

Abstract
The emergence of a new class of novel psychoactive substances, N-benzyl-substituted phenethylamine derivatives so-called “NBOMes” or “Smiles”, in the recreational drug market has forced the development of new sensitive analytical methodologies for their detection and quantitation. NBOMes’ hallucinogenic effects mimic those of the illegal psychedelic drug lysergic acid diethylamide (LSD) and are typically sold as LSD on blotter papers, resulting in a remarkable number of fatalities worldwide. In this article, four halide derivatives of NBOMe, namely, 2-(4-fluoro-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine, 2-(4-chloro-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine, 2-(4-bromo-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine, and 2-(4-iodo-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine, were detected and quantified simultaneously using a high-performance liquid chromatographic method, and two detection systems were compared: photodiode array detection (detection system I) and amperometric detection via a commercially available impinging jet flow-cell system incorporating embedded graphite screen-printed macroelectrodes (detection system II). Under optimized experimental conditions, linear calibration plots were obtained in the concentration range of 10–300 and 20–300 μg mL–1, for detection systems I and II, respectively. Detection limit (limit of detection) values were between 4.6–6.7 and 9.7–18 μg mL–1, for detection systems I and II, respectively. Both detectors were employed for the analysis of the four NBOMe derivatives in the bulk form, in the presence of LSD and adulterants commonly found in street samples (e.g. paracetamol, caffeine, and benzocaine). Furthermore, the method was applied for the analysis of simulated blotter papers, and the obtained percentage recoveries were satisfactory, emphasizing its advantageous applicability for the routine analysis of NBOMes in forensic laboratories.
Introduction
Over the last decade, the use of the so-called new psychoactive substances (NPSs) has escalated in an unprecedented rate worldwide, posing a significant risk to public health and to drug control agencies.1 NPSs are considered to be legal alternatives to illicit drugs and are synthetically designed to mimic their structure and euphoric effects but are not controlled under the Misuse of Drug Act.2 By the beginning of 2010, a new group of NPSs, commonly described as “NBOMes”, “N-Bombs”, “Smiles”, or “Solaris”, has emerged into the drug market and Internet vendors, resulting in several cases of intoxication and fatalities.3−8
NBOMes are N-benzylmethoxy-derivatives of the 2C–X series of psychoactive phenethylamines with methoxy substitutions at positions 2 and 5 and a substitution at position 4, often consisting of a halogen (i.e., fluoride, chloride, bromide, or iodide).9 The four most common NBOMe derivatives include (see Scheme 1) 2-(4-fluoro-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine (2a, 25F-NBOMe), 2-(4-chloro-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine (2b, 25C-NBOMe·HCl), 2-(4-bromo-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine (2c, 25B-NBOMe), and 2-(4-iodo-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethan-1-amine (2d, 25I-NBOMe), which is the most popular member of this family among drug abusers.9 They are potent agonist of the 5-HT2A receptor, and even doses in micrograms can produce psychoactive effects.10 NBOMes were first synthesized in the early 2000s by Ralf Heim as a pharmacological tool to study the 5-HT2A receptor.11 However, NBOMe’s use for recreational purposes had never been reported before 2010 when they started to be available through the Internet.9 They are considered as hallucinogenic stimulants that can induce euphoria, hallucination, agitation, tachycardia, serotonin-like syndrome, seizures, intoxication, and eventually death.5,12−14 These hallucinogenic properties resemble those of lysergic acid diethylamide (LSD) (Scheme 1); wherefore, they are commonly sold as LSD on blotter papers taken sublingually or by nasal insufflations.14−17
Scheme 1. Synthesis of 25F-, 25C-, 25B-, and 25I-NBOMe·HCl Derivatives (2a–d) from Their Corresponding Phenethylamine Hydrochlorides (1a–d),

Reagents and conditions: (i) 2-methoxybenzaldehyde/EtOH/room temperature (rt); (ii) NaBH4/EtOH/rt; (iii) HCl (3 M solution in cyclopentyl methyl ether).
(3) Chemical structure of illicit lysergic acid diethylamide (LSD).
In 2013, the U.S. Drug Enforcement Administration temporarily controlled 2b–d as schedule I illegal drugs, for 2 years under the Controlled Substances Act.18 In June 2014, the UK permanently controlled NBOMes and listed them as class A substances under the Misuse of Drugs Act (1971).19 Later, in November 2014, the World Health Organization in Geneva has placed NBOMe derivatives under international control.20 Currently, the U.S. is listing the previously mentioned NBOMe derivatives under schedule I controlled substances according to the Electronic Code of Federal Regulations (e-CFR) published in November 2018.21 Hence, there is a requirement for the development of simple analytical methodologies for the detection and quantification of such illicit drugs to restrict their commercialization and abuse. Analysis of NBOMe derivatives, by validated analytical procedures or in individual case reports, has been extensively performed using a range of analytical approaches (see Table 1 for a summary), such as liquid chromatography–tandem mass spectrometry (LC–MS/MS),11,22−31 ultraperformance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS),4,32−34 gas chromatography–mass spectrometry (GC–MS),15 and paper spray ionization-mass spectrometry (PSI-MS).35 In addition to attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR),36 high-performance liquid chromatography-photodiode array detection (HPLC-DAD)37 and high-performance thin-layer chromatography (HPTLC)38,39 have been previously introduced as standardized protocols for the qualitative and/or the quantitative analysis of NBOMes (Table 1). Other approaches have reported the use of electrochemical methodologies including differential pulse voltammetry (DPV) using screen-printed carbon electrodes (SPEs)9 and square wave voltammetry (SWV) utilizing glassy carbon12 and boron-doped diamond40,41 as working electrodes. These reported electrochemical methodologies have the limitation of quantifying NBOMes individually, and they are unable to detect NBOMes simultaneously in complex samples as present in forensic scenarios.
Table 1. Comparison of Previous Analytical Approaches for the Analysis of NBOMe Derivatives.
