

SUMMARY
A key feature of brain plasticity is the experience-dependent selection of optimal connections· implemented by a set of activity-regulated genes that dynamically adjust synapse strength and number. The activity-regulated gene cpg15/neuritin has been previously implicated in stabilization and maturation of excitatory synapses. Here· we combine two-photon microscopy with genetic and sensory manipulations to dissect excitatory synapse formation in vivo and examine the role of activity and CPG15 in dendritic spine formation, PSD95 recruitment, and synapse stabilization. We find that neither visual experience nor CPG15 is required for spine formation. However, PSD95 recruitment to nascent spines and their subsequent stabilization requires both. Further, cell-autonomous CPG15 expression is sufficient to replace experience in facilitating PSD95 recruitment and spine stabilization. CPG15 directly interacts with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors on immature dendritic spines, suggesting a signaling mode for this small extracellular molecule acting as an experience-dependent “selector” for spine stabilization and synapse maturation.
Graphical Abstract
In Brief
Experience plays a key role in formation and continuous optimization of brain circuits. Subramanian et al. show that the molecule CPG15/neuritin can replace experience in selecting which nascent contacts between neurons are retained, facilitating the recruitment of proteins that promote synapse maturation and stabilization.
INTRODUCTION
Use-dependent selection of optimal connections is a key feature of neural circuit development (Constantine-Paton et al., 1990; Goodman and Shatz, 1993; Shatz, 1990) and in the mature brain underlies functional adaptation of sensory maps as well as learning and memory (Buonomano and Merzenich, 1998; Caroni et al., 2012). Patterned activity selectively strengthens and stabilizes some connections while weakening and pruning others. Experience plays a critical role in biasing the formation and stabilization of excitatory synapses that transmit appropriately patterned activity (reviewed in Hua and Smith, 2004), and N-methyl-D-aspartate (NMDA)-type glutamate receptors mediate this activity-dependent synapse selection (Gomperts et al., 2000). NMDA receptor activation engages multiple cellular pathways required for both synaptic strengthening and establishing new synapses (reviewed in Cohen and Greenberg, 2008; West and Greenberg, 2011) ultimately implemented through synaptic insertion of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors (reviewed in Anggono and Huganir, 2012; Kerchner and Nicoll, 2008). While the myriad of proteins contained in these pathways may be considered the downstream mediators of experience at the synapse, no one molecule has been shown to be both required and sufficient in this respect.
The majority of excitatory connections in the brain are glutamatergic synapses located on specialized protrusions called dendritic spines. In vitro and in vivo studies suggest that newly formed spines typically lack PSD95 (Cane et al., 2014; De Roo et al., 2008a; Lambert et al., 2017; Villa et al., 2016), the scaffolding protein that clusters and stabilizes glutamate receptors at the synapse (reviewed in Kim and Sheng, 2004). In organotypic slice cultures, the recruitment of GFP-tagged PSD95 has been shown to be dependent on synaptic activity (De Roo et al., 2008a) and required for the activity-dependent stabilization of excitatory synapses and spines (Ehrlich et al., 2007), so much so that its presence is an excellent predictor of a spine’s future stability (De Roo et al., 2008a; Ehrlich et al., 2007) and mature synaptic function (Ehrlich and Malinow, 2004; Stein et al., 2003). Thus, PSD95 recruitment is a defining step in excitatory synapse maturation.
Cpg15/Nrn1 is an activity-regulated gene whose expression in the mammalian cortex is experience dependent (Harwell et al., 2005; Nedivi et al., 1996) and regulated by Ca2+ signaling via the NMDA receptor (Fujino et al., 2003). It is highly expressed at developmental times of synapse formation and maturation (Corriveau et al., 1999; Lee and Nedivi, 2002; Nedivi et al., 1996). Cpg15 knockout mice show developmental delays in synapse formation, with many dendritic spines initially lacking synaptic contacts (Fujino et al., 2011). These mice also show poor learning (Fujino et al., 2011) and aberrant plasticity in visual cortical networks (Picard et al., 2014). Cpg15/Nrn1 encodes a small glycosylphosphatidylinositol (GPI)-anchored membrane protein (Naeve et al., 1997; Nedivi et al., 1996; Putz et al., 2005) that has been suggested to act non-cell autonomously as an extracellular ligand (Nedivi et al., 1998) and promotes synapse maturation through recruitment of AMPA receptors (Cantallops et al., 2000). However, it has no known receptor, and its mode of action in promoting synapse maturation has been unknown.
Here, we use multicolor two-photon imaging to delineate, in vivo, the steps in spine formation, PSD95 recruitment, and spine and synapse stabilization and determine which of these steps are experience dependent. We also test the hypothesis that the activity-dependent gene product CPG15/neuritin is both required and sufficient for the step in spine and synapse stabilization that is experience dependent. We find that postsynaptic CPG15 is required cell autonomously for the recruitment of PSD95 to newly formed spines, and its knockdown occludes the effect of experience on PSD95 recruitment. Further, induction of exogenous CPG15 is sufficient to rescue deficits in PSD95 recruitment during visual deprivation. These results identify CPG15 as an activity-dependent spine stabilization signal that acts downstream of spine initiation but upstream of PSD95 recruitment. To examine how CPG15, an extracellular GPI-linked protein, can recruit the intracellular synaptic PSD95 scaffold, we follow up on proteomic studies showing a potential interaction between CPG15 and AMPA receptors (Schwenk et al., 2012) and show that this interaction is direct. Through elucidation of CPG15’s mechanism of action, we also clarify a sequence of molecular events whereby AMPA receptor presence on immature spines precedes activity-dependent PSD95 recruitment and subsequent spine stabilization and synapse maturation.
RESULTS
Experience Instructs PSD95 Recruitment to Spines
To resolve in vivo the progression of synapse formation and how it might be influenced by experience, we used a strategy for sparsely labeling neurons with three fluorophores: enhanced yellow fluorescent protein (eYFP) to visualize cell morphology, including dendritic spines, PSD95 fused to mCherry to mark mature excitatory synapses, and Teal-gephyrin to mark inhibitory synapses (Villa et al., 2016). This allows discrimination of three spine types, spines without PSD95 (PSD95− spines), spines with only PSD95 (PSD95+ spines), and spines with both PSD95 and gephyrin, termed dually innervated spines (DISs) (Villa et al., 2016). The three fluorophores in Cre-dependent constructs were co-electroporated in utero at high concentration to achieve a high incidence of fluorophore co-expression, together with a limiting amount of Cre plasmid to guarantee sparse labeling (Figure 1A). Electroporation into the brain ventricles of embryonic day 15.5 (E15.5) mouse embryos and electrode positioning targeted neural progenitors that give rise to layer 2/3 pyramidal neurons of primary visual cortex. When pups reached adulthood, labeled neurons were imaged through a cranial window using a custom-built two-photon microscope (Villa et al., 2016). The first two imaging sessions were taken before and after 2 weeks of a normal light and dark cycle (Figure 1B). Mice were then transferred to total darkness for 2 weeks, followed by a third imaging session. After 2 weeks of recovery under a normal light and dark cycle, cells were imaged a fourth time (Figure 1B).
Figure 1. Simultaneous In Vivo Tracking of Dendritic Spines and Their Synapses during Normal Visual Experience and Visual Deprivation.
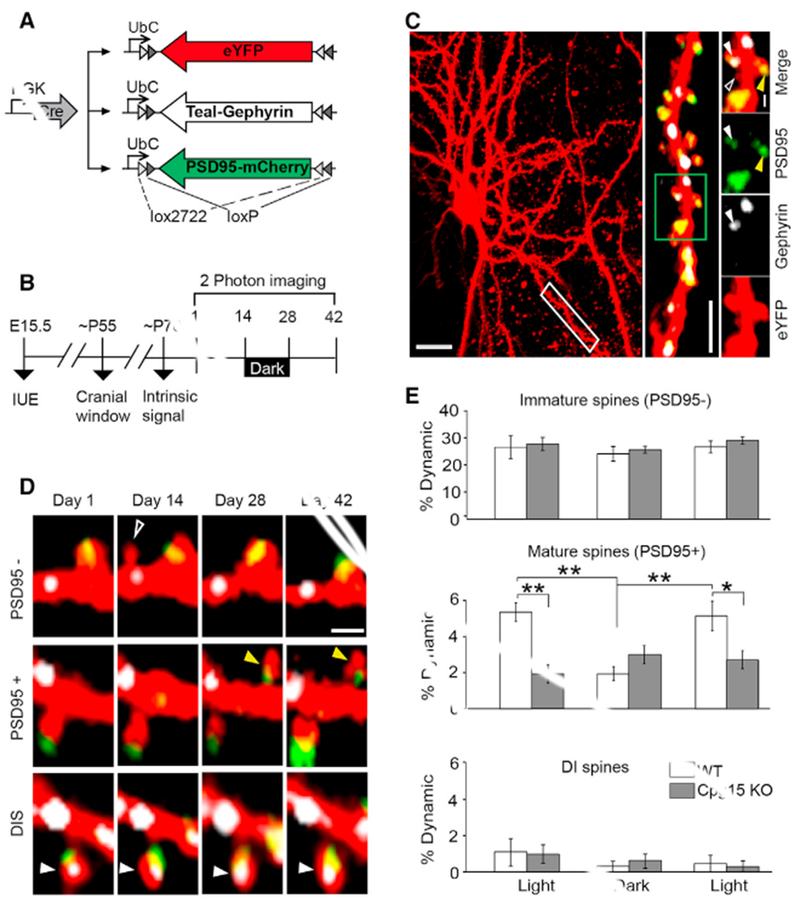
(A) Plasmid combination for co-expression of cellular and synaptic labels in sparsely labeled L2/3 pyramidal neurons of primary visual cortex.
(B) Experimental timeline.
(C) Maximum Z-projection of an eYFP cell fill (pseudocolored red); scale bar, 20 μm. The dendritic segment in the white box is magnified (middle; scale bar, 10 μm) and further magnified (green box) to reveal different spine classes (rightmost panels): PSD95− (open white arrow), PSD95+ (yellow arrow), and dually innervated spines containing both PSD95 and gephyrin (DISs; filled white arrow). Scale bar, 2 μm.
(D) Examples of dynamic spines of different categories. Open white arrow points to a PSD95− spine that formed and disappeared (top). Yellow arrow points to a new PSD95+ spine that formed and persisted (middle). Filled white arrow points to a stable DIS (bottom). Scale bar, 2 –m.
(E) Percentage of dynamic spines (combined gains and losses) of different classes after 2 weeks of normal visual experience (light) or 2 weeks in the dark, represented as mean ± SEM (n = 6 mice; *p ≤ 0.05 **p ≤ 0.01 by two-way ANOVA with Tukey’s post hoc test for multiple comparisons). IUE, in utero electroporation.
See also Figure S1 for separate gains and losses per spine category.
