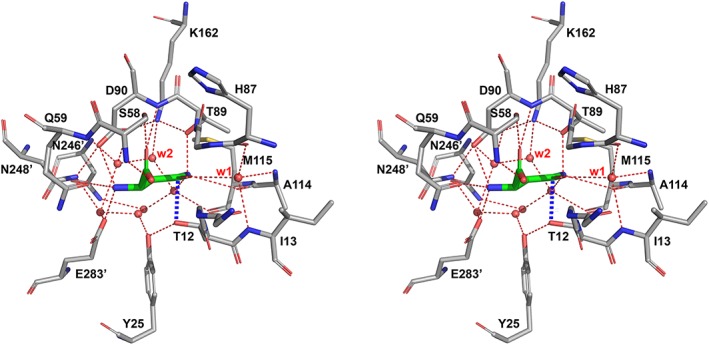
Active site of EcAII in complex with l‐Asp. Structure No. 2 (complex between EcAII (wt) and l‐Asp at pH 5.6), was determined at 1.6 å and refined to R work (R free) = 0.130 (0.172). The stabilizing interaction (2.6 å) between Thr12 and the substrate is marked with thick dashed blue line. Also, well‐defined position of Tyr25 (in the closed state of N‐terminal hairpin) is shown. Two, invariant water molecules are labeled as w1 and w2
Finally, it is instructive to confront this scenario with the distribution of rBf's (Table 4). Their values for the carboxyamide (or carboxylate) groups of substrates, as well as for the side chain of Thr12 are comparable to the averages calculated for the whole enzyme. Positional stability of the side chain of Thr12 is particularly remarkable, since that residue is contributed by the most dynamic fragment of the enzyme. In contrast, rBf values of CT and OH, contributed by Tyr25 that is, like Thr12, also located within the N‐terminal flexible hairpin, indicate significantly more dynamic properties of Tyr25. A comparison of rBf's values for Thr12 and Tyr25 clearly indicates a different degree of stabilization of these residues by the environment of the active site. Although the values of rBf's provide only modest support for the nucleophilic role of n1, their meaning becomes more significant when the two remaining scenarios of a putative catalytic mechanism are discussed below.
The geometric and stereochemical relationships between Thr12 and the adjacent components of active sites of l‐asparaginases in complex with substrates are fully consistent with the possibility that this residue could act as the primary nucleophile in the catalytic reaction. More importantly, this conclusion applies not just to a specific enzyme (i.e., EcAII) or a narrow subset of l‐asparaginases (i.e., ASNase II) but it is in good agreement with all structurally related enzymes, including Type I l‐asparaginases (i.e., from guinea pig), or enzymes from extremophile bacteria.
2.7. Double‐displacement mechanism with Thr89 as the primary nucleophile
Similarly to Thr12, the side chain of Thr89 is positioned close to the plane of carboxyamide (or carboxylate) of the substrate, but on the opposite side (Figure 3). The average distance of n2 (the Oγ1 atom of Thr89) from the RC, d2, for 99 available active sites is 2.98 å and it is also significantly less than the sum of van der Waals radii (3.25 å). However, analysis of a trajectory projecting n2 toward RC, described by the αBD2 and αFL2 angles, indicates their significant deviations from the expected optimal values (Figure 2c). While the average value of αBD2 of 106.1° appears to be close to that proposed by Bűrgi and Dunitz,14 it is quite different from the updated values observed for both proteolytic enzymes17 and for small molecules.18 The average αFL2 angle appears to be even less favorable (expected to be close to 0°), which in nearly half of the analyzed active sites departs from the optimal value by more than 20°, and in an extreme case (PDB entry 1ho3, monomer A) reaches −45°.
