

Abstract
Central melanocortin signaling plays an important role in regulation of energy homeostasis by leptin and insulin. We investigated the interaction between leptin, insulin, and melanocortin-4 receptors (MC-4Rs) in the control of renal sympathetic nerve activity (RSNA) in mice. We compared the effects of intracerebroventricular (ICV) administration of leptin, insulin, MC-3/4R agonist (MTII), and corticotrophin-releasing factor (CRF) on RSNA in leptin receptor-deficient (db/db) mice, MC-4R knock-out mice, and their wild-type controls. ICV administration of leptin and MTII caused a significant and dose-dependent increase in RSNA in control mice. As expected, leptin had no significant effect on RSNA in the db/db mice. Interestingly, db/db mice exhibited markedly attenuated RSNA responses to ICV administration of MTII. However, the increase in RSNA induced by insulin and CRF was comparable between db/db and control mice. In the heterozygous and homozygous MC-4R knock-out mice, the RSNA response to MTII was attenuated and abolished, respectively. The RSNA response to ICV leptin and insulin was also attenuated and abolished in the heterozygous and homozygous MC-4R knock-out mice, respectively. In contrast, CRF induced a similar increase in RSNA in the MC-4R knock-out and wild-type mice. Our data demonstrate that in the absence of leptin receptors, the sympathoexcitatory effects of melanocortin system stimulation are attenuated. In addition, the renal sympathoexcitatory responses to leptin and insulin are dependent on the MC-4R, demonstrating an important role for the MC-4R in the regulation of renal sympathetic nerve outflow by leptin and insulin.
Keywords: obesity, melanocortin system, leptin, insulin, sympathetic nerve activity, hypothalamus
Introduction
Leptin and insulin act in the CNS as afferent signals, to regulate food intake and energy expenditure (Schwartz et al., 2000). The action of these two hormones in the CNS is also known to modulate several other functions, including sympathetic nerve activity (Muntzel et al., 1995; Dunbar et al., 1997; Haynes et al., 1997).
Receptors for leptin and insulin are expressed in several sites in the brain including the arcuate nucleus of the hypothalamus (Schwartz et al., 2000). The leptin receptor is a single transmembrane protein that belongs to the cytokine-receptor superfamily. Six different alternatively spliced isoforms of this receptor have been identified (designated Ob-Ra to Ob-Rf) (Tartaglia, 1997). Five isoforms (Ob-Ra to Ob-Rd and Ob-Rf) differ in the length of their intracellular domain, whereas Ob-Re, which lacks the transmembrane domain, is a soluble form of the receptor. The Ob-Rb form that encodes the full receptor, including the long intracellular domain, appears to mediate most of the biological effects of leptin (Chen et al., 1996; Tartaglia, 1997). Absence of the Ob-Rb in db/db mice leads to severe obesity and multiple metabolic and endocrine abnormalities (Chen et al., 1996; Chua et al., 1996). Central or peripheral administration of leptin has no effect on food intake or body weight in db/db mice (Campfield et al., 1995; Harris et al., 2001).
The physiological role of brain insulin receptors in the regulation of energy balance has been demonstrated by neuron-specific deletion of this receptor throughout the CNS, which results in a marked increase in fat mass (Bruning et al., 2000). Furthermore, downregulation of the insulin receptor in the arcuate nucleus of the hypothalamus, using insulin receptor anti-sense, leads to hyperphagia and increased fat mass (Obici et al., 2002). Prevention of the sympathetic nerve responses to systemic administration of insulin by ablation of the anteroventral third ventricule (Muntzel et al., 1995) suggests that the sympathoactivation to insulin also emanates from the hypothalamus.
Several observations suggest that the melanocortin system is an essential mediator for leptin and insulin action in the CNS (Schwartz et al., 2000; Benoit et al., 2002). The melanocortins, such as α-melanocyte-stimulating hormone (α-MSH), are peptides that are processed from the proopiomelanocortin (POMC). Five melanocortin receptors (MC-1R to MC-5R) have been identified (Mountjoy et al., 1992; Adan and Gispen, 2000). MC-3R and MC-4R are highly expressed in the CNS (Mountjoy et al., 1992, 1994; Roselli-Rehfuss et al., 1993). The critical role for the MC-4R in energy balance was demonstrated by target disruption of the MC-4R gene that induces hyperphagia and obesity in mice (Huszar et al., 1997). Furthermore, pharmacological studies have shown that blockade of MC-4R prevents the effects of leptin (Seeley et al., 1997; Satoh et al., 1998) and insulin (Benoit et al., 2002) on food intake. Thus, MC-4R seems to play a pivotal role in the regulation of energy homeostasis by leptin and insulin.
The implication of the MC-4R in the control of sympathetic nerve activity is more controversial, however. In rat, we have shown previously that the effects of leptin on renal sympathetic nerve activity (RSNA) are mediated by the melanocortin receptors because intracerebroventricular (ICV) administration of the MC-3/4R antagonist SHU9119 blocks the sympathoexcitatory effects of leptin to the kidney but not to brown adipose tissue (Haynes et al., 1999). In contrast, the agouti obese mice, in which the obesity is caused by ubiquitous overexpression of agouti protein that blocks hypothalamic MC-4R, are resistant to the appetite- and weight-reducing effects of leptin but have preservation of the increase in RSNA induced by leptin (Correia et al., 2002; Rahmouni et al., 2002), suggesting that the MC-4Rs are not essential for the control of RSNA by leptin. Thus, the relationship between leptin and MC-4R in control of sympathetic nerve activity is currently unclear. Furthermore, the involvement of the MC-4R in the control of sympathetic nerve traffic by insulin has not been reported.
