

Abstract
Carcinoembryonic antigen-like cell adhesion molecules (CEACAMs) are human cell-surface proteins that can exhibit increased expression on tumor cells, and are thus a potential target for novel tumor-seeking therapeutic delivery methods. We hypothesize that engineered nanoparticles containing a known interaction partner of CEACAM, the Neisseria gonorrhoeae outer membrane protein Opa, can be used to deliver cargo to specific cellular targets. In this study, the cell association and uptake of protein-less liposomes and Opa proteoliposomes into CEACAM-expressing cells was measured using imaging flow cytometry. A size-dependent internalization of liposomes into HeLa cells was observed through endocytic pathways. Opa-dependent, CEACAM1-mediated uptake of liposomes into HeLa cells was observed, with very little colocalization with endosomal and lysosomal trafficking compartments. Given the overexpression of CEACAM1 on several distinct cancers and the strong interest in using CEACAM1 as a component in treatment strategies, these results support further pursuit of investigating Opa-dependent specificity and internalization mechanism for therapeutic delivery.
Keywords: Proteoliposome, nanoparticle, targeted delivery, imaging flow cytometry, CEACAM, Opa protein
Introduction
Carcinoembryonic antigen-like cell adhesion molecules (CEACAMs) are a family of glycoproteins in the immunoglobulin (Ig) superfamily. At least twenty-seven human splice variants exist across twelve unique members; eleven members are found on the cell surface and one member is secreted1. Members of the CEACAM family have wide-ranging tissue expression as well as homo- and heterotypic interactions1. CEACAM1 (formerly Bgp/CD66a) is the most widely distributed CEACAM member and is found on a broad range of epithelial cells, endothelial cells, and immune cells, such as neutrophils and lymphocytes1–2. CEACAM1 exists as 12 splice variants, with most containing a transmembrane domain and up to four extracellular Ig-domains. Long (L) splice variants in humans contain a 71 amino acid intracellular region with an immunoreceptor tyrosine-based inhibitory motif (ITIM), while short (S) splice variants contain only ten intracellular residues and lack phosphorylation sites2. In addition to participating in cell adhesion through homophilic and heterotypic interactions, CEACAM1 can modulate cell proliferation and differentiation2, promote insulin internalization3–4 and neovascularization5, as well as inhibit T cell and Natural Killer cell activity 6–7.
Perhaps due to its role in proliferation and differentiation, CEACAM1 is overexpressed on several types of cancers. Compared to corresponding healthy cells, in which approximately 1–10% of patient samples show CEACAM1 expression, CEACAM1 is significantly overexpressed in melanoma (97% of metastatic tumor cells express CEACAM1), colorectal cancer (94% expression on metastatic cells), lung cancer (81% expression on metastatic cells), pancreatic cancer (71% expression in early carcinoma cells), and bladder cancer (nearly 100% expression on metastatic cells)8. Importantly, CEACAM1 drives progression of both lung adenocarcinomas and melanomas as well as serve as a marker of poor prognosis 8–9. As a result of the significant role of CEACAM1 in driving cancer progression and its overexpression on several carcinomas, CEACAM1 is proposed to be a target for receptor-mediated therapeutic delivery8.
Neisseria meningitidis and Neisseria gonorrhoeae are bacterial pathogens that trigger their engulfment into human cells through binding to CEACAMs. Neisserial Opacity-associated (Opa) proteins bind to several members of the CEACAM family. Opa proteins are eight-stranded beta barrels that span the outer membrane. To date, 345 unique opa alleles have been identified, which generate substantial Opa sequence diversity on the Neisserial surface10. Engagement of the N-terminus of CEACAM1, CEACAM3, CEACAM5, or CEACAM6 with Opa extracellular hypervariable regions (HV1 and HV2) induces bacterial phagocytosis into human host cells11–12. Subsequent to binding, the Opa-CEACAM interaction induces bacterial uptake into non-phagocytic cells, including HeLa cells stably transfected to express CEACAMs13. Given the overexpression of CEACAM1 on several distinct cancers and the strong interest in using CEACAM1 as a component in treatment strategies, the specificity and internalization mechanism of the Opa – CEACAM1 interaction is of interest for therapeutic delivery. Although full-length Opa is likely not the final design of this therapeutic, the requirement of different regions of the protein (HV1 and HV2)11 prevents single HV-derived peptide designs and requires an understanding of the structure and function of the system. The ability to investigate the surface adherence and internalization facilitates these investigations and provides a comparison for future therapeutic designs. For these reasons, the Opa-CEACAM interaction was investigated as a platform for liposomal delivery.
Liposomes can be used to encapsulate hydrophilic and hydrophobic compounds, facilitating their use as vehicles for drug delivery. By incorporating receptor ligands on the liposomal surface, such as peptides14, antibodies15–16, or small molecule ligands17, liposomes can target specific receptors18–19. Receptor binding may subsequently lead to liposome internalization and delivery of encapsulated drugs to the interior of target cells. Recombinantly-expressed Opa proteins can be folded into unilamellar liposomes20. Opa60 proteoliposomes bind the soluble N-terminus of CEACAM1 and CEACAM321, indicating that liposomal Opa proteins retain their interaction with CEACAMs.
In this study, the binding and internalization of Opa-reconstituted liposomes by CEACAM-expressing human cells was investigated using liposome internalization assays and imaging flow cytometry. Using imaging flow cytometry, a method was developed to differentiate surface-bound liposomes from internalized liposomes with high confidence. The high-throughput method described to measure liposome internalization is applicable to studying uptake of other types of particles and in other cellular contexts. This approach was used to investigate the internalization of liposomes and nonspecific uptake of liposomes and Opa60 proteoliposomes into HeLa cells. A size-dependent internalization of unilamellar vesicles (diameters approximately 50–300 nm) into HeLa cells was observed. Minimizing non-specific uptake with 300 nm liposomes facilitated the detection of CEACAM1 mediated uptake of Opa60 liposomes into HeLa cells.
Experimental Methods
Propagation of HeLa cells.
HeLa cells stably transfected to express CEACAM1, CEACAM3 or a control plasmid (generously provided by Scott Gray-Owen, University of Toronto)13 were cultured in a 37°C incubator with 5% CO2 in Dulbecco’s Modified Eagle Media (DMEM) (Gibco, 11965–092) supplemented with 10% fetal bovine serum (VWR, 97068–085), 1x Anti-anti (Gibco, 15240–062), and 1x Glutamax (Gibco, 35050–061). Cells were split using 0.25% trypsin- ethylenediaminetetraacetic acid (EDTA) (Gibco, 25200–056) when ~80% confluent and discarded by 25 passages in order to limit the potential for endogenous CEACAM expression in control cells. HeLa CEACAM expression was monitored through staining of surface CEACAM using a polyclonal CEACAM antibody (Dako, A0115) and imaging flow cytometry.
Staining of HeLa cells for surface CEACAM.
HeLa cells were allowed to grow to ~60% confluence before dissociation with 2 mM EDTA in phosphate buffered saline (PBS). Cells were centrifuged at 300 x g and fixed in 4% paraformaldehyde (PFA) in PBS for 15 minutes before being centrifuged again and washed with PBS containing 10% normal-goat serum (NGS) to block non-specific antibody binding. Antibody staining was done with a rabbit polyclonal pan-CEACAM antibody (Dako, A0115) diluted in PBS-NGS for 1 hour. Following two rounds of washing with 10% NGS in PBS, the cells were stained with an Alexa-647 goat anti-rabbit antibody diluted in PBS-NGS (ThermoFisher, A-21245). Cells were washed with PBS and stored at 4°C for imaging.