The n2 atom forms a strong H‐bond with the adjacent Lys162 NZ, as indicated by the value of dy (2.73 å). This notion is also supported by the values of angles αC2n2NZ (119.3°), and αCLNZn2 (118.7°). As mentioned earlier, all three parameters are highly conserved in all structures (Table 4). Therefore, to attain an optimal position for efficient nucleophilic attack, either the substrate molecule would have to shift along the direction of its side chain, or the side chain of Thr89 should reorient. Either of these alterations, however, presents energetic challenges. The first alteration is prevented by an interaction of the carboxyamide group with the structurally rigid main chain carbonyl of Ala114 (Figure 4). Reorientation of the Thr89 side chain would break a strong H‐bond with the Lys162 NZ. Additionally, an analysis of rBf's values for Thr89 and Lys162 (Table 4) shows that both side chains form one of the most rigid motifs in the whole enzyme. Such analysis does not completely eliminate Thr89 as a candidate for the role of the primary nucleophile, but indicates that the residue would have to undergo significant conformational changes throughout the reaction. In such a case, however, even if the catalytic events occur in just a small fraction on the time between subsequent binding events, any significant conformational changes taking place should be recorded in the form of elevated Bf's. Yet, this effect is consistently not seen in l‐asparaginases. Therefore, based purely on the stereochemical analysis, it may be concluded that n2 (Thr89) is a less likely candidate for a role of the primary nucleophile than n1 (Thr12).
Evaluation of the nucleophilic potential of Thr89 needs to be extended beyond the strictly geometric considerations. In all l‐asparaginases, Thr89 participates in an arrangement suggestively similar to the catalytic triad of serine proteases5 (here Asp‐Lys‐Thr (Figure 4), compared to Asp‐His‐Ser in proteases). In contrast to Thr12, n2 is adjacent to a potentially basic residue, Lys162. A basic role of Lys162, however, is manifested only when its amino group is unprotonated. Under physiological conditions, such a state is more likely in a water‐restricted environment. However, Lys162 NZ in l‐asparaginases is well hydrated, even if a substrate occupies the active site (Figure 4). Therefore, it is most plausible assuming that the amino group of Lys162 is charged, thus unable to extract a proton from the adjacent n2 (or enhance its nucleophilic properties). Support for this conclusion is further strengthened by the fact that EcAII retains about 25% of its maximum catalytic activity even at pH 4.5.7 Under such conditions, deprotonation of Lys162 would be equivalent to a change of its pKa value from the standard 10.526 by over six log units, which is highly unlikely.27, 28 There are two additional possibilities. The first one involves Asp90, the remaining component of the putative triad, which potentially might accept a proton. Yet, this scenario would still require Lys162 to transition through an uncharged state. The other option could be transfer of the proton from n2 to the leaving group of a substrate (NH2 or OH); however, the considered leaving groups do not have the basic properties needed to deprotonate n2, so this option is also is not favorable. These additional considerations again suggest that Thr89 is a less likely candidate for the role of the primary nucleophile in a reaction catalyzed by l‐asparaginases.
Analysis of three additional parameters, d3, α1, and αC2n2L, with the average values of 2.74 å, 9.4°, and 120.7°, respectively, correlating n2 with the leaving group of the substrate, L (the Nδ2 atom in case of l‐Asn or the Oδ2 atom in l‐Asp), suggests the presence of a potential H‐bonded interaction between both atoms, or maybe even proton transfer from n2 to L. In the Michaelis complex of l‐asparaginase, the angular relationship between n2 and L (best described by a small value of the angle α1) does not seem to support a strong H‐bond. However, during the catalytic process, that is, beginning with a nucleophilic attack by Thr12(Oγ1), hybridization of the Cγ atom of a substrate changes from sp2 to sp3. As a result, presentation of the leaving group L toward n2 may become suitable for near‐optimal H‐bond (possibly proton transfer), without any need for rearrangement of the Thr89 side chain. Being engaged in a persistent H‐bond with Lys162, Thr89 may play a central role in conveying a proton to L, a process that is critical to catalysis. Based on geometric relationships linking Thr89 with Lys162 and a substrate (Table 4), it appears plausible that this residue, while not in the optimal location for nucleophilic attack, may play a central role in supplying L with the necessary proton.