In the present study, we evaluated the interaction between leptin, insulin, and the MC-4R in the regulation of sympathetic nerve outflow to the kidney. First, to evaluate the consequences of absence of leptin receptors on the RSNA response to stimulation of MC-4R and insulin, we compared the effects of stimulation of the MC-4R on RSNA in leptin-deficient db/db mice and their control littermates. Second, we examined the role of the MC-4R in mediating the RSNA response to leptin and insulin. For this, we studied the effects of leptin and insulin on RSNA in the MC-4R knock-out mice and their wild-type littermates.
Materials and Methods
Animals. db/db mice (C57BL/KsJ-Lep db) and their littermate controls were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice of mixed 129/Sv × C57BL/6J background heterozygous for the disrupted MC-4R allele were first obtained from Dr. Dennis Huszar (Millenium Pharmaceuticals, Cambridge, MA) and then bred to generate male and female MC4-R (-/-), MC-4R (+/-) and wild-type mice. Genotyping was performed by PCR as described (Marsh et al., 1999). All mice were maintained on a 12 hr light/dark cycle and fed standard mouse chow and water ad libitum. The University of Iowa Animal Research Committee approved all procedures.
Cannula implantation. Adult mice were anesthetized by intraperitoneal injection of ketamine (91 mg/kg) and xylazine (9.1 mg/kg) and placed in a stereotactic device (Kopf Instruments). A sterile guide cannula 9 mm in length was implanted into the lateral brain ventricle (0.3 mm posterior and 1 mm lateral relative to bregma, and 3 mm below the surface of the skull). The cannula was then fixed in place using dental cement. After surgery, a 33 gauge wire plug was inserted into each cannula to prevent blockage. The position of the cannulas was verified at the end of the experiments by dye administration before animals were killed and by histological analysis.
Recording of hemodynamic and renal sympathetic nerve activity. After 5–7 d of postoperative recovery from ICV cannulation, mice were anesthetized using an intraperitoneal ketamine/xylazine mixture and a catheter inserted in the jugular vein for maintenance of anesthesia with α-chloralose (25 mg · kg -1 · hr -1). The trachea was cannulated, allowing the mice to breath spontaneously oxygen-enriched air. Rectal temperature was maintained at 37.5°C using a temperature-controlled surgical table and a lamp. Hemodynamic parameters (arterial pressure and heart rate) were monitored through a pressure transducer connected to the catheter inserted in the left carotid artery. RSNA was measured by multi-fiber recording. Briefly, the left kidney was exposed retroperitoneally through a flank incision, and a renal nerve was carefully dissected free and placed on a bipolar 36 gauge platinum–iridium electrode (Cooner Wire). When optimum recording of RSNA was obtained, the electrode was covered with silicone adhesive gel (World Precision Instruments).
The nerve electrode was attached to a high-impedance probe (HIP-511, Grass Instruments). The nerve signal was amplified 10,000 times with a Grass P5 AC pre-amplifier, filtered at a 100 and 1000 Hz cutoff with a nerve traffic analysis system (model 706C, University of Iowa Bioengineering), and led to an oscilloscope (model 54501A, Hewlett-Packard) with a cursor that was positioned precisely above the background noise. The nerve traffic analyzer counted the action potentials that exceeded this threshold voltage. Both the counted action potentials and the renal neurogram were routed to a MacLab analog-to-digital converter (model 8S, AD Instruments) for permanent recording and data analysis on a Macintosh 9500 computer. To ensure that electrical noise was excluded in the assessment of sympathetic outflow, RSNA was corrected for postmortem background activity.
Experimental protocol. After surgery for RSNA recording was completed, animals were allowed to stabilize for 20 min. Baseline RSNA, mean arterial pressure (MAP), and heart rate (HR) were then collected on two occasions during a 10 min control period and averaged to obtain a single value for this control period. Each mouse received one ICV injection, over 1 min, of 2 μl of vehicle (0.9% NaCl) or one of the following agents: leptin (R & D Systems, Minneapolis, MN) at a dose of 0.5, 1, or 2 μg; MC-3/4R agonist MTII (Phoenix Pharmaceuticals) at a dose of 0.5 or 2 μg; 20 μU of insulin (Novo Nordisk Pharmaceuticals); or 5 μg of corticotrophin releasing factor (CRF) (Sigma Aldrich). Because it has been reported that the rise in RSNA induced by leptin may result from its peripheral action (Tanida et al., 2000), in one experiment the effects of intravenous leptin (100 μg) were compared between db/db and control mice. After administration of experimental agents, hemodynamic and RSNA were recorded continuously during the next 4 hr, and data were collected every 15 min. At the end of the experiment, artery blood was collected for leptin and insulin assay. Mice were then killed by methohexital overdose. In the MC-4R knock-out mice and their wild-type littermates, we measured at kill the weights of three fat pads: the interscapular brown adipose tissue (BAT), epididymal fat, and renal fat.
Plasma assay. Blood obtained from mice was immediately centrifuged at 5000 rpm for 5 min at room temperature. The collected plasma was frozen at -20°C. Plasma concentration of leptin and insulin were measured by radioimmunoassay using commercially available kits (both from Linco).
Data analysis. Because there is significant inter-individual variation in baseline RSNA, the data for RSNA are expressed as percentage change from baseline with 0% as baseline. The RSNA in the fourth hour represents the average of the four measurements obtained in the last hour of the experiment (maximal response). All results are expressed as mean ± SEM and analyzed using Student's t test, one-way ANOVA, or two-way ANOVA. When ANOVA reached significance, post hoc comparisons were made using Bonferroni test. p < 0.05 was considered to be statistically significant.