| analytical method | target analytes | limit of detection (LOD) | limit of quantitation (LOQ) | linearity range | ref |
|---|---|---|---|---|---|
| LC–MS/MS | 25I-NBOMe | Δa | 25 pg mL–1 | 25–2000 pg mL–1 | (11) |
| LC–MS/MS | 25C-, 25B-, 25I-NBOMe | Δ | 1 ng mL–1 | 1–100 ng mL–1 | (22) |
| LC–MS/MS | 25C-, 25B-, 25I-, 25D-, 25H-NBOMe | 0.02–0.05 ng mL–1 | 0.08–0.1 ng mL–1 | 0.1–5.0 ng mL–1 | (23) |
| LC–MS/MS | 25C-, 25B-, 25I-NBOMe | urine: 5–25 pg mL–1 | urine: 50 pg mL–1 | urine: 0.1–100 ng mL–1 | (24) |
| hair: 3–5 pg mg–1 | hair: 6.25–12.5 pg mg–1 | hair: 0.025–2.5 ng mg–1 | |||
| LC–MS/MS | 25B-NBOMe | Δ | plasma: 0.1 mg L–1 | plasma:0.1–10 mg L–1 | (25) |
| urine: 1 mg L–1 | urine: 1–200 mg L–1 | ||||
| LC–MS/MS | 25C-, 25B-, 25I-, 25D-, 25H-, 25T2-NBOMe | 0.005–0.01 ng mL–1 | 0.01–0.02 ng mL–1 | 0.01–20 ng mL–1 | (26) |
| LC–MS/MS | 25I-, 25G-, 25B-, 25D-, 25H-NBOMe | 0.1 ng mL–1 | 1 ng mL–1 | 1–100 ng mL–1 | (27) |
| LC–MS/MS | 25I-NBOMe | 10 pg mL–1 | 30 pg mL–1 | 30–2000 pg mL–1 | (28) |
| LC–MS/MS | 25I-NBOMe | 0.09 ng mL–1 | 0.1 ng mL–1 | 0.1–0.5 ng mL–1 | (29) |
| LC–MS/MS | 25C-, 25B-NBOMe | qualitative analysis | qualitative analysis | qualitative analysis | (30) |
| LC–MS/MS | 25B-NBOMe | 10 pg mL–1 | 25 pg mL–1 | 25–2000 pg mL–1 | (31) |
| UPLC–MS/MS | 25B-, 25I-NBOMe | 0.2 ng mL–1 | 0.5 ng mL–1 | 0.5–20 ng mL–1 | (4) |
| UPLC–MS/MS | 25I-, 25C-, 25H-NBOMe | Δ | Δ | 1–500 ng mL–1 | (32) |
| UPLC–MS/MS | 25B-NBOMe | 5.3 pg mL–1 | 15.9 pg mL–1 | 10–1000 pg mL–1 | (33) |
| UPLC–MS/MS | 25C-NBOMe | 0.02 μg kg–1 | 0.08 μg kg–1 | 0.1–10 μg kg–1 | (34) |
| GC–MS | 25I-NBOMe | qualitative analysis | qualitative analysis | qualitative analysis | (15) |
| PSI-MS | 25C-, 25B-, 25I-NBOMe | qualitative analysis | qualitative analysis | qualitative analysis | (35) |
| ATR-FTIR | 25C-, 25B-, 25I-NBOMe | qualitative analysis | qualitative analysis | qualitative analysis | (36) |
| HPLC-DAD | 25C-, 25B-, 25I-NBOMe | 5 μg mL–1 | Δ | 20–330 μg mL–1 | (37) |
| HPTLC | 25B-NBOMe | 7.12 μg per band | 21.56 μg per band | 19.18–115.00 μg per band | (38) |
| HPTLC | 25C-NBOMe | 7.1 μg per band | 21.63 μg per band | 19.72–118.28 μg per band | (39) |
| differential pulse voltammetry | 25B-, 25I-NBOMe | 0.011 and 0.004 mg mL–1 | 0.034 and 0.012 mg mL–1 | 0.01–0.08 mg mL–1 | (9) |
| square wave voltammetry | 25H-NBOMe | 1.28 × 10–6 mol L–1 | 4.25 × 10–6 mol L–1 | 4.25 × 10–6–4.96 × 10–5 mol L–1 | (12) |
| square wave voltammetry | 25C-, 25B-, 25I-NBOMe | 0.05–0.1 μmol L–1 | Δ | 0.2–340 μmol L–1 | (40) |
| square wave voltammetry | 25C-, 25B-, 25I-NBOMe | 0.1–0.27 μmol L–1 | Δ | 1–555 μmol L–1 | (41) |
| this work HPLC-DAD (detection system I) | 25F-, 25C-, 25B-, 25I-NBOMe | 4.56–6.65 μg mL–1 | 10 μg mL–1 | 10–300 μg mL–1 | |
| this work HPLC-AD (detection system II) | 9.65–17.98 μg mL–1 | 20 μg mL–1 | 20–300 μg mL–1 |
Δ: not disclosed; LC–MS/MS: liquid chromatography–tandem mass spectrometry; UPLC–MS/MS: ultraperformance liquid chromatography–tandem mass spectrometry; GC–MS: gas chromatography–mass spectrometry; PSI-MS: paper spray ionization-mass spectrometry; ATR-FTIR: attenuated total reflection-Fourier transform infrared spectroscopy; HPLC-DAD: high-performance liquid chromatography-photodiode array detection; HPTLC: high-performance thin-layer chromatography.
Herein, for the first time, we report a potential high-performance liquid chromatographic method using two detection systems: photodiode array detection (HPLC-DAD) (detection system I) and amperometric detection (HPLC-AD) utilizing commercially available impinging jet flow cell incorporating SPEs (detection system II) for the simultaneous quantification of four NBOMe derivatives (2a–d; Scheme 1). The proposed analytical methodology was validated according to the International Conference on Harmonization (ICH) guidelines for the quantitative analysis of the target analytes and demonstrated to be suitable for the routine analysis of NBOMe derivatives in pure forms, in the presence of LSD, and common adulterants typically found in street samples (e.g. paracetamol, caffeine, and benzocaine), as well as within simulated blotter papers offering excellent selectivity and specificity.
Results and Discussion
First, the cyclic voltammetric profiles of each NBOMe derivative were explored using SPEs (electrode size: 3.1 mm diameter) in a 0.04 M Britton–Robinson (B–R) buffer at pH 7.0, which is the optimum pH previously reported by Andrade et al. for the electrochemical characterization and quantification of NBOMe derivatives.9Figure 1 depicts an overlay of the cyclic voltammograms obtained for the four NBOMe derivatives, and, as can be seen in the figure, each derivative develops two oxidation peaks (peak I and peak II) at the following potentials (vs Ag/Ag/Cl): Ep1 ≈ +0.815 V and Ep2 ≈ +0.996 V for 25F-NBOMe (2a), Ep1 ≈ +0.817 V and Ep2 ≈ +1.027 V for 25C-NBOMe (2b), Ep1 ≈ +0.829 V and Ep2 ≈ +1.031 V for 25B-NBOMe (2c), and Ep1 ≈ +0.80 V and Ep2 ≈ +1.013 V for 25I-NBOMe (2d) (Table 2). Andrade et al. have described the oxidation mechanism of these drugs occurring on the SPE surface as follows (see Scheme 2):9 the first observed oxidation peak (peak I) develops as a result of the oxidation of the secondary amine present in the NBOMe structure into a primary amine, which will be attached to the electrode surface, in addition to the generation of an aldehyde. This process involves the removal of one electron from the amino-nitrogen of the NBOMe secondary amine. The second oxidation peak (peak II) occurs due to the replacement of the halogen atom in the NBOMe compounds by a hydroxyl group, which afterwards produces a ketone. The latter oxidation step happens via an electron transfer from the highest-filled molecular orbital of organic halide to the electrode, leading to the formation of cation radical [RX]+•; this is followed by the fission of a carbon–halogen bond to form a carbocation, which will be attacked nucleophilically by the hydroxyl group and its subsequent oxidation to a ketone (quinone). Due to the overlap of the voltammetric signatures of the four NBOMe derivatives, it is impossible to quantify each drug separately if present together in a mixture by conventional electrochemical methods when in a forensic scenario. Therefore, efforts were next directed toward using high-performance liquid chromatography for the separation of the target analytes, and quantification was carried out by comparing the sensitivity of two detectors, which are photodiode array (detection system I) and amperometric detectors (detection system II).
Figure 1.
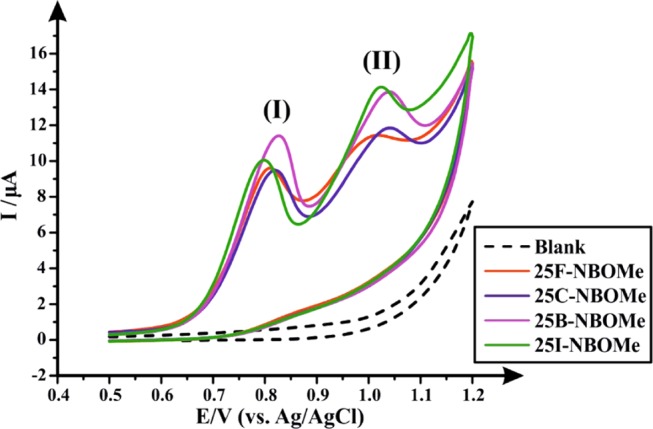
Cyclic voltammograms of 100 μg mL–1 of each of 25F-NBOMe (2a), 25C-NBOMe (2b), 25B-NBOMe (2c), and 25I-NBOMe (2d) in a 0.04 M B–R buffer (pH 7.0) using SPEs. Scan rate: 50 mV s–1 vs Ag/AgCl reference electrode. (I) First NBOMes’ oxidation peak; (II) second NBOMes’ oxidation peak.
Table 2. Summary of the Anodic Peak Potentials (Ep1 and Ep2) of NBOMe Derivatives (2a–d) Obtained Using SPEs vs Ag/AgCl.
| peak
potential |
||
|---|---|---|
| NBOMe derivatives | Ep1 (V) | Ep2 (V) |
| 25F-NBOMe (2a) | +0.815 | +0.996 |
| 25C-NBOMe (2b) | +0.817 | +1.027 |
| 25B-NBOMe (2c) | +0.829 | +1.031 |
| 25I-NBOMe (2d) | +0.800 | +1.013 |
Scheme 2. Electrochemical Oxidation Mechanism of NBOMe Halide Derivatives Previously Reported by Andrade et al.9.