Expression and purification of recombinant Opa proteins.
opa60 and opa(HV−) genes for the His6-tagged mature protein were subcloned into pET28b vectors and transformed into a BL21 (DE3) E. coli strain in order to produce Opa proteins as described previously20–21. Opa(HV−) (described in Supplemental Figures) was generated in the pET-28b plasmid by General Biosystems (Morrisville, NC). Briefly, cells were grown in LB supplemented with kanamycin until they reached an OD600 ≈ 0.8, when protein expression was induced with isopropyl-β-D-thiogalactoside. Following Opa expression, which expresses into inclusion bodies, cells were centrifuged and then resuspended in lysis buffer [50 mM Tris (pH 8.0), 150 mM NaCl, Complete protease inhibitor tablet] before being lysed. The insoluble protein fraction was pelleted (5,000 x g) and resuspended in extraction buffer (lysis buffer with 8 M urea) overnight. The remaining insoluble fraction was removed through centrifugation, and soluble Opa proteins were purified using Co2+-immobilized metal affinity chromatography and eluted [20 mM sodium phosphate (pH 7.0), 150 mM NaCl, 680 mM imidazole, 8 M urea]. The eluted fractions containing Opa were concentrated (Molecular Weight Cutoff = 10 kDa) and the final Opa concentration was determined by A280 (MW= (29367.5 Da), ε=41830 M−1 cm−1 for Opa60; MW=22487.8 Da, ε=37360 M−1 cm−1 for Opa(HV−)). Protein purity was assessed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Preparation of fluorescent liposomes and size determination by dynamic light scattering.
A fluorescent lipid mixture composed of 62 mol% 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 16 mol% 1,2-dimyristoyl-sn-glycero-3-phospho(1’-rac-glycerol) (sodium salt) (DMPG), 16 mol% cholesterol, 5 mol% 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethylene glycol)-1000] (ammonium salt) (DMPE PEG 1000), and 1 mol% (DiI) (ThermoFisher, D282) was dried under nitrogen gas and resuspended in 10 mM HEPES in Hank’s Balanced Salt Solution (HBSS, components purchased from Sigma-Aldrich). The resulting lipid mixture was vortexed for 5 minutes and shaken at 500 rpm overnight before being hand-extruded through a Nucleopore track-etched membrane with pore sizes of 0.03 µm, 0.1 µm, 0.2 µm, or 0.4 µm (Whatman). Liposome sizes after extrusion were determined by dynamic light scattering (DLS) using a Wyatt DynaPro Plate Reader II and Dynamics V7 software (Supplemental Table S1 and Fig. S1). Ten repeats were measured for each condition and the average liposome size was reported along with the polydispersity.
Preparation of fluorescent Opa proteoliposomes.
Opa protein folding was adapted from previously published protocols20–21. 1,2-didecanoyl-sn-glycero-3-phosphocholine (diC10PC, Avanti Polar Lipids) dissolved in chloroform was dried under nitrogen gas and resuspended in borate buffer [10 mM sodium borate (pH 12.0) and 1 mM EDTA], then sonicated for 30 minutes at 40% amplitude (Q500, Q Sonica) in order to form liposomes. Following sonication, 4 M urea was added and 50 nm unfolded Opa60 or Opa(HV−) was aliquoted and mixed. The Opa/diC10PC-liposome mixture was incubated for 4 days at 37°C, after which folding was confirmed by SDS-PAGE (Fig S5). Following Opa folding, diC10PC-proteoliposomes were pelleted through ultracentrifugation (142,400 x g for 2 hrs at 12°C), resuspended in resuspension buffer [10 mM HEPES (pH 7.4) in HBSS], and mixed with dried fluorescent lipids (DMPC, DMPG, cholesterol, DMPE-PEG-1000) as described above. The lipid mixture was vortexed for 5 minutes and shaken at 500 rpm for several hours before extrusion through a Nucleopore track-etched membrane with a 0.4 or 0.2 µm pore size (Whatman). Opa60 reconstituted into liposomes are referred to as Opa60 proteoliposomes.
Proteoliposome incubation with pre-fixed HeLa cells.
Prior to Opa60 proteoliposome exposure, 2.0 × 106 CEACAM1 HeLa cells per condition were dissociated and fixed with 4% paraformaldehyde in PBS for 10 minutes. Cells were centrifuged (300 x g, 10 min) and washed with PBS and then exposed to Opa60 proteoliposomes in 10 mM HEPES/HBSS (pH 7.4) at a final phospholipid concentration of 0.2 mM for two hours. Cells were then centrifuged and washed with PBS before being fixed with PFA as described above. Cells were washed in PBS and then incubated with 1:1000 DAPI in PBS for one hour before being washed once more and stored in PBS at 4°C for imaging.
Timecourse of Opa proteoliposome uptake.
Approximately 20 hours prior to liposome exposure, 2.0 × 106 HeLa cells expressing CEACAM1, CEACAM3, or the vector control line were seeded onto 60 × 15 mm cell culture plates (Cellstar, 628160). Proteoliposomes were produced as described above and extruded through a 0.4 µm membrane. Before the experiment, the liposomal phospholipid concentration was determined by a colorimetric assay according to established protocols22. HeLa cells were exposed to liposomes at a concentration of 0.2 mM total phospholipid for 20 minutes at 37°C in serum-free DMEM. Following liposome exposure, cells were washed and allowed to incubate further for 0, 1, 2, or 3 hrs before being washed and lifted by 2mM EDTA in PBS (pH 7.4). Cells were pelleted by centrifugation at 300 x g for 10 minutes and then fixed in 4% PFA in PBS for 15 minutes before being pelleted at 400 x g for 10 minutes. The cell pellet was resuspended in PBS and stained with 1:1000 DAPI in PBS for one hour before being centrifuged and washed with PBS. Cells were stored at 4°C prior to imaging.
Inhibition of cells with inhibitors.
2.0 × 106 HeLa cells per plate were seeded the day before the experiment. Opa proteoliposomes were prepared as described previously. The day of the experiment, cells were pre-incubated for 30 minutes at 37°C with DMEM and 10 mM sodium azide and 100 mM 2-deoxyglucose (Sigma) or with staurosporine (Sigma) ranging from 0 to 400 nM. Opa proteoliposomes were given at a concentration of 0.1 mM total phospholipids for 1 hour at 37°C, after which the cells were washed, lifted, and fixed as described previously. Cells were stained with 1:1000 DAPI in PBS before centrifugation at 400 g. Cells were washed with PBS and stored at 4°C prior to imaging.
Imaging flow cytometry.
