

Abstract
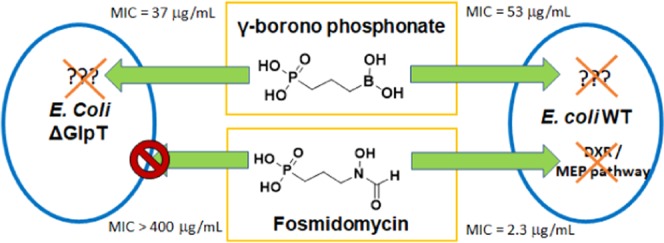
Drug resistance in bacteria is a serious threat, and drugs with novel modes of action are constantly needed. Fosmidomycin is a naturally occurring antibiotic that inhibits the nonmevalonate pathway via inhibition of the enzyme 1-deoxylulose-5-phosphate reductoisomerase (DXR). This work is the first report in which a boronic acid is evaluated as an isostere of the retrohydroxamate moiety of fosmidomycin. We report the novel synthesis of a γ-borono phosphonate analog of fosmidomycin and its corresponding prodrugs. We evaluate the inhibition of DXR and the antimicrobial activity of γ-borono phosphonate compounds against Escherichia coli wild type, E. coli Δglycerol-3-phosphate transporter, and Mycobacterium smegmatis. Despite its structural similarities, the γ-borono phosphonate compound shows antimicrobial activity against E. coli with a mechanism of action that is different from fosmidomycin. This was proven with an underutilized method for studying in vitro inhibition of the MEP pathway in E. coli via isopentenyl pyrophosphate chemical rescue. These results indicate that these compounds may serve as a promising scaffold for developing a new class of antimicrobial agents.
Introduction
Antimicrobial drug resistance is a worldwide threat to public health.1 According to the Centers for Disease Control and Prevention (CDC), two million people contract antibiotic-resistant infections and 23 000 die every year in the U.S. alone.2 An infectious disease that affects people worldwide is tuberculosis (TB). According to the World Health Organization (WHO), 10 million people died worldwide in 2017 due to Mycobacterium tuberculosis (Mtb) infections.3 Even though TB is typically detectable and treatable, the development of drug-resistant and multidrug-resistant strains of Mtb requires the continuous development of new drugs and the discovery of new drug targets.4,5
A novel target that has been proposed recently is the nonmevalonate pathway (MEP) (Figure 1).6 This pathway is essential because it leads to the synthesis of isopentenyl pyrophosphate (IPP), an isoprenoid building block that is required for the biosynthesis of many biomolecules essential to the life cycle of many pathogens such as Mtb and Escherichia coli.7,8 Additionally, the MEP pathway is not present in humans since mammals use the mevalonate pathway for the synthesis of IPP. Therefore, enzymes within the MEP pathway make attractive targets for the development of new antimicrobial agents.9
Figure 1.

MEP pathway and inhibition of 1-deoxylulose-5-phosphate reductoisomerase (DXR) with fosmidomycin.
A natural product able to inhibit the MEP pathway is fosmidomycin (1, Figure 1).10 Fosmidomycin is a low nanomolar inhibitor of 1-deoxy-d-xylulose-5-phosphate reductoisomerase (DXR), the first committed step of the MEP pathway.11 In the DXR active site, the phosphonate group of fosmidomycin interacts through noncovalent interactions with serine and lysine residues, while the retrohydroxamic acid portion is coordinated in a bidentate fashion with the divalent metal Mg2+.6 Although it shows potent in vitro activity, the polar character of fosmidomycin renders it membrane-impermeable.12,13 However, in pathogens such as E. coli, the active uptake of fosmidomycin is mediated by the glycerol-3-phosphate transporter (GlpT) and fosmidomycin resistance in E. coli can be caused by a mutation in GlpT.14,15 Fosmidomycin effectively inhibits mycobacterial DXR, yet it is not active against these organisms due to the highly hydrophobic mycobacterial cell wall and the absence of a corresponding GlpT transporter.16 Several studies have reported on the synthesis of various analogs of fosmidomycin to address its unfavorable pharmacokinetic properties.17−20 One approach carried out by Larhed et al. was to replace the retrohydroxamic acid moiety of fosmidomycin with alternative metal-coordinating groups.21 Particularly, they investigated the use of catechol and hydroxypyridinone as hydroxamic acid isosteres. This interaction is important for establishing the inhibition of DXR since it coordinates with the divalent metal Mg2+ present in the DXR active site. Even though these compounds showed modest inhibition against MtbDXR, they exhibited no activity against the whole organism.21
To date, isosteric replacement of the retrohydroxamic acid group with a boronic acid has not been investigated. In this work, we evaluate the substitution of the retrohydroxamate group with a boronic acid moiety (Figure 2). Boronic acids possess a similar geometry to retrohydroxamic acids and can act as metal-binding chelators. Thus, we hypothesize that the boronic acid can act as an alternate metal chelator in the DXR active site. Additionally, boronic acids can be masked as labile, lipophilic esters, which may provide an avenue to produce novel, membrane-permeable fosmidomycin analogs.
Figure 2.

Core structure of the γ-borono phosphonate library. MIDA = N-methyl iminodiacetic acid. γ-Borono phosphonate 4, dibenzyl γ-borono MIDA phosphonate 5, γ-borono MIDA phosphonate 6, pivaloyloxymethyl (POM) γ-borono MIDA phosphonate 7.
Recently, boronic acids have been gaining the attention of the pharmaceutical industry. Currently, there are five FDA-approved drugs containing boron on the market: Kerydin, Velcade, Ninlaro, Vabomere, and Eucrisa.22−27 Kerydin is an antifungal, which contains a benzoxaborole ring. It acts as an inhibitor of cytoplasmic leucyl-tRNA synthetase, thus blocking the protein synthesis.22 Vabomere is a drug cocktail, and the active ingredient that contains boron is vaborbactam, which can act as a reversible covalent inhibitor of KPC-2 β-lactamase.23 Velcade and Ninlaro are antitumor drugs. They both contain the boronic acid group and act as proteasome inhibitors.24,25
Eucrisa (crisaborole) is used for the treatment of atopic dermatitis and is of particular interest in this respect since the boronic acid moiety acts as a metal chelator when inhibiting phosphodiesterase 4 (PDE4).26 It contains a benzoxaborole ring, and the free hydroxyl group interacts in the hydrate form with the divalent metals, Mn2+ and Zn2+, present in the active site of PDE4.27 Another example of a boron-containing compound where the boronic acid moiety acts as a metal-binding inhibitor is 2-S-amino-6-boronohexanoic acid (ABH).28 Specifically, ABH is an inhibitor of arginase I where the boronic acid coordinates with Mg2+ in the active site of arginase I. ABH-based boronic acid inhibitors are currently in phase I and II clinical trials.27,28
With the γ-borono phosphonate compound, we have the possibility to enhance the lipophilic character of the library by modification of both the negatively charged phosphonate group and the boronic acid moiety. The boronic acid can be masked with N-methyl iminodiacetic acid (MIDA), which is known for its ability to slowly release the free boronic acid via aqueous hydrolysis (compounds 5–7, Figure 2).29 Incorporation of the pivaloyloxymethyl (POM) ester group is a commonly used approach for phosphonate prodrugs (compound 7, Figure 2).30 Labile phosphonate ester prodrugs are present in several FDA-approved drugs, e.g., Hepsera (adefovir dipivoxil) and Viread (tenofovir disoproxil fumarate).31,32 These phosphonate ester moieties are cleaved by nonspecific esterases to produce the free phosphonic acids.33 An alternate approach that we evaluated is the esterification of the phosphonate group as a bisbenzyl ester (compound 5, Figure 2). In contrast with the POM groups, the hydrolysis of the bisbenzyl group is spontaneous and pH-dependent.34
Herein, we describe the results of incorporating a boronic acid group as a metal-chelating, isosteric replacement for the retrohydroxamic acid moiety for the inhibition of DXR. We achieved the synthesis of a γ-borono phosphonate analog (4) of fosmidomycin and evaluated its inhibition of E. coli DXR. Additionally, to increase the lipophilic character of 4, we synthesized several prodrugs via lipophilic protection of both the phosphonate and boronic acid moieties (Figure 2). We evaluated the antimicrobial activity against E. coli WT (BW25113), E. coli ΔGlpT (JW2234-2), and M. smegmatis (as a surrogate organism for Mtb).35−37 Finally, we investigated the mechanism of action of these unique compounds.