Results
Effects of leptin on RSNA in control and db/db mice
As shown in Table 1, the obese db/db mice had higher levels of MAP measured at baseline under anesthesia. Obese mice also had significantly higher baseline RSNA. The circulating levels of leptin and insulin were dramatically elevated in the db/db mice as compared with the control mice.
Table 1.
Body weight, hemodynamics, RSNA, and endocrine data obtained from db/db mice and their wild-type controls
|
Parameter |
Control mice |
db/db mice |
|---|---|---|
| Body weight (g) | 31 ± 1 | 53 ± 1* |
| MAP (mmHg) | 84 ± 1 | 96 ± 2* |
| HR (bpm) | 308 ± 5 | 352 ± 28 |
| RSNA (v × sec/min) | 1.3 ± 0.03 | 1.8 ± 0.10* |
| Leptin (ng/ml) | 7 ± 1 | 220 ± 48* |
| Insulin (ng/ml)
|
1.08 ± 0.15
|
19.6 ± 3.00*
|
Body weight, MAP, HR, and RSNA were obtained at baseline, whereas endocrine parameters (plasma leptin and insulin) were measured at the end of the protocol. HR, Heart rate; MAP, mean arterial pressure; RSNA, renal sympathetic nerve activity. Data are mean ± SEM of 49 control mice and 48 db/db mice. *p<0.0001 versus control mice.
To determine whether the sympathoexcitatory effects of leptin in mice were receptor mediated, we compared the RSNA response to leptin in control and leptin-deficient db/db mice. In control mice, ICV administration of different doses of leptin caused a significant and dose-dependent increase in RSNA (p = 0.001) (Fig. 1A), with a 297 ± 97% increase in the fourth hour after the 2 μg dose (p < 0.01 vs vehicle). Despite this robust increase in RSNA induced by ICV administration of 2 μg of leptin, arterial pressure did not change significantly (MAP was 80 ± 4 mmHg at baseline and 79 ± 8 mmHg in the fourth hour). In all groups, there was a tendency for HR to increase during the study (from 301 ± 18 bpm at baseline to 466 ± 70 bpm in the fourth hour after ICV administration of 2 μg of leptin), but the magnitude of the increase did not differ significantly between leptin and vehicle groups. As expected, the leptin receptor-deficient db/db mice exhibited markedly blunted RSNA responses to ICV administration of leptin (p = 0.81) (Fig. 1B).
Figure 1.
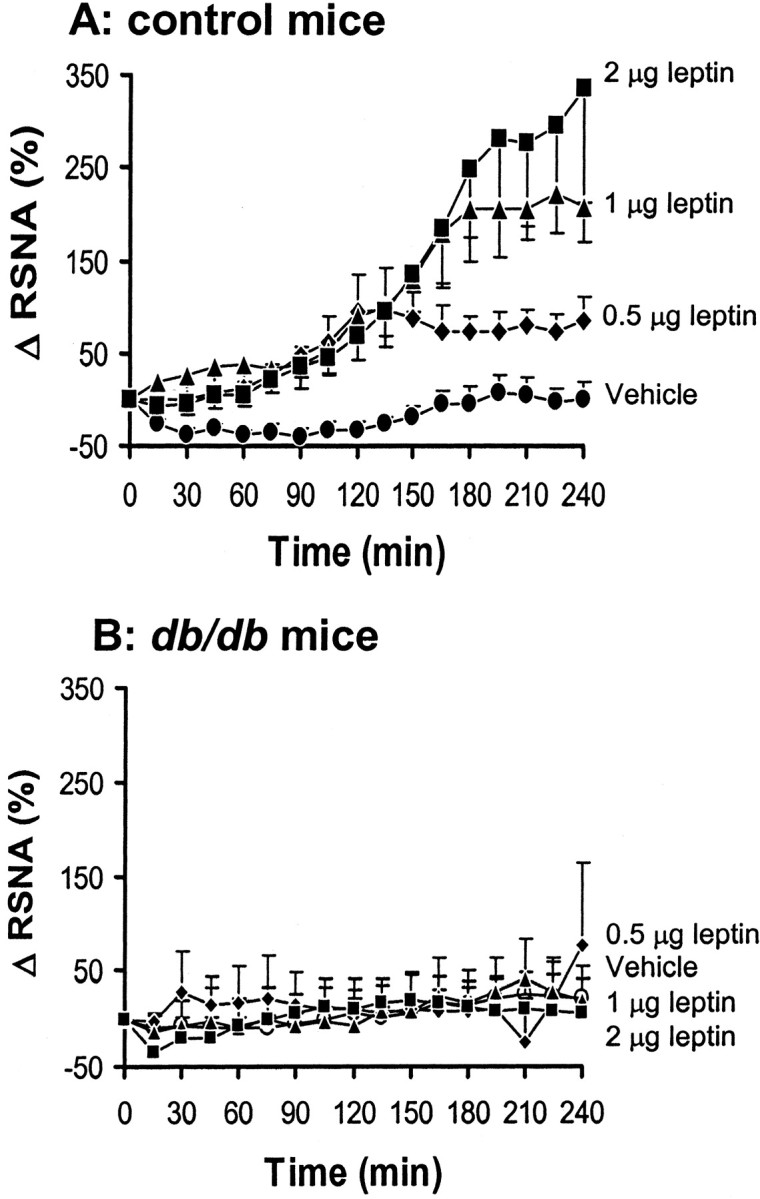
Effect of ICV administration of leptin on renal sympathetic nerve activity (RSNA) in control and db/db mice. A, Dose–response of RSNA to ICV administration of leptin in control mice. B, Absence of RSNA response to ICV administration of leptin in db/db mice. Δ RSNA represents the percentage change in RSNA from baseline. Data represent means ± SEM; n = 7–9 mice for each group.