Cell imaging was performed on an ImageStreamX Mark II imaging flow cytometer (Amnis Corporation). DAPI fluorescence was excited with a 405 nm laser set to 40.0 mW intensity and emission was collected with a 420–505 nm filter (Ch 7). TMR-dextran fluorescence was excited with a 488 nm laser set to 100.0 mW intensity and emission was collected with a 595–660 nm filter (Ch 4). DiI fluorescence was excited with a 488 nm laser set to 100.0 mW intensity and a 561 laser set to 100.0 mW intensity and read using a 560–595 nm filter (Ch 3). Alexa-647 fluorescence was excited using a 642 nm laser set to 40.0 mW intensity and collected with a 660–740 nm filter (Ch 11). Brightfield images were collected on Ch 1 (camera 1) and Ch 9 (camera 2). Images were captured using a 60X, 0.9 NA objective. Approximately 4000–8000 in-focus, nucleated cells were captured for each sample. Single-label controls were imaged using the same settings to generate a compensation matrix.
Image processing.
Images were analyzed using IDEAS V. 6.2.64.0 software (Amnis Corporation). For each file, a compensation matrix created using single-label controls was applied to reduce spectral overlap between channels. In-focus cells were selected using a Brightfield RMS gradient cutoff above 55, while single cells were gated on by plotting the brightfield area against the aspect ratio. An internalization mask was created by an Adaptive Erode algorithm (100–75%, with 75% Adaptive Erode chosen in experiments to define the internal mask) applied to a brightfield mask in order to exclude fluorescence at the membrane. A surface mask was designed by subtracting a 90% Adaptive Erode mask from the full brightfield mask in order to capture fluorescence only at the cell surface. For DiI fluorescence measurements, a mask was applied to each cell to select for Ch3 (DiI) fluorescence intensity between 100–4095 greyscale value in order to exclude low-level background fluorescence (background threshold). An internalization or cell surface mask was combined with a DiI background threshold mask in order to quantify above-background DiI fluorescence either within or at the surface of the cell. For E. coli, a spot count algorithm was used instead of intensity in order to quantify the average number of spots (bacteria) per cell. To measure internalized TMR-dextran fluorescence, a background threshold mask was created to exclude Ch4 TMR intensity outside a 70–4095 greyscale value range. Ch4 (TMR) fluorescence intensity was quantified within the TMR background threshold mask and the Internalization mask.
Results and Discussion
Development of an imaging flow cytometric method to measure liposome internalization.
Distinguishing surface-bound from internalized particles in cells is an important but non-trivial task with numerous publications describing a variety of methods. Surface washes23, non-cell penetrating fluorophores24, and low-temperature incubations25–26 have all been used, among other techniques, in an attempt to differentiate between surface and internal fluorescent signals. Often, confocal fluorescence microscopy is used to analyze particle localization with respect to brightfield images27, internal stains23, or membrane stains28, with one proposed method combining confocal fluorescence imaging of cells with flow cytometry in an attempt to generate a high-throughput method for determining nanoparticle internalization29. Recently, imaging flow cytometry, which combines fluorescence microscopy with flow cytometry, has gained attention as a technique enabling the high-throughput evaluation of thousands of microscopic images when determining internalization and surface binding30 of exogenous particles such as bacteria24, 31 or exosomes32.
In order to distinguish membrane-associated (also referred to as surface) from internalized liposomes, a masking method was developed using the ImageStreamX Mark II Imaging Flow Cytometer and IDEAS image processing software. Brightfield gradient RMS, DAPI intensity, cell area, and cell aspect ratio were used to select images of cells that were in focus, singlets, and nucleated (Fig. S2). Fluorescently-labelled liposomes, fluorescently-labelled CEACAM antibody and fluorescently-labelled antibodies to endosomal proteins were individually evaluated using brightfield masks at varying Adaptive Erode values (95–75%) of the full brightfield mask (100%). Internal and surface fluorescence was measured for fluorescent Opa60 proteoliposomes in pre-fixed CEACAM1 HeLa cells (Fig. 1A). Fixation prior to liposome exposure prevents liposome internalization33; thus, any liposome fluorescence is exclusively membrane-associated, and accurate internal masks should measure low internal liposomal fluorescence in pre-fixed cells. Surface masks were defined as full brightfield mask minus the internal mask for each Adaptive Erode percent value, and are expected to report high fluorescence values for non-internalized liposomes on pre-fixed cells. When defining internal fluorescence as all fluorescence within the full (100%) brightfield mask, all liposomal fluorescence for pre-fixed cells is incorrectly identified as internalized. The percent of internal fluorescence decreases and percent surface fluorescence increases as the internal mask defined as a percent of the full brightfield mask is reduced from 95% to 75% (Fig. 1B). An internal mask defined as 75% of the full brightfield mask after Adaptive Erode reported nearly all fluorescence on pre-fixed cells as surface-associated, with only 6.2% of liposomal fluorescence measured as internal. Similar results were obtained when the same masking strategy was applied to non-permeabilized CEACAM1 HeLa cells stained with an anti-CCM antibody to mark the cell membrane (Fig. S3). These findings support the use of an Adaptive Erode 75% mask to delineate the internal and surface areas for determining particle internalization.
Figure 1.

Fluorescence values of surface versus internalized proteoliposomes determined in pre-fixed CEACAM1 HeLa cells. CEACAM1 HeLa cells were dissociated and fixed with 4% PFA prior to incubation with Opa60 proteoliposomes (yellow). (A) Surface and internalization masks were used to determine adhered versus internalized liposome fluorescence. Internalization masks were defined as being a set percent of the full brightfield mask (75–95%) while surface masks were defined as being fluorescence outside of each internalization mask. White scale bar is set to 7 μm. (B) When used to quantify internal and surface proteoliposome fluorescence in pre-fixed cells, which are not expected to internalize liposomes following binding, high surface and low internal (6.2% of total) liposome fluorescence was measured with an Adaptive Erode mask set to 75% of full brightfield. Error bars represent 95% C.I.
Additional refinement of the masking strategy was accomplished by staining intracellular markers, such as Early Endosomal Antigen 1 (EEA1), and comparing whether this intracellular fluorescence appears in surface masks (Fig. 2). When the cell surface mask is defined as the cell area outside the 90% Adaptive Erode mask, low EEA1 fluorescence was measured within the surface mask consistent with the expected intracellular staining of EEA1 within early endosomes. However, EEA1 localization was inaccurately quantified as within the surface mask when less restrictive surface masks covering a larger cell area were used, such as when the surface was defined as the cell area outside the 75% Adaptive Erode mask (Fig. 2). Therefore, in all subsequent experiments, cell surface fluorescence is defined as all fluorescence within the full brightfield mask that is not also within the 90% Adaptive Erode mask, and internal fluorescence was defined as fluorescence only within the 75% Adaptive Erode mask. Using these two definitions results in a zone within each cell between the edge of 75% and 90% Adaptive Erode in which fluorescence cannot be completely categorized as internal or surface fluorescence as both surface and intracellular fluorescence was observed within this area. Fluorescence from surface-particles appearing in this zone, which can appear by eye as internalized, may be due to uneven ruffling at the cell membrane, as has been observed for extracellular bacteria24. Fluorescence intensity within this zone is therefore not used when calculating fluorescence specific to the internal or surface compartment.
Figure 2.