Results
Synthesis of the γ-Borono Phosphonate Analogs
The synthetic route for the γ-borono phosphonate 4 is described in Scheme 1. The boron electrophile is synthesized by the hydroboration of 3-bromopropene in an efficient, one-pot method developed in our lab.38 Boron trichloride and triethyl silane allow the in situ production of dichloroborane for hydroboration. Exposure of this intermediate to water followed by potassium hydrogen fluoride provides for hydrolysis to the boronic acid and subsequent conversion to the trifluoroborate salt. Without further purification, compound 2 was used as an electrophile for the introduction of the boronic acid in the final product. Substitution on 2 with dibenzyl phosphonate yielded 3 in a 50% yield. Next, deprotection of the phosphonate esters with boron trichloride provided the desired γ-borono phosphonate 4 in a 63% yield.
Scheme 1. Synthesis of 4.

Reagents and conditions: (a) BCl3, Et3SiH, −78 °C to room temperature (rt), 3h; (b) H2O, 0 °C to rt, 30 min; (c) KHF2, H2O, ether, rt, 1h, 38%; (d) dibenzyl phosphonate, NaH, dimethylformamide (DMF), 0 °C to rt, overnight , 50%; (e) BCl3, dichloromethane (DCM), −78 °C to rt, 4 h, 63%.
In an attempt to enhance membrane permeability and, potentially, activity against M. smegmatis, we synthesized lipophilic prodrugs of 4 (Scheme 2). The synthetic route for prodrug 7 (Scheme 2) starts with compound 3, where the boron functionality is protected with MIDA to form 5. Hydrogenolysis of compound 5 allowed the elimination of the benzyl ester functionality and the synthesis of compound 6 in a 72% yield. Without further purification, 6 was coupled with pivaloyloxymethyl chloride to give 7 in a 42% yield.
Scheme 2. Synthesis of 6 and 7.

Reagents and conditions: (a) methyl iminodiacetic acid, dimethyl sulfoxide (DMSO), toluene, 130 °C, overnight, 83%; (b) H2, Pd-C, MeOH, rt, 4 h, 72%; (c) Et3N, DMF, rt, 30 min; (d) POM-Cl, from rt to 60 °C, overnight, 42%.
Compound 4 Is a Relatively Poor Inhibitor of E. coli DXR
Compound 4 was evaluated as an inhibitor of recombinant E. coli DXR. Different concentrations of analog 4 were incubated with DXR and NADPH at 37 °C. The reaction was initiated with the addition of the natural substrate DXP, and the disappearance of NADPH was monitored at 340 nm.11 From this data, initial rates were determined and plotted vs the inhibitor concentration. We determined that the fosmidomycin IC50 is 0.049 ± 0.007 μM. This result is consistent with the literature value (IC50 0.032 μM).11 The IC50 for compound 4 was determined to be 128 ± 3 μM (Figure 3).
Figure 3.
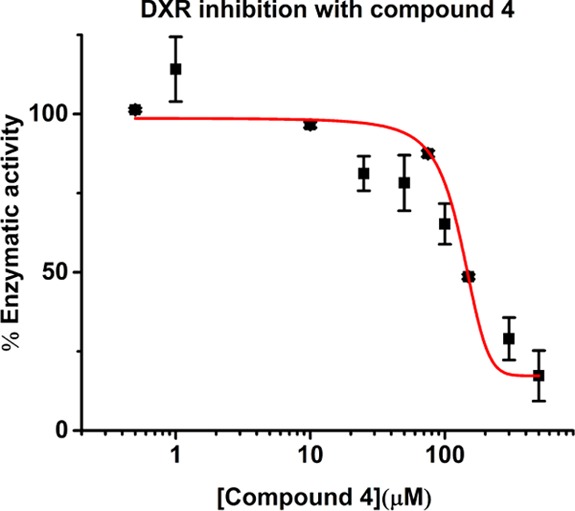
Evaluation of E. coli DXR inhibition by compound 4.
Compound 4 and the Borono Prodrug 6 Showed Significant Antimicrobial Activity against Both E. coli WT and ΔGlpT Strains
The antimicrobial activity for all of the γ-borono phosphonate analogs was evaluated by the microdilution assay against E. coli strains BW25113 and JW2234-2 and M. smegmatis.39 In general, each strain was grown overnight and then diluted to an initial OD600 of 0.05. Different concentrations of the analogs were incubated at 37 °C, for 18 h in E. coli and for 30 h in M. smegmatis. The concentration of the inhibitor, which reduced bacterial growth by 90%, was determined to be the MIC90 (Table 1). None of the compounds tested, including our lipophilic prodrugs, showed any activity against M. smegmatis. However, our antimicrobial evaluation of compounds 4 and 6 against E. coli WT and E. coli ΔGlpT showed that they have a reasonable activity against both strains and a mechanism of cellular penetration that is independent of the glycerol-3-phosphate transporter and thus are unique from fosmidomycin.
Table 1. Minimum Inhibitory Concentration 90% (MIC90) and Determination of the clog Pa,c.
|
E. coli (μg/mL) |
M. smegmatis (μg/mL) | clog P | ||
|---|---|---|---|---|
| WT | ΔGlpT | |||
| 1 | 2.3 ± 5 | >400 | >400 | –2.21 |
| 4 | 53 ± 6 | 37 ± 4 | >1000 | –1.53 |
| 5 | >1000 | >1000 | >1000 | 4.02 |
| 6 | 63 ± 13 | 52 ± 6 | >500 | –0.99 |
| 7 | >750 | >750 | >750 | 4.28 |
| POM-FR900098(48) | >200 | N.D. | 50–100b | 2.44 |
For comparison, MIC for fosmidomycin in E. coli K12 is 12.5 μg/mL; in M. tuberculosis (H37Rv), it is >500 μg/mL.48
Reported MIC in M. tuberculosis (H37Rv); NR = not reported. The unionized clog P values for each compound were calculated with the use of MarvinSketch v.19.11 by ChemAxon.