Optimization of the Experimental and Chromatographic Conditions
Experimental Setup and Configuration of HPLC-AD System
Since the analytical quantification of NBOMe halide derivatives was achieved herein by comparing two detection systems, namely, DAD and AD, wherefore, the order of the detectors and the experimental setup is important for the optimization of the proposed analytical method. The most suitable arrangement of the two detectors was made following Zuway et al. who used the same commercial impinging jet flow cell for the HPLC amperometric detection of synthetic cathinones.42 They reported that the optimum experimental setup can be accomplished by placing the flow-cell amperometric detection system after the photodiode array detector and connecting them via poly(tetrafluoroethylene) (PTFE) tubing (230 × 1.6 mm2, i.d. 0.3 mm, internal volume: 16.25 μL). This configuration was proven to be better as it reduced the system back-pressure and thereby decreased the flow-cell leakages observed when the amperometric detector precedes the UV detector.42 In addition, Zuway et al. reported using two flow cells of different designs, which were the commercially available impinging jet flow cell and custom-made iCell channel flow cell. In this work, only the impinging jet flow cell was used, as Zuway et al. demonstrated that iCell flow cell has reduced sensitivity as a result of its large internal chamber volume used with the SPE, which increases the sample dispersion, diluting the analytes, thus reducing the SPE’s sensor sensitivity via mass transfer/diffusion to the electrode surface.42−45 Full optimization of all of the experimental parameters, affecting the separation and quantitation of the analytes of interests, is described in detail in the Supporting Information (SI) with the chromatographic conditions chosen for this study summarized in Table 3.
Table 3. Summary of the Optimized Experimental Parameters Chosen for the Separation and Quantitation of NBOMe Derivatives.
| studied experimental parameter | optimized parameter |
|---|---|
| analytical column | ACE 5 C18-AR column (150 × 4.6 mm2 i.d., particle size: 5 μm) |
| mobile phase | isocratic (5 mM ammonium formate + 100 mM KCl:acetonitrile 70:30% v/v) |
| ionic strength of ammonium formate buffer | 5 mM |
| pH of the aqueous phase | pH 7 |
| linear velocity of the mobile phase | 2.5 mL min–1 |
| column temperature | 60 °C |
| applied potential for amperometric detection (AD) | +1.0 V |
| detection wavelength λ for photodiode array detection (DAD) | 205 nm |
After optimization of all of the experimental conditions, typical HPLC-DAD chromatogram and HPLC-AD amperogram are presented in Figure 2A,B, respectively. The order of elution of the drugs (from the chromatographic column) depends on their degree of polarity, the more polar derivative, which is the fluoride; 25F-NBOMe (2a) elutes first (tR = 3.83 ± 0.02 min) followed by the chloride; 25C-NBOMe (2b) (tR = 5.96 ± 0.02 min) and then the bromide; 25B-NBOMe (2c) (tR = 7.01 ± 0.03 min) and eventually the iodide derivative; 25I-NBOMe (2d) elutes last (tR = 9.45 ± 0.05 min). The amperometric peaks of the target analytes were slightly delayed by 0.39 s in comparison with their corresponding peaks in the HPLC-DAD chromatogram; this is due to the PTFE connection tubing between the HPLC-DAD and the flow cell accommodating the SPE sensing platform. The retention times of the analytes of interest within the amperogram were as follows: 25F-NBOMe (2a) (tR = 3.84 ± 0.02 min), 25C-NBOMe (2b) (tR = 5.97 ± 0.02 min), 25B-NBOMe (2c) (tR = 7.02 ± 0.03 min), and 25I-NBOMe (2d) (tR = 9.46 ± 0.05 min).
Figure 2.

(A) Representative HPLC-DAD chromatogram of a solution containing 100 μg mL–1 of each of “F”: 25F-NBOMe (2a), “Cl”: 25C-NBOMe (2b), “Br”: 25B-NBOMe (2c), and “I”: 25I-NBOMe (2d). (B) Representative HPLC-AD amperogram of a solution containing 100 μg mL–1 of each of F: 25F-NBOMe (2a), Cl: 25C-NBOMe (2b), Br: 25B-NBOMe (2c), and I: 25I-NBOMe (2d). Experimental parameters include ACE C18-AR column (150 × 4.6 mm2 i.d., particle size: 5 μm), mobile phase: [5 mM ammonium formate + 100 mM KCl (pH 7.0): acetonitrile 70:30 (v/v)], flow rate: 2.5 mL min–1, column temperature: 60 °C, detector wavelength (UV): 205 nm, and potential: +1.0 V.
Validation of the Proposed Method
Validation of the proposed method was performed in accordance with the International Conference on Harmonization (ICH) guidelines for the validation of analytical procedures.46 The linearity of the proposed HPLC-DAD (detection system I) and HPLC-AD (detection system II) was evaluated by analyzing a series of different concentrations of calibration standards (n = 6). Calibration curves were constructed by plotting the average of peak areas (for detection system I) or the average of peak heights (current, μA) (for detection system II) for each concentration level of each investigated drug against its corresponding concentration. The linear regression equations were generated by the least-squares treatment of the calibration data and are presented in Tables 4 and 5. Good linearity of the tested analytical methodology is demonstrated by the high values of coefficient of regression (r2), which is ≥0.999, for detection system I, and ≥0.997, for detection system II. When comparing the sensitivity of the two detectors, it is apparent that detection system I is more sensitive than detection system II. This can be confirmed by the higher slope (b) values of the calibration curves of the HPLC-DAD (b = 9.08–23.12) in contrast to those obtained by the HPLC-AD system (b = 2.37 × 10–3–5.70 × 10–3). Furthermore, the sensitivity of the accessible linear range of detection system I (10–300 μg mL–1) slightly surpasses that of detection system II (20–300 μg mL–1). However, this difference in sensitivity is negligible in exchange for the advantage of the amperometric detector being less expensive and more economic than photodiode array detector. In both detection systems, HPLC-DAD (detection system I) and HPLC-AD (detection system II), the limit of detection (LOD) for each drug was determined using the following formula: LOD= (3Sy/x)/b, where Sy/x is the standard deviation of residuals and b is the slope of the regression line of the calibration curve of each drug. The values of LOD range from 4.56 to 6.65 and 9.65 to 17.98 μg mL–1, for detection system I and detection system II, respectively (Tables 4 and 5). These small values of LOD confirm the sensitivity of the proposed method. However, by comparing the LOD of both detectors, we concluded that HPLC-DAD (detection system I) is more sensitive than HPLC-AD (detection system II). The repeatability (intraday precision) of the developed methodology, utilizing both detectors, was assessed by analyzing each concentration level six times (n = 6), within the same day, in the same laboratory, by the same analyst using the same equipment. The percentage relative standard deviation (RSD %) of the obtained results, for both detectors, was calculated and is presented in Tables 4 and 5. The RSD % values were <2%, indicating the good agreement between the individual test results and confirming the precision of the proposed method. The system suitability testing is used to evaluate the suitability of the chromatographic system prior to using and is considered as an integral part of method validation.47 All system suitability parameters are listed in Tables 4 and 5, and it is evident from the tables that all of the analytes fulfilled the ideal range of system suitability parameters, proving the high-quality performance of the proposed chromatographic system and ensuring confidence in the analytical method. Next, the robustness of the proposed method was verified by introducing slight changes in the method parameters such as column temperature (60 ± 2 °C), molarity of formate buffer (5.0 ± 2.0 mM), and pH of buffer (7.0 ± 0.2 pH units). These variations did not have any significant effect on the measured responses (peak area or peak heights) as the calculated RSD % for the measured responses did not exceed 2% in all of the cases (Tables S1 and S2). Furthermore, the retention time of all of the drugs and the total run time were not affected when changing the aforementioned three parameters (Table S1). The selectivity of the proposed method was examined by testing the possibility of interference of LSD with the determination of NBOMEs, as NBOMes are commonly sold on blotter papers represented as LSD.22 As seen in Figure 3, the LSD did not interfere with any of the four NBOMe derivatives, as it eluted early at the beginning of the chromatographic run with retention times (tR) = 2.77 min in HPLC-DAD (Figure 3A) and 2.78 min in HPLC-AD (Figure 3B). The resolution (Rs) between the LSD peak and the next peak in the chromatogram (25F-NBOMe, 2a) is equivalent to 3.73, which proves the good selectivity and the successfulness of the proposed protocol. Further selectivity and specificity testing employing adulterants commonly found in street samples and pharmaceutical excipients are found in the Supporting Information (SI).