Because it has been shown that the renal sympathoexcitatory action of leptin could be caused by its systemic action (Tanida et al., 2000), we tested the effects of intravenous administration of 100 μg of leptin on RSNA in control and db/db mice. We have shown previously that this dose of leptin is effective in increasing RSNA in mice (Correia et al., 2002). A significant increase in RSNA (as compared with the ICV vehicle-treated mice) was observed in control mice (154 ± 26% in the fourth hour; n = 7; p = 0.0002) but not in the db/db mice (16 ± 9% in the fourth hour; n = 6; p = 0.76).
Effects of insulin and CRF on RSNA in control and db/db mice
To determine whether absence of leptin receptors affects the sympathoexcitatory effects of stimulation of the melanocortin system, we compared the RSNA response to the MC-3/4R agonist MTII in control mice and db/db mice. As depicted in Figure 2A, ICV administration of MTII caused a significant (p = 0.0083) and dose-dependent increase in RSNA, with a 291 ± 68% (p < 0.01 vs vehicle) increase from baseline in the fourth hour at the highest dose. In db/db mice, MTII at a dose of 0.5 and 2 μg had no significant effect on RSNA (p = 0.83) (Fig. 2B). A 10-fold higher dose (20 μg) of MTII was required to induce a significant increase in RSNA (170 ± 40% in the fourth hour; p < 0.05) in db/db mice.
Figure 2.

Effect of ICV administration of MC-3/4R agonist (MTII) on renal sympathetic nerve activity (RSNA) in control mice and db/db mice. A, Dose–response of RSNA to ICV administration of MTII in control mice. B, Attenuated RSNA response to ICV administration of MTII in db/db mice. Δ RSNA represents the percentage change in RSNA from baseline. Data represent mean ± SEM; n = 6–9 mice for each group.
To examine whether the RSNA response to other stimuli were attenuated in db/db mice, we assessed the effects of ICV administration of insulin (20 μU) and CRF (5 μg) in db/db mice and their littermate controls. As shown in Figure 3, both insulin and CRF caused a significant (p < 0.01) increase in RSNA in control and db/db mice with the same time course and magnitude.
Figure 3.

Effect of intracerebroventricular (ICV) administration of 20 μU of insulin (A) and 5 μg of CRF (B) on renal sympathetic nerve activity (RSNA) in control mice and db/db mice. Δ RSNA represents the percentage change in RSNA from baseline. Data represent means ± SEM; n = 6–9 mice for each group.
There was no significant change in MAP and HR after ICV administration of MTII, insulin, or CRF as compared with vehicle (data not shown).
Effects of MTII and CRF on RSNA in MC-4R knock-out mice
As shown in Table 2 and as expected from a previous report (Huszar et al., 1997), graded knock-out of the MC-4R caused graded obesity and a graded increase in BAT, epididymal fat, and renal fat. Plasma levels of leptin and insulin were also elevated in the homozygous MC-4R knock-out mice and intermediate in the heterozygous mutant mice. Baseline MAP, HR, and RSNA did not differ between the MC-4R knock-out mice and their wild-type controls when measured in the anesthetized state.
Table 2.
Different physiological parameters obtained from heterozygous and homozygous MC4R knock-out mice and their wild-type controls
|
Parameter |
Wild type |
MC4R(+/−) |
MC4R(−/−) |
|---|---|---|---|
| Body weight (g) | 23±1 | 31±2* | 37±2† |
| BAT (g) | 0.067±0.003 | 0.113±0.016 | 0.140±0.027* |
| Epididymal fat (g) | 0.209±0.038 | 0.956±0.259* | 1.610±0.276* |
| Renal fat (g) | 0.100±0.022 | 0.293±0.101 | 0.528±0.064* |
| MAP (mmHg) | 74±3 | 72±2 | 79±2 |
| HR (bpm) | 259±31 | 227±41 | 213±21 |
| RSNA (v × sec/min) | 1.2±0.2 | 1.2±0.1 | 1.5±0.1 |
| Leptin (ng/ml) | 3.1±0.7 | 8.6±2.6 | 24.1±5.6† |
| Insulin (ng/ml)
|
0.8±0.2
|
1.1±0.3
|
2.6±0.7*
|
Body weight, MAP, HR, and RSNA were obtained at baseline, and fat pads and endocrine parameters (plasma leptin and insulin) were measured at the end of the protocol. BAT, Brown adipose tissue; HR, heart rate; MAP, mean arterial pressure; RSNA, renal sympathetic nerve activity. Data are mean ± SEM of 20-27 mice per group. *p < 0.05 versus MC-4R (+/+); †p < 0.05 versus MC-4R (+/+) and MC-4R (+/−).
To assess whether mice lacking the MC4-R exhibited an alteration in the RSNA response to MTII, we compared the effects of MTII on RSNA between homozygous and heterozygous mutant mice and their wild-type controls. As expected, ICV administration of 2 μg of MTII caused a significant increase in RSNA in the wild-type mice (299 ± 48% in the fourth hour; p < 0.001 vs vehicle), but MTII had no significant effect on RSNA in the homozygous MC-4R knock-out mice. The RSNA response to MTII in the heterozygous mutant mice was intermediate between the wild-type and homozygous mutant siblings [153 ± 31% in the fourth hour; p < 0.001 vs vehicle and p < 0.05 vs MC-4R (+/+) and MC-4R (-/-)]. However, lack of the MC4-R did not alter the RSNA response to ICV administration of 5 μg of CRF, because the renal sympathoactivation induced by CRF was comparable in wild-type and MC4-R knock-out mice (p = 0.78) (Fig. 4).