Exogenous Isopentenyl Pyrophosphate (IPP) Has a Dose-Dependent Effect on the Rescue of E. coli WT Growth Inhibited by the MEP Pathway Inhibitor Fosmidomycin
The concentration of isopentyl pyrophosphate (IPP) necessary for the rescue experiment was determined for E. coli WT (BW25113). This strain was grown overnight and then diluted to an initial OD600 of 0.05. The E. coli WT was incubated for 18 h at 37 °C with varied concentrations of IPP in the presence of two fixed concentrations of fosmidomycin. An effective concentration of IPP for the rescue of fosmidomycin-inhibited E. coli growth was determined to be 125 μM (Figure 4). This experiment proves that E. coli is capable of utilizing IPP from the media to relieve MEP pathway inhibition. With this tool, we next developed a chemical rescue assay that will prove useful in the study of MEP pathway inhibitors in vitro in E. coli.
Figure 4.
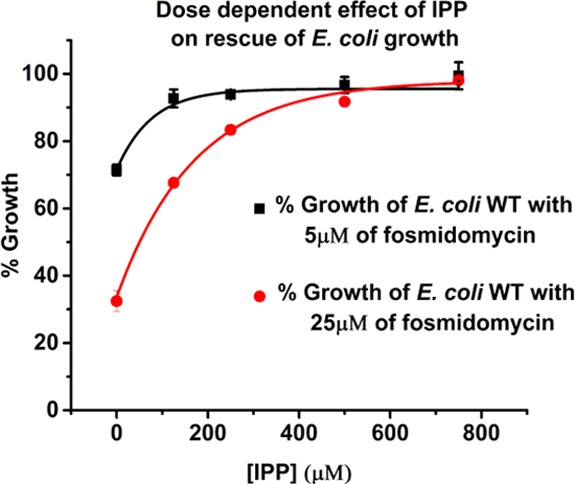
Determination of concentration-dependent rescue of fosmidomycin-inhibited E. coli growth by the addition of exogenous IPP. IPP concentration of 125 μM is used in the E. coli rescue assay (n = 3).
E. coli Chemical Rescue Experiments Reveal That Compounds 4 and 6 Do Not Inhibit the MEP Pathway
IPP chemical rescue experiments for E. coli growth inhibited by fosmidomycin or our compounds were completed for the E. coli WT and E. coli ΔGlpT (strains BW25113 and JW2234-2, respectively).40 Each strain was grown overnight and then diluted to an initial OD600 of 0.05. Different concentrations of fosmidomycin, 4, or 6 were incubated with the bacterium in the presence or absence of 125 μM IPP for 18 h at 37 °C. In the presence of fosmidomycin, the addition of IPP led to the restoration of E. coli growth (Figure 5A). However, in the presence of 4 or 6, the addition of IPP did not restore bacterial growth (Figures 5B,C and S19). These results indicate that the γ-borono phosphonate analogs are acting on a target outside of the nonmevalonate pathway and thus have a mechanism of action that is unique from fosmidomycin.
Figure 5.

Chemical rescue of E. coli inhibition with IPP. (A) E. coli WT was treated with different concentrations of fosmidomycin for 18 h with and without the addition of 125 μM IPP in LB media. (B, C) E. coli WT and E. coli ΔGlpT were treated with different concentrations of 4 for 18 h with and without the addition of 125 μM IPP in LB media (n = 3). These rescue experiments were also performed with compound 6, and similar results were obtained (Figure S19).
Discussion
Compound 4 was evaluated as an inhibitor of recombinant E. coli DXR but showed only modest inhibition (128 μM), unlike fosmidomycin (0.049 μM). However, in comparison to most of the analogs previously synthesized by Larhed et al.,21 compound 4 is a stronger inhibitor of DXR. The best retrohydroxamic acid replacement reported in their work, the catechol substitution, had a 41 μM EC50 against MtbDXR. Even though the boronic acid and the retrohydroxamate group have similar geometries, our result indicates that the boronic acid is a weaker metal chelator than the retrohydroxamic acid group. This effect can be explained by the different resonance characteristics for the boronic acid and retrohydroxamate moieties. The retrohydroxamic acid possesses a double-bond character that favors the coordination between the lone pair of the oxygen and the metal, whereas the boron atom has an empty p orbital, which makes it a strong Lewis acid.41 This characteristic renders the boron–oxygen bonds partially double-bonded; however, the lone-pair electrons of the oxygens are less available to coordinate with the metal because they are also interacting with the empty p orbital on the boron center. It is expected that the anionic, boronate form would be much more strongly metal-chelating as is observed with crisaborole.
Since modest DXR inhibition was observed, we evaluated the antimicrobial activity of 4 against E. coli WT, E. coli ΔGlpT, and M. smegmatis (Table 1). It is known that M. smegmatis is not a perfect analog for M. tuberculosis, yet metabolic pathways (including MEP) and cell wall properties are similar in both species.42,43 The DXR sequence is ∼79% identical between these two species, and sensitivity to fosmidomycin is similar.44−46
Our results show that 4 inhibited in vitro growth of both E. coli WT and E. coli ΔGlpT, unlike fosmidomycin. E. coli ΔGlpT is inherently resistant to fosmidomycin due to compromised membrane transport, yet this strain is not resistant to 4. This indicates that compound 4 is crossing the cell wall of E. coli by passive diffusion or is able to utilize a different transporter. Despite these promising results, 4 is inactive against M. smegmatis, probably due to its inability to cross the mycobacterial cell wall. The mycobacterial cell wall is different from the cell wall of Gram-negative bacteria, such as E. coli, since they possess a high concentration of lipids that make the passive diffusion of polar compounds more difficult.47
In an attempt to enhance membrane permeability and, potentially, activity against M. smegmatis, we synthesized lipophilic prodrugs of 4 and evaluated their antimicrobial effects and determined their calculated log P values (Scheme 2 and Table 1). With these boronic acid-containing compounds, we have the ability of introducing a double prodrug by masking both the highly charged phosphonate moiety and the boronic acid group. Phosphonates can be modified with the use of pivaloyloxymethyl (POM) or bisbenzyl ester functionalities. The use of the POM group in the synthesis of phosphonate prodrugs is already established.30 The cleavage of POM in vivo is driven by nonspecific esterases, while the hydrolysis of the bisbenzyl ester group is pH-dependent and naturally faster in an acidic environment.33,34 The boronic acid can be masked using methyl iminodiacetic acid (MIDA). MIDA is typically used as a protecting group for boronic acids because it is known for its capacity to slowly release free boronic acid.29
First, we wanted to evaluate the protection of the boronic acid group in isolation. Antimicrobial evaluation of 6 confirmed the results obtained with 4 (Table 1). Compounds 4 and 6 are equipotent against E. coli WT and E. coli ΔGlpT, while neither have any relevant activity against M. smegmatis. Therefore, the introduction of MIDA neither enhanced nor hindered the activity or membrane permeability. We would like to note, however, that the incorporation of the MIDA group may be useful for formulation purposes since it improved the solubility of the analog and should provide increased chemical stability.29
Next, the effects of phosphonate-masking in combination with boronic acid protection were evaluated. Compounds 5 and 7 show no antimicrobial activity against either strain of E. coli or M. smegmatis. These results are consistent with the work of Ponaire et al. when they investigated the antimicrobial effects of propyloxymethyl ester prodrugs of fosmidomycin and FR900098.44 Their fosmidomycin/FR900098 prodrugs were unable to inhibit the growth of E. coli WT at all and only inhibited the growth of M. smegmatis at the very highest concentrations evaluated. Dowd et al. tested the hypothesis that efflux pumps were responsible for the export of these lipophilic prodrugs and their subsequent lack of activity.48 They tested POM-FR900098 against E. coli with a deletion in the function of the efflux pump (E. coli tolC). They reported an increase in antimicrobial activity for POM-FR900098 against E. coli tolC compared to the MIC value against E. coli WT. Therefore, compounds 5 and 7, with no significant activity against E. coli WT, E. coli ΔGlpT, or M. smegmatis, are likely suffering from the same limitations or a complete inability to release the active compound under the assay conditions.