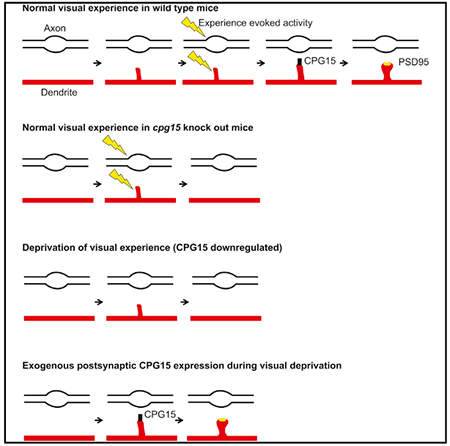
Imaging a 200-μm cube, we captured the full dendritic arbor of well-isolated neurons with distinctly resolved dendritic spines, Teal-gephyrin puncta, and PSD95-mCherry puncta (Figure 1C). Individual spines were tracked for their appearance or disappearance and categorized based on the presence or absence of a PSD95 punctum (Figures 1D and S1). Dynamics of PSD95− spines were similar in both light and dark conditions (Figure 1E, top panel). PSD95+ spine dynamics were significantly reduced in the dark but recovered when animals were returned to normal light and dark conditions (Figure 1E, middle panel). For both light and dark conditions, dynamics equally represented gains and losses (data not shown), with percent gain of PSD95+ spines reduced from 5.6 ± 0.7 in light to 1.6 ± 0.6 in dark (Figure S1, p < 0.01 repeated-measures ANOVA Tukey’s post hoc test for multiple comparisons). DISs were extremely stable in both conditions (Figure 1E, bottom panel), consistent with our prior findings showing their baseline stability and their stability in response to monocular deprivation (Villa et al., 2016). Since new spines are typically PSD95−, these results indicate that visual experience does not influence spine formation, but rather, their conversion to PSD95+ spines.
CPG15 Removal Occludes the Effect of Experience on PSD95+ Spine Dynamics
We hypothesized that CPG15, whose visual cortical expression is responsive to light-driven activity (Nedivi et al., 1996), and is required for spine stabilization (Fujino et al., 2011) and synaptic maturation (Cantallops et al., 2000), might act as a molecular signal linking experience with PSD95 recruitment. If this were the case, Cpg15 removal should occlude the effect of dark on the remodeling of PSD95+ spines, but not PSD95− spines, so that spine remodeling in Cpg15 knockout (KO) mice would be equivalent in light and dark. Further, spine remodeling in the Cpg15 KO should also be equivalent to spine remodeling in wild-type (WT) animals in the dark, when endogenous CPG15 expression is downregulated (Nedivi et al., 1996). To test these predictions, we applied to Cpg15 KO mice the same labeling and imaging strategy described above for WT mice (Figures 1A–1C). We found that in Cpg15 KO mice, dynamics of PSD95− spines were similar in both light and dark conditions (Figure 1E, top panel), indicating that neither experience nor CPG15 is required for the emergence and elimination of transient spines. As predicted, in the case of PSD95+ spines, loss of CPG15 severely reduced spine dynamics under normal light conditions (Figure 1E, middle panel), so much so as to occlude the effect of visual deprivation. In fact, dynamics of PSD95+ spines in the CPG15 KO in the light were similar to those of control animals in the dark (Figure 1E, middle panel). In the case of DISs, similar to WT, Cpg15 KO mice had no effect on dynamics (Figure 1E, bottom panel). These results show that loss of visual experience or CPG15 has no effect on the remodeling of immature spines. However, gain and loss of PSD95+ spines require both experience and CPG15.
CPG15 Recruits PSD95 to Nascent Spines
To further examine the link between PSD95 recruitment to newly formed spines, spine stabilization, and the potential role of CPG15 in this process, we tracked the formation and the subsequent fate of new spines by imaging neurons daily for up to 9 days in both WT and Cpg15 KO animals (Figure 2A). As seen for the 2-week imaging intervals, the daily rate of spine emergence was identical in WT and Cpg15 KO mice (Figure 2B), further demonstrating that CPG15 is not required for new spine formation.
Figure 2. CPG15 Is Required for PSD95 Recruitment and Stabilization of Nascent Spines.
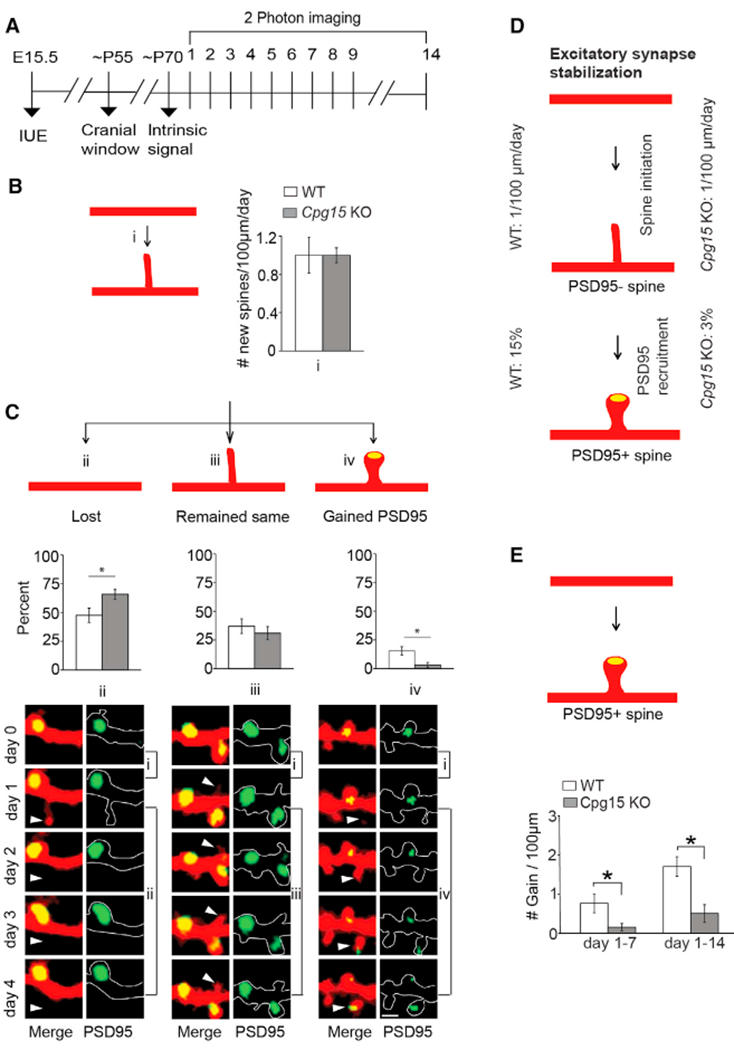
(A) Experimental timeline. Neurons labeled as in Figure 1 were imaged daily for up to 9 days and again on day 14. For a subset, imaging was done on day 1 and 14.
(B) Rate of spine initiation (i).
(C) Quantification of newly formed spine fate as lost (ii), remained same (iii), or gained PSD95 (yellow oval; iv), over a 4-day period. Representative examples are shown (bottom). Scale bar, 2 μm. Arrows point to newly formed spines (n = 8 mice each for wild-type (WT; 159 new spines) and cpg15 KO (145 new spines).
(D) Schematic of excitatory synapse stabilization. Roughly one new spine emerges per 100 μm of dendrites every day in both WT and cpg15 KO. However, only 15% of WT and 3% of Cpg15 KO spines recruit PSD95 within a 4-day period.
(E) Schematic of mature spine gain (top). Gain of PSD95+ spines over a 6- and 13-day period (mean ± SEM; n = 8 and 6 mice for WT and cpg15 KO, respectively). *p < 0.05 by unpaired t test.
See Figure S1 for gains and losses per every spine and synapse category.
We next tracked the fate of newly formed spines within four days of their emergence. In WT, most newly formed spines were either lost within four days (48%) or remained without PSD95 (37%), with only 15% gaining PSD95 (Figures 2C and 2D). New spines were nearly 20% more likely to be lost in Cpg15 KO than in WT (66% in KO versus 48% in WT). This was concomitant with a 5-fold reduction in the rate of PSD95 acquisition (3% in KO versus 15% in WT) (Figures 2C and 2D). Therefore, CPG15 is required for PSD95 recruitment to nascent spines, a critical step for spine stabilization and synaptic maturation (De Roo et al., 2008a; Taft and Turrigiano, 2013).
To test directly whether PSD95 recruitment to nascent spines influences their long-term stability, we looked at cumulative gain or loss of PSD95+ spines in WT and Cpg15 KO mice over a 14-day period. In WT mice, we observed 0.76 ± 0.24 (day 7) and 1.9 ± 0.4 (day 14) new PSD95+ spines per 100 μm dendritic segment as compared to 0.15 ± 0.09 (day 7) and 0.5 ± 0.2 (day 14) in the Cpg15 KO (Figure 2E). Thus, similar to light deprivation in WT mice, Cpg15 KO does not affect new spine initiation but reduces recruitment of PSD95 to nascent spines, resulting in their eventual loss rather than stabilization. This results in a lower overall spine density in the adult Cpg15 KO mice that can be entirely accounted for by a reduction in the number of PSD95+ spines (Figures S2A and S2B). Cpg15 KO mice also exhibited a reduction in the loss of PSD95+ spines (Figure S2B), likely as a homeostatic adaptation to reduced PSD95+ spine gain. Due to the reduction in gain and loss of PSD95+ spines, Cpg15 KO had more stable PSD95+ spines than WT (percentage of stable PSD95+ spines: WT, 88.9 ± 1.1 versus KO, 95.6 ± 1.8; p = 0.01 by unpaired t test). PSD95− spines, DISs, and inhibitory synapses are unaffected in the Cpg15 KO (Figures S2A and S2C–S2E). These data suggest that the reduced spine density in Cpg15 KO mice is due not to a deficit in initial spine formation but rather to a specific failure of conversion from nascent PSD95− to mature PSD95+ spines. Further, the requirement for CPG15 seems specific to excitatory synapse maturation.
Postsynaptic CPG15 Is Acutely Required for PSD95 Recruitment
These studies were conducted in adult Cpg15 KO mice, which are known to have several plasticity-related developmental deficits (Fujino et al., 2011; Picard et al., 2014). To confirm that Cpg15 KO deficits in PSD95 recruitment and spine stabilization were not secondary to other developmental issues, we used a floxed Cpg15 mouse line to acutely delete Cpg15 using an inducible Cre. This sparse CPG15 KO strategy also allowed testing for a pre- or postsynaptic requirement for CPG15, something not possible in the KO, where both pre- and postsynaptic cells are CPG15 deficient. Prior studies on the developmental timing of CPG15 expression profiles in the visual pathway suggested a presynaptic role for this protein (Corriveau et al., 1999). Consistent with this idea, overexpression of CPG15 in Xenopus tectal neurons leads to an increase in dendritic arbor elaboration in nearby WT neurons (Nedivi et al., 1998). Furthermore, CPG15 mRNA and protein have been shown to localize to axons (Cantallops and Cline, 2008; Merianda et al., 2013; Nedivi et al., 2001). Despite such strong evidence for a presynaptic role for CPG15, it has also been shown that postsynaptic overexpression of CPG15 in Xenopus tectal neurons can influence the branching of presynaptic retinal axons (Cantallops et al., 2000). Given the uncertainty regarding whether CPG15 pre- or post-synaptic expression might be required for PSD95 recruitment, we used inducible Cre to acutely KO CPG15 not only in postsynaptic neurons but also in some of the presynaptic inputs to these neurons.
