

Abstract
The Gram-negative Acinetobacter genus has several species of clear medical relevance. Many fully sequenced genomes belonging to the genus have been published in recent years; however, there has not been a recent attempt to infer the evolutionary history of Acinetobacter with that vast amount of information. Here, through a phylogenomic approach, we established the most up-to-date view of the evolutionary relationships within this genus and highlighted several cases of poor classification, especially for the very closely related species within the Acinetobacter calcoaceticus–Acinetobacter baumannii complex (Acb complex). Furthermore, we determined appropriate phylogenetic markers for this genus and showed that concatenation of the top 13 gives a very decent reflection of the evolutionary relationships for the genus Acinetobacter. The intersection between our top markers and previously defined universal markers is very small. In general, our study shows that, although there seems to be hardly any universal markers, bespoke phylogenomic approaches can be used to infer the phylogeny of different bacterial genera. We expect that ad hoc phylogenomic approaches will be the standard in the years to come and will provide enough information to resolve intricate evolutionary relationships like those observed in the Acb complex.
Keywords: phylogenomics, Acinetobacter, bacterial species, evolutionary biology
Introduction
The biological species concept creates challenges to many organisms, from large mammals to bacteria (Hey 2001); but bacteria are particularly affected because the phenotypic characters that can be used for their classification are limited. Nonetheless, bacterial species designation has a vital role in clinical environments, food industry, agriculture, bioremediation, public health, environmental sciences, and biosafety (Gevers et al. 2005). From a clinical point of view, the description and classification of bacteria are of great importance when it comes to identifying pathogenic agents, which determines epidemiological characteristics useful to its treatment and prevention (Godreuil et al. 2005). Also, cataloging species can reveal clues about the evolutionary forces behind the emergence, transformation, and extinction of bacterial lineages; and even the role of different mechanisms of genetic differentiation and the course of adaptation to new niches (Fraser et al. 2009).
The genus Acinetobacter, which belongs to the order Pseudomonadales within the γ-Proteobacteria, is a genus of Gram-negative, oxidase-negative, and strictly aerobic bacteria. The genus includes pathogenic and nonpathogenic species (de Berardinis et al. 2009). Acinetobacter spp. have been increasingly recognized as important nosocomial pathogens involved in hospital outbreaks; particularly in intensive care units, where they quickly develop resistance even to the most potent antimicrobials (Turton et al. 2010; Antunes et al. 2014). Furthermore, they are very abundant in natural environments, including soils, water, oceans, sediments, polar regions, and contaminated sites (Al Atrouni et al. 2016). Additionally, these species have physiological characteristics associated with important microbiological aspects such as biofilm formation, quorum sensing, oxidative stress, and resistance to antibiotics (Jung and Park 2015). At the time of writing, 60 species with valid names could be found in the List of Prokaryotic Names with Standing in Nomenclature (LPSN, http://www.bacterio.net/; last accessed August 22, 2019), and more are waiting for validation (https://apps.szu.cz/anemec/Classification.pdf; last accessed August 22, 2019). Although the genus description dates back to 1954 (Brisou and Prevot 1954), most species have been described in the last 10 years, at which time many genomospecies have been resolved and named; this goes to show the rapid development of the Acinetobacter taxonomy, which in turn reflects the methodological improvements in bacterial systematics over the last years (Caputo et al. 2019). Several species circumscription methods have been used for the Acinetobacter genus both phenotypic and genotypic. The problem of bacterial species identification affects this versatile genus to a large extent, as it includes a large number of named species but no simple technique for their proper identification (Dijkshoorn and Nemec 2008). At the phenotypic level, MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass spectrometry) is currently becoming the method of choice for the rapid identification of bacterial species in routine hospital diagnoses. However, this method cannot reliably differentiate between some closely related species, including those from the Acinetobacter calcoaceticus–Acinetobacter baumannii complex (Acb complex) (Šedo et al. 2013). Regarding the genotypic methods, the most prominent are DNA–DNA hybridization methods (DDH), different phylogenetic markers such as 16S rRNA, rpoB (La Scola et al. 2006), recA (Krawczyk et al. 2002), gyrB (Yamamoto et al. 1999; Teixeira et al. 2017) and, lately, whole genome sequencing (WGS) along with average nucleotide identity (ANI) methodology have been applied (Nemec et al. 2017; Hu et al. 2018). Of note, from a clinical point of view, the proper identification of a species is of paramount importance, given that very closely related species can have very different antibiotic resistance phenotypes. Thus, to ensure an adequate antimicrobial treatment a reliable species assignation can be very useful.
Numerous completed genome sequences are now available for Acinetobacter spp. opening up the possibility of using whole genome approaches to infer species relationships. Moreover, next-generation sequencing technologies have enabled sequencing genomes of multiple strains within a species or even a population, making it possible to untangle the level of intraspecies variation in the genus (Graña-Miraglia et al. 2017). Despite the great number of genomes available for this genus, no recent study has tried to infer the evolutionary relationships for the species within the Acinetobacter genus.
Here, we use a phylogenomic approach to provide the latest view of the phylogenetic relationships for the Acinetobacter spp., highlighting several cases of misclassification, and to produce a list of well-suited genes for species assignation in the genus. This set of genes was chosen for meeting the most valuable requirements in a phylogenetic marker; high genetic diversity, universality across the genus, no signs of recombination, and a genomic stable context.