Table 4. Summary of HPLC-DAD (Detection System I) Validation Data for the Quantification of NBOMe Halide Derivatives (2a–d) Using ACE C18-AR Column (150 × 4.6 mm2 i.d., Particle Size: 5 μm), Mobile Phase: [5 mM Ammonium Formate + 100 mM KCl (pH 7.0): Acetonitrile 70:30% (v/v)], Flow Rate: 2.5 mL min–1, Column Temperature: 60 °C, and Detector Wavelength (UV): 205 nm.
| drug
of abuse |
||||
|---|---|---|---|---|
| parameters | 25F-NBOMe (2a) | 25C-NBOMe (2b) | 25B-NBOMe (2c) | 25I-NBOMe (2d) |
| r2 a | 0.999b | 0.999c | 0.999d | 0.999e |
| af | –13.52 | –29.79 | –41.17 | –34.88 |
| bg | 9.08 | 16.38 | 23.12 | 15.58 |
| LOD (μg mL–1)h | 4.56 | 5.90 | 5.14 | 6.65 |
| Precision (% RSD, n = 6) | ||||
| 10 (μg mL–1) | 0.70 | 0.98 | 1.14 | 0.51 |
| 20 (μg mL–1) | 0.58 | 1.84 | 1.78 | 0.01 |
| 50 (μg mL–1) | 1.26 | 1.42 | 1.00 | 0.28 |
| 100 (μg mL–1) | 1.08 | 0.76 | 0.65 | 0.98 |
| 150 (μg mL–1) | 0.49 | 0.31 | 0.38 | 0.82 |
| 200 (μg mL–1) | 0.93 | 0.30 | 0.36 | 0.79 |
| 300 (μg mL–1) | 0.27 | 0.40 | 0.47 | 0.58 |
| System Suitability Parameters | ||||
| tR ± SD (min)i (n = 6) | 3.83 ± 0.02 | 5.96 ± 0.02 | 7.01 ± 0.03 | 9.45 ± 0.05 |
| RRTj | 0.41 | 0.63 | 0.74 | 1 |
| k′k | 5.18 | 8.61 | 10.31 | 14.24 |
| N (plates)l | 8000 | 8629 | 8967 | 8670 |
| HETP (m)m | 1.88 × 10–5 | 1.74 × 10–5 | 1.67 × 10–5 | 1.73 × 10–5 |
| Rsn | 9.97 | 3.82 | 6.95 | |
| Aso | 0.85 | 0.82 | 0.85 | 0.94 |
| αp | 1.66 | 1.20 | 1.38 | |
r2: coefficient of regression.
y = 9.08x – 13.52.
y = 16.38x – 29.79.
y = 23.12x – 41.17.
y = 15.58x – 34.88.
a: intercept of the regression line of the calibration curve.
b: slope of the regression line of the calibration curve.
LOD: limit of detection using the formula (3Sy/x)/b.
tR: retention time in minutes for drugs eluted from the chromatographic column (Detection System I).
RRT: relative retention time (determined with respect to 25I-NBOMe, 2d, retention time obtained from Detection System I).
k′: capacity factor.
N: number of theoretical plates expressed in plates per meter.
HETP: height equivalent to the theoretical plate expressed in meter.
Rs: resolution between two successive eluted peaks.
As: asymmetry factor.
α: relative retention factor.
Table 5. Summary of HPLC-AD (Detection System II) Validation Data for the Quantification of NBOMe Halide Derivatives (2a–d) Using ACE C18-AR Column (150 × 4.6 mm2 i.d., Particle Size: 5 μm), Mobile Phase: [5 mM Ammonium Formate + 100 mM KCl (pH 7.0): Acetonitrile 70:30% (v/v)], Flow Rate: 2.5 mL min–1, Column Temperature: 60 °C, and Potential: +1.0 V.
| drug
of abuse |
||||
|---|---|---|---|---|
| parameters | 25F-NBOMe (2a) | 25C-NBOMe (2b) | 25B-NBOMe (2c) | 25I-NBOMe (2d) |
| r2 a | 0.997b | 0.998c | 0.999d | 0.999e |
| af | 14.57 × 10–2 | 8.93 × 10–2 | 8.52 × 10–2 | 4.62 × 10–2 |
| bg | 5.70 × 10–3 | 2.85 × 10–3 | 3.68 × 10–3 | 2.37 × 10–3 |
| LOD (μg mL–1)h | 17.98 | 16.52 | 9.65 | 10.54 |
| Precision (% RSD, n=6) | ||||
| 20 (μg mL–1) | 0.74 | 0.45 | 1.84 | 0.83 |
| 50 (μg mL–1) | 0.57 | 0.84 | 0.97 | 1.54 |
| 100 (μg mL–1) | 0.79 | 0.77 | 0.69 | 1.12 |
| 150 (μg mL–1) | 0.87 | 0.63 | 0.97 | 0.07 |
| 200 (μg mL–1) | 0.72 | 0.24 | 0.27 | 0.97 |
| 300 (μg mL–1) | 0.32 | 0.95 | 0.53 | 0.63 |
| System Suitability Parameters | ||||
| tR ± SD (min)i (n = 6) | 3.84 ± 0.02 | 5.97 ± 0.02 | 7.02 ± 0.03 | 9.46 ± 0.05 |
| RRTj | 0.41 | 0.63 | 0.74 | 1 |
r2: coefficient of regression.
y = 0.0057x + 0.1457.
y = 0.0029x + 0.0893.
y = 0.0037x + 0.085.
y = 0.0024x + 0.0462.
a: intercept of regression line of the calibration curve.
b: slope of regression line of the calibration curve.
LOD: limit of detection using the formula (3Sy/x)/b.
tR: retention time in minutes for drugs eluting from the flow cell Detection System II).
RRT: relative retention time (determined with respect to 25I-NBOMe, 2d, retention time obtained from Detection System II).
Figure 3.

(A) Representative HPLC-DAD chromatogram of a solution containing 100 μg mL–1 of each of LSD, F: 25F-NBOMe (2a), Cl: 25C-NBOMe (2b), Br: 25B-NBOMe (2c), and I: 25I-NBOMe (2d). (B) Representative HPLC-AD amperogram of a solution containing 100 μg mL–1 of each of LSD, F: 25F-NBOMe (2a), Cl: 25C-NBOMe (2b), Br: 25B-NBOMe (2c), and I: 25I-NBOMe (2d). Experimental parameters include ACE C18-AR column (150 × 4.6 mm2 i.d., particle size: 5 μm), mobile phase: [5 mM ammonium formate + 100 mM KCl (pH 7.0): acetonitrile 70:30% (v/v)], flow rate: 2.5 mL min–1, column temperature: 60 °C, detector wavelength (UV): 205 nm, and potential: +1.0 V.
Forensic Application
Application of the Proposed Method to Simulated Blotter Papers
Most recreated NBOMe blotter papers contain higher doses of 500–800 μg per blotter,22,27 and the dimensions of the seized blotters usually are in the range of [(0.5 × 0.5)–(1 × 1) cm2].40,41 Therefore, three different simulated blotter papers were laboratory-prepared (each blotter was prepared in triplicate) using concentrations of 500 and 600 μg per blotter of NBOMes, and LSD was added in two blotters to simulate real samples. After the extraction of the drugs from the blotters with methanol, the dilution of the samples was done to obtain concentrations within the linearity ranges and then analyses were carried out; each blotter was analyzed in triplicate. Depicted in Table 6, each drug eluted at its specified retention time (tR) when utilizing detection systems I and II, and the recovery% (R%) and error% (Er%) ranged from 99 to 101% and −1 to (+)1%, respectively, emphasizing the high accuracy of the proposed method. The good precision of the method was demonstrated by the low RSD% values, which did not exceed 3%. Furthermore, the standard addition procedure was applied and the calculated recoveries% ranged from 98 to 102 and 98 to 100% for detection systems I and II, respectively, confirming the accuracy of the developed method (Table 7).