Fluorescence values of anti-Early Endosomal Antigen-1 (EEA1) staining in CEACAM1 HeLa cells as determined using internalization adaptive erode masks. (A) CEACAM1 HeLa cells were dissociated, fixed with 4% PFA, and stained with an αEEA1 antibody (green). Internalization masks were defined as being a set percent of the full brightfield mask (75–95%) while surface masks were defined as being fluorescence outside of a set internalization mask. Masks were used to determine adhered versus internalized fluorescence. White scale bar is set to 7 μm. (B) When varying internalization and surface masks are used to quantify EEA1 fluorescence, which should only be within cells, fluorescence was considered internalized and not surface as long as surface masks were set to quantify fluorescence outside of at least the 90% Adaptive Erode internal mask. Error bars represent 95% C.I.
Opa-independent HeLa cell uptake of different size liposomes.
HeLa cells are generally reported to be non-phagocytic and often do not internalize large particles unless promoted by particle binding to cell surface proteins34–36. There are, however, several reports of liposomes without receptor-specific targeting gaining entry into HeLa cells37–39. Many endocytic processes are limited based on the size of the cargo entering the cell40; therefore, to limit non-specific liposome uptake, optimization of liposome size was investigated in order to determine how liposome size affects non-specific uptake of liposomes into CEACAM1, CEACAM3, and control HeLa cells. CEACAM expression across cell lines was monitored by imaging flow cytometry and the presence of CEACAM on the surface of HeLa cells transfected to express CEACAM1 or CEACAM3 was visualized and quantified (Fig 3).
Figure 3.

Surface CEACAM expression on HeLa cells. (A) Control (black), CEACAM1 (red), and CEACAM3 (blue) HeLa cells were dissociated, fixed in 4% PFA, and stained with a pan-CEACAM antibody. Compared to control cells, histograms of fluorescence intensity show high fluorescence in CEACAM1 and CEACAM3 cells, with highest staining on CEACAM1 cells. (B) Merged images of pan-CEACAM antibody (red) staining and brightfield images of control, CEACAM1, and CEACAM3 cells show CEACAM localization at the cell surface.
To investigate non-specific liposome uptake, CEACAM1, CEACAM3, and control cells were incubated with non-proteinaceous liposomes between 50 and 300 nm diameter. Internal fluorescence of non-proteinaceous liposomes was observed within cells, with an inverse correlation observed between liposome size and non-specific liposome internalization into cells (Fig. 4). This non-specific, size-dependent internalization of liposomes likely results from cellular processes that are CEACAM-independent, due to the absence of Opa proteins on the liposomes. Uptake pathways that may be capable of internalizing liposomes without Opa/CEACAM interactions include clathrin-mediated endocytosis (CME)41, clathrin-independent carriers (CLIC)42, and macropinocytosis43–44. There is evidence macropinocytosis is occurring in these cells as fluorescent dextran, a fluid-phase marker of macropinocytosis45, was internalized into control and CEACAM expressing HeLa cells (Fig S4). CEACAM1 expressing cells had higher dextran internalization than control or CEACAM3 cells, implying CEACAM1 may promote macropinocytic processes as has been shown for other cell surface adhesion molecules, such as cadherins46. Uptake of fluorescent dextran into HeLa cells was decreased by treatment with the macropinocytic inhibitor ethylisopropyl amiloride (EIPA)47 in control and CEACAM1 HeLa cells, but not CEACAM3 (Fig S4). These results demonstrate the importance of correlating the size of nanoparticles with their propensity for non-specific uptake into cells in targeted internalization experiments and that overexpression of surface adhesion proteins can impact internalization processes. Because high levels of non-specific internalization could obscure CEACAM-mediated uptake, Opa proteoliposomes in all subsequent experiments are with 300 nm, the size with the lowest non-specific internalization.
Figure 4.
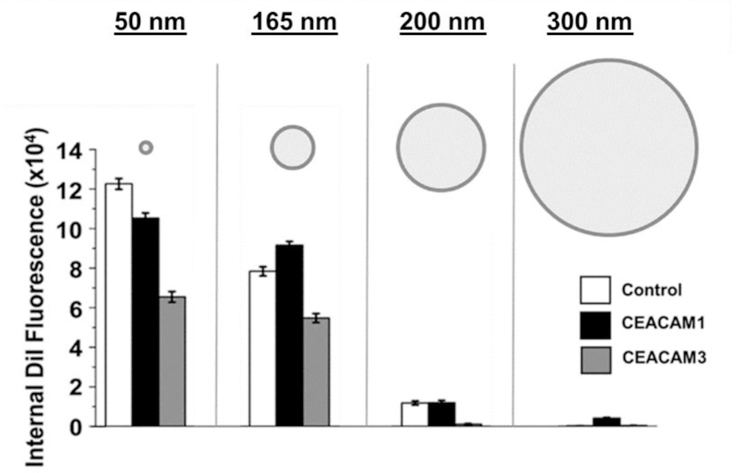
Opaless liposome size inversely correlates with Opa-independent uptake. Control (white), CEACAM1 (black), and CEACAM3 (grey) HeLa cells were incubated with 0.2 mM [phospholipid] of non-proteinaceous liposomes that ranged in diameter from approximately 30 to 400 nm. The highest Opa-independent uptake occurred in cells exposed to 30 nm liposomes, with liposome uptake decreasing as liposome size increases. Error bars represent 95% C.I.
CEACAM-mediated uptake of Opa60 proteoliposome into HeLa cells.
Opa-expressing Neisseria bacteria induce phagocytosis of the bacteria into HeLa cells displaying CEACAM113. Proteoliposomes containing refolded recombinant Opa60 bind the soluble N-terminus of CEACAM1 and CEACAM3 in vitro with approximately 50% of Opa proteins facing outward and 50% facing into the liposomes as determined by trypsin cleavage assays21. Opa(HV−) has the hypervariable regions required to engage CEACAM11–12 removed (Fig. S5) and are internalized by non-CEACAM mediated internalization as was demonstrated for liposomes. Opa(HV−) proteoliposomes may better mimic the physical properties of Opa60 proteoliposomes such as membrane rigidity compared to liposomes without protein48.
To determine the contribution of the Opa-CEACAM interaction to proteoliposomal uptake, proteoliposomes containing folded (Fig. S6) Opa60 or Opa(HV−) protein were exposed for fifteen minutes to control HeLa cells and HeLa cells expressing CEACAM1 or CEACAM3. After washing, the cells were imaged immediately (0 hrs) and at 1, 2, and 3-hour incubation time to monitor liposomes uptake (representative images are provided in Fig. S7). The amount of internal and surface DiI fluorescence was quantified (Fig. S8) using Adaptive Erode masks (Fig. 2 and 3 and S3). In order to capture binding and internalization into one measurement, the ratio of internal fluorescence to surface fluorescence was used to evaluate uptake efficiency at each time point (Fig. 5). A ratio greater than one indicates there are more internalized liposomes than surface bound liposomes, and a ratio less than one indicates there are more liposomes on the surface of the cell than exist internalized.
Figure 5.

Opa60 promotes proteoliposome uptake into HeLa cells. HeLa cells were pulsed for 15 min with proteoliposomes, then chased in fresh medium without liposomes for 0, 1, 2, or 3 hrs. Liposome internalization efficiency (internal/surface fluorescence) was determined for control (A), CEACAM1 (B), and CEACAM3 (C) HeLa cells following exposure to Opa60 (black) and Opa(HV−) (red) proteoliposomes. Opa60 enhances proteoliposome uptake into HeLa cells compared to Opa(HV−), shown as higher Internal/Surface values. The increase or decrease in proteoliposome internalization efficiency from Opa(HV−) to Opa60 is shown as a percent of baseline Opa(HV−) proteoliposome efficiency on each graph. These values were plotted in (D), showing that as time increases, efficiency of Opa60 internalization when compared to Opa(HV−) internalization increases only within the context of CEACAM1 HeLa cells. Error bars represent 95% C.I.