2.8. Direct displacement mechanism
Compared to both scenarios analyzed above, a putative direct displacement mechanism does not involve chemical engagement of the enzyme during catalysis. A water molecule participating in the reaction must acquire nucleophilic properties, most likely via activation by a basic residue of the enzyme, and it must have at least transient access to the reaction center along an appropriate trajectory. Such a nucleophilic water may putatively approach RC from either side of the carboxyamide plane, and the mode of its activation could be similar to those discussed earlier for either n1 or n2. It is important, however, to stress that no water molecule positioned for nucleophilic attack has been observed in any of the structures published to date. Therefore, to consider a direct displacement mechanism, it is necessary to evaluate a possibility of rearrangements necessary for placing a water molecule in a site suitable for a nucleophilic attack. Figure 5 depicts a representative active site of l‐asparaginase with several atoms surrounding the reaction center, represented by van der Waals spheres. Three water molecules shown in this figure are observed in multiple structures, although not in some due to mutations of the active site residues or because of lower resolution of the structural models. It is quite clear, however, that in the case illustrated here, water molecules occupy some of the accessible positions closest to RC. Therefore, it is quite apparent that for a water molecule to assume a position suitable for nucleophilic substitution, some active site side chains and/or the substrate molecule would have to undergo significant conformational changes. Although such changes have not been so far observed in crystal structures of l‐asparaginases, one may argue that such states could be transient in nature and too short‐lived to be seen in static X‐ray reconstructions. However, even such an argument is unconvincing, since the required conformational changes would most likely be reflected by elevated values of Bf's. Yet, the values of rBf's for Thr89 and Lys162, residues defining the most probable direction from which a nucleophilic attack by a water molecule could take place, are among the lowest for the whole enzyme. In the context of experimental structures, a water molecule closest to RC is the one labeled w2 in Figure 4 (also depicted in Figure 5). This water molecule is highly conserved among all l‐asparaginases and it may be critical for hydrolysis of the covalent intermediate in a double displacement mechanism. However, this molecule is not in a suitable position for nucleophilic attack according to the direct displacement scenario and has no obvious access to such a position without pronounced conformational changes of Thr89, substrate, or both. In conclusion, it appears that, based on the available structural data and on geometrical considerations, a possibility of direct displacement mechanism of catalysis by l‐asparaginases is unlikely.
Figure 5.
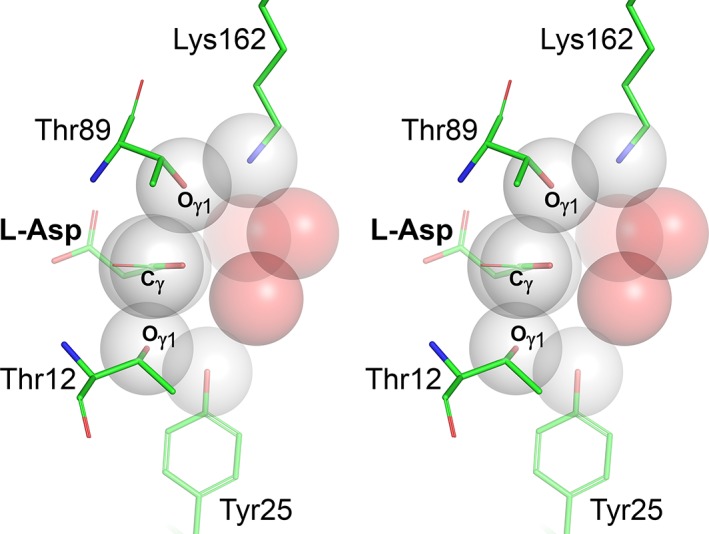
Access of water molecules to a preferred trajectory of the nucleophilic attack in the active sites of l‐asparaginases. Several active site atoms (Oγ1 atoms in Thr12 and Thr89, Cβ and Cγ atoms in a substrate, OH atom in Tyr25, and NZ atom in Lys162) are represented by grey semi‐transparent Van der Waals spheres. Three water molecules (oxygen atoms), found in crystals structures in positions closest to the reaction center (atom Cγ of substrate) are shown as red transparent Van der Waals spheres. It is quite apparent that no water molecule can reach the site suitable for a nucleophilic attack (i.e., below or above the carboxyamide plane near the reaction center) without major structural rearrangement of at least one threonine residue, residue adjacent to it (i.e., Tyr25 or Lys162) and the side chain of a substrate
3. DISCUSSION
In this report, we provided an analysis of a set of distances and angles relating a substrate/product molecule to the active site residues of l‐asparaginase. Our analyses were conducted for all structures, as well as for narrower subsets (i.e., ASNases II, EcAII, etc.). Our first goal was to determine how consistent (or divergent) are the active sites of l‐asparaginases in complex with substrates (or products). Moreover, we aimed to determine whether general stereochemical relationships within the active site of such complexes could point towards a particular enzymatic mechanism. Whereas catalytic reactions are principally governed by chemical properties of the groups involved, it is clear that most enzymatic processes, including nucleophilic substitutions, require favorable stereochemical arrangements for their efficient progression. Furthermore, the use of crystal structures in evaluating enzymatic mechanisms may suffer from such limitations as the static nature of results, potential effects of crystal packing, or conditions of crystal growth. Despite such limitations, X‐ray crystallography at high‐to‐atomic resolution is still a gold standard for description of interatomic interactions. While the effects of crystal packing and crystallization conditions can sometimes lead to misinterpretation of structural data, they can be filtered out in a global analysis involving structures obtained from crystals grown under widely different conditions and characterized by different packing of molecules, as is the case in this study.