Table 6. Comparison between Quantitative Data Obtained by HPLC-DAD (Detection System I) and HPLC-AD (Detection System II) for the Analysis of NBOMe Derivatives (2a–d) in Simulated Blotter Papers.
| |
detection system I (HPLC-DAD) |
detection system II (HPLC-AD) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| detection
system |
drug |
||||||||
| sample | parameter | LSD | 25C-NBOMe (2b) | 25B-NBOMe (2c) | 25I-NBOMe (2d) | LSD | 25C-NBOMe (2b) | 25B-NBOMe (2c) | 25I-NBOMe (2d) |
| tR (min.) ± SD | 2.8 ± 0 | 6.0 ± 0.1 | 7.0 ± 0 | 9.5 ± 0.1 | 2.8 ± 0 | 6.0 ± 0.1 | 7.0 ± 0 | 9.5 ± 0.1 | |
| blotter paper I | mean % R ± SDa | + | – | – | 99 ± 0.8 | + | – | – | 100 ± 2.7 |
| RSD %b | + | – | – | 1 | + | – | – | 3 | |
| Er (%)c | + | – | – | –1 | + | – | – | 0 | |
| blotter paper II | mean % R ± SDa | + | 100 ± 0.1 | – | 100 ± 0.1 | + | 101 ± 0.5 | – | 100 ± 1.4 |
| RSD %b | + | 1 × 10–1 | – | 1 × 10–1 | + | 1 | – | 1 | |
| Er (%)c | + | 0 | – | 0 | + | 1 | – | 0 | |
| blotter paper III | mean % R ± SDa | – | – | 99 ± 1.5 | – | – | – | 99 ± 2.2 | – |
| RSD %b | – | – | 2 | – | – | – | 2 | – | |
| Er (%)c | – | – | –1 | – | – | – | –1 | – | |
Mean % recovery ± standard deviation (SD) of three determinations for each drug detected in each blotter paper.
% relative standard deviation of three determinations for each drug detected in each blotter paper.
% relative error; (+) = present in the blotter paper sample; (−) absent from the blotter paper sample.
Table 7. Comparison between Standard Addition Statistical Analysis Obtained by HPLC-DAD (Detection System I) and HPLC-AD (Detection System II) for the Quantitation of NBOMe Derivatives (2a–d) in Simulated Blotter Papers.
| |
detection system I (HPLC-DAD) |
detection system II (HPLC-AD) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| detection
system |
drug |
||||||||
| sample | parameter | LSD | 25C-NBOMe (2b) | 25B-NBOMe (2c) | 25I-NBOMe (2d) | LSD | 25C-NBOMe (2b) | 25B-NBOMe (2c) | 25I-NBOMe (2d) |
| tR (min.) ± SD | 2.8 ± 0 | 6.0 ± 0.1 | 7.0 ± 0 | 9.5 ± 0.1 | 2.8 ± 0 | 6.0 ± 0.1 | 7.0 ± 0 | 9.5± 0.1 | |
| blotter paper I | mean % R ± SDa | + | – | – | 102 ± 1.0 | + | – | – | 100 ± 0.3 |
| RSD %b | + | – | – | 1 | + | – | – | 3 × 10–1 | |
| Er (%)c | + | – | – | 2 | + | – | – | 0 | |
| blotter paper II | mean % R ± SDa | + | 102 ± 0.5 | – | 98 ± 1 | + | 98 ± 1.5 | – | 99 ± 2.1 |
| RSD %b | + | 1 | – | 1 | + | 2 | – | 2 | |
| Er (%)c | + | 2 | – | –2 | + | –2 | – | –1 | |
| blotter paper III | mean % R ± SDa | – | – | 101 ± 1.7 | – | – | – | 99 ± 2 | – |
| RSD %b | – | – | 2 | – | – | – | 2 | – | |
| Er (%)c | – | – | 1 | – | – | – | –1 | – | |
Mean % recovery ± SD of three determinations for each drug detected in each blotter paper.
% relative standard deviation of three determinations for each drug detected in each blotter paper.
% relative error; (+) = present in the blotter paper sample; (−) absent from the blotter paper sample.
Conclusions
This work presents, for the first time, a simple rapid reliable chromatographic method utilizing both photodiode array (detection system I) and amperometric (detection system II) detectors for the simultaneous qualitative and quantitative analyses of four NBOMe derivatives (25F-, 25C-, 25B-, and 25I-NBOMe) (2a–d) in a single run. Both detection systems were reproducible and sensitive, as the detection limits (LOD) reported herein are low enough for the detection of the target analytes in confiscated blotter papers. Also, the availability of instrumentation and the cost-effectiveness are a noted advantage if compared with expensive mass detectors, as normal UV detectors can be used in the case of detection system I because quantification was done at a single wavelength (λmax = 205 nm), while detection system II employs commercially available impinging jet flow cell incorporating SPEs, which are single shot (disposable), mass-produced, and of economic prices. However, hyphenated methodologies coupled to MS (e.g., LC–MS and GC–MS) are indispensable tools that cannot be substituted with the developed method for the analysis of seized blotters in forensic laboratories. This is due to the fact that these blotters usually contain untargeted compounds, which can be primarily screened and detected with mass spectroscopy, while the presented methodology can be used as a final conclusive confirmation of the presence of NBOMe in blotter papers. The selectivity and the specificity of the proposed methodology were tested by analyzing the studied drug mixture in the presence of LSD, common adulterants found in street samples (i.e., caffeine, paracetamol, and benzocaine), and pharmaceutical excipients, and none of them interfered with the determination of the analytes of interest, demonstrating the high selectivity of the method. Moreover, the proposed method was successfully applied for the analysis of laboratory-prepared (simulated) blotter papers, and the percentage recoveries obtained showed acceptable levels of accuracy and precision, which can make this protocol the one of choices for the routine analysis of the studied drugs in forensic and quality control labs after prior screening by mass spectroscopy.
Experimental Section
Chemicals and Materials
All reagents (2C-F·HCl, 2C-C·HCl, 2C-B·HCl, 2C-I·HCl, 2-methoxybenzaldehyde, triethylamine, and sodium borohydride) were of commercial quality (Sigma-Aldrich, Gillingham, U.K., or Fluorochem Limited, Hadfield, U.K.) and used without further purification. Lysergic acid diethylamide (LSD, 3) was obtained from LGC GmbH (Luckenwalde, Germany) and used without any further purification. Solvents (Fisher Scientific, Loughborough, U.K.) were dried, where necessary, using standard procedures.48 The target compounds (2a–d) were synthesized from their corresponding phenethylamine hydrochlorides (1a–d), using an adaptation of the method reported by Hansen et al.49 and obtained as stable (confirmed by 1H NMR and GC–MS), off-white powders. The hydrochloride salts were determined to be soluble (10 mg mL–1) in deionized water, methanol, and dimethyl sulfoxide. To ensure authenticity of the materials utilized in this study, the synthesized samples were structurally characterized (see the Supporting Information) by high-field NMR, FTIR, and GC–EI-MS. The purity of all samples was confirmed by both NMR and GC–EI-MS (>99.5% in all cases). 1H NMR and 13C NMR spectra (10 mg/600 μL in MeOH-d4) were acquired on a JEOL AS-400 (JEOL, Tokyo, Japan) NMR spectrometer operating at a proton resonance frequency of 400 MHz and referenced to the residual solvent peak (MeOH-d4: 1H NMR δ = 3.31 ppm, 13C NMR δ = 49.00 ppm50). All samples were filtered prior to analysis. Infrared spectra were obtained in the range of 4000–400 cm–1 using a Thermo Scientific Nicolet iS10ATR-FTIR instrument (Thermo Scientific, Rochester). GC–MS analysis was performed using an Agilent 7890B GC and an MS5977B mass selective detector (Agilent Technologies, Wokingham, U.K.). The mass spectrometer was operated in the electron ionization mode at 70 eV. Separation was achieved with a capillary column (HP5 MS, 30 mÅ, ∼0.25 mm, i.d. 0.25 μm) with helium as the carrier gas at a constant flow rate of 1.2 mL min–1. A 2 μL aliquot of the samples was injected with a split ratio of 50:1. Both the injector and the GC interface temperatures were maintained at 280 °C. Oven temperature program: 50 °C (hold for 3 min) → 50–320 °C (30 °C min–1) → 320 °C (hold for 6 min). The MS source and quadrupole temperatures were set at 230 and 150 °C, respectively. Mass spectra were obtained in full-scan mode (50–550 amu). All samples were prepared as 1 mg mL–1 solutions in methanol with no derivatization and analyzed individually. Eicosane (1 mg mL–1) was used as an internal standard, and each sample was injected six times.