As anticipated from the Opa-independent uptake of liposomes presented in Figure 4, CEACAM-independent binding and internalization of Opa(HV−) proteoliposomes was observed with control HeLa cells (Fig. S8 and S9). More total fluorescence was observed for Opa60 compared to Opa(HV−) (Fig. S9) indicating the amount of endogenous CEACAM1 expressed by the control cells (Fig. 3A) was sufficient to promote uptake. Approximately 50% of Opa60 proteoliposome internalization into control cells was not inhibited by treatment with the metabolic inhibitors sodium azide and 2-deoxyglucose (Fig. 6), however, suggesting non-active uptake processes contribute to liposome uptake into control HeLa cells.
Figure 6.
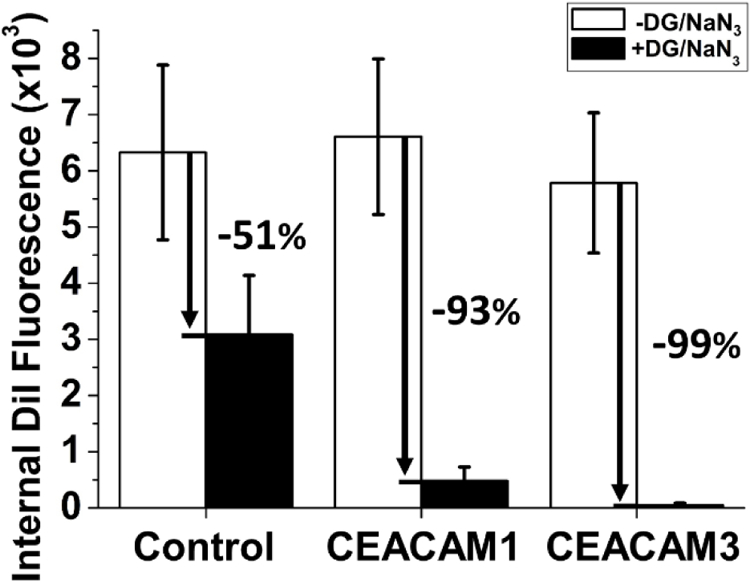
Cellular metabolic energy promotes internalization of Opa60 proteoliposomes into HeLa cells. Control, CEACAM1, and CEACAM3 HeLa cells were treated with metabolic inhibitors (black) in order to deplete available cellular ATP compared to untreated cells (white). Following treatment, cells were exposed to 0.2 mM [phospholipid] Opa60 proteoliposomes for one hour before being washed, lifted, and fixed in 4% PFA. Depletion of cellular ATP inhibits internalization of Opa60 proteoliposomes into cells, with ATP depletion strongly inhibiting liposome uptake into HeLa cells expressing CEACAM1 or CEACAM3. A smaller effect was seen on internalization into control cells, which could be due to non-active uptake processes present in these cells. Error bars represent 95% C.I.
In CEACAM1 expressing cells, immediately after liposome exposure (0 h) Opa60 proteoliposomes have a higher surface fluorescence than Opa(HV−) proteoliposomes (Fig. S8), suggesting higher initial binding of Opa60 proteoliposomes to CEACAM1 cells. In addition, Opa60 proteoliposomes already demonstrate a higher internal fluorescence than Opa(HV−) proteoliposomes immediately after liposome exposure, indicating internalization for Opa60 proteoliposomes can be rapid. Higher internal and surface fluorescence of Opa60 proteoliposomes compared to Opa(HV−) proteoliposomes results in a higher total fluorescence for Opa60 proteoliposomes than Opa(HV−) proteoliposomes with CEACAM1 cells (Fig. S9). As time extends beyond the initial liposome exposure, proteoliposomes on the cell surface decrease and internalized proteoliposome fluorescence increases up to 2 hours following exposure. Starting at 2 hours, the surface fluorescence of Opa60 proteoliposomes is lower than Opa(HV−) surface fluorescence (Fig. S8), suggesting Opa60 proteoliposomes are internalized faster than Opa(HV−). Over the time investigated, the internal fluorescence is higher for Opa60 proteoliposomes in CEACAM1 cells compared to Opa(HV−) and correlates with the decrease in fluorescence observed on the cell surface (Fig. S8). The ratio of internal to surface captures these trends and indicates Opa60 proteoliposomes are more efficiently internalized than Opa(HV−) (Fig 5B). The internalization of Opa60 proteoliposomes was reduced by 93% with the metabolic inhibitors sodium azide and 2-deoxyglucose (Fig. 6), suggesting that unlike in control cells, Opa60 proteoliposome internalization into CEACAM1 cells is almost entirely dependent on active uptake processes that require ATP.
In order to probe further the dependence of CEACAM on liposomal uptake, the correlation between CEACAM expression levels and Opa60 proteoliposome internalization was investigated. A positive correlation was observed between surface CEACAM1 expression levels on CEACAM1+ cells and Opa60 proteoliposome uptake after one hour (Fig 7A), suggesting that CEACAM1+ HeLa cells with higher CEACAM1 expression levels have more internalized proteoliposomes than CEACAM1+ cells with less CEACAM1 expression. Previously published experiments using Neisseria bacteria have demonstrated that staurosporine, a broad-spectrum serine kinase inhibitor, decreases uptake of Opa-expressing bacteria into human cells through CEACAM1 without affecting bacterial surface adhesion13. To determine the effect of staurosporine treatment on CEACAM-dependent proteoliposomal uptake, CEACAM1 cells were pre-treated with increasing concentrations of staurosporine before proteoliposome exposure. Similar to results using Opa-expressing Neisseria, pre-treatment of CEACAM1 HeLa cells with increasing concentrations of staurosporine inhibits Opa60 proteoliposomal uptake while leaving surface binding of proteoliposomes unaffected (Fig. 7B). The combined results suggest that Opa60 liposomes are internalized through a CEACAM1-dependent uptake pathway.
Figure 7.
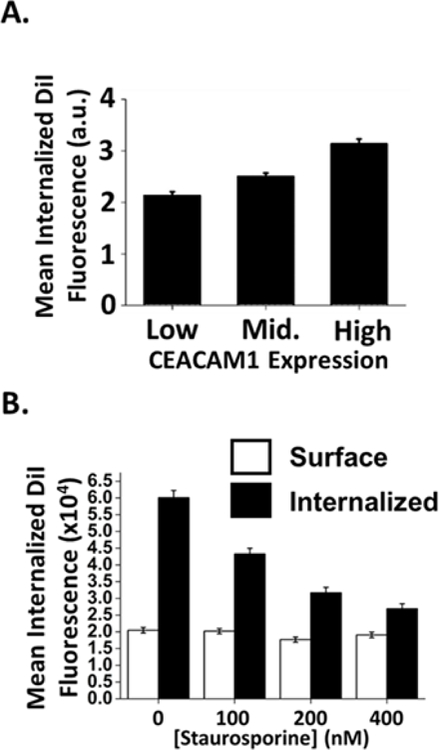
Opa60 proteoliposome uptake correlates with increased CEACAM expression on cell surface and is inhibited by treatment with staurosporine. (A) CEACAM1 cells were gated in to lowest 25%, middle 50%, and highest 25% expressing cells after staining with a pan-CEACAM antibody and the proteoliposome fluorescence for each sub-population was measured. A positive correlation was seen between CEACAM expression and Opa60 proteoliposome uptake. Error bars represent 95% C.I. (B) Treatment of CEACAM1 HeLa cells with the serine kinase inhibitor staurosporine inhibits Opa60 proteoliposome internalization into cells. Binding of proteoliposomes to the cell surface was unaffected by treatment. Error bars represent 95% C.I.