Based on our analysis, we found that two active site residues, Thr89 and Lys162, exhibit very low mobility (low relative Bf's) and both their mutual spatial relationship as well as that with a substrate/product molecule is highly conserved across all l‐asparaginases. In the case of two other active site residues, Thr12 and Tyr25, these properties are not as highly conserved. This effect is a result of several factors. First, in a majority of l‐asparaginases included in this analysis, both Thr12 and Tyr25 are contributed by a flexible N‐terminal hairpin, one of the most labile fragments in this enzyme. As a result, structural description of these residues may be somewhat less accurate, particularly in the case of Tyr25. Second, in some l‐asparaginases (from guinea pig and from P. furiosus), Tyr25 is contributed to the active site in a different mode than in the case of ASNases II. Finally, as discussed in Section 2, geometric relationships between both residues and the substrate/product molecule are dependent on the step of the enzymatic process. Since the latter likely varies from structure to structure, the effects are reflected by inter‐atomic distances and the associated angular relations. In general, however, we found that the four active site residues mentioned above and the substrate/product molecule form a remarkably invariant system in terms of their stereochemical relationships. This observation itself suggests a common enzymatic mechanism for all common‐fold l‐asparaginases.
An evaluation of three scenarios describing a putative catalytic mechanism, based mainly on geometric characterization of the active sites of l‐asparaginases, suggests that direct displacement is the least likely. No single currently available structure supports a possibility for an approach of a water molecule (or OH−) to a position suitable for nucleophilic substitution. Even if such an event would take place within the timeframe that is undetectable by crystallographic methods, due to the required significant conformational changes its remnants would be recorded by elevated Bf's of adjacent residues. Such an effect, however, is not observed. While this report focuses exclusively on geometric relations, we also indicated in quite general terms that the chemistry required by the direct displacement mechanism is unlikely in the l‐asparaginase active site. This observation disagrees with a recent proposal6 whose authors postulated a single‐displacement mechanism because of an inability to observe the evidence of a double displacement mechanism (i.e., burst kinetics or a covalent intermediate). Since with the current technology it is rather difficult to find direct evidence for direct displacement, a conclusion of such a scenario is reached indirectly, by an exclusion of an alternative.
Of the two other scenarios, both representing a double displacement reaction, the analysis presented here suggests Thr12 to be the primary nucleophile. Such stereochemical and geometric considerations as the directional presentation of the nucleophilic hydroxyl oxygen towards the reaction center (the Cγ atom of substrate) and the distance between both atoms clearly favor Thr12 over Thr89 to be involved in the first step of a chemical reaction. While neither of the two threonine residues is a strong nucleophile, experiments and analyses going beyond the scope of this report are necessary to clarify the chemical aspects of the reaction.
Finally, it is important to note that l‐asparaginases represent a quite unique case within a larger family of hydrolases. Except for a few examples of enzymes that utilize N‐terminal, self‐activated threonine residue as a nucleophile and are structurally completely unrelated to l‐asparaginases,29 no other hydrolases with a nucleophilic threonine are presently known. The lack of a familiar catalytic triad (well characterized for serine proteases5) further emphasizes the unique properties of l‐asparaginases. An availability of continuously increasing number of high‐resolution structures of these enzymes from quite divergent organisms bodes well for the possibility of a very accurate description of their catalytic mechanism, which is even more valuable taking into account over 40‐year history of their therapeutic service.