Given the low affinity to DXR and the structural similarity of our compound to other metabolites within the MEP pathway, we wanted to independently evaluate compound 4 as a potential inhibitor for any step within the MEP pathway. This was achieved with a chemical rescue experiment utilizing IPP, the end product of the MEP pathway.40 Recent work provided some evidence that IPP, a multiply-charged diphosphate species, is able to cross the membrane in E. coli via an unknown mechanism.49 We confirmed these findings by observing that fosmidomycin-induced growth inhibition was relieved by the supplementation of the E. coli growth media with IPP. Exogenous IPP was able to restore E. coli growth in a concentration-dependent manner (Figure 4). With fosmidomycin, which targets the DXR enzyme within the MEP pathway, the presence of IPP in the media results in the rescue of E. coli’s growth (Figure 5A). This experiment thus provides a ready assay for the rapid determination of the mechanism of action for MEP pathway inhibitors in E. coli. To evaluate the mechanism of action of compound 4, this rescue experiment was done on both E. coli WT and E. coli ΔGlpT (Figure 5B,C, respectively). In both sets of experiments, the addition of IPP to the media did not restore growth. These results indicate that the target of 4 is not any of the enzymes that are involved in the MEP pathway. Similar results were obtained for the borono prodrug 6 (Figure S19).
In conclusion, this work describes the synthesis and the antimicrobial evaluation of a novel class of antimicrobial agents, γ-borono phosphonates. These compounds were rationally designed analogs of the natural product fosmidomycin. This work is the first report of evaluating a boronic acid as an isostere of the metal-chelating retrohydroxamate moiety. DXR inhibition by 4 was considerably weaker than with fosmidomycin, indicating that this alkyl boronic acid group is suboptimal in this application, which requires strong metal chelation. None of the compounds tested, including our lipophilic prodrugs, showed any activity against M. smegmatis. However, our antimicrobial evaluation of these compounds against E. coli WT and E. coli ΔGlpT showed that they have a reasonable activity against both strains and a mechanism of action that is unique from fosmidomycin. We have proven that these compounds are independent of the glycerol-3-phosphate transporter and are acting on a target outside of the nonmevalonate pathway. In addition, we have confirmed that E. coli is capable of utilizing exogenous IPP to relieve MEP pathway inhibition. Using this finding, we developed a chemical rescue assay that will prove useful in the study of MEP pathway inhibitors in vitro in E. coli. Elucidation of the mechanism of action and further structure–activity relationship studies are ongoing to improve the antimicrobial activity of this class of γ-borono phosphonate compounds.
Materials and Methods
General Procedures
All reactants and reagents were purchased commercially and used without further purification unless otherwise indicated. Reactions were carried out under a dry, inert atmosphere of argon or nitrogen unless otherwise indicated. “Concentrated” refers to the removal of a solvent with a rotary evaporator followed by further evacuation with a two-stage mechanical pump. Yields refer to chromatographically and spectroscopically pure (>95%) compounds. Thin-layer chromatography was performed using silica gel 200 μM precoated polyester backed plates with a fluorescent indicator. Developed TLC plates were visualized with UV light (254 nm), iodine, KMnO4, or p-anisaldehyde staining. Flash column chromatography was conducted with the indicated solvent system using normal-phase silica gel 40–63 μM, 230–400 mesh. 1H NMR spectra were recorded at 400 MHz, 13C NMR spectra were recorded at 100 MHz, 11B NMR spectra were recorded at 128 MHz, and 31P NMR spectra were recorded at 161 MHz. Chemical shifts are reported in δ values (ppm) relative to an internal reference (0.05% v/v) of tetramethyl silane or the residual solvent signal. Peak-splitting patterns in the 1H NMR are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. 13C NMR experiments were conducted with the continuous pulse decay sequence. Mass spectra were obtained on a Thermo-Fisher Exactive Orbitrap mass spectrometer using matrix-assistant inlet ionization (MAII) as an ionization source (3-nitrobenzonitrile matrix).50 Compound purity was determinate with quantitative one-dimensional 1H NMR (qNMR).51 An E. coli BL21 strain containing a plasmid for the overexpression of His-tagged E. coli DXR was provided by Meyers.52E. coli strains BW25113 (E. coli WT, CGSC# 7636, parent strain for the GlpT deletion) and JW2234-2 (E. coli ΔGlpT, CGSC# 11875) were purchased from The Coli Genetic Stock Center (CGSC) at Yale.35,36M. smegmatis strain was purchased from (Trevisan) Lehmann and Neumann (ATCC 700084).37
Synthesis of the (3-(Bis(benzyloxy)phosphoryl) propyl) Boronic Acid (3)
A dry, Ar-flushed round-bottom flask in a glovebox was charged with pure sodium hydride (1.10 equiv, 15.79 mmol). The flask was removed from the glovebox, and then dibenzyl phosphonate (1 equiv, 14.35 mmol) was dissolved in dry DMF (6 mL) and transferred into the reaction vessel at 0 °C. The suspension was stirred for 2 h at rt. Next, the reaction vessel was cooled to 0 °C, and a solution of 3-bromo propyl trifluoroborate potassium salt (1 equiv, 14.35 mmol) in dry DMF (3 mL) was added. The solution was warmed to rt and stirred overnight. The following day, the reaction was quenched with water at 0 °C and extracted three times with ethyl acetate. The organic layers were dried over Na2SO4, filtered, and concentrated. The product was purified via column chromatography (95:5 EtOAc/EtOH) to give a white solid with a 50% yield.
1H NMR (400 MHz, CDCl3): δ 7.37–7.26 (m, 10H), 5.05–4.91 (m, 4H), 1.85–1.72 (m, 4H), 0.93 (t, 2H, J = 8.0 Hz). 13C NMR (100 MHz, CDCl3): δ 136.24, 128.61, 128.45, 127.92, 67.33, 28.21, 26.84, 17.14. 11B NMR (128 MHz, CDCl3): δ 33.74. 31P NMR (161 MHz, CDCl3): δ 35.06. HRMS (MAII) m/z calcd for C17H22BO5P + Na (M + Na)+: 371.1190, found 371.1196. Mp > 250 °C.