The neuronal labeling strategy was similar to the one described in Figure 1, except that fluorophore expression was rendered Flp recombinase (Flp) rather than Cre dependent, because Cre expression would delete Cpg15 immediately after electroporation in the floxed Cpg15 background (Figure 3A). To temporally control Cre expression, we used a Cre version that is induced only upon binding to 4-hydroxytamoxifen (4-OHT). To identify neurons that received inducible Cre, the construct bearing the inducible Cre (ERT2-Cre-ERT2, labeled OHT-Cre) also carried Synaptophysin-TdTomato expressed from the same promoter (Figures 3A and S3). This construct was co-electroporated into floxed Cpg15 pups at E15.5 along with eYFP and PSD95-Teal in dio-cassettes conditional to Flp, and limiting amounts of Flp (Figures 3A and 3B). All neurons that receive Flp, eYFP, and PSD95-Teal also receive Synaptophysin-TdTomato/OHT-Cre, and therefore, they are conditional postsynaptic KOs for CPG15. However, Synaptophysin-TdTomato/OHT-Cre expression is independent of Flp and therefore, unlike PSD95-Teal and eYFP, will not be restricted to Flp-expressing neurons. Consequently, the presynaptic inputs from these neurons to the imaged neurons will be conditional presynaptic KOs for CPG15 (Figure 3C). Synaptophysin-TdTomato labeling is restricted to cell bodies and axons, allowing confirmation of inducible Cre expression without interfering with dendritic labeling of eYFP cell fill and PSD95-Teal (Figures 3D and 3E). Before 4-OHT injection, imaged postsynaptic neurons and all of their inputs (labeled or unlabeled with Synaptophysin-TdTomato) are WT (Figure 3F, top). After 4-OHT injection, Cpg15 will be deleted in all imaged postsynaptic neurons as well as some of their presynaptic inputs marked with Synaptophysin-TdTomato. Spines without apposing Synaptophysin-TdTomato will remain presynaptic WT even after 4-OHT injection (Figure 3F, bottom).
Figure 3. Postsynaptic CPG15 Is Acutely Required for PSD95+ Recruitment to Spines.
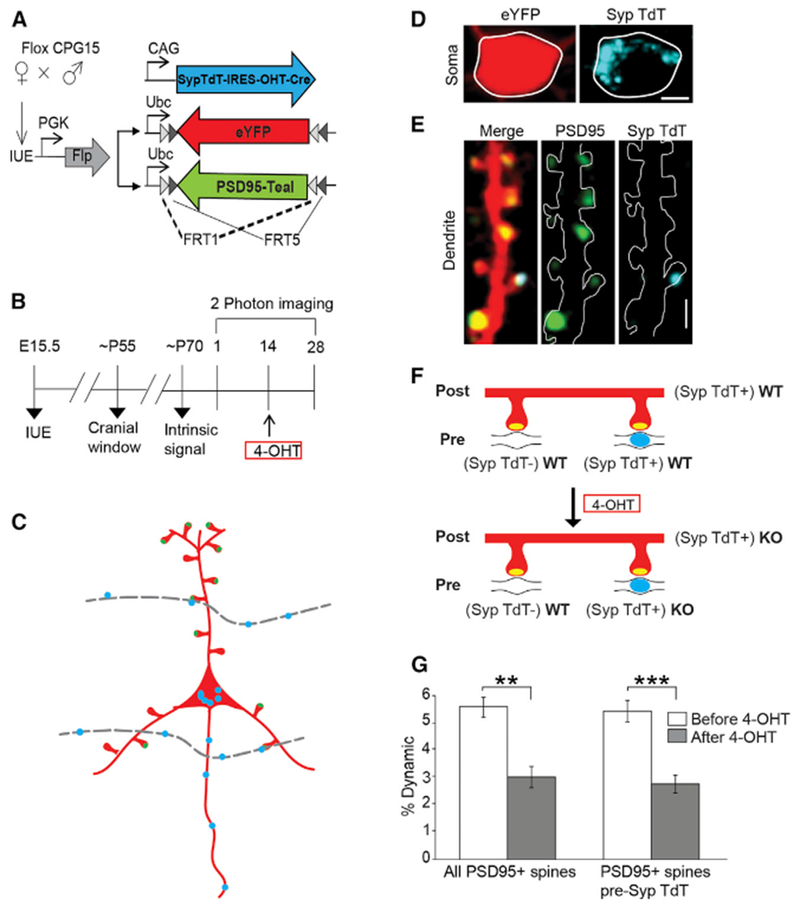
(A) Plasmids for conditional removal of Cpg15 and neuronal and synaptic labeling in floxed Cpg15 mice. Upon 4-OHT injection, OHT-Cre expressed from pCAG-Synaptophysin-TdTomato-IRES-OHT-Cre (SypTdT-IRES-OHT-Cre) will translocate to the nucleus and delete Cpg15. Presynaptic terminals of Cre expressing cells will be labeled with Synaptophysin-TdTomato (Syp-TdT) and will be WT before OHT injection and KO after. Cell and synaptic labeling utilize Flp- rather than Cre-dependent eYFP and PSD95-Teal expression due to the floxed background.
(B) Experimental timeline.
(C) Scheme illustrating the labeling and KO strategy. An imaged neuron expressing a cell fill (red), PSD95 (green oval), and Syp-TdT (blue oval) in its soma and OHT-Cre. Since the SypTdT-IRES-OHT-Cre is not Flp dependent, it will express more widely in neighboring layer 2/3 neurons. Boutons (blue ovals) on their axons (dotted gray line) will be labeled with Syp-TdT and synapse onto some spines of the labeled neuron. Since they also express OHT-Cre, these presynaptic contacts will be WT before OHT injection and KO after. All other spines will receive unlabeled WT presynaptic contacts regardless of OHT injection.
(D) Representative images of somal eYFP fill (left), and Syp-TdT (right) for labeled neuron such as shown in (C). Scale bar, 5 μm.
(E) Dendrites of soma shown in (D) (merged (left), PSD95 (middle), and Syp-TdT (right)). Scale bars, 2.5 μm.
(F) Schematic of dendritic spines (red) and PSD95 (yellow oval) opposed to an unlabeled (Syp TdT−) axon (black line) or Syp-TdT+ axon (black line with blue oval). After 4-OHT, the postsynaptic cell and the Syp-TdT labeled axons turn KO.
(G) Percent dynamics (gains plus losses) of PSD95+ spines on Syp-TdT labeled postsynaptic neurons is reduced after 4-OHT injection. This is true, even when counting only PSD95+ spines not apposed by Syp-TdT (presynaptic WT), showing that CPG15 is required postsynaptically (PSD95+, Pre Syp-TdT−; n = 7 mice) **p < 0.01, ***p < 0.001 by paired t test. Data are represented as mean ± SEM. 4-OHT, 4-hydroxytamoxifen; IUE, in utero electroporation.
See also Figures S3 and S4.
The density and dynamics of PSD95− and PSD95+ spines were comparable to experiments described in Figure 1, despite the transition to the Flp system for fluorophore expression and from PSD95-mCherry to PSD95-Teal (Figure S4A). Also, 4-OHT had no influence on PSD95+ spine dynamics (Figures S4B–S4D).
To test whether acute deletion of Cpg15 diminishes representation of PSD95+ spine dynamics as seen in Cpg15 KO mice, we quantified gain in PSD95+ spines over a 2-week period in neurons expressing YFP, PSD95-Teal, and OHT-Cre (Synaptophysin-TdTomato) before and after 4-OHT injection (Figures 3B–3F). We found that gain in PSD95+ spines was significantly reduced after 4-OHT injection (Figure 3G), suggesting that CPG15 is acutely required for PSD95+ spine remodeling. Surprisingly, when spines receiving a Synaptophysin-TdTomato contact (from other Cpg15 KO neurons) were excluded from analysis, PSD95+ spine dynamics were comparable to pre-exclusion numbers, indicating an unequivocal postsynaptic requirement for CPG15 in PSD95+ spine remodeling (Figure 3G). To test whether postsynaptic CPG15 is also sufficient to rescue PSD95+ spine dynamics without a potential presynaptic contribution, we exogenously expressed CPG15 only in the imaged neuron in a Cpg15 KO mouse. A double inverted Cre-dependent CPG15 construct was co-electroporated at E15.5 together with the Cre-dependent fluorophores into pups from Cpg15 KO and Cpg15+/− crosses, restricting CPG15 expression to the sparsely labeled neurons (Figure 4A). Electroporated Cpg15 KO pups were imaged twice, with a 2-week interval between sessions (Figure 4B). Overexpression of CPG15 in the imaged cell rescued PSD95+ spine dynamics to WT levels (Figure 4C), demonstrating that cell-autonomous CPG15 expression is both required and sufficient for PSD95+ recruitment to spines and their subsequent stabilization.
Figure 4. Cell-Autonomous CPG15 Expression Is Both Necessary and Sufficient for Experience-Dependent PSD95+ Spine Remodeling.
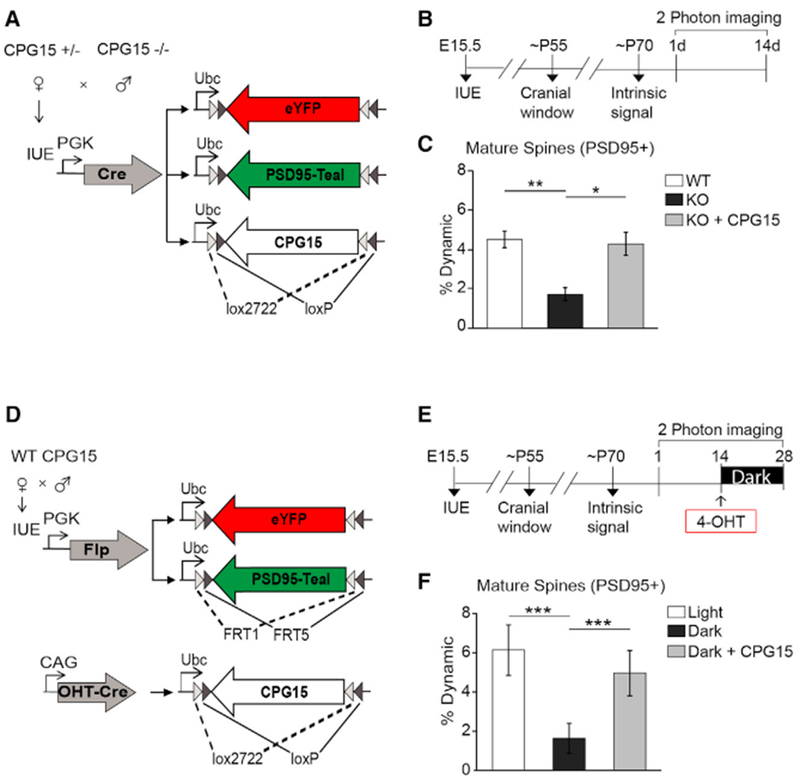
(A) Plasmid combination for co-expression of CPG15 and cellular and synaptic labels.
(B) Experimental timeline.
(C) Percent dynamics (combined gains and loss) of PSD95+ spines in WT mice (n = 6 mice), KO mice (n = 6 mice), and KO mice with CPG15 overexpression (KO+CPG15 n = 3 mice).
(D) Constructs for conditional expression of Cpg15 in neurons labeled with cell fill and PSD95. Neuronal and synaptic labeling was the same as described for Figure 3 but combined with a plasmid expressing CPG15 within a Cre-dependent dio-cassette and an inducible OHT-Cre plasmid. Upon OHT injection, CPG15 will be induced in the imaged neuron.
(E) Experimental timeline.
(F) Percent dynamics (combined gains and loss) of PSD95+ spines in light or dark (n = 6 mice) or dark with exogenous CPG15 expression (OHT injection) (n = 5 mice). *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA with Tukey’s post hoc test for multiple comparisons. Data are represented as mean ± SEM. 4-OHT, 4-hydroxytamoxifen; IUE , in utero electroporation.
See Figure S5 for controls and Figure S6 for separate PSD95+ gains and losses.