Materials and Methods
Database
We built one of the most comprehensive genome databases for the genus Acinetobacter to date. A total of 230 genomes of almost all of the Acinetobacter species described were downloaded from NCBI in November 2018. For each species, we included a maximum of ten either draft or complete genomes and, when, possible at least one type strain. Also, variation within species was taken into account including different sequence types (ST) when possible. We analyzed the completeness and contamination of each genome with CHECKM (Parks et al. 2015), and the genomes that did not meet the requirements of less than 5% contamination and more than 95% completeness were excluded from the database. Species with few genomes at NCBI, Acinetobacterpuyangensis, Acinetobacterqinfengensis, Acinetobacterpragensis, Acinetobacterbohemicus, Acinetobacterkyonggiensis, and Acinetobactermarinus did not meet the quality requirements and were left out of the study. These analyses led to a database of 214 genomes representing 51 different Acinetobacter species (see supplementary table 1, Supplementary Material online), which were (re) annotated using PROKKA version 1.12 (Seemann 2014).
Homologous Groups and Phylogenetic Reconstruction
To construct orthologous groups, we run BlastP of A. baumannii ACICU proteome against the whole database as described in Graña-Miraglia et al. (2018). We selected hits with ≥40% identity, ≥60% of alignment coverage, and with an e-value cutoff of 1.0e-30, as we did in a previous study analyzing the genus Staphylococcus (Graña-Miraglia et al. 2018). We created homologous groups (HG) that had only one gene per genome, which we will refer to as single gene families (SGF) from now on; we found 305 SGFs. Each SGF was aligned with Fast Statistical Alignment version 1.15.9 employing the option –nucprot to align in frame (Bradley et al. 2009). We further tested every SGF for recombination using PhiTest implemented via PhiPack (Bruen et al. 2006), setting the window parameter to 50 nucleotides, and 50 SGFs showed recombination signals. We concatenated all the nonrecombinant SGF alignments and built a tree with RAxML version 8.2.4 with 20 independent inferences from 20 different maximum parsimony trees using GTRGAMMAIX model (Stamatakis 2014), this was our Species Tree. Similarly, we constructed trees as described above but with ten different randomized maximum parsimony trees for each SGF, for the top-ranked genes concatenated alignments and for the concatenated randomly chosen genes. The Shimodaira–Hasegawa (SH) topology test and Robinson–Fould distance (RFD) against the Species Tree were implemented via RAxML version 8.2.4 with the options –f H and –f r, respectively.
Ranking of Phylogenetic Markers, ANI Analysis, and Screening in Environmental Samples
The SGF were ranked according to the percentage of shared bipartitions (ShBip) with the Species Tree and to their nucleotide diversity (π) values, which were obtained with “pegas” in R (Paradis 2010). SGF were ranked in decreasing order according to ShBip and then according to π. Functional annotation for each gene was corroborated in UniProt. We also performed an ANI analysis to estimate the relatedness of the genomes. For this purpose, we ran pyani with MUMmer (ANIm) (Pritchard et al. 2016). The genome pairs with more than 95% of identity were considered to belong to the same species (Goris et al. 2007). The results were visualized with Pheatmap R library. To evaluate the utility of our top marker for species assignation in metagenomes, we downloaded 32 samples from a large freshwater metagenome (The Anacostia river data set, PRJNA498951), reported to have the presence of Acinetobacter species. We used TrimGalore (to trim the reads) and fastqc (to check the quality) to process the data. This shotgun metagenomic data set has 571,885,812 pair-end reads on which we screened for the presence of two Acinetobacter spp. using our top markers and employing the SRST2 tool (Inouye et al. 2014).
Gene Composition Analysis
We used ROARY (Page et al. 2015) to build a matrix of gene content for all species in the genus. We modified default identity (45%) and coverage (60%) parameters to fit a genus analysis. We built a Euclidean distance matrix using the dist function in R and from this, a Neighbor-Joining (NJ) tree using “ape” package (Paradis et al. 2004). We also built a Bray–Curtis dissimilarity index matrix that was analyzed using principal coordinates analysis (PCoA) with “vegan” in R as in Tu and Lin (2016). A correlation matrix was created employing the cor() function in R based on an initial gene content matrix; where the gene content matrix was constructed as in our previous study (Graña-Miraglia et al. 2017).
Results
Establishing the True Evolutionary Relationships for Acinetobacter spp.
The genus Acinetobacter comprises 60 species with validly published names and at least five more could be added soon (http://apps.szu.cz/anemec/Classification.pdf). As of November 2018, at least 55 species had one or more genomes publicly available. The number of Acinetobacter species described has grown exponentially in recent years but the number of publicly available genomes for each species is still very uneven. Therefore, in creating our Acinetobacter genome database, for species with a large number of sequenced strains we only included ten genomes and when the number of genomes for a species was lower than ten, we included as many as available. Type strains were included, when possible, and also different genotypes as per Multi Locus Sequence Typing scheme for A. baumannii and Acinetobacterhaemolyticus. Only high-quality genomes were included (see Materials and Methods) and in total 214 complete genome sequences, comprising 51 species (see supplementary table 1, Supplementary Material online), were considered for the analyses. To reconstruct the phylogenetic relationships of the Acinetobacter species, we used SGF without recombination signals (see Materials and Methods) (Castillo-Ramírez and González 2008) as a proxy of orthologous genes. We found 255 nonrecombinant SGFs and built a phylogeny on the concatenated alignment of these 255 SGFs and rooted the phylogeny using Moraxella atlantae and Moraxella catarrhalis, two species of a closely related genus. Figure 1A shows this phylogeny, which had very good support for most of the clades, as most of the bootstrap values were higher than 80%.
Fig. 1.