All aqueous solutions were prepared with Milli-Q deionized water of resistivity ≥18.2 Ω cm (Millipore system). All solutions (unless stated otherwise) were vigorously degassed, for 10 min, with highly pure nitrogen to remove oxygen prior to analysis.
Instrumentation
High-Performance Liquid Chromatography-Photodiode Array Detection (HPLC-DAD) (Detection System I)
Reverse-phase HPLC was performed with an Agilent HP series 1100 liquid chromatography instrument (Agilent Technologies, Wokingham, U.K.). It consisted of an Agilent 1100 series quaternary pump G1310A (serial DE80301064), which comprises a solvent cabinet, an Agilent 1100 series vacuum degasser G1322A (serial JP73017007), and an Agilent 1100 series photodiode array detector G1315A (serial DE74603601), which was set at 205 nm. The LC system was equipped with an Agilent 1100 series thermostated column compartment G1316A (serial DE91810205) set to 60 °C and a 100-place autoinjector G1313A (serial DE54901543), with an injection volume of 10.0 μL. The analytical column used was an ACE C18-AR column (150 × 4.6 mm2 i.d., particle size: 5 μm), Hichrom Ltd., U.K. The mobile phase consisted of 30:70% (v/v) of 5 mM ammonium formate and 100 mM potassium chloride buffer (pH 7.0): acetonitrile, flowing at a rate of 2.5 mL min–1. The total run time was 10 min. The LC system was controlled by Agilent Chemstation (ver. 10.02) software (Agilent Technologies, Wokingham, U.K.) for data analysis.
High-Performance Liquid Chromatography-Amperometric Detection (HPLC-AD) (Detection System II)
The HPLC was coupled, in sequence, to a flow cell obtained from Metrohm UK, Runcorn, U.K. (impinging jet flow cell; product code: DRP-FLWCL-TEF-71306; 3.3 × 6.0 × 3.3 cm3, flow chamber volume = 8 μL), housing the SPE to give the HPLC-AD system. The connection between the DAD and the flow cell was achieved via PTFE tubing (230 × 1.6 mm2, i.d. 0.3 mm, internal volume: 16.25 μL). The SPEs utilized in this part of the study were fabricated in-house, as previously described,42,51 and consisted of a 3.1 mm diameter graphite working electrode, a graphite counter electrode, and a Ag/AgCl reference electrode. Amperometric measurements were carried out using an EmStat 3 potentiostat/galvanostat (PalmSens BV, The Netherlands) and controlled by PSTrace (version 4.4) software (PalmSens, The Netherlands). All of the amperometric measurements were carried out using the following parameters: (i) potential (E, +1.0 V); (ii) equilibration time (tequibriation, 10.0 s); (iii) data interval (tinterval, 0.08 s); (iv) current range (100 nA to 1 mA); and (iv) total run time (trun, 5000 s). A new SPE was used for each experiment performed. Buffer pH measurements were made using a “SevenCompact pH/Ion S220” (Mettler-Toledo AG, Switzerland) pH meter.
Preparation of the Mobile Phase [5 mM Ammonium Formate–100 mM Potassium Chloride Buffer (pH 7.0): Acetonitrile, 70:30% v/v]
The 5 mM ammonium formate–100 mM potassium chloride buffer was prepared in a 2.0 L volumetric flask, by dissolving 0.6306 g of ammonium formate and 14.91 g of potassium chloride into ultrapure deionized water, and then the pH was adjusted to 7.0 (±0.02) with 0.2 M NaOH. Afterwards, appropriate proportions of each of the aqueous phase (1.4 L) and the organic modifier (0.6 L) were mixed to obtain a 2.0 L mobile phase of the desired ratio. Finally, the mobile phase was vacuum-filtered through a 0.45 mm pore filter paper.
Preparation of Standard Stock Solution and Calibration Curve Working Solutions for HPLC
Each NBOMe derivative (10.0 mg, 2a–d) was weighted accurately into one 20.0 mL glass volumetric flask and diluted to volume with the mobile phase to give a stock solution (S) containing 0.5 mg mL–1 of each drug. Working solutions for calibration standards were prepared by further dilution of the latter solution (S) with the mobile phase to obtain concentrations of 10, 20, 50, 100, 150, 200, and 300 μg mL–1 of each analyte. Working solutions were injected directly into the HPLC, and the peak areas (for the HPLC-DAD analysis) and peak heights (for the HPLC-AD analysis) of the target drugs were plotted against their corresponding concentrations to construct the calibration curves.
Selectivity Standards
Lysergic Acid Diethylamide (LSD)
LSD (5.0 mg) was weighed into a 10.0 mL glass volumetric flask and diluted to volume with the mobile phase to get a stock solution of concentration 0.5 mg mL–1 of the drug. The latter solution (1.0 mL) was transferred to a 5.0 mL volumetric flask, and the solution was made to the mark with the mobile phase and injected into the HPLC to monitor the retention time (tR) of LSD alone. Another 1.0 mL of the LSD stock solution was transferred to a 5.0 mL volumetric flask containing 1.0 mL of the NBOMe standard stock solution (S), and the flask was made to the mark with the mobile phase and injected into the HPLC to test the possibility of LSD interference with the determination of the four NBOMe derivatives.
Adulterants commonly found in street samples (paracetamol, caffeine, and benzocaine): refer to the Supporting Information (SI).
Specificity standards: refer to the Supporting Information (SI).
Forensic Application
Application of the Method to Simulated Blotter Papers
Three blotter papers (A4 white sheet blotters purchased from Amazon, U.K.) were laboratory-prepared and analyzed using the proposed method as follows: blotter papers were cut into pieces with dimensions 1 × 1 cm2; then, nine blotter papers were soaked into three different vials (each three blotters were soaked into one vial), and each vial contained 2.0 mL of the following solutions: vial 1 had 200 μg mL–1 LSD and 600 μg mL–1 25I-NBOMe (2d), vial 2 contained 100 μg mL–1 LSD, 500 μg mL–1 25I-NBOMe (2d), and 600 μg mL–1 25C-NBOMe (2b), and vial 3 had 500 μg mL–1 25B-NBOMe (2c). The blotter papers were kept submerged into the solutions in the vials overnight; the next day, the contents in the blotter papers were extracted by transferring each blotter paper to an Eppendorf containing 1.0 mL methanol and sonicated it in an ultrasonic bath for 20 min. Finally, the obtained solutions were diluted to half with the mobile phase to get concentrations of the drugs within the linearity ranges and injected into the HPLC. Standard addition solutions were prepared by a twofold dilution of the latter sample solutions with the mobile phase and then spiking them with known portions of 25I-NBOMe (2d) and/or 25C-NBOMe (2b) and/or 25B-NBOMe (2c) standard solutions to obtain total concentrations within the specified linearity ranges, and then the solutions were injected into the HPLC.
Acknowledgments
The authors sincerely thank Newton-Mosharafa Ph.D. Scholarship funded by both the Egyptian Ministry of Higher Education and the British Council for supporting this research. Funding from the Engineering and Physical Science Research Council (Reference: EP/N001877/1) and a British Council Institutional Grant Link (No. 172726574) is acknowledged. The authors thank Manchester Metropolitan University and Oxford Instruments for the provision of a match-funded studentship for J.M.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01366.
Instrumentation for cyclic voltammetry (CV); preparation of solutions for cyclic voltammetry (CV), and preparation of selectivity (adulterants commonly found in street samples) and specificity standards; optimization of the experimental conditions; selectivity (adulterants commonly found in street samples) and specificity study validation results; robustness tables (Tables S1 and S2); optimization of the experimental condition figures (Figures S1 and S2); selectivity and specificity study figures (Figures S3 and S4); and NMR and FTIR spectra of the NBOMe derivatives (PDF).