Figure 4.

Effect of ICV administration of 2 μg of MC-3/4R agonist (MTII) and 5 μg of CRF on renal sympathetic nerve activity (RSNA, mean of the fourth hour) in homozygous and heterozygous MC-4R knock-out mice as compared with their wild-type controls. Δ RSNA represents the percentage change in RSNA from baseline. Data represent mean ± SEM; n = 5–9 mice for each group. *p < 0.05 versus vehicle group; *p < 0.05 versus MC-4R (+/-) and MC-4R (-/-); #p < 0.05 versus MC-4R (-/-).
Effects of leptin and insulin on RSNA in MC-4R knock-out mice
To investigate the role of MC-4R in the RSNA response to leptin, we compared the effect of leptin on RSNA between wild-type mice and MC-4R knock-out mice. ICV administration of 2 μg of leptin increased RSNA in the wild-type mice (220 ± 45% in the fourth hour; p < 0.001 vs vehicle), but leptin had no significant effect on RSNA in the homozygous MC-4R knock-out mice (Fig. 5). A 10-fold higher dose (20 μg) of leptin did not affect RNSA in the null mutant mice (28 ± 22% in the fourth hour; n = 5; p = 0.58). As with the ICV injection of MTII, the RSNA response to ICV leptin in the heterozygous MC-4R knock-out mice was intermediate between the wild-type and the homozygous mutant mice [111 ± 19% in the fourth hour; p < 0.01 vs vehicle and p < 0.05 vs MC-4R (+/+)].
Figure 5.
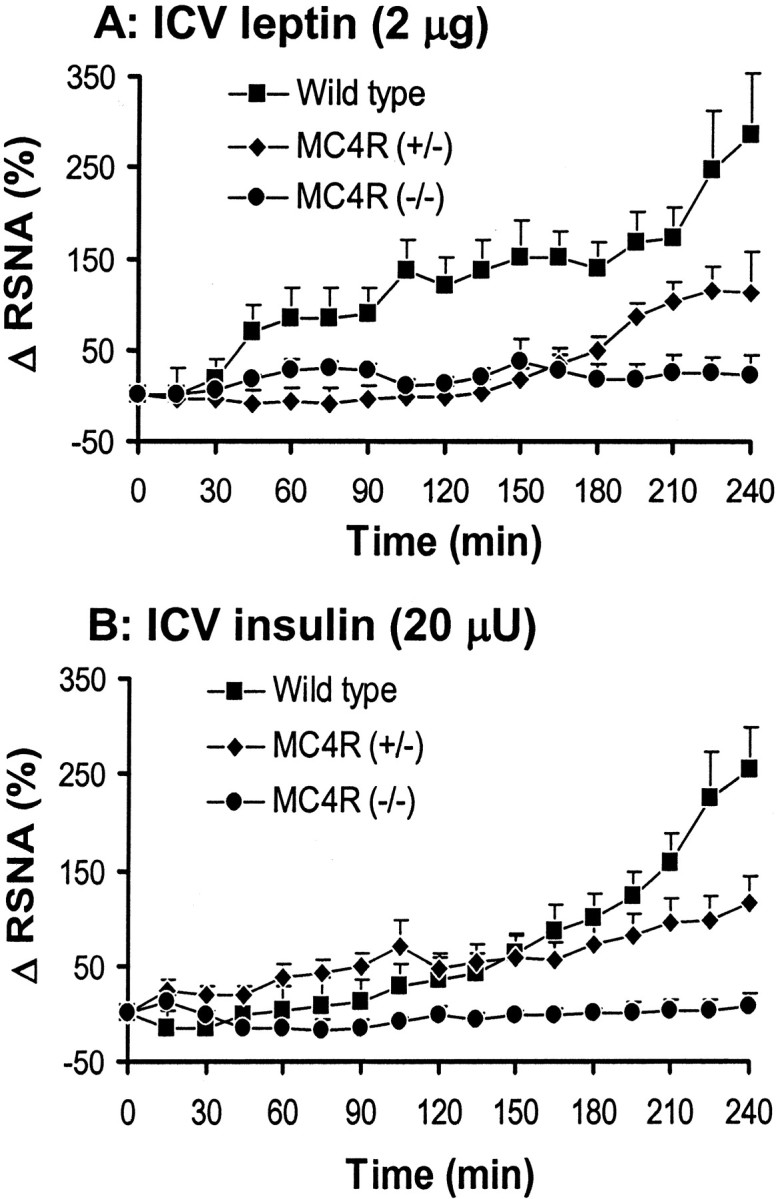
Effect of intracerebroventricular (ICV) administration of 2 μg of leptin (A) and 20 μU of insulin (B) on renal sympathetic nerve activity (RSNA) in homozygous and heterozygous MC-4R knock-out mice as compared with their wild-type controls. Δ RSNA represents the percentage change in RSNA from baseline. Data represent mean ± SEM; n = 6–11 mice for each group.
Next we examined the role of the MC4-R in the RSNA response to insulin. ICV administration of 20 μU of insulin induced a significant increase in RSNA in wild-type mice (190 ± 30% in the fourth hour; p < 0.001 vs vehicle) but not in the homozygous MC4-R knock-out mice (4 ± 11% in the fourth hour). Again, the heterozygous MC4-R knock-out mice exhibited an intermediate response to insulin [95 ± 22% in the fourth hour; p < 0.01 vs vehicle and p < 0.05 vs MC-4R (+/+) and MC-4R (-/-)].