Acute CPG15 Expression Is Sufficient to Replace Experience in Promoting PSD95+ Spine Formation
We next asked whether CPG15 is also sufficient to rescue the effects of visual deprivation on PSD95 recruitment. To test for CPG15 sufficiency, we induced expression of exogenous CPG15 specifically in the dark, when endogenous CPG15 in WT mice is downregulated and PSD95 recruitment is low. To express inducible CPG15 in the imaged neurons, we co-electroporated into WT mice the same Flp-dependent fluorophore and Flp plasmids used above together with a high concentration of plasmid expressing Cre-dependent CPG15, OHT-Cre (Figure 4D). Labeled neurons are similar to other WT neurons before 4-OHT administration but should express high levels of exogenous CPG15 after 4-OHT (Figure S5). Electroporated mice were imaged before and after a 2-week period of a normal light and dark cycle (Figure 4E). Immediately following the second imaging session, animals were injected with 4-OHT and transferred to 2 weeks in the dark, followed by a third imaging session (Figure 4E). We found that CPG15 expression was sufficient to rescue PSD95+ spine formation in the absence of visual experience (Figure 4F). WT cells in the dark show only 36% of PSD95+ spine dynamics as seen in the light condition. Induction of exogenous CPG15 in WT cells while animals were in the dark rescued PSD95+ spine dynamics to 81% of light dynamics (dark versus dark with CPG15 induction, p < 0.001 by one-way ANOVA with Tukey’s post hoc test for multiple comparisons) (Figure 4F). While the gain of PSD95+ spines could be fully rescued by CPG15, PSD95+ spine loss showed only a partial rescue (Figure S6). These results show that CPG15 can replace experience in recruiting PSD95 to spines.
CPG15 Interacts with GluA1 and Is Critical for the Recruitment of PSD95 to GluA1 Containing Synapses
Since CPG15 is an extracellular molecule, in order for it to initiate PSD95 recruitment intracellularly, it would potentially need to work through an intermediary that spans the membrane. Recently, antibody pull-down studies of the AMPA receptor identified CPG15 as a possible component of the AMPA receptor proteome (Schwenk et al., 2012). To confirm this finding and test if the CPG15-AMPA receptor interaction is direct, we co-expressed FLAG-tagged CPG15 and hemagglutinin (HA)-tagged GluA1 in a heterologous system. In HEK293T cells expressing only HA-GluA1, no AMPA receptor signal was detected after immunoprecipitation with anti-FLAG affinity beads (Figure 5A, left). In HEK293T cells co-expressing FLAG-tagged CPG15 and HA-tagged GluA1, immunoprecipitation with anti-FLAG affinity beads showed robust co-immunoprecipitation of HA-GluA1, suggesting a direct interaction between CPG15 and the AMPA receptor (Figure 5A, right).
Figure 5. CPG15 Interacts Directly with the GluAI AMPA Receptor Subunit and Is Critical for Recruitment of PSD95 to AMPA-Receptor-Containing Spines.
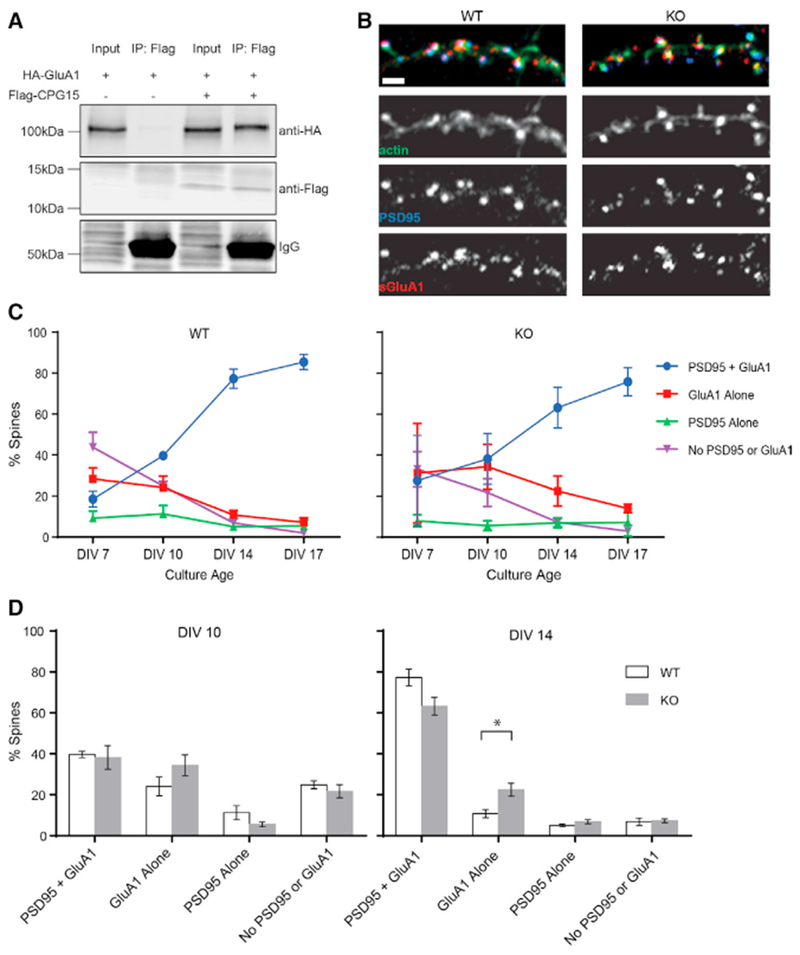
(A) Immunoprecipitation of FLAG-tagged CPG15 co-precipitates HA-tagged GluA1 from HEK293T cells expressing both tagged proteins. Immunoglobulin G (IgG) bands are shown to demonstrate equal amounts of anti-FLAG affinity beads added to the reaction.
(B) A section of dendrite from WT (left) and Cpg15 KO (right) cultured hippocampal neurons at DIV 14 stained for actin (Alexa Phalloidin-488, green), anti-PSD95 (blue), and anti-surface-GluA1 (red). Scale bar, 1 μm.
(C) Categories of spines containing PSD95 and GluA1, GluA1 alone, PSD95 alone, or neither PSD95 nor GluA1 as a percentage of total spines for each given time point.
(D) Comparison of spine categories in WT and Cpg15 KO cultures for DIV 10 (left) and DIV 14 (right). *p < 0.05 by unpaired t test. Data are represented as mean ± SEM.
If the interaction of CPG15 with AMPA receptors is required for experience-dependent recruitment of PSD95 to synapses, then one would expect that AMPA receptor presence at young synapses would precede PSD95 and be unaffected by removal of CPG15. To examine this sequence of events more closely, we cultured hippocampal neurons from E16 WT and Cpg15 KO mice and monitored synaptic localization of PSD95 and the GluA1 AMPA receptor subunit at four separate time points through culture maturation (Figures 5B and 5C). Our first observation at 7 days in vitro (DIV) showed that ~43% of spines in WT neurons contain neither PSD95 nor GluA1, 29% contain only GluA1, and 9% contain only PSD95, with as few as 19% containing both PSD95 and GluA1 (Figure 5C, left). In Cpg15 KO neurons, percentages of the different spine categories at DIV 7 were not significantly different at 33%, 31%, 9%, and 27%, respectively. As neurons matured, the percentage of spines containing both GluA1 and PSD95 markedly increased, with the largest increase occurring between DIV 10 and DIV 14, from 40% to 77%. This increase in mature spines was largely at the expense of spines containing GluA1 alone, which decreased from 24% to 10%. In the Cpg15 KO cultures, the same trends could be seen as in the WT cultures; a gradual reduction in spines containing neither PSD95 nor GluA1, paired with increased recruitment of both these proteins (Figure 5C, right). However, during the time interval when the majority of spines recruit PSD95, between DIV 10 and DIV 14, the proportion of spines containing GluA1 without PSD95 was twice as high in the KO as compared to WT cultures, recapitulating our in vivo finding that CPG15 is critical for PSD95 recruitment (Figure 5C, right; Figure 5D, right). These findings further suggest that initial recruitment of GluA1 precedes recruitment of PSD95 and is unaffected by Cpg15 KO.
DISCUSSION
In vitro, not only PSD95 recruitment (De Roo et al., 2008a) but also spine formation (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Nägerl et al., 2004) and spine stabilization (De Roo et al., 2008a, 2008b) have been shown to be influenced by neuronal activity. Recently, the introduction of genetically tagged synaptic scaffolding molecules has enabled researchers to separately and simultaneously track spine and synapse dynamics in vivo (Cane et al., 2014; Gray et al., 2006; Isshiki et al., 2014; Villa et al., 2016). These studies confirmed that although the presence of PSD95 is not entirely predictive of long-term spine stability (Cane et al., 2014), the step in excitatory synapse formation most closely associated with long-term stability is the presence of PSD95 (Cane et al., 2014; Villa et al., 2016). However, how experience facilitates PSD95 recruitment to synapses has not been established. Here, we show unequivocally that the recruitment of PSD95 is a definitive visual signature of synapse and spine stabilization. It is this step rather than spine initiation that is activity regulated. KO of the activity-regulated gene product, CPG15, mimics and occludes the effect of experience on PSD95 recruitment, and CPG15 expression can on its own replace experience in promoting PSD95 recruitment and synapse stabilization. No other effector molecule downstream of the NMDA receptor has been shown to be both required and sufficient for replacing experience in implementing synaptic structural or functional plasticity.
While excitatory synaptogenesis can occur in the absence of synaptic activity (Sando et al., 2017; Sigler et al., 2017; Varoqueaux et al., 2002; Verhage et al., 2000), PSD95 (Béïque et al., 2006; Migaud et al., 1998), or CPG15 (Fujino et al., 2011), in all cases, the synapses are functionally immature or deficient, and circuits are operationally suboptimal (Béïque et al., 2006; Fujino et al., 2011; Migaud et al., 1998; Sando et al., 2017; Sigler et al., 2017). This is consistent with a view that activity is not required to initiate synapse formation per se but rather is critical for selecting synapses for maturation and long-term stabilization.
How do these findings reconcile with previous studies showing activity-dependent spine formation in vitro (Engert and Bonhoeffer, 1999; Hill and Zito, 2013; Kwon and Sabatini, 2011; Maletic-Savatic et al., 1999; Nägerl et al., 2004) or even in vivo (reviewed in Holtmaat and Svoboda, 2009)? Without exception, these studies were all done prior to recent technical developments allowing the in vivo visualization and longitudinal tracking of both tagged PSD95 and spine dynamics (Cane et al., 2014; Villa et al., 2016). When examining the effect of activity or experience on new spine formation in the absence of a PSD95 label, it is impossible to resolve whether the increase in spine number is due to an increase in new spine initiation or transition of nascent spines to stable PSD95+ spines. In the case where spinogenesis has been shown in vitro to be directly elicited by glutamate uncaging (Hill and Zito, 2013; Kwon and Sabatini, 2011), although the induced spines acquire mature morphology and exhibit electrical responsiveness, their long-term stability beyond 30 min is not known, and whether or not they possess PSD95 is unclear. It is also possible that spinogenesis is not entirely stochastic but rather locations of glutamate release (preexisting boutons) favor new spine formation but long-term spine stability is dependent on experience-induced neuronal activity.