4. MATERIALS AND METHODS
4.1. Geometry of residues in the active sites of l‐asparaginases
Our analysis included all structures of l‐asparaginases currently available in the PDB that describe active sites of complexes with a substrate (i.e., l‐Asn, l‐Asp, and succinic acid, but not larger substrates such as l‐Gln). We identified 20 such structures (Table 1). Additionally, 11 new structures of such complexes of EcAII and its mutated forms, as well as ErA (Tables 2, 3) were determined. Altogether 120 independent active sites were included in the analysis. For each entry, up to five distances and 10 angles were measured and subsequently averaged over all entries and their specific subsets.
Definitions of selected distances and angles are provided in Figures 2a and 3. Due to mutations, not all parameters could be calculated for each active site. The resulting 15 parameters are represented by their average values, standard deviations, and the number of individual measurements used for averaging (Table 4).
4.2. Cloning, expression, and purification of EcAII and its variants
Preparation of plasmids and bacterial cell lines used for expression was described previously.30, 31 The identity and correctness of all plasmids used for heterologous expression of proteins were confirmed by DNA sequencing. Expression experiments were performed using E. coli cell line deficient in three endogenous l‐asparaginse genes (a triple knockout), thus assuring that all l‐asparaginase activity would originate from the recombinant gene. Whereas such a procedure is critical for kinetic and/or functional studies, it was not crucial to strictly structural experiments discussed here. In all cases l‐asparaginases were secreted to media in a general form Met‐Asp‐(His)6‐EcAII, and the affinity tag was not excised prior to structural studies.
The purification protocol consisted of two steps, Ni‐affinity chromatography in batch mode and size exclusion chromatography (for details see Supporting Information). On average about 15–20 mg of purified enzyme could be recovered from 1 L of E. coli cell culture. It is worth noting that Ni‐affinity chromatography resulted in high‐purity preparations (as monitored by SDS‐PAGE); however, subsequent gel‐filtration contributed to (a) buffer replacement, (b) increased purity, and (c) lower content of aggregates, all of which are significant for successful crystallization. In the case of ErA the protein sample used for crystallization was the same as the one described previously.32
4.3. Crystallization and collection of X‐ray data
All crystals of EcAII or its mutated forms included in this study were grown under very similar conditions using solutions of protein at 8–12 mg/ml in 50 mM Hepes buffer (pH 7.0) and 150 mM NaCl. In some instances, crystals were grown in the presence of l‐Asp or l‐Asn, at defined concentrations. Otherwise, crystals grown in the absence of a substrate were subsequently soaked in equivalent solutions enriched with substrates at defined concentrations. In two cases (both utilizing the EcAIIT89V/K162T mutant), crystals grown at pH 7.0 were subsequently transferred to a compatible precipitating solution buffered at pH 8.3. For more details, see Table S2. This table also shows a composition of the cryo‐protecting solutions for each type of crystals.
With an exception of crystal No. 5 (see Table 2), which describes the complex of the EcAII(T89 V,K162 T) mutant with l‐Asn at pH 7, all diffraction experiments were performed using an in‐house conventional X‐ray source, a Rigaku rotating anode MicroMax‐007 HF generator operated at 40 kV and 30 mA, with the CuKα wavelength of 1.5418 å. Images were recorded in a continuous mode with a Dectris Eiger 4 M pixel detector. Diffraction data for crystal No. 5 were collected on beamline 22‐ID at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). All diffraction experiments were conducted at 100 K. The images were processed and scaled using the program HKL3000.33 Details of data collection and the processing statistics are presented in Table 3.
4.4. Structure solution and refinement
Although a majority of crystals were isomorphous to those previously published, all structures were solved independently by molecular replacement with the program Phaser in order to minimize possible bias.34 As the search probe we used either the monomer A of EcAII (taken from PDB entry 3eca), or a monomer of ErA, extracted from the PDB entry 1o7j. In both cases, the N‐terminal flexible hairpins (residues 11–35 in EcAII or residues 14–38 in ErA) were removed from the search models and residues corresponding to Thr12, Thr89 and Lys162 were mutated to Ala. The search models were also “stripped” of all ligands and solvent. In all cases, easily identifiable molecular replacement solutions were first subjected to rigid‐body refinement at the resolution of 2.5 å with Refmac5,12 followed by several cycles of refinement of positions and isotropic atomic displacement parameters (Bf's) for non‐H atoms, using the same program. In subsequent rounds of crystallographic refinement, the resolution was gradually extended to reach the limits of experimental data. Models were regularly inspected using the program Coot35 and appropriate corrections were introduced, including proper modeling of residues 12, 89, and 162, as well as ordered sections of the N‐terminal hairpin. Ligand molecules and solvent were gradually incorporated in the structure based on difference electron density peaks. The near‐final models were evaluated by the MolProbity server36 and completed by applying additional corrections coupled with structural refinement. The statistics for the final structural models are shown in Table 3.