In the MC-4R knock-out and wild-type mice, ICV administration of MTII, CRF, leptin, or insulin did not alter MAP and HR as compared with vehicle-treated mice (data not shown).
Discussion
The present study demonstrates that cerebroventricular administration of leptin and insulin in mice caused a significant increase in sympathetic nerve activity to the kidney, thus confirming previous reports (Muntzel et al., 1995; Dunbar et al., 1997; Haynes, 2000; Rahmouni et al., 2002) that the sympathoexcitatory effects of leptin and insulin are caused by their action in the CNS. Sympathoactivation to leptin was absent in the leptin receptor-deficient db/db mice. Surprisingly, absence of the Ob-Rb in the db/db mice attenuates also the sympathetic response to MC-4R stimulation. This attenuation of the RSNA response in db/db mice seems to be specific to the stimulation of the melanocortin system because the sympathoexcitatory effects of insulin and CRF were comparable between the db/db mice and their control littermates. We also confirmed the role of MC-4R in leptin-induced renal sympathoexcitation. More surprisingly, our data show that the rise in RSNA induced by insulin is also dependent on the MC-4R because the sympathoexcitatory effect of insulin was absent in homozygous MC-4R knock-out mice and attenuated when the expression of the MC-4R gene was reduced in the heterozygous mutant mice.
The receptor-mediated sympathoexcitatory effect of leptin is also supported by the substantially decreased sympathetic response to leptin in the obese Zucker rats (Haynes et al., 1997), known to possess a mutation in the gene for the leptin receptor (Chua et al., 1996; Iida et al., 1996; Phillips et al., 1996). It was not clear which form of the leptin receptor was involved, however, because this mutation in leptin receptor observed in the obese Zucker rat results in glutamine 269 to proline 269 amino acid substitution in the extracellular domain common to all known isoforms of the leptin receptor. The db/db mice have a specific deficit in the Ob-Rb isoform because of a mutation in the intracellular domain (Chen et al., 1996; Chua et al., 1996; Tartaglia, 1997). Recently, Harris et al. (2001) reported that leptin can affect blood glucose in the db/db mice despite the resistance of these mice to the anorectic and weight-reducing effects of leptin. This suggests that the short form of the leptin receptor (Ob-Ra, Ob-Rc, or Ob-Rd) has signaling capability and could mediate some metabolic and perhaps sympathoexcitatory effects of leptin. Our data demonstrate that the effects of leptin on sympathetic out-flow to the kidney are mediated by the long-form Ob-Rb of the leptin receptor.
Our finding of a MC4-R gene dose effect on the RSNA response to MTII indicates the importance of this receptor in melanocortin control of sympathetic nerve activity. Furthermore, our results support a pivotal role for MC-4R in control of sympathetic nerve activity by leptin and insulin. Failure of leptin and insulin to increase RSNA in MC-4R knock-out mice cannot be attributed to obesity because the RSNA response to these hormones is preserved in obese mice such as the db/db mice for insulin (present study) and agouti obese mice for leptin (Correia et al., 2002; Rahmouni et al., 2002). The pivotal role of MC-4R in the control of RSNA is also supported by our previous report (Haynes et al., 1999), in which SHU9119, which antagonizes MC-4R at least 10-fold more potently than MC-3R (Fan et al., 1997), prevented the sympathoexcitatory effects of leptin to the kidneys but not to brown adipose tissue. This suggests that leptin controls sympathetic nerve activity in a tissue-specific manner through different pathways. Indeed, it appears that leptin-induced sympathoactivation to brown adipose tissue is mediated by CRF (Correia et al., 2001). POMC neurons in the arcuate nucleus are known to express receptors for leptin (Cheung et al., 1997) and insulin (Benoit et al., 2002), and binding of leptin and insulin increases POMC gene expression (Schwartz et al., 1997; Thornton et al., 1997; Benoit et al., 2002). It is likely that leptin and insulin increase the release of α-MSH, thereby leading to the activation of the MC-4R pathway and an increase in renal sympathetic nerve outflow. This pathway is also postulated to be a major mechanism by which leptin and insulin achieve their effects on energy balance (Schwartz et al., 2000), because blockade of MC-4R has been shown to inhibit the feeding responses to leptin (Seeley et al., 1997; Satoh et al., 1998) and insulin (Benoit et al., 2002).
The attenuated RSNA response to stimulation of the melanocortin system in db/db mice was unexpected because melanocortin signaling is considered downstream to leptin receptors. Absence of leptin receptor might be expected to produce an exaggerated response to MTII caused by upregulation of the melanocortin receptors. We have no explanation for this attenuated RSNA response to MTII in db/db mice. Nonetheless, consistent with our findings, Benoit et al. (2000) reported that the decrease in food intake induced by stimulation of MC-4R was slightly attenuated (38.5%) in the db/db mice as compared with their wild-type controls (50.5%). Thus, the presence of the long-form Ob-Rb of the leptin receptor seems to be required for the melanocortin signaling to function normally.