The authors declare no competing financial interest.
Supplementary Material
References
- Hillebrand J.; Olszewski D.; Sedefov R. Legal highs on the Internet. Subst. Use Misuse 2010, 45, 330–340. 10.3109/10826080903443628. [DOI] [PubMed] [Google Scholar]
- Cahal D. A. Misuse of Drugs Act 1971. Br. Med. J. 1974, 1, 70–72. 10.1136/bmj.1.5897.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakou C.; Marinelli E.; Frati P.; Santurro A.; Afxentiou M.; Zaami S.; Busardo F. P. NBOMe: new potent hallucinogens-pharmacology, analytical methods, toxicities, fatalities: a review. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3270–3281. [PubMed] [Google Scholar]
- Shanks K. G.; Sozio T.; Behonick G. S. Fatal Intoxications with 25B-NBOMe and 25I-NBOMe in Indiana During 2014. J. Anal. Toxicol. 2015, 39, 602–606. 10.1093/jat/bkv058. [DOI] [PubMed] [Google Scholar]
- Lowe L. M.; Peterson B. L.; Couper F. J. A Case Review of the First Analytically Confirmed 25I-NBOMe-Related Death in Washington State. J. Anal. Toxicol. 2015, 39, 668–671. 10.1093/jat/bkv092. [DOI] [PubMed] [Google Scholar]
- Kueppers V. B.; Cooke C. T. 25I-NBOMe related death in Australia: A case report. Forensic Sci. Int. 2015, 249, e15–e18. 10.1016/j.forsciint.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Yoshida K.-i.; Saka K.; Shintani-Ishida K.; Maeda H.; Nakajima M.; Hara S.-i.; Ueno M.; Sasaki K.; Iwase H.; Sakamoto T. A case of fatal intoxication due to the new designer drug 25B-NBOMe. Forensic Toxicol. 2015, 33, 396–401. 10.1007/s11419-015-0276-7. [DOI] [Google Scholar]
- Bersani F. S.; Corazza O.; Albano G.; Valeriani G.; Santacroce R.; Bolzan Mariotti Posocco F.; Cinosi E.; Simonato P.; Martinotti G.; Bersani G.; Schifano F. 25C-NBOMe: Preliminary Data on Pharmacology, Psychoactive Effects, and Toxicity of a New Potent and Dangerous Hallucinogenic Drug. BioMed Res. Int. 2014, 2014, 734749 10.1155/2014/734749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade A. F. B.; Mamo S. K.; Gonzalez-Rodriguez J. Rapid Screening Method for New Psychoactive Substances of Forensic Interest: Electrochemistry and Analytical Determination of Phenethylamines Derivatives (NBOMe) via Cyclic and Differential Pulse Voltammetry. Anal. Chem. 2017, 89, 1445–1452. 10.1021/acs.analchem.6b02426. [DOI] [PubMed] [Google Scholar]
- Nikolaou P.; Papoutsis I.; Stefanidou M.; Spiliopoulou C.; Athanaselis S. 2C-I-NBOMe, an “N-bomb” that kills with “Smiles”. Toxicological and legislative aspects. Drug Chem. Toxicol. 2015, 38, 113–119. 10.3109/01480545.2014.911882. [DOI] [PubMed] [Google Scholar]
- Poklis J. L.; Devers K. G.; Arbefeville E. F.; Pearson J. M.; Houston E.; Poklis A. Postmortem detection of 25I-NBOMe [2-(4-iodo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine] in fluids and tissues determined by high performance liquid chromatography with tandem mass spectrometry from a traumatic death. Forensic Sci. Int. 2014, 234, e14–e20. 10.1016/j.forsciint.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oiye É. N.; Toia Katayama J. M.; Muzetti Ribeiro M. F.; de Oliveira M. F. Electrochemical analysis of 25H-NBOMe by Square Wave Voltammetry. Forensic Chem. 2017, 5, 86–90. 10.1016/j.forc.2017.07.001. [DOI] [Google Scholar]
- Kristofic J. J.; Chmiel J. D.; Jackson G. F.; Vorce S. P.; Holler J. M.; Robinson S. L.; Bosy T. Z. Detection of 25C-NBOMe in Three Related Cases. J. Anal. Toxicol. 2016, 40, 466–472. 10.1093/jat/bkw035. [DOI] [PubMed] [Google Scholar]
- Al-Imam A. 25b-NBOMe: A Case Report of Sudden Death and Insightful View of Google Trends Data. Iran. J. Psychiatry Behav. Sci. 2018, 12, e9870 10.5812/ijpbs.9870. [DOI] [Google Scholar]
- Zheng H.; Zhao Y.; Yang H.; Gao L.; Zheng H. Seized Blotters Containing One Regioisomer of 25I-NBOMe. J. Forensic Biomech. 2018, 9, 138 10.4172/2090-2697.1000138. [DOI] [Google Scholar]
- Nichols D. E. Psychedelics. Pharmacol. Rev. 2016, 68, 264–355. 10.1124/pr.115.011478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski L. K.; Elbakoush F.; Calvo J.; Exantus-Bernard G.; Fong J.; Poklis J. L.; Poklis A.; Nelson L. S. Evolution of the NBOMes: 25C- and 25B-Sold as 25I-NBOMe. J. Med. Toxicol. 2015, 11, 237–241. 10.1007/s13181-014-0445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United States Drug Enforcement Administration . Three More Synthetic Drugs Become Illegal For At Least Two Years, 2013. https://www.dea.gov/press-releases/2013/11/15/three-more-synthetic-drugs-become-illegal-least-two-years.
- Home Office and The Rt Hon Norman Baker. Ban on NBOMe and Benzofurans Comes into Force, June 2014. https://www.gov.uk/government/news/ban-on-nbome-and-benzofurans-comes-into-force) (accessed Nov 2018).
- World Health Organisation Recommendation. http://www.who.int/medicines/areas/quality_safety/Letter_toUNSG_onECDD_June20_2014.pdf (accessed Nov 2018).
- Electronic Code of Federal Regulations. https://www.ecfr.gov/cgi-bin/text-idx?SID=b632b274cf6322a0450af69d7c7a4f46&node=pt21.9.1308&rgn=div5#se21.9.1308_111 (accessed Nov 2018).