Synthesis of 3-Borono Propyl Phosphonic Acid (4)
A dry, Ar-flushed round-bottom flask was charged with 3 (1 equiv, 0.92 mmol) and dry DCM (1 mL). To this mixture was added a solution of 1 M boron trichloride in DCM (10 equiv, 1.92 mmol) slowly at −78 °C. The solution was stirred for 4 h and gently warmed up to rt over this time. The reaction was quenched with water (10 mL) at 0 °C, and the biphasic solution was stirred for 30 min at rt. Then, the solution was transferred to a separatory funnel, and the aqueous phase was washed three times with DCM and then frozen and lyophilized. This crude product was triturated with methanol, and the organic phase was concentrated to give the pure product as a white solid in a 63% yield.
1H NMR (400 MHz, D2O-d6): 1.75–1.56 (m, 4H), 0.82 (t, 2H, J = 8.0 Hz). 13C NMR (100 MHz, D2O-d6): δ 28.92, 27.61, 17.16 (d, J = 5.0 Hz). 11B NMR (128 MHz, D2O-d6): δ 31.03. 31P NMR (161 MHz, D2O-d6): δ 30.67. Mp > 250 °C. HRMS (MAII) m/z calcd for C9H19BO5P – H (M – H)−: 249.1058, found 249.1032. HRMS for compound 4 was determined as the pinacol ester via derivatization with 2,3-dimethyl butane diol.53
Synthesis of the (3-(Bis(benzyloxy)phosphoryl) propyl) MIDA Boronate (5)
A dry, Ar-flushed round-bottom flask was charged with 3 (1 equiv, 0.35 mmol), toluene (10 mL), and DMSO (1 mL). Methyl iminodiacetic acid (2 equiv, 0.70 mmol) was added to the solution. The flask was fitted with a Dean-Stark trap and condenser, and the resulting suspension was refluxed overnight. The suspension was then filtered. The filtrate was diluted with ethyl acetate and washed with a saturated solution of ammonium chloride in water. The organic layer was dried over Na2SO4, filtered, and concentrated to provide the pure product as colorless oil in an 83% yield.
1H NMR (400 MHz, ((CD3)2CO)-d6): δ 7.25–7.16 (m, 10H), 4.94–4.80 (m, 4H), 3.79 (d, 2H, J = 16 Hz), 3.52 (d, 2H, J = 16 Hz), 2.62 (s, 3H), 1.78–1.72 (m, 2H), 1.70–1.59 (m, 2H), 0.56 (t, 2H, J = 8 Hz). 13C NMR (100 MHz, CDCl3-d6): δ 167.18, 136.52, 128.62, 128.38, 127.95, 67.05, 61.83, 45.63, 28.75, 27.38, 17.20. 11B NMR (128 MHz, ((CD3)2CO)-d6): δ 12.94. 31P NMR (161 MHz, ((CD3)2CO)-d6): 32.92. HRMS (MAII) m/z calcd for C22H28BNO7P + H (M + H)+: 460.1690, found 460.1691.
Synthesis of the Phosphonate Propyl MIDA Boronate (6)
A dry, Ar-flushed round-bottom flask was charged with Pd/C (0.2 equiv, 0.07 mmol) and methanol (5 mL). To the mixture was added a solution of 5 (1 equiv, 0.35 mmol) in methanol (5 mL). The reaction vessel was charged with H2 and stirred under 1 atm H2 (balloon) for 4 h at rt. Next, the mixture was filtered through celite, and the filtrate was concentrated to provide the pure product in a 72% yield as a white solid. 1H NMR (400 MHz, CD3OD-d6): δ 4.18 (d, 2H, J = 16 Hz), 3.98 (d, 2H, J = 16 Hz), 2.82 (s, 3H), 1.53–1.46 (m, 4H), 0.59 (t, 2H, J = 8 Hz). 13C NMR (100 MHz, CD3OD-d6): δ 169.54, 61.50, 44.97, 30.40, 29.05, 17.60. 11B NMR (128 MHz, DMSO-d6): δ 13.14. 31P NMR (161 MHz, CD3OD): δ 25.86. HRMS (MAII) m/z calcd C8H15BNO7P + H (M + H)+: 280.0750, found 280.0752. Mp > 250 °C.
Synthesis of Pivaloyloxymethyl Phosphonate Propyl MIDA Boronate (7)
A dry, Ar-flushed round-bottom flask was charged with 6 (1 equiv, 0.95 mmol) and dry DMF (20 mL). Dry triethylamine (2 equiv, 1.88 mmol) was added, and the solution was stirred at rt for 30 min. Following the addition of pivaloyloxymethyl chloride (10 equiv, 9.5 mmol), the reaction was stirred overnight at 60 °C. DMF was removed under vacuum, and the residue was taken up in ethyl acetate and extracted with water. The organic layer was dried over Na2SO4 and concentrated to obtain a crude product. The crude product was purified via column chromatography (1:1 EtOAc/acetone) to afford the pure product as a white solid in a 42% yield. 1H NMR (400 MHz, ((CD3)2CO)-d6): δ 5.69–5.65 (m, 4H), 4.22 (d, 2H, J = 16 Hz), 4.05 (d, 2H, J = 16 Hz), 3.10 (s, 3H), 2.06−1.89 (m, 2H), 1.89−1.66 (m, 2H), 1.22 (s, 18H), 0.75 (t, 2H, J = 8 Hz). 13C NMR (100 MHz, ((CD3)2CO)-d6): δ 176.26, 167.76, 81.25, 61.72, 45.39, 38.37, 26.18, 16.96. 11B NMR (128 MHz, ((CD3)2CO)-d6): δ 12.82. 31P NMR (161 MHz, ((CD3)2CO)-d6): δ 31.97. HRMS (MAII) m/z calcd for C20H35BNO11P + Na (M + Na)+: 530.1930, found 530.1933. Mp > 250 °C.
Enzyme Assay of E. coli DXR (11)
His-tagged E. coli DXR obtained was overexpressed and purified as reported.52 The DXR enzymatic activity was determined at 37 °C in HEPES (50 mM, pH 8.0), MgCl2 (2 mM), NADPH (0.15 mM), TCEP (0.5 mM), BSA (1 mg/mL), DXR (0.001 μM), and different concentrations of inhibitor (from 0.5 to 500 μM) in a final volume of 1 mL. The reaction mixture was incubated for 1 min at 37 °C prior to initiation with the substrate DXP (0.08 mM). Initial rates were monitored at 340 nm (Cary 300 UV–vis, Agilent) following the decrease of NADPH over 2.5 min. Data were fitted using Origin 2016 64Bit.
E. coli Growth Inhibition Assay (33)
Minimal inhibitory concentrations 90% (MIC90) were determined for the E. coli strains BW25113 and JW2234-2 by microdilution. Each strain was grown overnight and then diluted to an initial OD600 (Eppendorf BioPhotomer) of 0.05. A concentration gradient of each inhibitor (from 5 to 1000 μM) was created and aliquoted into the assay plate (Corning, 96-well clear flat-bottom polystyrene not-treated microplate, with low evaporation lid, sterile). In general, the 96-well plates were prepared by dispensing 135 μL of inoculum, sterile water, and inhibitor to a final volume of 150 μL. Compounds not soluble in water were dissolved in DMSO. The plates included a sterility check (water, medium, and DMSO), negative control (medium, water, DMSO, and inoculum), positive control (medium, water, DMSO, inoculum, 25 and 50 μg/mL of kanamycin, and 25 and 50 μg/mL of ampicillin). The plates were incubated at 37 °C, shaking at 300 rpm for 18 h and then read at 600 nm (Synergy H1 Microplate Readers Biotek). The concentration of the inhibitor, which reduced bacterial growth by 90%, was determined to be the MIC90. Data were fitted using Origin 2016 64Bit.