We have reported previously that yellow obese agouti mice, in which it has been postulated that blockade of α-MSH effects on MC-4R by the agouti protein is a primary cause of obesity, have a preserved RSNA response to leptin despite the resistance to the anorectic and weight-reducing actions of leptin (Correia et al., 2002; Rahmouni et al., 2002). This contrasting effect of leptin on RSNA between agouti mice and MC-4R knock-out mice is intriguing. The redundancy in leptin signaling pathways could explain the preservation of the sympathoexcitatory action of leptin in the agouti mice. However, the absence of RSNA response to leptin in the MC-4R knock-out mice suggests that these redundant pathways that mediate the sympathetic effects of leptin in the agouti mice depend on the presence of the MC-4R. The feeding (Fan et al., 1997) and RSNA (our unpublished data) responses induced by MTII in agouti obese mice indicate that the MC-4Rs are functional in these mice. Further studies are needed to assess the mechanisms for the differences in the renal sympathetic nerve response to leptin between agouti obese mice and MC-4R knock-out mice.
In contrast to the high level of arterial pressure described in the agouti mice (Rahmouni et al., 2002), we found that the MC-4R knock-out mice are not hypertensive; however, the db/db mice have higher baseline levels of arterial pressure and RSNA. Similar results concerning arterial pressure and RSNA were reported in the obese Zucker rats as compared with their lean controls (Morgan et al., 1995). The preservation of the sympathoexcitatory effects of insulin in the presence of hyperinsulinemia could explain the higher levels of arterial pressure and RSNA in the db/db mice. However, in db/db mice the high increase in plasma insulin (∼20-fold) is not followed by a similar increase in baseline RSNA, perhaps because of the reduced efficiency of CNS insulin uptake from plasma. Indeed, insulin is known to cross the blood–brain barrier by a specific and saturable mechanism, which appears to limit the access of this hormone to the brain when its circulating levels are increased (Figlewicz et al., 1985;
Kaiyala et al., 2000). In the so-called insulin hypothesis of hyper-tension (Mark and Anderson, 1995; Landsberg, 2001), the sympathetic effects of chronic hyperinsulinemia have been suggested to link insulin resistance, cardiovascular mortality, and high blood pressure. In the present studies, however, the values of arterial pressure in the MC-4R knock-out mice and db/db mice were obtained when the mice were anesthetized and may not be representative of values in the conscious state.
Our focus was on RSNA because renal nerves are the communication link between the CNS and the kidney, which is known to play a major role in the control of cardiovascular function (DiBona and Kopp, 1997). Furthermore, long-term renal sympathetic stimulation caused by leptin and insulin could raise arterial pressure by causing peripheral vasoconstriction and by increasing renal tubular sodium reabsorption. The increase in RSNA induced by leptin and insulin was not associated with a change in arterial pressure, perhaps because of the presence of anesthesia. Alternatively, a longer recording might be required to reveal the effect of leptin and insulin on arterial pressure. For example, in rat we found that 6 hr of recording, after leptin administration, is necessary to observe an increase in arterial pressure (Correia et al., 2001). The prohypertensive effect of leptin and insulin was demonstrated by long-term infusion in animals (Huang et al., 1998; Shek et al., 1998).
In conclusion, we have shown that intracerebroventricular administration of leptin, insulin, and MC-3/4R agonist (MTII) caused a significant increase in RSNA. Absence of the long form of the leptin receptor Ob-Rb in the db/db mice abolished the increase in RSNA induced by leptin and attenuated the sympathoexcitatory effect of MTII. This suggests that leptin activation of hypothalamic nuclei is required for renal sympathoactivation by the melanocortin system. Homozygous MC-4R knock-out mice have an absent RSNA response to MTII, leptin, and insulin, whereas the heterozygous MC-4R knock-out had an attenuated response to these stimuli. These findings indicate an important physiologic role for MC-4R in the regulation of renal sympathetic traffic by both leptin and insulin.
Footnotes
This work was supported by Grants HL 44546 and HL 14388 from the National Heart, Lung, and Blood Institute and by research funds from the Department of Veterans Affairs. K.R. was supported by a postdoctoral fellowship award (0120606Z) from the Heartland Affiliate of American Heart Association. W.G.H. was the recipient of the Pharmaceutical Research Manufacturers of America Faculty Development Award.
Correspondence should be addressed to Dr. Kamal Rahmouni, University of Iowa, Cardiovascular Center, 524 Medical Research Center, Iowa City, IA 52242. E mail: kamal-rahmouni@uiowa.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235998-07$15.00/0
References
- Adan RA, Gispen WH ( 2000) Melanocortins and the brain: from effects via receptors to drug targets. Eur J Pharmacol 405: 13–24. [DOI] [PubMed] [Google Scholar]
- Benoit SC, Schwartz MW, Lachey JL, Hagan MM, Rushing PA, Blake KA, Yagaloff KA, Kurylko G, Franco L, Danhoo W, Seeley RJ ( 2000) A novel selective melanocortin-4 receptor agonist reduces food intake in rats and mice without producing aversive consequences. J Neurosci 20: 3442–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ, Seeley RJ, Woods SC ( 2002) The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci 22: 9048–9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR ( 2000) Role of brain insulin receptor in control of body weight and reproduction. Science 289: 2122–2125. [DOI] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P ( 1995) Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269: 546–549. [DOI] [PubMed] [Google Scholar]
- Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP ( 1996) Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84: 491–495. [DOI] [PubMed] [Google Scholar]