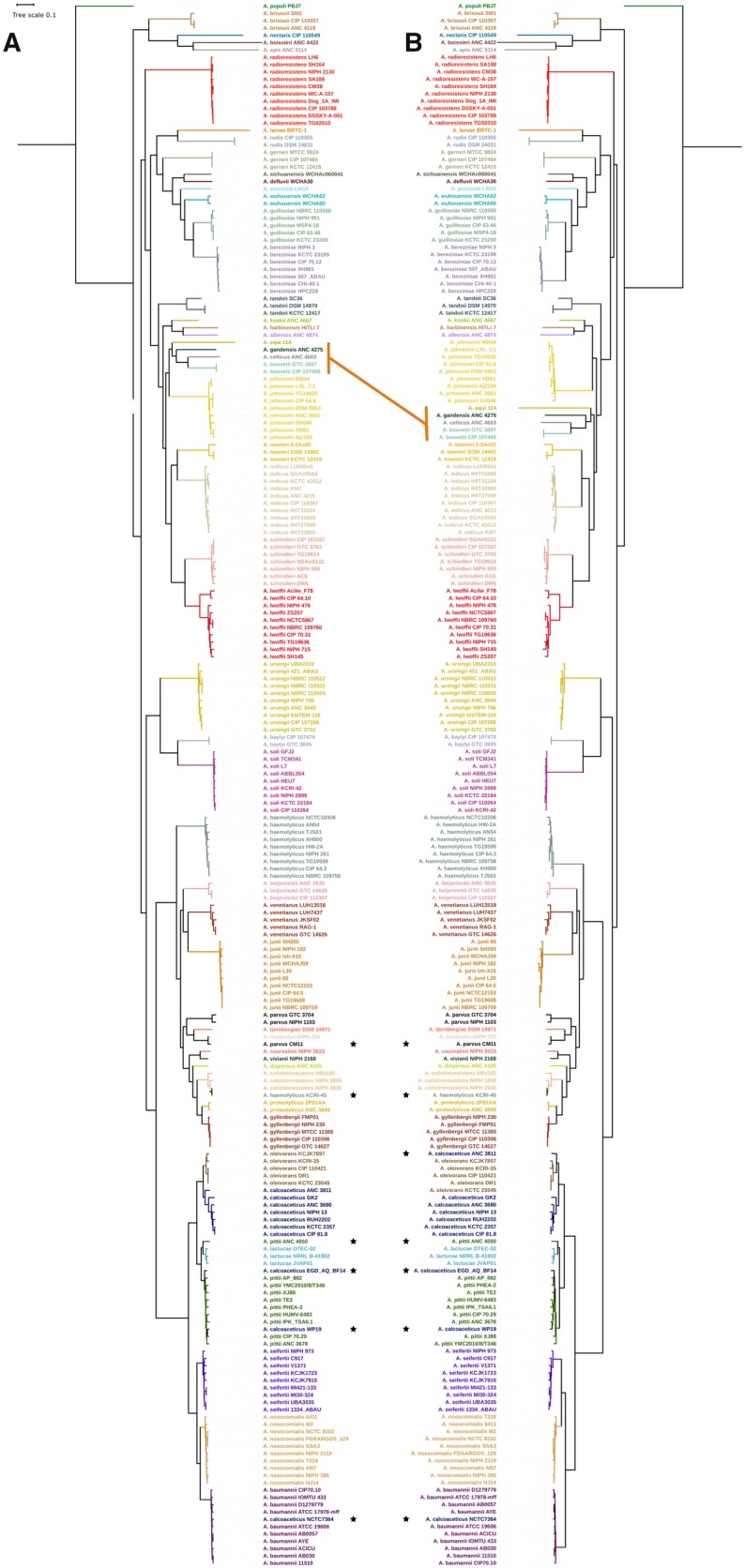
—(A) Maximum likelihood phylogenetic reconstruction of the species tree for the genus Acinetobacter based on the concatenated alignment of 255 nonrecombinant SGF. (B) Maximum likelihood phylogenetic reconstruction based on the 13 top-ranked genes for species assignation (table 1). (A and B) Strains belonging to the same species are colored equally, asterisks show misclassified strains. The orange line highlights one major topological difference between the two trees; this difference involves the clade composed by Acinetobacter equis, Acinetobacter gandensis, Acinetobacter celticus, and Acinetobacter bouvetii. Tree scale shows substitutions per site.
Most of the strains previously assigned to a given species were grouped into monophyletic clades yet some clear exceptions were poorly classified (see asterisks, fig. 1A). Particular cases are highlighted below, but it is worth mentioning that most of them correspond to strains whose genomic features like G + C content or genome size did not match those of the assigned species. We obtained similar clades to those found in a previous study using just a few genomes (Touchon et al. 2014); however, the clades in our study also include other species whose kinship with the rest of the genus was not shown previously. The species branching deeper in the genus is Acinetobacterpopuli and there is also a basal clade formed by Acinetobacterapis, Acinetobacterboissieri, Acinetobacternectaris, and Acinetobacterbrisouii. Furthermore, Acinetobacterradioresistens is also placed in a basal position. The Acb complex is a taxonomic group defined by the inability to properly distinguish phenotypically the species that conform it (Gerner-Smidt et al. 1991) and these species are A. baumannii, Acinetobacterpittii, Acinetobacternosocomialis, A. calcoaceticus, and Acinetobacterseifertii. The group has been previously described as a monophyletic clade (Touchon et al. 2014) and our phylogeny shows that Acinetobacteroleivorans and Acinetobacterlactucae, quite recently described (Kang et al. 2011; Rooney et al. 2016), cluster within the Acb complex even though were supposed to cluster outside the Acb complex. We found various misclassified strains within the Acb complex (see asterisks in fig. 1A). For instance, two A. calcoaceticus strains (WP19 and EGD_AQ_BF 14) were classified as A. pittii according to our phylogeny; additionally, A. calcoaceticus NCTC 7364 clustered within A. baumannii. This is not surprising given that classification issues are common in the Acb complex. Also the A.pittii-like strain ANC 4050 was grouped with the closely related species A. lactucae, but judging by the long-branch length, A. pittii-like strain ANC 4050 could be classified as a new, different species within the Acb complex. Another case of misclassification is A. haemolyticus strain KCRI-45, which according to our phylogeny the strain actually belongs to Acinetobactercolistiniresistens species. Furthermore, a strain previously classified as Acinetobacterparvus CM11 was shown to be Acinetobactermodestus species. This strain (A. parvus CM11) was classified using 16S rRNA, rpoB, and gyrB (Saffarian et al. 2015), but identity percentages estimated from gyrB and rpoB alignments of A. parvus CM11 and the A. parvus type strain indicate that the two strains cannot belong to the same species (85.47% and 95%, respectively). The previous cases demonstrate that several cases of poor classification have occurred within this genus.