Studying the role of experience and CPG15 in excitatory synapse stabilization also sheds light on the sequence of events related to excitatory synapse formation in vivo. Excitatory synapse formation is a multistep process that begins with spine formation, followed by recruitment of a plethora of synaptic proteins, including adhesion molecules, the NMDA- and AMPA-type glutamate receptors, the scaffolding molecules thought to anchor them at the synapse, as well as adaptor proteins linking the synaptic apparatus to an array of downstream second messenger signaling pathways (reviewed in McAllister, 2007; Waites et al., 2005). While there is a rich literature describing the molecular composition of excitatory synapses and specific roles of its individual components (reviewed in Sheng and Kim, 2011; Li and Sheng, 2003; Scannevin and Huganir, 2000; Sheng and Hoogenraad, 2007; Südhof, 2018), few of the steps in synapse assembly have been examined in vivo. Adhesion molecules, specifically neuroligins, were initially identified by in vitro assays as synaptogenic (Chih et al., 2005; Scheiffele et al., 2000), but their in vivo examination suggested their up- and downregulation does not influence synapse initiation but rather insertion of NMDA receptors (Chubykin et al., 2007; Varoqueaux et al., 2006). The NMDA receptor has been considered an early synaptic inhabitant, given the preponderance of NMDA only “silent synapses” early in development and the low AMPA to NMDA ratios at young synapses (reviewed in Hanse et al., 2013; Kerchner and Nicoll, 2008). Insertion of AMPA receptors at these synapses has been associated with activity-dependent synapse maturation (Isaac et al., 1995; Liao et al., 1995), and the molecule shown to be most important for AMPA receptor accrual at synapses is PSD95 (Bats et al., 2007; Béïque et al., 2006; Chen et al., 2011; Ehrlich and Malinow, 2004; Schnell et al., 2002). In turn, cell adhesion molecules such as neuroligin-I, netrin-G-ligand-I, and ephrin-B3 have been shown to physically interact with PSD95, facilitating its recruitment to synapses (reviewed in Han and Kim, 2008). The common logical sequence to these events would therefore be that once spines form, adhesion molecules help the recognition and alignment of pre- with postsynaptic partners in an activity-independent manner (Scheiffele et al., 2000). NMDA receptors and early scaffolding molecules, such as Sap102 and SAP97 (Elias et al., 2008; Lambert et al., 2017), are then recruited, facilitating insertion of a small number of rather unstable AMPA receptors (reviewed in Groc et al., 2006). This young immature synapse can form within minutes of spine formation and support synaptic transmission (Lambert et al., 2017; Zito et al., 2009). By virtue of the NMDA receptor’s property as a coincidence detector, salient activity can then trigger its maturation through recruitment of PSD95 and AMPA receptors (Ashby and Isaac, 2011; De Roo et al., 2008a; Wu et al., 1996; Zhang et al., 2015). This last step in particular can now be revisited in light of our findings. We propose a model (Figure 6) whereby new spines are initiated across the dendritic arbor in an experience-independent manner, although glutamate release may bias their emergence sites. These nascent spines acquire an immature synaptic structure with a small number of unstable AMPA receptors that can support transmission. Such spines that have an immature synapse lacking PSD95 are transient in nature and are likely to be eliminated within a few days (Cane et al., 2014; Villa et al., 2016), except in the presence of salient activity. Coincident pre- and postsynaptic activity induces expression, and/or perhaps insertion, of CPG15 downstream of NMDA receptor activation. Since CPG15 is an extracellular GPI-linked protein, its effect on PSD95 recruitment is implemented through an interaction with AMPA receptors, suggesting that early AMPA receptors at immature synapses are critical to the recruitment of PSD95. This is consistent with the accepted view that PSD95 recruitment, in turn, promotes further insertion of AMPA receptors to generate not only structurally stable but also functionally mature synapses.
Figure 6. A Model for Experience-Dependent Synapse Stabilization by CPG15.
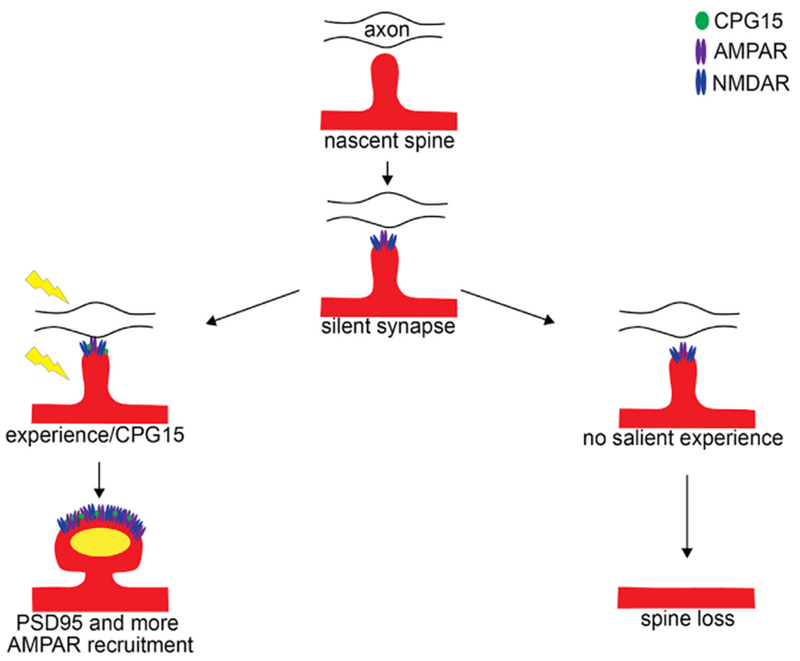
From top: a nascent spine emerging from a dendrite (red) tests an opportunity to synapse with an axonal (black line) bouton (open oval). This transient spine acquires immature synaptic machinery, including NMDA-type (blue) and some AMPA-type glutamate receptors (purple ovals) that are unstable, anchored by a non-PSD95 scaffold. In the absence of salient experience, these spines are lost (right fork). In the presence of experience-dependent correlated activity (yellow lightning bolt), GPI-anchored CPG15 (green oval) expressed in postsynaptic neurons interacts with AMPA receptors. This interaction recruits PSD95 (yellow oval) to immature, transient spines and stabilizes them. PSD95 then recruits additional AMPA receptors, leading to mature and stable synapses.
Recently, small extracellular proteins have emerged as important players in synaptogenesis through their interactions with larger transmembrane proteins (Pandya et al., 2018; Singh et al., 2016; Uemura et al., 2010). Similar to these proteins, CPG15 does not behave like a traditional ligand with its own cognate receptor. Rather, CPG15 directly interacts with the AMPA receptor to facilitate its downstream interaction with PSD95. Unlike these other extracellular synaptic mediator molecules that are secreted, the cell-autonomous postsynaptic requirement for CPG15 suggests that it acts in a membrane-bound form and that its GPI membrane anchoring enables its interaction with AMPA receptors. GPI-anchored proteins are known to be enriched in lipid rafts (reviewed in Mayor and Riezman, 2004), and both PSD95 and AMPA receptors have been shown to be part of lipid rafts at synapses (Hering et al., 2003; Hou et al., 2008).
CPG15/neuritin has been shown to activate the insulin receptor and fibroblast growth factor pathways (Shimada et al., 2016; Yao et al., 2012). Its expression has been shown to positively affect axonal regeneration in neuromuscular disorders (Akten et al., 2011) and neurite outgrowth after ischemia (Zhao et al., 2017) and ameliorate depression associated with chronic stress (Kojima et al., 2005; Son et al., 2012. Outside the nervous system, CPG15 has been implicated in hepatocyte maturation and melanoma migration (Bosserhoff et al., 2017). It would be interesting to consider that given its ubiquitous expression, CPG15’s role in synapse stabilization may be critical for synaptic regulation by extracellular factors other than activity and that it may serve as an adaptor to receptors other than AMPA-type glutamate receptors to perform diverse cell-type-specific functions.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Elly Nedivi (nedivi@mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal procedures were approved by the Massachusetts Institute of Technology Committee on Animal Care and meet the NIH guidelines for the use and care of vertebrate animals. Animals were housed in the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) accredited facility at MIT. The Cpg15 KO mouse line (Fujino et al., 2011) was maintained by breeding heterozygous (Het) males and females. WT males and females (to obtain WT male pups for cranial windows) or Het males and KO females (to obtain KO male pups for cranial windows) were crossed to generate WT and Het timed pregnant females for in utero electroporation. For acute Cpg15 deletion experiments, floxed Cpg15 males and females were used to generate floxed Cpg15 pups. Breeding cages typically had one male and two female mice. Males and females were housed separately after weaning with a maximum of five mice in a standard home cage. Ai6 mice (Jackson # 007906) were used for testing inducible Cre mediated recombination at a genomic locus. All mice were of C57BL/6 background. In utero electroporations were performed on E15.5–E16.5 embryos from 3-5 months old female mice. Cranial windows were performed on ~2 months old male mice. Following cranial window surgery mice were individually housed.
Primary neuron cultures
Hippocampal neurons were prepared from littermates of E16 cpg15 WT and KO embryos obtained from a Het x Het crosses. Embryos of both sexes were genotyped while hippocampi from individual mice were prepared for culture by digestion with Trypsin, followed by cell trituration, counting, and seeding onto 12mm glass coverslips pre-coated with poly-L-lysine. Cells were plated in Neurobasal A media containing 1% Glutamax and 10% fetal calf serum. After 3h, media was changed to Neurobasal A containing 2% B27 supplement and 1% Glutamax. After 7 days in vitro, 1 mM arabinofuranosylcytosine was added to stop glial growth, and cells were cultured for up to 17 days.
HEK293T cells
HEK293T cells (female) were cultured on 10cm or 6cm tissue culture dishes (Falcon) in DMEM high glucose media (HyClone) containing penicillin/streptomycin, L-glutamine, 1mM pyruvate and 10% fetal bovine serum.
METHOD DETAILS
DNA constructs
The Cre dependent eYFP, Teal-gephyrin and PSD95-mCherry plasmids (pFUdioeYFPW, pFudioTealgephyrinW and pFudioPSD95mCherryW, respectively) and the Cre plasmid (pSIN-W-PGK-Cre) have been previously described (Chen et al., 2012; Subramanian et al., 2013; Villa et al., 2016). Faithful expression of the fluorescently labeled gephyrin and PSD95 synaptic markers was previously validated, and shown to have no effect on spine, inhibitory synapse, or PSD95 densities or dynamics (Chen et al., 2012; Subramanian et al., 2013; Villa et al., 2016). The plasmids expressing OHT-Cre (pCAG-ERT2CreERT2; Addgene plasmid # 13777 (Matsuda and Cepko, 2007)), and Flp recombinase (pPGKFLPobpA; Addgene plasmid # 13793 (Raymond and Soriano, 2007)) were gifts from Connie Cepko and Phillippe Soriano, respectively. To generate Flp dependent constructs in the same backbone as the Cre constructs, we first replaced mKate2 with eYFP (from pFudioeYFP) between Asc1 and Nhe1 sites of pAAV-EF1a-fio-H2B-LSS-mKate2, a plasmid with FRT sites in a double inverted orientation (a gift from Jerry Chen). Next, the entire cassette containing FRT sites and eYFP was used to replace loxP sites and eYFP between BamHI and EcoRI sites in pFudioeYFPW. The eYFP between the AscI and NheI sites in the resultant plasmid, pFudioFRTeYFPW, was replaced with PSD95-Teal, which was amplified with additional NheI and MluI sites, to generate pFudioFRTPSD95TealW.
The pCAG-Synaptophysin-TdTomato-IRES2-OHT-Cre plasmid was made as follows. ERT2CreERT2 (OHT-Cre) was amplified from pCAG-ERT2CreERT2 with BstXI and Not1 site containing primers and cloned between the BstXI and Not1 sites of pCMV-Cpg15-IRES2-GFP (a gift from Tadahiro Fujino), after removing GFP, to generatepCMV-Cpg15-IRES2-OHT-Cre. Next, the CAG promoter fragment between Ase1and EcoR1 sites from pCAG-ERT2CreERT2 was used to replace the CMV promoter between the same sites in pCMV-Cpg15-IRES2-OHT-Cre to generatepCAG-Cpg15-IRES-OHT-Cre. Finally, CPG15 between the EcoRI and XmaI sites was replaced with Synaptophysin-TdTomato, which was amplified from the plasmid Ai34 (Addgene plasmid# 34881 (Madisen et al., 2012), a gift from Hongkui Zheng) with primers containing MfeI and XmaI sites, to generate pCAG-Synaptophysin-TdTomato-IRES2-OHT-Cre.