In CEACAM3 expressing HeLa cells, both the surface and internal fluorescence of Opa60 proteoliposomes was higher than for Opa(HV−) proteoliposomes (Fig S8). As time extends (1 to 3 hrs) beyond the initial liposome exposure to cells (0 hrs), the internal fluorescence of Opa60 liposomes increases while surface fluorescence decreases (Fig. S8). Over the time investigated, Opa60 proteoliposomes were more efficiently internalized than Opa(HV−) proteoliposomes (Fig. 5C). Similar to CEACAM1 cells, the internalization of Opa60 proteoliposomes into CEACAM3 cells was almost entirely reduced when cells were treated with metabolic inhibitors (Fig. 6).
Based on the combined results, the different HeLa cells are difficult to compare. The cells differ in CEACAM expression levels and different internalization pathways. The difference in the ratio of internal to surface fluorescence between Opa60 and Opa(HV−) allows a comparison of uptake efficiency between the different cells that is independent of CEACAM expression levels (Fig. 5D). Over the entire time investigated, Opa60 proteoliposomes show greater uptake efficiency than Opa(HV−) proteoliposomes; however, the difference between the Opa60 and Opa(HV−) proteoliposomes over time was different between the three types of HeLa cells. The difference in uptake efficiency between Opa60 and Opa(HV−) proteoliposomes decreases over time in CEACAM3-expressing cells, while CEACAM1 expressing cells were the only cell line for which the uptake efficiency of Opa60 proteoliposomes increases over time compared to Opa(HV−) (Fig. 5D). The differences in uptake observed between CEACAM1 and CEACAM3 could be due to different signal transduction processes that initiate internalization, such as the use of an immunoreceptor tyrosine-based inhibition motif (ITIM) on the intracellular region of CEACAM1, and an immunoreceptor tyrosine-based activation motif (ITAM) on CEACAM3, which recruits Src kinases and Rac1 prior to bacterial internalization13. It is interesting to note that N. gonorrhoeae bacteria expressing Opa57 had similar trends in the percent of intracellular bacteria49 with CEACAM1 and CEACAM3 expressing HeLa cells. In that study, the percent of bacteria internalized in CEACAM1 expressing HeLa cells increased over 3 hours, while CEACAM3 expressing HeLa cells had a maximum uptake by 1–2 hours. In experiments performed using E. coli expressing OpaI (Opa60 expressed recombinantly in E. coli), a similar internalization was measured at the one-hour time point for CEACAM1 and CEACAM3 expressing HeLa cells (Fig. S10), as determined using a spot count algorithm.
Cellular-fate of Opa60 proteoliposomes.
To begin to evaluate the cellular fate of internalized Opa60 proteoliposomes, cells were fluorescently stained with markers for early endosomes and lysosomes. Early in the endocytic process, primary endocytic vesicles fuse with low pH (~5.5) early endosomes using Rab5, a small GTP binding protein that also recruits the effector protein early endosomal antigen 1 (EEA1) to early endosomes50–52. Because of the specific localization, EEA1 is used as an early endosome marker. Following early endosomes, cargo may proceed to late endosomes and eventually lysosomes. Lysosomes are low pH (<5.0) degradative vesicles containing numerous hydrolytic enzymes, and the trafficking of internalized particles to degradative lysosomes has significant implications for cargo survival53–54. The major membrane protein constituents of lysosomes, lysosome-associated membrane protein (LAMP)-1 and LAMP2, help maintain lysosomal integrity, and LAMP1 is a common lysosomal marker, although it may also be found on the cell surface53, 55. In order to explore Opa60 proteoliposomal trafficking through the endocytic pathway, the colocalization of liposomal fluorescence with antibodies to EEA1 or LAMP1 was determined over the course of one hour following exposure to cells (Fig. S11). At 60 minutes following initial proteoliposome exposure, colocalization between DiI and LAMP1 is highest while colocalization between DiI and EEA1 is lowest. Since cargo processed through endocytic pathways encounters early endosomes before lysosomes, this may indicate a small fraction of proteoliposomes are processed through the endocytic pathway and have trafficked out of early endosomes toward lysosomes by this timepoint. Nonetheless, average bright detail similarity scores less than 2.0 indicate very little DiI fluorescence colocalizes with either EEA1 or LAMP1, suggesting that the majority of proteoliposomes do not colocalize with either of these markers over the time surveyed. Trafficking through endosomes to lysosomes is associated with cargo degradation, and a lack of significant colocalization between proteoliposomes and endocytic markers suggests that Opa proteoliposomes and associated cargo may be able to access the cell cytosol following cell internalization.
Conclusion
Here, we report a method to differentiate internalized from cell surface-adhered liposomes in HeLa cells using imaging flow cytometry. This method is high-throughput, allowing for the quantification of signals from thousands of cells, and avoids human subjectivity in defining particle internalization and selection of cells. The approach presented here for measuring nanoparticle internalization into cells can be applied to the cellular uptake of other nanoparticles across many cellular contexts. In this study, non-specific liposome internalization was quantified in HeLa cells and is inversely correlated with liposome size. Non-specific liposome internalization into cells increases with smaller particles, underscoring the importance of considering nanoparticle size in these kinds of targeted uptake experiments.
In addition to the non-specific uptake observed, ATP-dependent CEACAM-mediated uptake of Opa60 proteoliposomes was observed in CEACAM1 and CEACAM3 expressing HeLa cells. CEACAM1 is overexpressed on several cancers and, therefore, has the potential to serve as a target receptor in treatment strategies. Targeting CEACAM with Opa requires both HV1 and HV2 regions, which are in different extracellular loops of the protein. Therefore, a single Opa-derived peptide will not be sufficient to accomplish CEACAM mediated uptake and full length Opa proteins currently remain our best tool to understand how to target CEACAM. In order to design a CEACAM-targeting liposome that avoids the use of a full length bacterial protein, the Opa-CEACAM interaction needs to be determined and assays of internalization are needed to correlate the molecular determinants of the interaction and cellular uptake. These combined results support further investigation of Opa proteoliposome cell entry as models for therapeutic delivery mechanisms.
Supplementary Material
Acknowledgements
This research was supported by National Institutes of Health (NIH) Grants R01 GM087828 (LC) and R01 AI097312 (AKC), a Cottrell Scholar Award, RCSA (LC), and the nanoSTAR Institute at the University of Virginia (LC and AKC)). We acknowledge TCS Keller for feedback on the manuscript. Additionally, we acknowledge Joanne Lannigan and the staff at the University of Virginia’s Flow Cytometry Facility for helpful advice and discussions.