M. smegmatis Inhibition Assay (33)
Minimal inhibitory concentrations 90% (MIC90) were determined for M. smegmatis by microdilution. M. smegmatis was grown at 37 °C overnight in 7H9 medium and supplemented with OADC and 1% Tween 20 at 37 °C. The overnight culture was diluted to an initial OD600 (Eppendorf BioPhotomer) of 0.05 with 7H9 and 1.5% Tween 80. A concentration gradient of each inhibitor (from 5 to 1000 μM) was created and aliquoted into the assay plate (Corning, 96-well clear flat-bottom polystyrene not-treated microplate, with low evaporation lid, sterile). Compounds not soluble in water were dissolved in DMSO. In general, the 96-well plates were prepared by dispensing 135 μL of inoculum, sterile water, and inhibitor to a final volume of 150 μL. The plates included a sterility check (water, medium, and DMSO), negative control (medium, water, DMSO, and inoculum), positive control (medium, water, DMSO, inoculum, 5 μg/mL of isoniazid, and 10 μg/mL of isoniazid). The plates were incubated at 37 °C, shaking at 300 rpm for 30 h and then read at 600 nm (Synergy H1 Microplate Reader Biotek). The concentration of the inhibitor, which reduced bacterial growth by 90%, was determined to be the MIC90. Data were fitted using Origin 2016 64Bit.
Dose-Dependent Effect of Isopentenyl Pyrophosphate (IPP) on Rescue of E. coli WT Growth
The concentration of isopentyl pyrophosphate (IPP) for the rescue experiment was determined for E. coli WT. E. coli WT was grown overnight and diluted to an initial OD600 (Eppendorf BioPhotometer) of 0.05. A concentration gradient of IPP (from 125 to 750 μM) and two concentrations for fosmidomycin (5 and 25 μM) were created and aliquoted into the assay plate (Corning, 96-well clear flat-bottom polystyrene not-treated microplate, with low evaporation lid, sterile). In general, the 96-well plates were prepared by dispensing 135 μL of inoculum, 2 μL of fosmidomycin, IPP, and sterile water to a final volume of 200 μL. The plates included a sterility check (water and medium), negative control (medium, water, and inoculum), positive control (medium, water, inoculum, 25 and 50 μg/mL of kanamycin, and 25 and 50 μg/mL of ampicillin). The plates were incubated at 37 °C, shaking at 300 rpm for 18 h and then read at 600 nm (BioTek Synergy Microplate Reader). Data were fitted using Origin 2016 64Bit.
E. coli Chemical Rescue Assay
These experiments were adapted from those previously reported for malaria.54,55 The IPP chemical rescue experimental conditions for E. coli inhibited by fosmidomycin or our compounds were determined for the E. coli WT and E. coli ΔGlpT (strains BW25113 and JW2234-2, respectively). Each strain was grown overnight and then diluted to an initial OD600 (Eppendorf BioPhotomer) of 0.05. A concentration gradient of each inhibitor (from 0.05 to 100 μM for fosmidomycin in E. coli WT; from 25 to 500 μM for compound 4 in E. coli WT; from 50 to 500 μM for compound 4 in E. coli ΔGlpT) was created and aliquoted into the assay plate (Corning, 96-well clear flat-bottom polystyrene not-treated microplate, with low evaporation lid, sterile). In general, the 96-well plates were prepared by dispensing 135 μL of inoculum, sterile water, inhibitor, and 3.5 μL of IPP to a final volume of 200 μL. Compounds not soluble in water were dissolved in DMSO. The plates included a sterility check (water, medium, and DMSO), negative control (medium, water, DMSO, and inoculum), positive control (medium, water, DMSO, inoculum, 25 and 50 μg/mL of kanamycin, and 25 and 50 μg/mL of ampicillin). The plates were incubated at 37 °C, shaking at 300 rpm for 18 h and then read at 600 nm (Synergy H1 Microplate Reader Biotek). Data were fitted using Origin 2016 64Bit.
Acknowledgments
The authors would like to thank the University of the Sciences for financial support of this work. G.M. and J.M.G. were supported by Robert D. Spiers Graduate Research Fellowships. The authors would also like to thank Adetoun Adeniji-Adele and Amber Gunderwala for their helpful discussion.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01774.
Characterization data: 1H NMR and 13C NMR spectra for compounds 3−7 and qNMR (internal standard DMSO2) for compounds 4−7. Data for compound 4 additionally includes 11B NMR, 31P NMR, COSY, and HSQC spectra. IPP chemical rescue of E. coli growth inhibition for compound 6 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. High Levels of Antibiotic Resistance Found Worldwide, 2019.
- Center for Disease Control and Prevention. About Antimicrobial Resistance, 2019.
- World Health Organization. Global Tuberculosis Report, 2018.
- Koch A.; Cox H.; Mizrahi V. Drug-Resistant Tuberculosis: Challenges and Opportunities for Diagnosis and Treatment. Curr. Opin. Pharmacol. 2018, 42, 7–15. 10.1016/j.coph.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomino J. C.; Martin A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317–340. 10.3390/antibiotics3030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale I.; O’Neill P. M.; Berry N. G.; Odom A.; Sharma R. The MEP Pathway and the Development of Inhibitors as Potential Anti-Infective Agents. Med. Chem. Commun. 2012, 3, 418–433. 10.1039/c2md00298a. [DOI] [Google Scholar]
- Eisenreich W.; Bacher A.; Arigoni D.; Rohdich F. Biosynthesis of Isoprenoids via the Non-Mevalonate Pathway. Cell. Mol. Life Sci. 2004, 61, 1401–1426. 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuston S.; Begley M.; Gahan C. G.; Hill C. Isoprenoid Biosynthesis in Bacterial Pathogens. Microbiology 2012, 158, 1389–1401. 10.1099/mic.0.051599-0. [DOI] [PubMed] [Google Scholar]