- Chua Jr SC, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL ( 1996) Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 271: 994–996. [DOI] [PubMed] [Google Scholar]
- Correia ML, Morgan DA, Mitchell JL, Sivitz WI, Mark AL, Haynes WG ( 2001) Role of corticotrophin-releasing factor in effects of leptin on sympathetic nerve activity and arterial pressure. Hypertension 38: 384–388. [DOI] [PubMed] [Google Scholar]
- Correia MLG, Haynes WG, Rahmouni K, Morgan DA, Sivitz WI, Mark AL ( 2002) Selective leptin resistance in agouti yellow obese mice. Diabetes 51: 439–442. [DOI] [PubMed] [Google Scholar]
- Cheung CC, Clifton DK, Steiner RA ( 1997) Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology 138: 4489–4492. [DOI] [PubMed] [Google Scholar]
- DiBona GF, Kopp UC ( 1997) Neural control of renal function. Physiol Rev 77: 75–197. [DOI] [PubMed] [Google Scholar]
- Dunbar JC, Hu Y, Lu H ( 1997) Intracerebroventricular leptin increases lumbar and renal sympathetic nerve activity and blood pressure in normal rats. Diabetes 46: 2040–2043. [DOI] [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD ( 1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385: 165–168. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Dorsa DM, Stein LJ, Baskin DG, Paquette T, Greenwood MR, Woods SC, Porte Jr D ( 1985) Brain and liver insulin binding is decreased in Zucker rats carrying the “fa” gene. Endocrinology 117: 1537–1543. [DOI] [PubMed] [Google Scholar]
- Harris RB, Mitchell TD, Yan X, Simpson JS, Redmann SM Jr ( 2001) Metabolic responses to leptin in obese db/db mice are strain dependent. Am J Physiol 281: R115–R132. [DOI] [PubMed] [Google Scholar]
- Haynes WG ( 2000) Interaction between leptin and sympathetic nervous system in hypertension. Curr Hypertens Rep 2: 311–318. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI ( 1997) Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest 100: 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL ( 1999) Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension 33: 542–547. [DOI] [PubMed] [Google Scholar]
- Huang WC, Fang TC, Cheng JT ( 1998) Renal denervation prevents and reverses hyperinsulinemia-induced hypertension in rats. Hypertension 32: 249–254. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F ( 1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88: 131–141. [DOI] [PubMed] [Google Scholar]
- Iida M, Murakami T, Ishida K, Mizuno A, Kuwajima M, Shima K ( 1996) Substitution at codon 269 (glutamine → proline) of the leptin receptor (OB-R) cDNA is the only mutation found in the Zucker fatty (fa/fa) rat. Biochem Biophys Res Commun 224: 597–604. [DOI] [PubMed] [Google Scholar]
- Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW ( 2000) Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 49: 1525–1533. [DOI] [PubMed] [Google Scholar]
- Landsberg L ( 2001) Insulin-mediated sympathetic stimulation: role in the pathogenesis of obesity-related hypertension (or, how insulin affects blood pressure, and why). J Hypertens 19: 523–528. [DOI] [PubMed] [Google Scholar]
- Mark AL, Anderson EA ( 1995) Genetic factors determine the blood pressure response to insulin resistance and hyperinsulinemia: a call to refocus the insulin hypothesis of hypertension. Proc Soc Exp Biol Med 208: 330–336. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD ( 1999) Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet 21: 119–122. [DOI] [PubMed] [Google Scholar]
- Morgan DA, Anderson EA, Mark AL ( 1995) Renal sympathetic nerve activity is increased in obese Zucker rats. Hypertension 25: 834–838. [DOI] [PubMed] [Google Scholar]
- Mountjoy KG, Robbins LS, Mortrud MT, Cone RD ( 1992) The cloning of a family of genes that encode the melanocortin receptors. Science 257: 1248–1251. [DOI] [PubMed] [Google Scholar]
- Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD ( 1994) Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol 8: 1298–1308. [DOI] [PubMed] [Google Scholar]
- Muntzel MS, Anderson EA, Johnson AK, Mark AL ( 1995) Mechanisms of insulin action on sympathetic nerve activity. Clin Exp Hypertens 17: 39–50. [DOI] [PubMed] [Google Scholar]
- Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L ( 2002) Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 5: 566–572. [DOI] [PubMed] [Google Scholar]
- Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ, Hess JF ( 1996) Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet 13: 18–19. [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Haynes WG, Morgan DA, Mark AL ( 2002) Selective resistance to central neural administration of leptin in agouti obese mice. Hypertension 39: 486–490. [DOI] [PubMed] [Google Scholar]
- Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD ( 1993) Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci USA 90: 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh N, Ogawa Y, Katsuura G, Numata Y, Masuzaki H, Yoshimasa Y, Nakao K ( 1998) Satiety effect and sympathetic activation of leptin are mediated by hypothalamic melanocortin system. Neurosci Lett 249: 107–110. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG ( 1997) Leptin increases hypothalamic proopiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes 46: 2119–2123. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DG ( 2000) Central nervous system control of food intake. Nature 404: 661–671. [DOI] [PubMed] [Google Scholar]
- Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW ( 1997) Melanocortin receptors in leptin effects. Nature 390: 349. [DOI] [PubMed] [Google Scholar]
- Shek EW, Brands MW, Hall JE ( 1998) Chronic leptin infusion increases arterial pressure. Hypertension 31: 409–414. [DOI] [PubMed] [Google Scholar]
- Tanida M, Iwashita S, Ootsuka Y, Terui N, Suzuki M ( 2000) Leptin injection into white adipose tissue elevates renal sympathetic nerve activity dose-dependently through the afferent nerves pathway in rats. Neurosci Lett 293: 107–110. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA ( 1997) The leptin receptor. J Biol Chem 272: 6093–6096. [DOI] [PubMed] [Google Scholar]
- Thornton JE, Cheung CC, Clifton DK, Steiner RA ( 1997) Regulation of hypothalamic proopiomelanocortin mRNA by leptin in ob/ob mice. Endocrinology 138: 5063–5066. [DOI] [PubMed] [Google Scholar]