Footnotes
Declarations of Interest: None
References
- 1.Sadarangani M; Pollard AJ; Gray-Owen SD, Opa proteins and CEACAMs: pathways of immune engagement for pathogenic Neisseria. FEMS Microbiol Rev 2011, 35 (3), 498–514. [DOI] [PubMed] [Google Scholar]
- 2.Kuespert K; Pils S; Hauck CR, CEACAMs: their role in physiology and pathophysiology. Curr Opin Cell Biol 2006, 18 (5), 565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poy MN; Ruch RJ; Fernström MA; Okabayashi Y; Najjar SM, Shc and CEACAM1 interact to regulate the mitogenic action of insulin. Journal of Biological Chemistry 2002, 277 (2), 1076–1084. [DOI] [PubMed] [Google Scholar]
- 4.Poy MN; Yang Y; Rezaei K; Fernström MA; Lee AD; Kido Y; Erickson SK; Najjar SM, CEACAM1 regulates insulin clearance in liver. Nature genetics 2002, 30 (3), 270. [DOI] [PubMed] [Google Scholar]
- 5.Rueckschloss U; Kuerten S; Ergün S, The role of CEA-related cell adhesion molecule-1 (CEACAM1) in vascular homeostasis. Histochemistry and cell biology 2016, 146 (6), 657–671. [DOI] [PubMed] [Google Scholar]
- 6.Morales VM; Christ A; Watt SM; Kim HS; Johnson KW; Utku N; Texieira AM; Mizoguchi A; Mizoguchi E; Russell GJ, Regulation of human intestinal intraepithelial lymphocyte cytolytic function by biliary glycoprotein (CD66a). The Journal of Immunology 1999, 163 (3), 1363–1370. [PubMed] [Google Scholar]
- 7.Markel G; Lieberman N; Katz G; Arnon TI; Lotem M; Drize O; Blumberg RS; Bar-Haim E; Mader R; Eisenbach L, CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. The Journal of Immunology 2002, 168 (6), 2803–2810. [DOI] [PubMed] [Google Scholar]
- 8.Dankner M; Gray-Owen SD; Huang Y-H; Blumberg RS; Beauchemin N, CEACAM1 as a multi-purpose target for cancer immunotherapy. OncoImmunology 2017, 6 (7), e1328336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thies A; Moll I; Berger J; Wagener C; Brümmer J; Schulze H-J; Brunner G; Schumacher U, CEACAM1 Expression in Cutaneous Malignant Melanoma Predicts the Development of Metastatic Disease. Journal of Clinical Oncology 2002, 20 (10), 2530–2536. [DOI] [PubMed] [Google Scholar]
- 10.Bilek N; Ison CA; Spratt BG, Relative Contributions of Recombination and Mutation to the Diversification of the opa Gene Repertoire of Neisseria gonorrhoeae. Journal of Bacteriology 2009, 191 (6), 1878–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bos MP; Kao D; Hogan DM; Grant CC; Belland RJ, Carcinoembryonic antigen family receptor recognition by gonococcal Opa proteins requires distinct combinations of hypervariable Opa protein domains. Infect Immun 2002, 70 (4), 1715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hauck CR; Meyer TF, ‘Small’ talk: Opa proteins as mediators of Neisseria-host-cell communication. Curr Opin Microbiol 2003, 6 (1), 43–9. [DOI] [PubMed] [Google Scholar]
- 13.McCaw SE; Liao EH; Gray-Owen SD, Engulfment of Neisseria gonorrhoeae: Revealing Distinct Processes of Bacterial Entry by Individual Carcinoembryonic Antigen-Related Cellular Adhesion Molecule Family Receptors. Infection and Immunity 2004, 72 (5), 2742–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mei L; Fu L; Shi K; Zhang Q; Liu Y; Tang J; Gao H; Zhang Z; He Q, Increased tumor targeted delivery using a multistage liposome system functionalized with RGD, TAT and cleavable PEG. Int J Pharm 2014, 468 (1–2), 26–38. [DOI] [PubMed] [Google Scholar]
- 15.Kirpotin D; Park JW; Hong K; Zalipsky S; Li W-L; Carter P; Benz CC; Papahadjopoulos D, Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry 1997, 36 (1), 66–75. [DOI] [PubMed] [Google Scholar]
- 16.Scindia Y; Deshmukh U; Thimmalapura PR; Bagavant H, Anti-alpha8 integrin immunoliposomes in glomeruli of lupus-susceptible mice: a novel system for delivery of therapeutic agents to the renal glomerulus in systemic lupus erythematosus. Arthritis Rheum 2008, 58 (12), 3884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shmeeda H; Mak L; Tzemach D; Astrahan P; Tarshish M; Gabizon A, Intracellular uptake and intracavitary targeting of folate-conjugated liposomes in a mouse lymphoma model with up-regulated folate receptors. Mol Cancer Ther 2006, 5 (4), 818–24. [DOI] [PubMed] [Google Scholar]
- 18.Park JW; Hong K; Kirpotin DB; Colbern G; Shalaby R; Baselga J; Shao Y; Nielsen UB; Marks JD; Moore D; Papahadjopoulos D; Benz CC, Anti-HER2 Immunoliposomes. Clinical Cancer Research 2002, 8 (4), 1172. [PubMed] [Google Scholar]
- 19.Cagle PT; Zhai QJ; Murphy L; Low PS, Folate Receptor in Adenocarcinoma and Squamous Cell Carcinoma of the Lung: Potential Target for Folate-Linked Therapeutic Agents. Archives of Pathology & Laboratory Medicine 2012, 137 (2), 241–244. [DOI] [PubMed] [Google Scholar]
- 20.Dewald AH; Hodges JC; Columbus L, Physical determinants of beta-barrel membrane protein folding in lipid vesicles. Biophys J 2011, 100 (9), 2131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin JN; Ball LM; Solomon TL; Dewald AH; Criss AK; Columbus L, Neisserial Opa Protein–CEACAM Interactions: Competition for Receptors as a Means of Bacterial Invasion and Pathogenesis. Biochemistry 2016, 55 (31), 4286–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ames BN, [10] Assay of inorganic phosphate, total phosphate and phosphatases. In Methods in Enzymology, Academic Press: 1966; Vol. 8, pp 115–118. [Google Scholar]
- 23.Khalil IA; Kogure K; Futaki S; Harashima H, High Density of Octaarginine Stimulates Macropinocytosis Leading to Efficient Intracellular Trafficking for Gene Expression. Journal of Biological Chemistry 2006, 281 (6), 3544–3551. [DOI] [PubMed] [Google Scholar]
- 24.Smirnov A; Solga Michael D; Lannigan J; Criss Alison K, High-Throughput Particle Uptake Analysis by Imaging Flow Cytometry. Current Protocols in Cytometry 2017, 80 (1), 11.22.1–11.22.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnstone SA; Masin D; Mayer L; Bally MB, Surface-associated serum proteins inhibit the uptake of phosphatidylserine and poly(ethylene glycol) liposomes by mouse macrophages. Biochimica et Biophysica Acta (BBA) - Biomembranes 2001, 1513 (1), 25–37. [DOI] [PubMed] [Google Scholar]
- 26.Straubinger RM; Hong K; Friend DS; Papahadjopoulos D, Endocytosis of liposomes and intracellular fate of encapsulated molecules: encounter with a low pH compartment after internalization in coated vesicles. Cell 1983, 32 (4), 1069–1079. [DOI] [PubMed] [Google Scholar]