To generate Cre dependent Cpg15, the Cpg15-IRES2 fragment was amplified using primers with added AgeI and Nhe1 sites and cloned between the corresponding sites in pFudio-AscI-AgeI-Nhe1W (Villa et al., 2016)to create pFudio-Cpg15-IRES2W. Next, TdTomato was amplified using primers with added AgeI and AscI sites and cloned between these sites in pFudio-Cpg15-IRES2W to create pFudio-Cpg15-IRES2-TdTomatoW.
Plasmids for GluA1 expression in co-immunprecipitation experiments were generated by cutting prk5-GluA1-EGFP (gift from R. Huganir) with MluI, and replacing the EGFP tag after the GluA1 signal sequence with a HA tag. Prk5-CPG15-Flag was generated by cutting prk5-GluA1-EGFP (gift from R. Huganir) with MluI and HindIII and replacing EGFP-GluA1 with CPG15-Flag amplified amplified from pIRES-EGFP-CPG15-Flag (Putz et al., 2005).
Surgical Procedures
In utero electroporation was performed as described previously (Villa et al., 2016). E15.5 embryos from WT, Het, floxed Cpg15 or Ai6 mice were injected with 1 μL of appropriate plasmids mixed with 1% fast green into the right lateral ventricle using a 32 gauge Hamilton syringe (Hamilton company). A pair of platinum electrodes (Protech International) placed to target visual cortex was used to provide 5 pulses of 36 V (50 ms duration at 1 Hz) from a square wave electroporator (ECM830, Harvard Apparatus). To achieve sparse labeling optimal for single neuron imaging with high incidence of fluorophore co-expression, we used high molar ratios of fluorophores and limiting amounts of recombinase. For labeling both PSD95 and gephyrin in WT and KO neurons, each embryo was injected with 0.7 μg pFudioeYFPW, 0.8 μg pFudioPSD95mCherryW, 0.4 μg pFudioTealgephyrinW and 0.06 μg of pSIN-W-PGK-Cre plasmids. For acute Cpg15 deletion experiments, each embryo was injected with 0.7 μg pFudioFRTeYFPW, 0.4 μg pFudioFRTPSD95TealW, 0.03 μg pPGKFLPobpA and 1 μg pCAG-Synaptophysin-TdTomato-IRES2-OHT-Cre plasmids. For inducible CPG15 experiments, each embryo received 0.7 μg pFudioFRTeYFPW, 0.4 μg pFudioFRTPSD95TealW, 0.03 μg pPGKFLPobpA, 0.8 μg each of pCAG-Ert2-Cre-Ert2 and pFudioCPG15IRESTdTomatoW plasmids.
Adult male pups (~P50-60) born after in utero electroporation were implanted with a 5 μm glass coverslip replacing a skull area over the occipital cortex in the right hemisphere as described (Villa et al., 2016). Mice were housed individually with Sulfamethoxazole (1 mg/ml) and trimethoprim (0.2 mg/ml) in the drinking water to retain optical clarity of the implanted windows (Lee et al., 2006).
Optical intrinsic signal imaging
~ 10-14 days after cranial window surgery, visual cortex was identified through intrinsic signal imaging as described previously (Villa et al., 2016). Briefly, animals were mildly anesthetized with 0.75%–1% isoflurane, restrained using a head mount, and placed facing a high refresh rate monitor at a distance of 25 cm with a horizontal bar (5° in height and 73° in width) drifting upward with a periodicity of 12 s for 60 s. Images were obtained continuously under 610 nm illumination with an intrinsic imaging system (LongDaq Imager, Optical Imaging Inc.) through a 2.5X/0.075 NA objective (Zeiss). Cortical intrinsic signal was computed by extracting the Fourier component of light reflectance changes matched to stimulus frequency from 4×4 spatially binned images. The fractional change in reflectance represents response magnitude, and the magnitude maps were thresholded at 30% of the peak-response amplitude.
In vivo two photon imaging
Following cranial window surgery, mice were allowed to recover for 2 weeks and then screened for labeled neurons in the visual cortex that were expressing all fluorophores. Well-isolated neurons were imaged daily or every two weeks depending on the experimental timeline. Mostly, one cell per mouse was imaged from anesthetized (1%–1.25% isoflurane) head fixed mice using a custom built two photon microscope. For inducible CPG15 experiments, synapses from more than one cell within the same imaging field were analyzed for each mouse. For neurons expressing YFP, Teal-gephyrin and PSD95-mCherry, the three fluorophores were simultaneously excited with a Mai Tai HP Ti:Sapphire laser (Spectra-Physics) at 915 nm to excite eYFP and Teal and a Chameleon compact OPO (Coherent) at 1,085 nm to excite mCherry. For neurons expressing YFP and PSD95-Teal with or without Synaptophysin-TdTomato, excitation was delivered by Mai Tai HP alone at 915nm. In experiments with Synaptophysin-TdTomato expression, cells were carefully chosen to have no overexpression of Synaptophysin, as judged by weak somal expression and the lack of dendritic synaptophysin puncta. A 192×192×200 μm neuronal volume at 250 nm/pixel XY was acquired for each cell by scanning the laser beams using galvanometric XY-scanning mirrors (6215H, Cambridge Technology). 0.9 μm/frame Z-resolution was achieved using a piezo actuator (Piezosystem, Jena). The output power from the 20X/1.0 NA water immersion objective (W Plan-Apochromat, Zeiss) was set to 50 mW. The emission signals were collected using the same objective, passed through an IR blocking filter (E700SP, Chroma Technology), and spectrally separated using dichroic mirrors at 520 nm and 560 nm. Emission signals were simultaneously collected with three independent PMTs after passing through the appropriate bandpass filters (485/70, 550/100 and 605/75).
Data analysis
Two photon raw data was processed for spectral linear unmixing as described previously (Villa et al., 2016) and the images were converted to a RGB image Z stack using MATLAB and ImageJ. Dendritic spines, PSD95, and gephyrin in dendritic segments located ~30mm or farther from the cell soma were scored manually with a custom written 4D point tracking system implemented in Fiji using a modified version of ObjectJ plugin (Villa et al., 2016). Analysis was performed blind to genotype or experimental conditions. Spines projecting in the Z axis were excluded from analysis. Gephyrin puncta were scored as synapses if they were at least 3×3 pixels or 8-9 clustered pixels and PSD95 puncta were scored as synapses if they were at least 2×2 pixels or 4-5 clustered pixels. In both cases, signal had to be present in two consecutive Z frames with average signal intensity at least four times above shot noise. We have previously confirmed using electron microscopy that our scoring of gephyrin and PSD95 faithfully represents inhibitory and excitatory synapses, respectively (Villa et al., 2016).
Percent dynamics were calculated as the fraction of synapses that appeared or disappeared between two consecutive sessions and calculated as ((Ngained+Nlost)/(N1total+N2total)) X100, where N1 and N2 represent total number of the same category of synapses in previous and next sessions, respectively. Ngained represents number of new synapses and Nlost represents the number of preexisting synapses that disappeared. Percentage gain was calculated as (Ngained/N2total) X 100 and percentage loss was calculated as (Nlost/N1total) X 100. Gain or loss per 100 μm was calculated as (Ngained/total dendritic length) X100 and (Nlost/total dendritic length) X 100, respectively. For the light deprivation experiments, a total of 1152 spines (697 PSD95+, 130 DIS and 325 PSD95 negative spines) from 2304 μm of dendrites from 6 WT mice and 853 spines (346 PSD95+, 187 DIS and 320 PSD95 negative spines) from 2322 μm of dendrites from 6 KO mice were analyzed. For the daily imaging experiments to calculate daily or weekly dynamics, 1330 spines (796 PSD95+, 204 DIS and 330 PSD95 negative spines) from 2960 μm of dendrites from 8 WT mice and 1067 spines (527 PSD95+, 225 DIS and 315 PSD95 negative spines) from 3193 μm of dendrites from 8 KO mice were analyzed. For acute Cpg15 deletion experiments, 1460 PSD95+ spines were analyzed from 3464 μm of dendrites from 7 floxed Cpg15 mice. 20% of these spines overlapped with Synaptophysin-TdTomato and were excluded from analysis to isolate the role of postsynaptic CPG15. For exogenous CPG15 expression, 1108 PSD95+ spines were analyzed from 2508 μm of dendrites from 5 WT mice.
Wide-field fluorescence images of 100 μm dendritic sections from cultured hippocampal neurons were obtained using a Nikon epifluorescent microscope using a 40x objective and Spot Software. Quantification of spine density and presence of surface GluA1 and PSD95 was conducted using ImageJ software. Dendritic spines labeled with Phalloidin-488 were identified and determined to contain surface-labeled GluA1 or PSD95 by the presence of puncta above background > 5×5 pixels. Three or 4 separately prepared cultures per time point were imaged. For WT cultures: 246,990,2004 and 843 spines were analyzed for DIV7,10,14 and 17 time points, respectively. For KO cultures: 213, 606, 2593 and 1054 spines were analyzed for DIV 7, 10, 14 and 17 time points, respectively.
Statistical analyses were performed using t test, repeated-measures ANOVA, or ANOVA with Tukey’s posthoc test for multiple-comparisons. Sample size for in vivo experiments represents number of mice and for in vitro experiments represents number of independently prepared cultures. Sample sizes were estimated based on comparable previously published literature. p value < 0.05 is considered significant. *, **, *** represents p values < 0.05, 0.01 and 0.001, respectively.
4-hydroxytamoxifen (4-OHT) preparation and administration
4-OHT (Sigma) was dissolved in 100% ethanol by shaking at 37°C to make a 20 mg/ml stock. For administering 50 mg/kg body weight of 4-OHT, the appropriate stock volume was mixed with ~200-250 μl corn oil (Sigma) by vortexing. Ethanol was removed using speedvac and the drug was delivered through intraperitoneal injection.
Immunohistochemistry
Brains were fixed with 4% Paraformaldehyde through transcardial perfusion and sectioned at 50 μm thickness using a vibratome. Sections were blocked with 10% Normal goat serum in 1% Triton X-100/PBS for 2 hours, then incubated with rabbit Dsred antibody (Clontech; 1:500 dilution; Cat# 632496; RRID:AB_10013483) overnight, and washed with PBS. After further incubation with Alexa Fluor 555 conjugated goat-anti-rabbit secondary antibodies (Thermo Fisher Scientific; 1:400; Cat # A21428; RRID:AB_2535849), and washing, the slices were mounted on slides using Fluoromount-G (Southern Biotech). Images were acquired using an upright fluorescence microscope (Nikon).