Supporting information
Table S1 Equivalent residues in the active sites of l‐asparaginases from different organisms
Table S2. Details of the crystallization conditions for structures determined in this study
ACKNOWLEDGMENTS
We would like to thank Dr. Matthias Bochtler for his insightful discussions, Drs. Phillip Lorenzi and Waikin Chan for providing cells expressing EcAII and its mutated forms, and Dr. Di Zhang for help with purification of EcAII preparations. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Lubkowski J, Wlodawer A. Geometric considerations support the double‐displacement catalytic mechanism of l‐asparaginase. Protein Science. 2019;28:1850–1864. 10.1002/pro.3709
REFERENCES
- 1. Clementi A. La désamidation enzymatique de l'asparagine chez les différentes espéces animales et la signification physiologique de sa presence dans l'organisme. Arch Int Physiol. 1922;19:369–398. [Google Scholar]
- 2. Kidd JG. Regression of transplanted lymphomas in vivo by means of normal Guinea pig serum. I course of transplanted cancers of various kinds in mice and rats given Guinea pig serum, horse serum or rabbit serum. J Exp Med. 1953;98:565–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broome JD. Evidence that the L‐asparaginase activity of Guinea pig serum is responsible for its antilymphoma effects. Nature. 1961;191:1114–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swain AL, Jaskólski M, Housset D, Rao JKM, Wlodawer A. Crystal structure of Escherichia coli L‐asparaginase, an enzyme used in cancer therapy. Proc Natl Acad Sci USA. 1993;90:1474–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem Sci. 1998;23:347–352. [DOI] [PubMed] [Google Scholar]
- 6. Schalk AM, Antansijevic A, Caffrey M, Lavie A. Experimental data in support of a direct displacement mechanism for type I/II L‐asparaginases. J Biol Chem. 2016;291:5088–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Röhm KH, Van Etten RL. The 18O isotope effect in 13C nuclear magnetic resonance spectroscopy: Mechanistic studies on asparaginase from Escherichia coli . Arch Biochem Biophys. 1986;244:128–136. [DOI] [PubMed] [Google Scholar]
- 8. Wehner A, Derst C, Specht V, Aung H‐P, Röhm KH. The catalytic mechanism of Escherichia coli asparaginase II. Hoppe Seylers Z Physiol Chem. 1994;375:S108–S108. [Google Scholar]
- 9. Palm GJ, Lubkowski J, Derst C, Schleper S, Röhm KH, Wlodawer A. A covalently bound catalytic intermediate in Escherichia coli asparaginase: Crystal structure of a Thr‐89‐Val mutant. FEBS Lett. 1996;390:211–216. [DOI] [PubMed] [Google Scholar]
- 10. Egler RA, Ahuja SP, Matloub Y. L‐asparaginase in the treatment of patients with acute lymphoblastic leukemia. J Pharmacol Pharmacother. 2016;7:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maggi M, Mittelman SD, Parmentier JH, et al. A protease‐resistant Escherichia coli asparaginase with outstanding stability and enhanced anti‐leukaemic activity in vitro. Sci Rep. 2017;7:14479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murshudov GN, Skubak P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. 2011;D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burgi HB, Dunitz JD, Shefter E. Geometrical reaction coordinates II nucleophilic addition to a carbonyl group. J Am Chem Soc. 1973;95:5065–5067. [Google Scholar]
- 14. Burgi HB, Dunitz JD, Lehn JM, Wipff G. Sterochemistry of reaction paths at carbonyl centres. Tetrahedron. 1974;30:1563–1572. [Google Scholar]
- 15. Heathcock CH, Flippin LA. Acyclic stereoselection. 16. High diastereofacial selectivity in Lewis acid mediated additions of enol silanes to chiral aldehydes. J Am Chem Soc. 1983;105:1667–1668. [Google Scholar]
- 16. Lodge EP, Heathcock CH. Steric effects, as well as sigma.*‐orbital energies, are important in diastereoface differentiation in additions to chiral aldehydes. J Am Chem Soc. 1987;109:3353–3361. [Google Scholar]
- 17. Radisky ES, Koshland DE Jr. A clogged gutter mechanism for protease inhibitors. Proc Natl Acad Sci USA. 2002;99:10316–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rzepa H. The Bürgi–Dunitz angle revisited: A mystery? Winnower. 2015;6:e143149.99424. [Google Scholar]
- 19. Schleper S (1999) PhD thesis, Chemistry Dept., University of Marburg, Germany.