The species assignation recovered from the phylogeny was confirmed with an ANI analysis (see supplementary fig. 1, Supplementary Material online). The strains grouped as monophyletic clades in the Species Tree show ANI values above 95%, which is the threshold for species designation (Richter and Rosselló-Móra 2009). Moreover, misclassification cases are also corroborated by ANI values; for instance, the A.pittii-like strain, ANC 4050, does not group with any other species and the highest ANI value observed for this strain is 94% with an A. lactucae strain, supporting the idea that this could be a different species also part of the Acb complex. In summary, we obtained a robust phylogeny for this genus and found some misclassified strains, which show that misclassification usually occurs when nonreliable methods for species assignation are used.
Gene Content Dissimilarity Is Not Useful for Species Delineation in Acinetobacter
It has been proposed that shared gene content between genomes is quantitatively determined by phylogeny (Snel et al. 1999) and that genomic fluidity is linked with microbial taxonomy; therefore, gene content dissimilarity can distinguish between closely related bacterial species (Tu and Lin 2016). Also, the identification of unique genes specific to each taxonomic rank has been used for assigning the bacterial taxonomy (Gupta and Sharma 2015). We calculated the gene repertoire for the Acinetobacter species and obtained a total of 39,595 HG (26,221 excluding unique genes) and built a gene composition profile for every genome. These data were analyzed with two different methodologies; on the one side, a Euclidean distance matrix was estimated from these data and the relationships were determined through an NJ tree (fig. 2A). On the other side, the Bray–Curtis dissimilarity index was estimated (excluding unique genes) and grouping was established through a PCoA as in Tu and Lin (2016) (fig. 2B).
Fig. 2.

—(A) NJ approach analysis of gene composition. Strains belonging to the same species are colored equally, asterisks show misclassified strains and strains that are grouped differently in the Species Tree phylogeny. (B) PCoA approach. Biplot Dots share color code with (A) Acb complex is denoted with a black rectangle.
Clearly, the grouping obtained with the NJ approach reflects kinship between strains, as it recovered the monophyletic groups for most of the species, although the topology depicting the evolutionary relationships between species appears to be significantly different to the Species Tree topology as per the SH test (P value <0.05). Some of the poorly classified strains detected with the Species Tree and ANI analysis could also be reassigned with the NJ topology but notably the relationships in the Acb complex are very dissimilar. The PCoA analysis also revealed issues with the Acb complex species, as the dots representing Acb complex strains are completely overlapped. Furthermore, the gene content dissimilarity values of these strains were lower than 0.2, which is the suggested cutoff value for species assignation by Tu and Lin (2016). Furthermore, two strains (AB030 and 11510) belonging to A. baumannii (fig. 1A) were placed within the A. nosocomialis cluster in the NJ approach. Notably, we have previously observed that the AB030 strain has a gene composition radically different from the rest of A. baumannii genomes (Graña-Miraglia et al. 2017). Moreover, A. pittii strains did not form a monophyletic group and were located in three different points in the tree (see fig. 2A dark green branches). To better understand the gene content dissimilarity across the genus, we created a gene content matrix considering all the genomes; this matrix was visualized using a heat map (see supplementary fig. 3, Supplementary Material online). From this analysis is clear that many species had considerable variation in gene content; one exception is A.radioresistens (small rectangle, supplementary fig. 3, Supplementary Material online), which actually show a very similar gene content. On the other hand, from this analysis is clear that species from the Acb complex (big rectangle, supplementary fig. 3, Supplementary Material online) have a rather similar gene content, which might help to explain why this group is not well differentiated in PCoA analysis. To sum up, the use of gene content variation to conduct taxonomic assignation is not reliable for this genus; this is especially true for the Acb complex, where species are not well defined by gene composition.
Adequate Phylogenetic Markers for the Genus
To have an idea of the individual gene histories of the SGFs and see how they compare with the Species Tree, we built individual gene trees for all the 255 SGF used in the Species Tree and compared their topologies with the Species Tree topology through the SH test and RFD. None of the SGF topologies differ significantly from the species tree according to the SH test (P < 0.05), but RFDs were found to vary widely, between 176 and 366. The RFD is the number of bipartitions that are different between the two topologies being compared and it depends on the tree size (number of bipartitions). Here, to standardize RFD we use the percentage of shared bipartitions as a measure of similarity between two topologies. The set of SGFs showed on average a 43.91% of shared bipartitions with the species tree, being the lowest 13.27% and the highest 58.29%. There was no gene tree topology identical to that of the Species Tree. We also estimated the nucleotide diversity (π) of the nonrecombinant SGFs and found that the mean π of all the SGFs was 0.19 ± 0.05; furthermore, tree topologies of genes with high levels of π show higher similarity with the Species Tree (Spearman correlation = 0.4523) (fig. 3). This has been previously observed in the genus Staphylococcus (Graña-Miraglia et al. 2018) and it is due to the resolution improvement that comes with increasing levels of genetic diversity; along those lines, the smallest levels of topological congruence with the Species Tree were produced by SGFs with π values below 0.19. The nucleotide diversity (π) of the SGF can and has been used as a measure of the phylogenetic power of the genes (Cooper and Feil 2006).
Fig. 3.

—The nucleotide diversity of every SGF plotted against the corresponding percentage of ShBip of the gene tree topology with the Species Tree. In turquoise, the top 13 genes in the ranked list of SGF are highlighted. Spearman correlation was estimated in 0.45.