Immunocytochemistry of cultured neurons was performed by first isolating coverslips containing hippocampal neurons at DIV 7, 10, 14 or 17. Cells were surfaced-labeled with 10 μg/mL rabbit anti-GluA1 primary antibody (Calbiochem; Cat# PC246; RRID:AB_564636), or chicken anti-MAP2 (negative control; Novus Biologicals; Cat# NB300-213; RRID:AB_2138178) for 15 min at 37C, then washed with PBS and fixed in 4% paraformaldehyde, 4% sucrose solution for 4 min at room temperature. Cells were then permeabilized with 0.25% Triton X-100 for 4 min at room temperature, washed with PBS, and blocked in 1% BSA for 1 hour. Coverslips were incubated in 1% BSA containing 2 mg/mL mouse anti-PSD95 (ThermoFisher; Cat# MA1-046; RRID:AB_2092361) for 2 hours at room temperature, washed in PBS, and incubated in 1% BSA containing Alexa Fluor Phalloidin-488 (1:500; ThermoFisher Cat # A12379; RRID:AB_2315147), Alexa Fluor anti-rabbit 555 (1:500; ThermoFisher Cat # A21428; RRID:AB_2535849) or anti-chicken 555; (1:500; ThermoFisher Cat# A21437; RRID:AB_2535858), and Alexa Fluor anti-mouse 647 (ThermoFisher Cat # A21235; RRID:AB_2535804) for 1 hour at room temperature. Coverslips were washed and mounted in Fluoro-mount G (Southern Biotech). Images were acquired using an upright fluorescence microscope (Nikon).
Co-Immunoprecipitation
HEK293T cells were transfected with calcium-phosphate for expression of HA-tagged GluA1 (prk5-HA-GluA1) and Flag-tagged CPG15 (prk5-CPG15-Flag). HEK293T cells were plated on 6cm culture dishes at a medium density (2×106 in 5mL) and allowed to grow for 24 hours. One microgram of the HA-GluA1 plasmid and a molar equivalent of Flag-CPG15 plasmid was transfected in a mixture of 0.1XTE (pH8.0), 2x HBSS (pH 7.40) and CaCl2. AGFP-expression vector was included to visualize transfection efficiency. HEK293T cells either received HA-GluA1 alone or with Flag-CPG15, with varying amounts of GFP-expression vector to keep molar ratios equal. Cells were lysed in buffer containing 100mM NaCl and 1% Triton X-100 for 20 min, lysates were spun at 14,000rpm at 4C for 30 min, and lysates were coupled to 40 mL EZviewanti-Flag M2 Affinity Gel (Sigma) for 2 hours at 4C. Beads were washed 3×5 min in lysis buffer and eluted in 1XSDS sample buffer for 10 min at 65C. Samples were run using SDS-PAGE and probed using rabbit antiHA (Covance; 1:2000; RRID:AB_2565335) and mouse anti-Flag (Sigma; 1:2000; RRID:AB_259529). Blots were developed using an Odyssey (Li-Cor) infrared scanner after staining with secondary anti-mouse IgG antibody DyLight 800 (Rockland; 1:10 000; RRID:AB_11182794), or respectively, anti-rabbit IgG antibody DyLight 680 (Rockland; 1:10 000; RRID:AB_11181436).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical methods are specified in each figure legend, in the Methods Details section, and in relevant places in the results section. Statistical methods used are Student t test, repeated-measures ANOVA and one way ANOVA with post hoc Tukey test for multiple comparisons. SPSS, Graph pad Prism and Microsoft excel were used for statistical and data analyses.
DATA AND CODE AVAILABILITY
The datasets supporting the current study have not been deposited in a public repository but will be available from the corresponding author on request. The code for object 4D point tracking system implemented in Fiji can be obtained by contacting nedivi@mit.edu.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti dsRed | Clontech | Cat # 632496; RRID:AB_10013483 |
| Rabbit polyclonal anti GluR1 | EMD Millipore | Cat # PC246; RRID:AB_564636 |
| Chicken polyclonal anti MAP2 | Novus Biological | Cat # NB300–213; RRID:AB_2138178 |
| Mouse monoclonal anti PSD95 | ThermoFisher | Cat # MA1–046; RRID:AB_2092361 |
| Mouse monoclonal anti FLAG M2 | Sigma-Aldrich | Cat # F3165; RRID:AB_259529 |
| Mouse monoclonal anti HA.11 | Biolegend (previously Covance) | Cat # 901513; RRID:AB_2565335 |
| Goat anti Rabbit secondary antibody – Alexa Fluor 555 | ThermoFisher | Cat # A21428; RRID:AB_2535849 |
| Goat anti Mouse secondary antibody – Alexa Fluor 647 | ThermoFisher | Cat # A21235; RRID:AB_2535804 |
| Goat anti Chicken secondary antibody – Alexa Fluor 555 | ThermoFisher | Cat# A21437; RRID:AB_2535858 |
| Phalloidin-488 – Alexa Fluor | ThermoFisher | Cat # A12379; RRID:AB_2315147 |
| Anti Rabbit IgG Antibody DyLight 680 | Rockland | Cat # 610-144-002-0.5; RRID:AB_11181436 |
| Anti Mouse IgG Antibody DyLight 800 | Rockland | Cat # 610-145-002-0.5; RRID:AB_11182794 |
| EZview Red ANTI-FLAG® M2 Affinity Gel | Sigma | Cat# F2426-1ML; RRID: AB_2616449 |
| Bacterial and Virus Strains | ||
| One Shot STBL3 chemically competent E.coli | ThermoFisher | Cat # C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-hydroxy Tamoxifen | Sigma-Aldrich | H7904 |
| Experimental Models: Cell Lines | ||
| HEK293T Cells | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: cpg15flox/+ | Fujino et al., 2011 | N/A |
| Mice: cpg15−/−;cpg15+/−;cpg15+/+ | Fujino et al., 2011; The Jackson Laboratory | Stock No: 018402 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm6(CAG ZsGreen1)Hze/J | The Jackson Laboratory | Stock No: 007906 |
| Oligonucleotides | ||
| Primers for ERT2CreERT2: | This paper | N/A |
| 5′ ATAGCCCCACAACCATGGCTGGAGACATGAG3′ | ||
| 5′TAGGTCGCGGCCGCTATCAAGCTGTGGCAGG3′ | ||
| Primers for Synaptophysin TdTomato | This paper | N/A |
| 5′ACAGTACAATTGCCGGGTGAGCCGCCACCATGGACGTG3′ | ||
| 5′TACATCCCCGGGCTACTTGTACAGCTCG3′ | ||
| Primers for Cpg15-Ires2 | This paper | N/A |
| 5′ACAGTAGCTAGCGCCGCCACCATGGGACTTAAGTTGAACG3′ | ||
| 5′ATCCAGACCGGTATTATCATCGTGTTTTTCAAAGG3′ | ||
| Primers for TdTomato | This paper | N/A |
| ATCCAGACCGGTGCCACAACCATGGTGAGCAAGGGCGAGG | ||
| TACACTGGCGCGCCTTACTTGTACAGCTCG | ||
| Recombinant DNA | ||
| pFUdioeYFPW | Villa et al., 2016 | RRID:Addgene_73858 |
| pFudioTealgephyrinW | Chen et al., 2012 | RRID:Addgene_73918 |
| pFudioPSD-95mCherryW | Villa et al., 2016 | RRID:Addgene_73919 |
| pCAG-ERT2CreERT2 | Matsuda and Cepko; 2007 | RRID:Addgene_13777 |
| pPGKFLPobpA | Raymond and Soriano; 2007 | RRID:Addgene_13793 |
| pSIN-W-PGK-Cre | Subramanian and Morozov, 2011 | RRID:Addgene_101242 |
| pAAV-EF1a-fio-H2B-LSS-mKate2 | Jerry Chen | N/A |
| pFudioFRTPSD-95TealW | This paper | N/A |
| pCAG-Synaptophysin-TdTomato-IRES2-OHT-Cre | This paper | N/A |
| pCMV-Cpg15-IRES2-GFP | This paper | N/A |
| pCMV-Cpg15-IRES2-OHT-Cre | This paper | N/A |
| pCMV-Cpg15-IRES2-OHT-Cre | This paper | N/A |
| pCAG-Cpg15-IRES-OHT-Cre. | This paper | N/A |
| Ai34 | Madisen et al., 2012 | RRID:Addgene_34881 |
| pCAG-Synaptophysin-TdTomato-IRES2-OHT-Cre | This paper | N/A |
| pFudio-AscI-AgeI-Nhe1W | Villa et al., 2016 | N/A |
| pFudio-Cpg15-IRES2W | This paper | N/A |
| pFudio-Cpg15-IRES2-TdTomatoW | This paper | N/A |
| pRK5-HA-GluA1 | This paper | N/A |
| pRK5-Flag-CPG15 | This paper | N/A |
| Software and Algorithms | ||
| 4D point tracking system, ObjectJ | Villa et al., 2016 | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Neurolucida 360 | MBF Biosciences | https://www.mbfbioscience.com/neurolucida360 |
| Graphpad Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| SPSS | IBM | https://www.ibm.com/analytics/spss-statistics-software |
| Microsoft Excel | Microsoft | N/A |
Highlights.