- 20. Anishkin A, Vanegas JM, Rogers DM, et al. Catalytic role of the substrate defines specificity of therapeutic l‐Asparaginase. J Mol Biol. 2015;427:2867–2885. [DOI] [PubMed] [Google Scholar]
- 21. Arnold WD, Sanders LK, McMahon MT, et al. Experimental, hartree−fock, and density functional theory investigations of the charge density, dipole moment, electrostatic potential, and electric field gradients in l‐asparagine monohydrate. J Am Chem Soc. 2000;122:4708–4717. [Google Scholar]
- 22. Strohmeier M, Stueber D, Grant DM. Accurate (13)C and (15)N chemical shift and (14)N quadrupolar coupling constant calculations in amino acid crystals: Zwitterionic, hydrogen‐bonded systems. J Phys Chem A. 2003;107:7629–7642. [DOI] [PubMed] [Google Scholar]
- 23. Ortlund E, LaCount MW, Lewinski K, Lebioda L. Reactions of pseudomonas 7A glutaminase‐asparaginase with diazo analogues of glutamine and asparagine result in unexpected covalent inhibitions and suggests an unusual catalytic triad Thr‐Tyr‐Glu. Biochemistry. 2000;39:1199–1204. [DOI] [PubMed] [Google Scholar]
- 24. Aghaiypour K, Wlodawer A, Lubkowski J. Structural basis for the activity and substrate specificity of Erwinia chrysanthemi L‐asparaginase. Biochemistry. 2001;40:5655–5664. [DOI] [PubMed] [Google Scholar]
- 25. Yun MK, Nourse A, White SW, Rock CO, Heath RJ. Crystal structure and allosteric regulation of the cytoplasmic Escherichia coli L‐asparaginase I. J Mol Biol. 2007;369:794–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lide DR. Handbook of chemistry and physics. 72nd ed. Boca Raton, FL: CRC Press, Inc, 1991. [Google Scholar]
- 27. Ho MC, Menetret JF, Tsuruta H, Allen KN. The origin of the electrostatic perturbation in acetoacetate decarboxylase. Nature. 2009;459:393–397. [DOI] [PubMed] [Google Scholar]
- 28. Isom DG, Castaneda CA, Cannon BR, Garcia‐Moreno B. Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci USA. 2011;108:5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 å resolution. Science. 1995;268:533–539. [DOI] [PubMed] [Google Scholar]
- 30. Khushoo A, Pal Y, Singh BN, Mukherjee KJ. Extracellular expression and single step purification of recombinant Escherichia coli L‐asparaginase II. Protein Expr Purif. 2004;38:29–36. [DOI] [PubMed] [Google Scholar]
- 31. Cantor JR, Yoo TH, Dixit A, Iverson BL, Forsthuber TG, Georgiou G. Therapeutic enzyme deimmunization by combinatorial T‐cell epitope removal using neutral drift. Proc Natl Acad Sci USA. 2011;108:1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller M, Rao JKM, Wlodawer A, Gribskov MR. A left‐handed crossover involved in amidohydrolase catalysis. Crystal structure of Erwinia chrysanthemi L‐asparaginase with bound L‐aspartate. FEBS Lett. 1993;328:275–279. [DOI] [PubMed] [Google Scholar]
- 33. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL‐3000: The integration of data reduction and structure solution ‐ from diffraction images to an initial model in minutes. Acta Cryst. 2006;D62:859–866. [DOI] [PubMed] [Google Scholar]
- 34. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallograhic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 36. Chen VB, Arendall WB III, Headd JJ, et al. MolProbity: All‐atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Equivalent residues in the active sites of l‐asparaginases from different organisms
Table S2. Details of the crystallization conditions for structures determined in this study