- Poklis J. L.; Raso S. A.; Alford K. N.; Poklis A.; Peace M. R. Analysis of 25I-NBOMe, 25B-NBOMe, 25C-NBOMe and Other Dimethoxyphenyl-N-[(2-Methoxyphenyl) Methyl]Ethanamine Derivatives on Blotter Paper. J. Anal. Toxicol. 2015, 39, 617–623. 10.1093/jat/bkv073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morini L.; Bernini M.; Vezzoli S.; Restori M.; Moretti M.; Crenna S.; Papa P.; Locatelli C.; Osculati A. M. M.; Vignali C.; Groppi A. Death after 25C-NBOMe and 25H-NBOMe consumption. Forensic Sci. Int. 2017, 279, e1–e6. 10.1016/j.forsciint.2017.08.028. [DOI] [PubMed] [Google Scholar]
- Nisbet L. A.; Venson R.; Wylie F. M.; Scott K. S. Application of a Urine and Hair Validated LC–MS-MS Method to Determine the Effect of Hair Color on the Incorporation of 25B-NBOMe, 25C-NBOMe and 25I-NBOMe into Hair in the Rat. J. Anal. Toxicol. 2017, 41, 559–565. 10.1093/jat/bkx053. [DOI] [PubMed] [Google Scholar]
- Gee P.; Schep L. J.; Jensen B. P.; Moore G.; Barrington S. Case series: toxicity from 25B-NBOMe–a cluster of N-bomb cases. Clin. Toxicol. 2016, 54, 141–146. 10.3109/15563650.2015.1115056. [DOI] [PubMed] [Google Scholar]
- Johnson R. D.; Botch-Jones S. R.; Flowers T.; Lewis C. A. An evaluation of 25B-, 25C-, 25D-, 25H-, 25I- and 25T2-NBOMe via LC-MS-MS: method validation and analyte stability. J. Anal. Toxicol. 2014, 38, 479–484. 10.1093/jat/bku085. [DOI] [PubMed] [Google Scholar]
- Poklis J. L.; Clay D. J.; Poklis A. High-performance liquid chromatography with tandem mass spectrometry for the determination of nine hallucinogenic 25-NBOMe designer drugs in urine specimens. J. Anal. Toxicol. 2014, 38, 113–121. 10.1093/jat/bku005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poklis J. L.; Charles J.; Wolf C. E.; Poklis A. High performance liquid chromatography tandem mass spectrometry method for the determination of 2CC-NBOMe and 25I-NBOMe in human serum. Biomed. Chromatogr. 2013, 27, 1794–1800. 10.1002/bmc.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman W.; Kała M.; Lechowicz W.; Gil D.; Anand J. S. Severe clinical toxicity caused by 25I-NBOMe confirmed analytically using LC-MS-MS method. Acta Biochim. Pol. 2018, 65, 567–571. 10.18388/abp.2018_2627. [DOI] [PubMed] [Google Scholar]
- Tang M. H.; Ching C. K.; Tsui M. S.; Chu F. K.; Mak T. W. Two cases of severe intoxication associated with analytically confirmed use of the novel psychoactive substances 25B-NBOMe and 25C-NBOMe. Clin. Toxicol. 2014, 52, 561–565. 10.3109/15563650.2014.909932. [DOI] [PubMed] [Google Scholar]
- Poklis J. L.; Nanco C. R.; Troendle M. M.; Wolf C. E.; Poklis A. Determination of 4-bromo-2,5-dimethoxy-N-[(2-methoxyphenyl)methyl]-benzeneethanamine (25B-NBOMe) in serum and urine by high performance liquid chromatography with tandem mass spectrometry in a case of severe intoxication. Drug Test. Anal. 2014, 6, 764–769. 10.1002/dta.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellpflug S. J.; Kealey S. E.; Hegarty C. B.; Janis G. C. 2-(4-Iodo-2, 5-dimethoxyphenyl)-N-[(2-methoxyphenyl) methyl] ethanamine (25I-NBOMe): clinical case with unique confirmatory testing. J. Med. Toxicol. 2014, 10, 45–50. 10.1007/s13181-013-0314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiergowski M.; Aszyk J.; Kaliszan M.; Wilczewska K.; Anand J. S.; Kot-Wasik A.; Jankowski Z. Identification of novel psychoactive substances 25B-NBOMe and 4-CMC in biological material using HPLC-Q-TOF-MS and their quantification in blood using UPLC–MS/MS in case of severe intoxications. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2017, 1041–1042, 1–10. 10.1016/j.jchromb.2016.12.018. [DOI] [PubMed] [Google Scholar]
- Andreasen M. F.; Telving R.; Rosendal I.; Eg M. B.; Hasselstrøm J. B.; Andersen L. V. A fatal poisoning involving 25C-NBOMe. Forensic Sci. Int. 2015, 251, e1–e8. 10.1016/j.forsciint.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Carvalho T. C.; Oliveira I. F.; Tose L. V.; Vanini G.; Kill J. B.; Neto A. C.; Machado L. F.; Ambrosio J. C. L.; Lacerda V.; Vaz B. G.; Romão W. Qualitative analysis of designer drugs by paper spray ionisation mass spectrometry (PSI-MS). Anal. Methods 2016, 8, 614–620. 10.1039/C5AY01265A. [DOI] [Google Scholar]
- Coelho Neto J. Rapid detection of NBOME’s and other NPS on blotter papers by direct ATR-FTIR spectrometry. Forensic Sci. Int. 2015, 252, 87–92. 10.1016/j.forsciint.2015.04.025. [DOI] [PubMed] [Google Scholar]
- Edmunds R.; Donovan R.; Reynolds D. The analysis of illicit 25X-NBOMe seizures in Western Australia. Drug Test. Anal. 2018, 10, 786–790. 10.1002/dta.2260. [DOI] [PubMed] [Google Scholar]
- Duffau B. E.; Camargo C.; Cassels B. K.; Kogan M.; Fuentes E. Analysis of a new potent hallucinogen, 25-B-NBOMe, in blotters by High-Performance Thin-Layer Chromatography. J. Planar Chromatogr.—Mod. TLC 2015, 28, 395–397. 10.1556/1006.2015.28.5.9. [DOI] [Google Scholar]
- Duffau B.; Camargo C.; Kogan M.; Fuentes E.; Cassels B. K. Analysis of 25 C NBOMe in Seized Blotters by HPTLC and GC–MS. J. Chromatogr. Sci. 2016, 54, 1153–1158. 10.1093/chromsci/bmw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza G. A.; Arantes L. C.; Guedes T. J.; de Oliveira A. C.; Marinho P. A.; Muñoz R. A. A.; dos Santos W. T. P. Voltammetric signatures of 2,5-dimethoxy-N-(2-methoxybenzyl) phenethylamines on boron-doped diamond electrodes: Detection in blotting paper samples. Electrochem. Commun. 2017, 82, 121–124. 10.1016/j.elecom.2017.08.001. [DOI] [Google Scholar]
- Souza G. A.; Pimentel D. M.; Lima A. B.; Guedes T. J.; Arantes L. C.; de Oliveira A. C.; Sousa R. M. F.; Muñoz R. A. A.; dos Santos W. T. P. Electrochemical sensing of NBOMes and other new psychoactive substances in blotting paper by square-wave voltammetry on a boron-doped diamond electrode. Anal. Methods 2018, 10, 2411–2418. 10.1039/C8AY00385H. [DOI] [Google Scholar]
- Zuway K. Y.; Smith J. P.; Foster C. W.; Kapur N.; Banks C. E.; Sutcliffe O. B. Detection and quantification of new psychoactive substances (NPSs) within the evolved “legal high” product, NRG-2, using high performance liquid chromatography-amperometric detection (HPLC-AD). Analyst 2015, 140, 6283–6294. 10.1039/C5AN01106J. [DOI] [PubMed] [Google Scholar]
- Guidelli R. Diffusion toward planar, spherical, and dropping electrodes at constant potential: I. Theory. J. Electroanal. Chem. Interfacial Electrochem. 1971, 33, 291–302. 10.1016/S0022-0728(71)80118-3. [DOI] [Google Scholar]
- Guidelli R. Diffusion toward planar, spherical, and dropping electrodes at constant potential: II. Examples. J. Electroanal. Chem. Interfacial Electrochem. 1971, 33, 303–317. 10.1016/S0022-0728(71)80119-5. [DOI] [Google Scholar]
- Oldham K. B. Diffusive transport to planar, cylindrical and spherical electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1973, 41, 351–358. 10.1016/S0022-0728(73)80413-9. [DOI] [Google Scholar]
- ICH. In Q2(R1), Validation of Analytical Procedures: Text and Methodology, International Conference on Harmonization, Nov 2005, Geneva; ICH Secretariat, 2005.
- Tiryaki O.; Baysoyu D.; Aydin G.; Secer E. Setting system suitability parameters for performance optimization of GC-NPD detection for pesticide residue analysis. Gazi Univ. J. Sci. 2009, 22, 149–155. [Google Scholar]
- Williams D. B. G.; Lawton M. Drying of Organic Solvents: Quantitative Evaluation of the Efficiency of Several Desiccants. J. Org. Chem. 2010, 75, 8351–8354. 10.1021/jo101589h. [DOI] [PubMed] [Google Scholar]
- Hansen M.; Phonekeo K.; Paine J. S.; Leth-Petersen S.; Begtrup M.; Bräuner-Osborne H.; Kristensen J. L. Synthesis and structure-activity relationships of N-benzyl phenethylamines as 5-HT2A/2C agonists. ACS Chem. Neurosci. 2014, 5, 243–249. 10.1021/cn400216u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- Smith J. P.; Metters J. P.; Irving C.; Sutcliffe O. B.; Banks C. E. Forensic electrochemistry: the electroanalytical sensing of synthetic cathinone-derivatives and their accompanying adulterants in “legal high” products. Analyst 2014, 139, 389–400. 10.1039/C3AN01985C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.