It is well established that a tree topology built from a single locus is not likely to agree with that of the species tree (reflecting the evolutionary history of the species), but this probability increases when several loci are used (Pamilo and Nei 1988). For instance, a common practice when assigning strains to a species is to sequence a couple of phylogenetic markers, being a very frequent combination the 16S rRNA and a protein-coding gene such as rpoB or gyrB (Choi et al. 2013; Wang et al. 2015). Therefore, we ranked our 255 SGF according to the topology similarity with the Species Tree to have an idea of a decent set of phylogenetic markers for the genus Acinetobacter; π was also taken into account and genes with π above the SGF mean were preferred over those with π below the mean. Then, we concatenated the alignments of the top 3, 5, 6, 7, 8, 9, 10, 12, 13, 15, 17, 18, 20 SGFs, built phylogenies and compare them to the Species Tree (fig. 4). As expected, we observed an increase in topology similarity when increasing the number of loci used for the phylogenetic estimation. Furthermore, the concatenated alignments of randomly chosen (from the 255 SGFs) 3, 5, 6, 7, 8, 9, 10, 12, 13, 15, 17, 18, 20 genes were tested and even though the percentage of shared bipartitions increases with loci number, in all cases but one it never reaches the similarity obtained by the top-ranked genes (fig. 4). We noted that with just the 13 top SGFs a very good reflection of the evolutionary relationships was reached (75% of shared bipartitions); we acknowledge that settling on the minimum number of genes reaching 75% of shared bipartitions was a judgment call and, as such, is not meant to be definitive or exhaustive. The top 20 genes are described in table 1 and are not the most well-known phylogenetic markers for this genus. The top 13 genes in this ranking (table 1) had a percentage of shared bipartitions above 53, the mean π of the top 13 genes is 0.22, which is higher than the π estimated for the concatenated alignment of 255 SGF (mean = 0.201). The topology of the phylogenetic reconstruction based on their concatenated alignment retrieves most of the clades observed in the Species Tree (fig. 1B). Of note, there are three genes (rpoB, recA, and, gyrB) that have been extensively used for species delimitation in the genus Acinetobacter; however, out of the three only recA is part of the 255 SGFs but had low π value (0.179). rpoB is not within the 255 SGF as it did not fulfill the alignment length requirement in one strain (Acinetobactersoli L7 < 60% of the gene aligned), whereas gyrB could not be considered as single copy gene (40% similarity between gyrB and parE in Acinetobacterlarvae). Furthermore, both rpoB and gyrB had recombination signals. We did not analyze 16S rRNA performance in this study given as it has been previously shown for Acinetobacter that species delimitation with the current cutoff identity value (99%) is not possible (Chan et al. 2012; Wang et al. 2014).
Fig. 4.

—Percentage of shared bipartitions with the Species Tree for the trees estimated for the concatenated alignments of the top 3, 5, 6, 7, 8, 9, 10, 12, 13, 15, 17, 18, 20 genes (red dots) and for 3, 5, 6, 7, 8, 9, 10, 12, 13 15, 17, 18, 20 randomly chosen genes.
Table 1.
Top 20 Best Ranked Genes for Species Assignation in the Genus Acinetobacter
| Gene Name | RFD | %ShBip | π | UniProt Annotation | |
|---|---|---|---|---|---|
| 1 | NA | 176 | 58.2938 | 0.2681134 | Site-specific recombinase |
| 2 | miaA | 176 | 58.2938 | 0.2180549 | tRNA dimethylallyltransferase |
| 3 | pbp | 180 | 57.3460 | 0.2483083 | Penicillin-binding protein 1B |
| 4 | bamA | 182 | 56.8720 | 0.2117751 | Outer membrane protein assembly factor BamA |
| 5 | cbl | 188 | 55.4502 | 0.2042251 | Cys regulon transcriptional regulator Cbl |
| 6 | tqsA/AI-2 | 192 | 54.5024 | 0.2645531 | AI-2 transport protein TqsA |
| 7 | hemA | 192 | 54.5024 | 0.2266403 | Glutamyl-tRNA reductase |
| 8 | YidC | 188 | 55.4502 | 0.1895524 | Membrane protein insertase YidC |
| 9 | ppsA | 188 | 55.4502 | 0.1570536 | Phosphoenolpyruvate synthase/pyruvate phosphate dikinase |
| 10 | mfd | 194 | 54.0284 | 0.2317331 | Transcription-repair-coupling factor |
| 11 | dxs a | 194 | 54.0284 | 0.2309427 | 1-deoxy-d-xylulose-5-phosphate synthase |
| 12 | NA | 194 | 54.0284 | 0.1792528 | S1 RNA binding domain protein |
| 13 | rlmD | 196 | 53.5545 | 0.2753780 | 23S rRNA (uracil(1939)-C(5))-methyltransferase RlmD |
| 14 | gpsA | 196 | 53.5545 | 0.2131462 | Glycerol-3-phosphate dehydrogenase [NAD(P)+] |
| 15 | lpxK | 200 | 52.6066 | 0.3135703 | Tetraacyldisaccharide 4′-kinase |
| 16 | NA | 200 | 52.6066 | 0.2376158 | Class II glutamine amidotransferase |
| 17 | rluD b | 200 | 52.6066 | 0.2173651 | Pseudouridine synthase |
| 18 | cdsA | 202 | 52.1327 | 0.2674116 | Phosphatidate cytidylyltransferase |
| 19 | rsmH | 202 | 52.1327 | 0.2234929 | Ribosomal RNA small subunit methyltransferase H |
| 20 | NA | 202 | 52.1327 | 0.2165309 | Peptidase S49 family protein |
Shared with previously reported phylogenetic markers for Bacteria [26].
Shared with previously reported phylogenetic markers for the genus Staphylococcus [61].