- 27.Lee JS; Hwang SY; Lee E, Imaging-based analysis of liposome internalization to macrophage cells: Effects of liposome size and surface modification with PEG moiety. Colloids and Surfaces B: Biointerfaces 2015, 136, 786–790. [DOI] [PubMed] [Google Scholar]
- 28.Ducat E; Evrard B; Peulen O; Piel G, Cellular uptake of liposomes monitored by confocal microscopy and flow cytometry. Journal of Drug Delivery Science and Technology 2011, 21 (6), 469–477. [Google Scholar]
- 29.Gottstein C; Wu G; Wong BJ; Zasadzinski JA, Precise quantification of nanoparticle internalization. ACS nano 2013, 7 (6), 4933–4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dominical V; Samsel L; McCoy JP, Masks in imaging flow cytometry. Methods 2017, 112, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenner D; Ducker C; Clark G; Prior J; Rowland Caroline A, Using multispectral imaging flow cytometry to assess an in vitro intracellular Burkholderia thailandensis infection model. Cytometry Part A 2016, 89 (4), 328–337. [DOI] [PubMed] [Google Scholar]
- 32.Franzen CA; Simms PE; Van Huis AF; Foreman KE; Kuo PC; Gupta GN, Characterization of Uptake and Internalization of Exosomes by Bladder Cancer Cells. BioMed Research International 2014, 2014, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mastrobattista E; Storm G; van Bloois L; Reszka R; Bloemen PGM; Crommelin DJA; Henricks PAJ, Cellular uptake of liposomes targeted to intercellular adhesion molecule-1 (ICAM-1) on bronchial epithelial cells. Biochimica et Biophysica Acta (BBA) - Biomembranes 1999, 1419 (2), 353–363. [DOI] [PubMed] [Google Scholar]
- 34.Onuma H; Komatsu T; Arita M; Hanaoka K; Ueno T; Terai T; Nagano T; Inoue T, Rapidly rendering cells phagocytic through a cell surface display technique and concurrent Rac activation. Sci. Signal 2014, 7 (334), rs4–rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang MH; Yoo HJ; Kwon YH; Yoon HY; Lee SG; Kim SR; Yeom DW; Kang MJ; Choi YW, Design of multifunctional liposomal nanocarriers for folate receptor-specific intracellular drug delivery. Molecular pharmaceutics 2015, 12 (12), 4200–4213. [DOI] [PubMed] [Google Scholar]
- 36.Kobatake E; Yamano R; Mie M, Targeted delivery using immunoliposomes with a lipid-modified antibody-binding protein. Applied biochemistry and biotechnology 2011, 163 (2), 296–303. [DOI] [PubMed] [Google Scholar]
- 37.Gupta V; Gupta R; Grover R; Khanna R; Jangra V; Mittal A, Delivery of molecules to cancer cells using liposomes from bacterial cultures. Journal of nanoscience and nanotechnology 2008, 8 (5), 2328–2333. [DOI] [PubMed] [Google Scholar]
- 38.Miller CR; Bondurant B; McLean SD; McGovern KA; O’Brien DF, Liposome−Cell Interactions in Vitro: Effect of Liposome Surface Charge on the Binding and Endocytosis of Conventional and Sterically Stabilized Liposomes. Biochemistry 1998, 37 (37), 12875–12883. [DOI] [PubMed] [Google Scholar]
- 39.Przybylo M; Glogocka D; Dobrucki JW; Fraczkowska K; Podbielska H; Kopaczynska M; Borowik T; Langner M, The cellular internalization of liposome encapsulated protoporphyrin IX by HeLa cells. European Journal of Pharmaceutical Sciences 2016, 85, 39–46. [DOI] [PubMed] [Google Scholar]
- 40.Khalil IA; Kogure K; Akita H; Harashima H, Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev 2006, 58 (1), 32–45. [DOI] [PubMed] [Google Scholar]
- 41.Kaksonen M; Roux A, Mechanisms of clathrin-mediated endocytosis. Nature Reviews Molecular Cell Biology 2018, 19, 313. [DOI] [PubMed] [Google Scholar]
- 42.Howes MT; Kirkham M; Riches J; Cortese K; Walser PJ; Simpson F; Hill MM; Jones A; Lundmark R; Lindsay MR, Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. The Journal of cell biology 2010, 190 (4), 675–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swanson JA; Watts C, Macropinocytosis. Trends in Cell Biology 1995, 5 (11), 424–428. [DOI] [PubMed] [Google Scholar]
- 44.Bloomfield G; Kay RR, Uses and abuses of macropinocytosis. Journal of Cell Science 2016, 129 (14), 2697–2705. [DOI] [PubMed] [Google Scholar]
- 45.Wang JT; Teasdale RD; Liebl D, Macropinosome quantitation assay. MethodsX 2014, 1, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabatini PJ; Zhang M; Silverman-Gavrila RV; Bendeck MP, Cadherins at cell-autonomous membrane contacts control macropinocytosis. Journal of cell science 2011, jcs. 076901. [DOI] [PubMed]
- 47.Koivusalo M; Welch C; Hayashi H; Scott CC; Kim M; Alexander T; Touret N; Hahn KM; Grinstein S, Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. The Journal of cell biology 2010, 188 (4), 547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fowler PW; Hélie J; Duncan A; Chavent M; Koldsø H; Sansom MSP, Membrane stiffness is modified by integral membrane proteins Soft Matter 2016, 12 (37), 7792–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahumada H; Montecinos R; Tieleman DP; Weiss-Lopez BE, Orientation and dynamics of benzyl alcohol and benzyl alkyl ethers dissolved in nematic lyotropic liquid crystals. 2H NMR and molecular dynamics simulations. J Phys Chem A 2005, 109 (30), 6644–51. [DOI] [PubMed] [Google Scholar]
- 50.Jovic M; Sharma M; Rahajeng J; Caplan S, The early endosome: a busy sorting station for proteins at the crossroads. Histology and histopathology 2010, 25 (1), 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubino M; Miaczynska M; Lippé R; Zerial M, Selective Membrane Recruitment of EEA1 Suggests a Role in Directional Transport of Clathrin-coated Vesicles to Early Endosomes. Journal of Biological Chemistry 2000, 275 (6), 3745–3748. [DOI] [PubMed] [Google Scholar]
- 52.Simonsen A; Lippe R; Christoforidis S; Gaullier J-M; Brech A; Callaghan J; Toh B-H; Murphy C; Zerial M; Stenmark H, EEA1 links PI (3) K function to Rab5 regulation of endosome fusion. Nature 1998, 394 (6692), 494. [DOI] [PubMed] [Google Scholar]
- 53.Eskelinen E-L, Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Molecular aspects of medicine 2006, 27 (5–6), 495–502. [DOI] [PubMed] [Google Scholar]
- 54.Schulze H; Kolter T; Sandhoff K, Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2009, 1793 (4), 674–683. [DOI] [PubMed] [Google Scholar]
- 55.Baldeon M; Ceresa B; Casanova J, Expression of constitutively active Rab5 uncouples maturation of the Salmonella-containing vacuole from intracellular replication 2001; Vol. 3, p 473–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.