The defining step for synapse stabilization in vivo is recruitment of PSD95
Experience influences PSD95 recruitment, but not spine initiation
CPG15 is required and sufficient for experience-dependent synapse stabilization
CPG15 interacts with AMPA receptors
ACKNOWLEDGMENTS
We thank Dr. Aygul Balcioglu for help with data analysis, Kelly Flavahan for help with animal work, Dr. Dalila Ordonez for help with figure edits, and members of the Nedivi lab and Dr. Charles Jennings for comments on earlier versions of the manuscript. This work was supported by National Eye Institute grants RO1-EY011894 and RO1-EY025437 (E.N.) and the JBP Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.012.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akten B, Kye MJ, Hao T, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, et al. (2011). Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc. Natl. Acad. Sci. USA 108, 10337–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, and Huganir RL (2012). Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol 22, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, and Isaac JT (2011). Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron 70, 510–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C, Groc L, and Choquet D (2007). The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron 53, 719–734. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Lin DT, Kang MG, Aizawa H, Takamiya K, and Huganir RL (2006). Synapse-specific regulation of AMPA receptor function by PSD-95. Proc. Natl. Acad. Sci. USA 103, 19535–19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosserhoff AK, Schneider N, Ellmann L, Heinzerling L, and Kuphal S (2017). The neurotrophin Neuritin1 (cpg15) is involved in melanoma migration, attachment independent growth, and vascular mimicry. Oncotarget 8, 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonomano DV, and Merzenich MM (1998). Cortical plasticity: from synapses to maps. Annu. Rev. Neurosci 21, 149–186. [DOI] [PubMed] [Google Scholar]
- Cane M, Maco B, Knott G, and Holtmaat A (2014). The relationship between PSD-95 clustering and spine stability in vivo. J. Neurosci 34, 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantallops I, and Cline HT (2008). Rapid activity-dependent delivery of the neurotrophic protein CPG15to the axon surface of neurons in intact Xenopus tadpoles. Dev. Neurobiol 68, 744–759. [DOI] [PubMed] [Google Scholar]
- Cantallops I, Haas K, and Cline HT (2000). Postsynaptic CPG15 promotes synaptic maturation and presynaptic axon arborelaboration in v/Vo. Nat. Neurosci 3, 1004–1011. [DOI] [PubMed] [Google Scholar]
- Caroni P, Donato F, and Muller D (2012). Structural plasticity upon learning: regulation and functions. Nat. Rev. Neurosci 13, 478–490. [DOI] [PubMed] [Google Scholar]
- Chen X, Nelson CD, Li X, Winters CA, Azzam R, Sousa AA, Leapman RD, Gainer H, Sheng M, and Reese TS (2011). PSD-95 is required to sus-tain the molecular organization of the postsynaptic density. J. Neurosci 31, 6329–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Villa KL, Cha JW, So PT, Kubota Y, and Nedivi E (2012). Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron 74, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Engelman H, and Scheiffele P (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, and Sudhof TC (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, and Greenberg ME (2008). Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol 24, 183–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT, and Debski E (1990). Patterned activity, synaptic convergence, and the NMDA receptor in developing visual pathways. Annu. Rev. Neurosci 13, 129–154. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Shatz CJ, and Nedivi E (1999). Dynamic regulation of cpg15 during activity-dependent synaptic development in the mammalian visual system. J. Neurosci 19, 7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Mendez P, Poglia L, and Muller D (2008a). Activity-dependent PSD formation and stabilization of newly formed spines in hippocampal slice cultures. Cereb. Cortex 18, 151–161. [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, and Muller D (2008b). LTP promotes a selective long term stabilization and clustering of dendritic spines. PLoS Biol. 6, e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, and Malinow R (2004). Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J. Neurosci 24, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, and Malinow R (2007). PSD-95 is required for activity-driven synapse stabilization. Proc. Natl. Acad. Sci. USA 104, 4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, and Nicoll RA (2008). Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc. Natl. Acad. Sci. USA 105, 20953–20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, and Bonhoeffer T (1999). Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399, 66–70. [DOI] [PubMed] [Google Scholar]
- Fujino T, Lee WC, and Nedivi E (2003). Regulation of cpg15 by signaling pathways that mediate synaptic plasticity. Mol. Cell. Neurosci 24, 538–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T, Leslie JH, Eavri R, Chen JL, Lin WC, Flanders GH, Borok E, Horvath TL, and Nedivi E (2011). CPG15 regulates synapse stability in the developing and adult brain. Genes Dev. 25, 2674–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts SN, Carroll R, Malenka RC, and Nicoll RA (2000). Distinct roles for ionotropic and metabotropic glutamate receptors in the maturation of excitatory synapses. J. Neurosci 20, 2229–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CS, and Shatz CJ (1993). Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell 72 (Suppl), 77–98. [DOI] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, and Svoboda K (2006). Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 4, e370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Gustafsson B, and Hanse E (2006). AMPA signalling in nascent glutamatergic synapses: there and not there!. Trends Neurosci. 29, 132–139. [DOI] [PubMed] [Google Scholar]
- Han K, and Kim E (2008). Synaptic adhesion molecules and PSD-95. Prog. Neurobiol 84, 263–283. [DOI] [PubMed] [Google Scholar]
- Hanse E, Seth H, and Riebe I (2013). AMPA-silent synapses in brain development and pathology. Nat. Rev. Neurosci 14, 839–850. [DOI] [PubMed] [Google Scholar]
- Harwell C, Burbach B, Svoboda K, and Nedivi E (2005). Regulation of cpg15 expression during single whisker experience in the barrel cortex of adult mice. J. Neurobiol 65, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Lin CC, and Sheng M (2003). Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci 23, 3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TC, and Zito K (2013). LTP-induced long-term stabilization of individual nascent dendritic spines. J. Neurosci 33, 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, and Svoboda K (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci 10, 647–658. [DOI] [PubMed] [Google Scholar]
- Hou Q, Huang Y, Amato S, Snyder SH, Huganir RL, and Man HY (2008). Regulation of AMPA receptor localization in lipid rafts. Mol. Cell. Neurosci 33, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua JY, and Smith SJ (2004). Neural activity and the dynamics of central nervous system development. Nat. Neurosci 7, 327–332. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, and Malenka RC (1995). Evidence for silent synapses: implications for the expression of LTP. Neuron 15, 427–434. [DOI] [PubMed] [Google Scholar]
- Isshiki M, Tanaka S, Kuriu T, Tabuchi K, Takumi T, and Okabe S (2014). Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat. Commun 5, 4742. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, and Nicoll RA (2008). Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci 9, 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, and Sheng M (2004). PDZ domain proteins of synapses. Nat. Rev. Neurosci 5, 771–781. [DOI] [PubMed] [Google Scholar]
- Kojima N, Shiojiri N, Sakai Y, and Miyajima A (2005). Expression of neuritin during liver maturation and regeneration. FEBS Lett. 579, 4562–4566. [DOI] [PubMed] [Google Scholar]
- Kwon HB, and Sabatini BL (2011). Glutamate induces de novo growth of functional spines in developing cortex. Nature 474, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JT, Hill TC, Park DK, Culp JH, and Zito K (2017). Protracted and asynchronous accumulation of PSD95-family MAGUKs during maturation of nascent dendritic spines. Dev. Neurobiol 77, 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WCA, and Nedivi E (2002). Extended plasticity of visual cortex in dark-reared animals may result from prolonged expression of cpg15-like genes. J. Neurosci 22, 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Huang H, Feng G, Sanes JR, Brown EN, So PT, and Nedivi E (2006). Dynamic remodeling of dendritic arbors in GABAergic interneurons of adult visual cortex. PLoS Biol. 4, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, and Sheng M (2003). Some assembly required: the development of neuronal synapses. Nat. Rev. Mol. Cell Biol 4, 833–841. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, and Malinow R (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375, 400–404. [DOI] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ 3rd, Gu X, Zanella S, et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci 15, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, and Svoboda K (1999). Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 233, 1923–1927. [DOI] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2007). Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA 104, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, and Riezman H (2004). Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell Biol 5, 110–120. [DOI] [PubMed] [Google Scholar]
- McAllister AK (2007). Dynamic aspects of CNS synapse formation. Annu. Rev. Neurosci 30, 425–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merianda TT, Gomes C, Yoo S, Vuppalanchi D, and Twiss JL (2013). Axonal localization of neuritin/CPG15 mRNA in neuronal populations through distinct 5‘ and 3‘ UTR elements. J. Neurosci 33, 13735–13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, et al. (1998). Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396, 433–439. [DOI] [PubMed] [Google Scholar]
- Naeve GS, Ramakrishnan M, Kramer R, Hevroni D, Citri Y, and Theill LE (1997). Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc. Natl. Acad. Sci. USA 94, 2648–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, and Bonhoeffer T (2004). Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44, 759–767. [DOI] [PubMed] [Google Scholar]
- Nedivi E, Fieldust S, Theill LE, and Hevron D (1996). A set of genes expressed in response to light in the adult cerebral cortex and regulated during development. Proc. Natl. Acad. Sci. USA 93, 2048–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Wu GY, and Cline HT (1998). Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science 231, 1863–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Javaherian A, Cantallops I, and Cline HT (2001). Developmental regulation of CPG15 expression in Xenopus. J. Comp. Neurol 435, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya NJ, Seeger C, Babai N, Gonzalez-Lozano MA, Mack V, Lodder JC, Gouwenberg Y, Mansvelder HD, Danielson UH, Li KW, et al. (2018). Noelin1 affects lateral mobility of synaptic AMPA receptors. Cell Rep. 24, 1218–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard N, Leslie JH, Trowbridge SK, Subramanian J, Nedivi E, and Fagiolini M (2014). Aberrant development and plasticity of excitatory visual cortical networks in the absence of cpg15. J. Neurosci 34, 3517–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz U, Harwell C, and Nedivi E (2005). Soluble CPG15 expressed during early development rescues cortical progenitors from apoptosis. Nat. Neurosci 3, 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CS, and Soriano P (2007). High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE 2, e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sando R, Bushong E, Zhu Y, Huang M, Considine C, Phan S, Ju S, Uytiepo M, Ellisman M, and Maximov A (2017). Assembly of excitatory synapses in the absence of glutamatergic neurotransmission. Neuron 94, 312–321.e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, and Huganir RL (2000). Postsynaptic organization and regulation of excitatory synapses. Nat. Rev. Neurosci 1, 133–141. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, and Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, and Nicoll RA (2002). Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc. Natl. Acad. Sci. USA 99, 13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Brechet A, Zolles G, Berkefeld H, MGller CS, Bildl W, Baehrens D, Huber B, Kulik A, et al. (2012). High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron 74, 621–633. [DOI] [PubMed] [Google Scholar]
- Shatz CJ (1990). Impulse activity and the patterning of connections during CNS development. Neuron 5, 745–756. [DOI] [PubMed] [Google Scholar]
- Sheng M, and Hoogenraad CC (2007). The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem 76, 823–847. [DOI] [PubMed] [Google Scholar]
- Sheng M, and Kim E (2011). The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol 3, a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Yoshida T, and Yamagata K (2016). Neuritin mediates activity-dependent axonal branch formation in part via FGF signaling. J. Neurosci 36, 4534–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigler A, Oh WC, Imig C, Altas B, Kawabe H, Cooper BH, Kwon HB, Rhee JS, and Brose N (2017). Formation and maintenance of functional spines in the absence of presynaptic glutamate release. Neuron 94, 304–311.e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Stogsdill JA, Pulimood NS, Dingsdale H, Kim YH, Pilaz LJ, Kim IH, Manhaes AC, Rodrigues WS Jr., Pamukcu A, et al. (2016). Astrocytes assemble thalamocortical synapses by bridging NRX1a and NL1 via Hevin. Cell 164, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son H, Banasr M, Choi M, Chae SY, Licznerski P, Lee B, Voleti B, Li N, Lepack A, Fournier NM, et al. (2012). Neuritin produces antidepressant actions and blocks the neuronal and behavioral deficits caused by chronic stress. Proc. Natl. Acad. Sci. USA 109, 11378–11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, and Nicoll RA (2003). Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci 23, 5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian J, and Morozov A (2011). Erk½ inhibit synaptic vesicle exocytosis through L-type calcium channels. J. Neurosci 31, 4755–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian J, Dye L, and Morozov A (2013). Rap1 signaling prevents L-type calcium channel-dependent neurotransmitter release. J. Neurosci 33, 7245–7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2018). Towards an Understanding of Synapse Formation. Neuron 100, 276–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft CE, and Turrigiano GG (2013). PSD-95 promotes the stabilization of young synaptic contacts. Philos. Trans. R. Soc. Lond. B Biol. Sci 360, 20130134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, and Mishina M (2010). Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 141, 1068–1079. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, and Rosenmund C (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. USA 99, 9037–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Sudhof TC, and Brose N (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, et al. (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287, 864–869. [DOI] [PubMed] [Google Scholar]
- Villa KL, Berry KP, Subramanian J, Cha JW, Oh WC, Kwon HB, Kubota Y, So PT, and Nedivi E (2016). Inhibitory synapses are repeatedly assembled and removed at persistent sites in vivo. Neuron 89, 756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites CL, Craig AM, and Garner CC (2005). Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci 28, 251–274. [DOI] [PubMed] [Google Scholar]
- West AE, and Greenberg ME (2011). Neuronal activity-regulated genetranscription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol 3, a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Malinow R, and Cline HT (1996). Maturation of a central glutamatergic synapse. Science 274, 972–976. [DOI] [PubMed] [Google Scholar]
- Yao JJ, Gao XF, Chow CW, Zhan XQ, Hu CL, and Mei YA (2012). Neuritin activates insulin receptor pathway to up-regulate Kv4.2-mediated transient outward K+ current in rat cerebellar granule neurons. J. Biol. Chem 287, 41534–41545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cudmore RH, Lin DT, Linden DJ, and Huganir RL (2015). Visualization of NMDA receptor-dependent AMPA receptor synaptic plasticity in vivo. Nat. Neurosci 18, 402–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Hu JX, Lu DX, Ji CX, Qi Y, Liu XY, Sun FY, Huang F, Xu P, and Chen XH (2017). Soluble cpg15 from astrocytes ameliorates neurite outgrowth recovery of hippocampal neurons after mouse cerebral ischemia. J. Neurosci 37, 1628–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, and Svoboda K (2009). Rapid functional maturation of nascent dendritic spines. Neuron 61, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository but will be available from the corresponding author on request. The code for object 4D point tracking system implemented in Fiji can be obtained by contacting nedivi@mit.edu.