RFD, Robinson–Fould distance; π, nucleotide diversity; %ShBip, percentage of shared bipartitions with the species tree.
Finally, we tried out the utility of our 13 best markers for searching Acinetobacter species in metagenomic data using SRST2 (see Materials and Methods); we tested the markers on the Anacostia river metagenome data set in which the presence of Acinetobacter species has been previously reported Anacostia river (Cagle et al. 2019). Notably, we found that all the 13 markers were well covered (above 99.9 of their length, see supplementary table 3, Supplementary Material online) with very low levels of divergence for A. baumannii and Acinetobacterjunii. Therefore, it seems that our top 13 markers are also well suited for screening Acinetobacter species in metagenomes.
Discussion
The genus Acinetobacter is a very versatile group of species, for which it is important to provide a phylogenetic context for any evolutionary analysis. Using a robust phylogenomic approach we inferred the most up-to-date and accurate picture of the phylogenetic relationships for the genus and even singled out suitable phylogenetic markers. The Species Tree allowed us to identify several instances of poor classification, with incorrectly assigned strains, and even one taxon (A. pittii-like ANC 4050) that it is very likely to be a new species.
We did not find that any single gene tree matched the Species Tree. This problem in bacteria has been mainly attributed to widespread of Horizontal Gene Transfer (Wolf et al. 2001); however, given that we are dealing with SGF specifically chosen due to vertical inheritance, lack of recombination, and no duplications, it is more likely that topological differences are due to the reduced number of sites analyzed in the gene versus the SGF concatenated alignment (Pamilo and Nei 1988; Haggerty et al. 2009) or incomplete lineage sorting (Castillo-Ramírez and González 2008; Castillo-Ramirez et al. 2010).
Analyzing gene content of different strains and species is very important because gene gain/loss dynamics lies at the center of the theories about the origin and diversification of bacterial species (Cohan 2002); and great importance to gene content and its association with the phenotypic characteristics of a species is expected. Thus, it is highly desirable to have a species delimitation method directly linked to gene composition. We assessed the level of variation in gene composition between Acinetobacter spp., in order to establish if these differences can help in species delimitation. We found that using gene content to delineate species it is not reliable for Acinetobacter spp. When applying the NJ approach (Euclidean distance + NJ clustering), although we obtained monophyletic groups for many species, when compared with the Species Tree topology, the NJ topology could not retrieve accurately the evolutionary relationships between species. Furthermore, a very conflicting arrangement in the Acb complex species was observed. In a similar way, the PCoA approach did not allow us to distinguish properly between species not only in Acb complex species but also in other species (A. soli and Acinetobacterbaylyi for example). Our gene content matrix analysis shows that there is considerable variation in gene content within and between the species of this genus, explaining why the NJ approach and the PCoA might be not as useful as the Species Tree to infer the evolutionary history of the genus. Furthermore, the issues with the Acb complex are not unexpected as the species from this group have a rather similar gene content. Acb complex poses a major challenge to this genus, the short branches in the phylogeny and the high level of gene content similarity shown by Acb complex species suggest a very recent diversification of those species. Interestingly, boundaries for gene exchange appear to be very flexible within the Acb complex clade and probably homologous recombination events can be contributing to homogenize gene composition as well. The grouping established by the presence–absence of genes in the genomes under consideration can reflect the existence of genetic barriers to recombination and the rate of Horizontal Gene Transfer between species (Shapiro and Polz 2014); in this sense, gene content comparison is a very valuable tool for analyzing evolutionary processes underlying species diversification but it is not reliable for species assignation when those rates and bounds of genetic variation can be very different between species in the same genus.
Species assignation issues in Acinetobacter are evident, especially for the Acb complex clade. The rapid and precise identification of Acb species is very relevant from a clinical point of view; thus, it is very important to have rapid and reliable methods for the allocation of bacterial species. Species assignation methods based on whole genome surveys, such as DDH or ANI, are ideal but DDH is methodologically laborious and ANI requires WGS, which can be costly and nonpractical when dealing with large samples. The search for appropriate phylogenetic markers for species assignation in Bacteria has been going for decades. Among the most frequently used markers (not only for Acinetobacter but for many other bacterial species) are rpoB, gyrB, and recA. However, here we show that at least for this genus these are not the best candidates. The multi locus sequence analysis (MLSA) approach has been proposed to replace the DDH technique and even a similarity percentage based on the concatenated alignment of MLSA genes has been proposed (Rong et al. 2009). But this scheme has also been criticized mainly for two reasons: The arbitrariness in the choice of the genes and the possibility of great variation between genera (Chan et al. 2012). Our work, this study and a recent article about the best markers for the genus Staphylococcus (Graña-Miraglia et al. 2018), strongly supports those two issues. On the other hand, other phylogenetic markers proposed previously for Bacteria by Zeigler (2003) were found to be amongst the group of the 255 SGFs. These are, according to the functional annotation, 13 genes (recN, ruvB, dnaJ, ffh, atpA, tig, rho, recA, rpoA, lepA, ftsZ, dxs, and pgk) with very variable functions and widely spread in our ranking. However, only one of these genes is in the top 20 ranked genes (dxs, in 11th position). We also found that 10 of our 255 SGF are shared with a group of phylogenetic markers proposed for the 3 domains of life (Ciccarelli et al. 2006), most of them corresponding to ribosomal proteins. Moreover, when we compared our 255 SGFs with the recently described 177 best makers for the genus Staphylococcus by our group (Graña-Miraglia et al. 2018), we obtained just 11 genes in common (see supplementary table 2, Supplementary Material online). All these facts support the notion that there are very few potential universals markers. However, this study and our recent report study on the genus Staphylococcus clearly suggest that one can identify phylogenetic markers for optimal bacterial species classification in specific genera. Notably, these markers can be used for pathogen detection from environmental samples.