- Lange B. M.; Rujan T.; Martin W.; Croteau R. Isoprenoid Biosynthesis: The Evolution of Two Ancient and Distinct Pathways across Genomes. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13172–13177. 10.1073/pnas.240454797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhiman R. K.; Schaeffer M. L.; Bailey A. M.; Testa C. A.; Scherman H.; Crick D. C. 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (IspC) from Mycobacterium tuberculosis: Towards Understanding Mycobacterial Resistance to Fosmidomycin. J. Bacteriol. 2005, 187, 8395–8402. 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz L.; Tritsch D.; Grosdemange-Billiard C.; Hemmerlina A.; Willem A.; Bach T.; Rohmer M. Isoprenoid Biosynthesis as a Target for Antibacterial and Antiparasitic Drugs: Phosphonohydroxamic Acids as Inhibitors of Deoxyxylulose Phosphate Reducto-Isomerase. Biochem. J. 2005, 386, 127–135. 10.1042/BJ20041378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa T.; Hiroshi S.; Shigemi F.; Takao K.; Minoru N. Pharmacokinetics of Fosmidomycin, a New Phosphonic Acid Antibiotic. Antimicrob. Agents Chemother. 1982, 21, 224–230. 10.1128/AAC.21.2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na-Bangchang K.; Ruengweerayut R.; Karbwang J.; Chauemung A.; Hutchinson D. Pharmacokinetics and Pharmacodynamics of Fosmidomycin Monotherapy and Combination Therapy with Clindamycin in the Treatment of Multidrug Resistant Falciparum malaria. Malar. J. 2007, 6, 70 10.1186/1475-2875-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckenney E. S.; Sargent M.; Khan H.; Uh E.; Jackson E. R.; Jose S.; Couch R. D.; Dowd C. S.; van Hoek M. L. Lipophilic Prodrugs of FR900098 Are Antimicrobial against Francisella Novicida In Vivo and In Vitro and Show GlpT Independent Efficacy. PLoS One 2012, 7, e38167 10.1371/journal.pone.0038167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y.; Furukawa S.; Ogihara H.; Yamasaki M. Fosmidomycin Resistance in Adenylate Cyclase Deficient (Cya) Mutants of Escherichia coli. Biosci., Biotechnol., Biochem. 2003, 67, 2030–2033. 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- Brown A. C.; Parish T. Dxr Is Essential in Mycobacterium tuberculosis and Fosmidomycin Resistance Is Due to a Lack of Uptake. BMC Microbiol. 2008, 8, 78 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips A. M. F.; Nogueira F.; Murtinheria F.; Barros M. T. Synthesis and Antimalarial Evaluation of Prodrugs of Novel Fosmidomycin Analogues. Bioorg. Med. Chem. Lett. 2015, 25, 2112–2116. 10.1016/j.bmcl.2015.03.077. [DOI] [PubMed] [Google Scholar]
- Devreux V.; Wiesner J.; Goeman J. L.; Van Der Eycken J.; Jomaa H.; Van Calenbergh S. Synthesis and Biological Evaluation of Cyclopropyl Analogues of Fosmidomycin as Potent Plasmodium Falciparum Growth Inhibitors. J. Med. Chem. 2006, 49, 2656–2660. 10.1021/jm051177c. [DOI] [PubMed] [Google Scholar]
- Haemers T.; Wiesner J.; Busson R.; Jomaa H.; Van Calenbergh S. Synthesis of α -Aryl-Substituted and Conformationally Restricted Fosmidomycin Analogues as Promising Antimalarials. Eur. J. Inorg. Chem. 2006, 3856–3863. 10.1002/ejoc.200600202. [DOI] [Google Scholar]
- Jackson E. R.; San Jose G.; Brothers R. C.; Eldestein E. K.; Sheldon Z.; Haymond A.; Johny C.; Boshoff H. I.; Couch R. D.; Dowd C. S. The Effect of Chain Length and Unsaturation on Mtb Dxr Inhibition and Antitubercular Killing Activity of FR900098 Analogs. Bioorg. Med. Chem. Lett. 2014, 24, 649–653. 10.1016/j.bmcl.2013.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andaloussi M.; Lindh M.; Björkelid C.; Suresh S.; Wieckowska A.; Iyer H.; Karlén A.; Larhed M. Substitution of the Phosphonic Acid and Hydroxamic Acid Functionalities of the DXR Inhibitor FR900098: An Attempt to Improve the Activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2011, 21, 5403–5407. 10.1016/j.bmcl.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Sharma N.; Sharma D. An Upcoming Drug for Onychomycosis: Tavaborole. J. Pharmacol. Pharmacother. 2015, 6, 236–239. 10.4103/0976-500X.171870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Stapleton P.; Haider S.; Healy J. Boronic Acid Inhibitors of the Class A Beta-Lactamase KPC-2. Bioorg. Med. Chem. 2018, 26, 2921–2927. 10.1016/j.bmc.2018.04.055. [DOI] [PubMed] [Google Scholar]
- Baker S. J.; Ding C. Z.; Akama T.; Zhang Y. K.; Hernandez V.; Xia Y. Therapeutic Potential of Boron-Containing Compounds. Future Med. Chem. 2009, 1, 1275–1288. 10.4155/fmc.09.71. [DOI] [PubMed] [Google Scholar]
- Hu Q. H.; Liu R. J.; Fang Z. P.; Zhang J.; Ding Y. Y.; Tan M.; Wang M.; Pan W.; Zhou H. C.; Wang E. D. Discovery of a Potent Benzoxaborole-Based Anti-Pneumococcal Agent Targeting Leucyl-TRNA Synthetase. Sci. Rep. 2013, 3, 2475 10.1038/srep02475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akama T.; Baker S. J.; Zhang Y. K.; Hernandez V.; Zhou H.; Sanders V.; Freund Y.; Kimura R.; Maples K. R.; Plattner J. J. Discovery and Structure-Activity Study of a Novel Benzoxaborole Anti-Inflammatory Agent (AN2728) for the Potential Topical Treatment of Psoriasis and Atopic Dermatitis. Bioorg. Med. Chem. Lett. 2009, 19, 2129–2132. 10.1016/j.bmcl.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Chen A. Y.; Adamek R. N.; Dick B. L.; Credille C. V.; Morrison C. N.; Cohen M. S. Targetig Metalloenzymes for Therapeutic Intervention. Chem. Rev. 2019, 119, 1323–1455. 10.1021/acs.chemrev.8b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio R.; Elbaum D.; Kanyo Z. F.; Carroll P. J.; Cavalli R. C.; Ash D. E.; Christianson D. W. Inhibition of Mn 2 + 2 -Arginase by Borate Leads to the Design of a Transition State Analogue Inhibitor, 2 ( S ) -Amino-6-Boronohexanoic Acid Temple Uni V Ersity School of Medicine Rat Liver Arginase, Has Yielded an X-Ray Crystal Structure to Guide Site. J. Am. Chem. Soc. 1997, 119, 8107–8108. 10.1021/ja971312d. [DOI] [Google Scholar]
- Knapp D. M.; Gillis E. P.; Burke M. D. A General Solution for Unstable Boronic Acids: Slow-Release Cross-Coupling from Air-Stable MIDA Boronates. J. Am. Chem. Soc. 2009, 131, 6961–6963. 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemer A. J.; Wiemer D. F. Prodrugs of Phosphonates and Phosphates: Crossing the Membrane Barrier. Top. Curr. Chem. 2015, 360, 115–160. 10.1007/128_2014_561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellin P.; Chang T.; Lim S. G.; Tong M. J.; Ph D.; Sievert W.; Shiffman M. L.; Wulfsohn M. S.; Ph D.; Xiong S.; et al. Adefovir Dipivoxil for the Treatment of Hepatitis B e Antigen–Positive Chronic Hepatitis B. N. Engl. J. Med. 2003, 348, 808. 10.1056/NEJMoa020681. [DOI] [PubMed] [Google Scholar]