We have chosen our phylogenetic markers using explicit evolutionary criteria and just to be used as specific markers for the genus Acinetobacter, these genes probed to distinguish between different species with high fidelity. Undoubtedly, WGS is the best strategy to infer the evolutionary relationships; however, if WGS is not affordable, we propose to use various loci to assign species within the genus Acinetobacter, according to our study this approach offer high resolution for species assignation. In this regard, we proved that the concatenation of the top 13 markers is enough to increase topological similarity almost to the level of WGS. Clearly, this increase in phylogenetic power is not only because of the larger number of sites being analyzed but also due to the genes chosen, as we tried sets of randomly chosen genes and almost none of them showed the high similarity to the Species Tree as the top markers (fig. 4). Theoretically, these top 13 SGFs are the ideal candidates for developing an MLSA for the genus. However, practical issues (primer design, for instance) should be taken into account; nonetheless, even if some of these were to show experimental drawbacks, those can be replaced by the following genes in the list. Summarizing, we highlight decent phylogenetic markers for reconstructing the phylogeny of the genus and these markers appear to be good phylogenetic markers even for screening species of this genus in metagenomes.
Conclusion
On the whole, this study gives the latest view of the phylogenetic relationships for the Acinetobacter spp. (showing several cases of poor classification) and unveils a list of well-suited genes for species assignation in the genus Acinetobacter. We anticipate that this sort of bespoke phylogenomic strategies will become the norm for many other bacterial genera in the next decades.
Availability of Data and Materials
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to the staff of “Unidad de Administración de Teconología de la Información” of the Centro de Ciencias Genómicas, in particular Victor Manuel del Moral Chávez, Alfredo José Hernández Álvarez, Joel Gómez Espíndola, and Iván Uhthoff Aguilera. We also thank Dr Miguel Angel Cevallos for his comments on the study. This study was supported by CONACyT Ciencia Básica 2016 (grant no. 284276) and “Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica PAPIIT” from UNAM (grant no. IN206019 to S.C.R.). V.M.E. received a scholarship from DGAPA. L.G.M. is a doctoral student from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM), and also received a CONACYT doctoral fellowship (no. 585414).
Literature Cited
- Al Atrouni A, Joly-Guillou M-L, Hamze M, Kempf M.. 2016. Reservoirs of non-baumannii Acinetobacter species. Front Microbiol. 7:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes LCS, Visca P, Towner KJ.. 2014. Acinetobacter baumannii: evolution of a global pathogen. Pathog Dis. 71(3):292–301. [DOI] [PubMed] [Google Scholar]
- Bradley RK, et al. 2009. Fast statistical alignment. PLoS Comput Biol. 5(5):e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisou J, Prevot AR.. 1954. Studies on bacterial taxonomy. X. The revision of species under Acromobacter group. Ann Inst Pasteur. 86(6):722–728. [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D.. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172(4):2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagle R, et al. 2019. Microbiota of the hickey run tributary of the Anacostia river. Microbiol Resour Announc. 8:e00123–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A, Fournier P-E, Raoult D.. 2019. Genome and pan-genome analysis to classify emerging bacteria. Biol Direct 14(1):5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Ramírez S, González V.. 2008. Factors affecting the concordance between orthologous gene trees and species tree in bacteria. BMC Evol Biol. 8(1):300.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Ramirez S, Liu L, Pearl D, Edwards SV.. 2010. Bayesian estimation of species trees: a practical guide to optimal sampling and analysis In: Knowles LL, Kubatko LS, editors. Estimating species trees: practical and theoretical aspects. New Jersey: Wiley-Blackwell; p. 15–33. [Google Scholar]
- Chan J-M, Halachev MR, Loman NJ, Constantinidou C, Pallen MJ.. 2012. Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 12(1):302.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, et al. 2013. Acinetobacter kookii sp. nov., isolated from soil. Int J Syst Evol Microbiol. 63(Pt 12):4402–4406. [DOI] [PubMed] [Google Scholar]
- Ciccarelli FD, et al. 2006. Toward automatic reconstruction of a highly resolved tree of life. Science 311(5765):1283–1287. [DOI] [PubMed] [Google Scholar]
- Cohan FM. 2002. What are bacterial species? Annu Rev Microbiol. 56(1):457–487. [DOI] [PubMed] [Google Scholar]
- Cooper JE, Feil EJ.. 2006. The phylogeny of Staphylococcus aureus—which genes make the best intra-species markers? Microbiology 152(5):1297–1305. [DOI] [PubMed] [Google Scholar]
- de Berardinis V, Durot M, Weissenbach J, Salanoubat M.. 2009. Acinetobacter baylyi ADP1 as a model for metabolic system biology. Curr Opin Microbiol. 12(5):568–576. [DOI] [PubMed] [Google Scholar]
- Dijkshoorn L, Nemec A. 2008. The diversity of the genus Acinetobacter. In: Ulrike Gerischer, editor. Acinetobacter Molecular Biology. Poole, UK: Caister Academic Press 1–34.
- Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP.. 2009. The bacterial species challenge: making sense of genetic and ecological diversity. Science 323(5915):741–746. [DOI] [PubMed] [Google Scholar]
- Gerner-Smidt P, Tjernberg I, Ursing J.. 1991. Reliability of phenotypic tests for identification of Acinetobacter species. J Clin Microbiol. 29(2):277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, et al. 2005. Opinion: re-evaluating prokaryotic species. Nat Rev Microbiol. 3(9):733–739. [DOI] [PubMed] [Google Scholar]
- Godreuil S, Cohan F, Shah H, Tibayrenc M.. 2005. Which species concept for pathogenic bacteria? An E-debate. Infect Genet Evol. 5:375–387. [DOI] [PubMed] [Google Scholar]
- Goris J, et al. 2007. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 57(1):81–91. [DOI] [PubMed] [Google Scholar]
- Graña-Miraglia L, et al. 2017. Rapid gene turnover as a significant source of genetic variation in a recently seeded population of a healthcare-associated pathogen. Front Microbiol. 8:1817.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graña-Miraglia L, et al. 2018. Phylogenomics picks out the par excellence markers for species phylogeny in the genus Staphylococcus. PeerJ 6:e5839.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Sharma VK.. 2015. Using the taxon-specific genes for the taxonomic classification of bacterial genomes. BMC Genomics 16(1):396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggerty LS, Martin FJ, Fitzpatrick DA, McInerney JO.. 2009. Gene and genome trees conflict at many levels. Philos Trans R Soc Lond B Biol Sci. 364(1527):2209–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. 2001. The mind of the species problem. Trends Ecol Evol. 16(7):326–329. [DOI] [PubMed] [Google Scholar]
- Hu Y, et al. 2018. Acinetobacter wuhouensis sp. nov., isolated from hospital sewage. Int J Syst Evol Microbiol. 68(10):3212–3216. [DOI] [PubMed] [Google Scholar]
- Inouye M, et al. 2014. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6(11):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Park W.. 2015. Acinetobacter species as model microorganisms in environmental microbiology: current state and perspectives. Appl Microbiol Biotechnol. 99(6):2533–2548. [DOI] [PubMed] [Google Scholar]
- Kang Y-S, Jung J, Jeon CO, Park W.. 2011. Acinetobacter oleivorans sp. nov. is capable of adhering to and growing on diesel-oil. J Microbiol. 49:29–34. [DOI] [PubMed] [Google Scholar]
- Krawczyk B, Lewandowski K, Kur J.. 2002. Comparative studies of the Acinetobacter genus and the species identification method based on the recA sequences. Mol. Cell. Probes 16(1):1–11. [DOI] [PubMed] [Google Scholar]
- La Scola B, Gundi VAKB, Khamis A, Raoult D.. 2006. Sequencing of the rpoB gene and flanking spacers for molecular identification of Acinetobacter species. J Clin Microbiol. 44(3):827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec A, Radolfova-Krizova L, Maixnerova M, Sedo O.. 2017. Acinetobacter colistiniresistens sp. nov. (formerly genomic species 13 sensu Bouvet and Jeanjean and genomic species 14 sensu Tjernberg and Ursing), isolated from human infections and characterized by intrinsic resistance to polymyxins. Int J Syst Evol Microbiol. 67:2134–2141. [DOI] [PubMed] [Google Scholar]
- Page AJ, et al. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31(22):3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamilo P, Nei M.. 1988. Relationships between gene trees and species trees. Mol Biol Evol. 5:568–583. [DOI] [PubMed] [Google Scholar]
- Paradis E. 2010. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26(3):419–420. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290. [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW.. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25(7):1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK.. 2016. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods 8(1):12–24. [Google Scholar]
- Richter M, Rosselló-Móra R.. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 106(45):19126–19131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Guo Y, Huang Y.. 2009. Proposal to reclassify the Streptomyces albidoflavus clade on the basis of multilocus sequence analysis and DNA–DNA hybridization, and taxonomic elucidation of Streptomyces griseus subsp solvifaciens. Syst Appl Microbiol. 32(5):314–322. [DOI] [PubMed] [Google Scholar]
- Rooney AP, Dunlap CA, Flor-Weiler LB.. 2016. Acinetobacter lactucae sp. nov., isolated from iceberg lettuce (Asteraceae: Lactuca sativa). Int J Syst Evol Microbiol. 66:3566–3572. [DOI] [PubMed] [Google Scholar]
- Saffarian A, et al. 2015. Draft genome sequences of Acinetobacter parvus CM11, Acinetobacter radioresistens CM38, and Stenotrophomonas maltophilia BR12, isolated from murine proximal colonic tissue. Genome Announc. 3(5):e01089–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šedo O, Nemec A, Křížová L, Kačalová M, Zdráhal Z.. 2013. Improvement of MALDI-TOF MS profiling for the differentiation of species within the Acinetobacter calcoaceticus–Acinetobacter baumannii complex. Syst Appl Microbiol. 36(8):572–578. [DOI] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069.24642063 [Google Scholar]
- Shapiro BJ, Polz MF.. 2014. Ordering microbial diversity into ecologically and genetically cohesive units. Trends Microbiol. 22(5):235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA.. 1999. Genome phylogeny based on gene content. Nat Genet. 21(1):108–110. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira AB, Barin J, Hermes DM, Barth AL, Martins AF.. 2017. PCR assay based on the gyrB gene for rapid identification of Acinetobacter baumannii–calcoaceticus complex at specie level. J Clin Lab Anal. 31(3):e22046.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M, et al. 2014. The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol Evol. 6(10):2866–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Q, Lin L.. 2016. Gene content dissimilarity for subclassification of highly similar microbial strains. BMC Genomics 17(1):647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turton JF, Shah J, Ozongwu C, Pike R.. 2010. Incidence of Acinetobacter species other than A. baumannii among clinical isolates of Acinetobacter: evidence for emerging species. J Clin Microbiol. 48(4):1445–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. 2014. Species distribution of clinical Acinetobacter isolates revealed by different identification techniques. PLoS One 9(8):e104882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Coleman-Derr D, Chen G, Gu YQ.. 2015. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 43(W1):W78–W84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, Tatusov RL, Koonin EV.. 2001. Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol Biol. 1(1):8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Bouvet PJM, Harayama S.. 1999. Phylogenetic structures of the genus Acinetobacter based on gyrB sequences: comparison with the grouping by DNA–DNA hybridization. Int J Syst Bacteriol. 49(1):87–95. [DOI] [PubMed] [Google Scholar]
- Zeigler DR. 2003. Gene sequences useful for predicting relatedness of whole genomes in bacteria. Int J Syst Evol Microbiol. 53(6):1893–1900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files.