- Callebaut C.; Stepan G.; Tian Y.; Miller D. In Vitro Virology Profile of Tenofovir Alafenamide, a Novel Oral Prodrug of Tenofovir with Improved Antiviral Activity Compared to That of Tenofovir Disoproxil Fumarate. Antimicrob. Agents Chemother. 2015, 59, 5909–5916. 10.1128/AAC.01152-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen E. M.; Johnson R. J. Microbial Esterases and Ester Prodrugs: An Unlikely Marriage for Combating Antibiotic Resistance. Drug Dev. Res. 2019, 80, 33–47. 10.1002/ddr.21468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto J.; Westheimer F. H. The Hydrolysis of Mono- and Dibenzyl Phosphates. J. Am. Chem. Soc. 1955, 77, 2515–2518. 10.1021/ja01614a047. [DOI] [Google Scholar]
- Baba T.; Ara T.; Hasegawa M.; Takai Y.; Okumura Y.; Baba M.; Datsenko K. A.; Tomita M.; Wanner B. L.; Mori H. Construction of Escherichia Coli K-12 in-Frame, Single-Gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2006, 2, 0008 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K. A.; Wanner B. L. One-Step Inactivation of Chromosomal Genes in Escherichia Coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper S. B.; Melton R. E.; Mustafa S.; Kieser T.; Jacobs W. R. Jr. Isolation and Characterization of Efficient Plasmid Transformation Mutants of Mycobacterium smegmatis. Mol. Microbiol. 1990, 4, 1911–1919. 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- Burke S. J.; Gamrat J. M.; Santhouse J. R.; Tomares D. T.; Tomsho J. W. Potassium Haloalkyltrifluoroborate Salts: Synthesis, Application, and Reversible Ligand Replacement with MIDA. Tetrahedron Lett. 2015, 56, 5500–5503. 10.1016/j.tetlet.2015.08.012. [DOI] [Google Scholar]
- Zgoda J. R.; Porter J. R. A Convenient Microdilution Method for Screening Natural Products Against Bacteria and Fungi. Pharm. Biol. 2001, 39, 221–225. 10.1076/phbi.39.3.221.5934. [DOI] [Google Scholar]
- Nakagawa K.; Takada K.; Imamura N. Probable Novel MEP Pathway Inhibitor and Its Binding Protein, IspG. Biosci., Biotechnol., Biochem. 2013, 77, 1449–1454. 10.1271/bbb.130094. [DOI] [PubMed] [Google Scholar]
- Defrancesco H.; Dudley J.; Coca A. Boron Chemistry: An Overview. ACS Symp. Ser. 2016, 1236, 1–25. 10.1021/bk-2016-1236.ch001. [DOI] [Google Scholar]
- Zimhony O.; Vilche C.; Jacobs W. R. Characterization of Mycobacterium smegmatis Expressing the Mycobacterium tuberculosis Fatty Acid Synthase I (Fas1) Gene. J. Bacteriol. 2004, 186, 4051–4055. 10.1128/JB.186.13.4051-4055.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi J. S.; Sharma D. Mycobacterium smegmatis and Mycobacterium tuberculosis. Trends Microbiol. 2002, 10, 68–69. 10.1016/S0966-842X(01)02296-X. [DOI] [PubMed] [Google Scholar]
- Ponaire S.; Zinglé C.; Tritsch D.; Grosdemange-Billiard C.; Rohmer M. Growth Inhibition of Mycobacterium smegmatis by Prodrugs of Deoxyxylulose Phosphate Reducto-Isomerase Inhibitors, Promising Anti-Mycobacterial Agents. Eur. J. Med. Chem. 2012, 51, 277–285. 10.1016/j.ejmech.2012.02.031. [DOI] [PubMed] [Google Scholar]
- Jansson A. M.; Wi A.; Bjo C.; Yahiaoui S.; Sooriyaarachchi S.; Lindh M.; Bergfors T.; Dharavath S.; Desroses M.; Suresh S.; et al. DXR Inhibition by Potent Mono- and Disubstituted Fosmidomycin Analogues. J. Med. Chem. 2013, 56, 6190–6199. 10.1021/jm4006498. [DOI] [PubMed] [Google Scholar]
- Dhiman R. K.; Schaeffer M. L.; Bailey A. M.; Testa C. A.; Scherman H.; Crick D. C. 1-Deoxy- D -Xylulose 5-Phosphate Reductoisomerase ( IspC ) from Mycobacterium tuberculosis: Towards Understanding Mycobacterial Resistance to Fosmidomycin. J. Bacteriol. 2005, 187, 8395–8402. 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Barry C. E.; Besra G. S.; Nikaido H. Mycolic Acid Structure Determines the Fluidity of the Mycobacterial Cell Wall. J. Biol. Chem. 1996, 271, 29545–29551. 10.1074/jbc.271.47.29545. [DOI] [PubMed] [Google Scholar]
- Uh E.; Jackson E. R.; San Jose G.; Maddox M.; Lee R. E.; Lee R. E.; Boshoff H. I.; Dowd C. S. Antibacterial and Antitubercular Activity of Fosmidomycin, FR900098, and Their Lipophilic Analogs. Bioorg. Med. Chem. Lett. 2011, 21, 6973–6976. 10.1016/j.bmcl.2011.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George K. W.; Thompson M. G.; Kim J.; Baidoo E. E.; Wang G.; Benites V. T.; Petzold C. J.; Chan L. J. G.; Yilmaz S.; Turhanen P.; et al. Integrated Analysis of Isopentenyl Pyrophosphate (IPP) Toxicity in Isoprenoid-Producing Escherichia Coli. Metab. Eng. 2018, 47, 60–72. 10.1016/j.ymben.2018.03.004. [DOI] [PubMed] [Google Scholar]
- McEwen C. N.; Pagnotti V. S.; Inutan E. D.; Trimpin S. New Paradigm in Ionization: Multiply Charged Ion Formation from a Solid Matrix without a Laser or Voltage. Anal. Chem. 2010, 82, 9164–9168. 10.1021/ac102339y. [DOI] [PubMed] [Google Scholar]
- Pauli G. F.; Chen S.; Simmler C.; Lankin D. C.; Go T.; Jaki B. U.; Friesen J. B.; Mcalpine J. B.; Napolitano J. G. Importance of Purity Evaluation and the Potential of Quantitative 1 H NMR as a Purity Assay. J. Med. Chem. 2014, 57, 9220–9231. 10.1021/jm500734a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brammer L. A.; Smith J. M.; Wades H.; Meyers C. F. 1-Deoxy-D-Xylulose 5-Phosphate Synthase Catalyzes a Novel Random Sequential Mechanism. J. Biol. Chem. 2011, 286, 36522–36531. 10.1074/jbc.M111.259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longstaff C.; Rose M. E. Derivatization and Mass Spectrometric Investigations of Substituted Benzeneboronic Acids. The Use of Linked Scanning During Gas Chromatography Mass Spectrometry. Org. Mass Spectrom. 1982, 17, 508–518. 10.1002/oms.1210171010. [DOI] [Google Scholar]
- Yeh E.; DeRisi J. L. Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium Falciparum. PLoS Biol. 2011, 9, e1001138 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Edwards R. L.; Ball H.; Johnson C.; Haymond A.; Girma M.; Manikkam M.; Brothers R. C.; McKay K. T.; Arnett S. D.; et al. MEPicides: α,β-Unsaturated Fosmidomycin Analogues as DXR Inhibitors against Malaria. J. Med. Chem. 2018, 61, 8847–8858. 10.1021/acs.jmedchem.8b01026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.