

Abstract
The neuronal L-type calcium channels (LTCCs) Cav1.2α1 and Cav1.3α1 are functionally distinct. Cav1.3α1 activates at lower voltages and inactivates more slowly than Cav1.2α1, making it suitable to support sustained L-type Ca2+ inward currents (ICa,L) and serve in pacemaker functions. We compared the biophysical and pharmacological properties of human retinal Cav1.4α1 using the whole-cell patch-clamp technique after heterologous expression in tsA-201 cells with other L-type α1 subunits. Cav1.4α1-mediated inward Ba2+ currents (IBa) required the coexpression of α2δ1 and β3 or β2a subunits and were detected in a lower proportion of transfected cells than Cav1.3α1. IBa activated at more negative voltages (5% activation threshold; -39mV; 15 mm Ba2+) than Cav1.2α1 and slightly more positive than Cav1.3α1. Voltage-dependent inactivation of IBa was slower than for Cav1.2α1 and Cav1.3α1(∼50% inactivation after 5 sec; α2δ1 + β3 coexpression). Inactivation was not increased with Ca2+ as the charge carrier, indicating the absence of Ca2+-dependent inactivation. Cav1.4α1 exhibited voltage-dependent, G-protein-independent facilitation by strong depolarizing pulses. The dihydropyridine (DHP)-antagonist isradipine blocked Cav1.4α1 with ∼15-fold lower sensitivity than Cav1.2α1 and in a voltage-dependent manner. Strong stimulation by the DHP BayK 8644 was found despite the substitution of an otherwise L-type channel-specific tyrosine residue in position 1414 (repeat IVS6) by a phenylalanine. Cav1.4α1 + α2δ1 + β channel complexes can form LTCCs with intermediate DHP antagonist sensitivity lacking Ca2+-dependent inactivation. Their biophysical properties should enable them to contribute to sustained ICa,L at negative potentials, such as required for tonic neurotransmitter release in sensory cells and plateau potentials in spiking neurons.
Keywords: calcium channels, calcium-dependent inactivation, retina, calcium channel blockers, dihydropyridines, congenital stationary night blindness
Introduction
Many cellular functions are controlled by a depolarization-induced influx of Ca2+ ions from the extracellular space through voltage-gated Ca2+ channels. In neurons, Ca2+ influx through presynaptic N-, P/Q-, and, to a limited extent, R-type Ca2+ channels is tightly coupled to neurotransmitter release from nerve terminals (stimulus-secretion coupling) (Catterall, 2000). L-type Ca2+ channels (LTCCs) are primarily targeted to dendrites and the cell soma (Catterall, 2000) and are responsible for Ca2+ signals, which activate signaling pathways controlling gene transcription (Graef et al., 1999).
We (Koschak et al., 2001), and others (for review, see Lipscombe, 2002), have recently found that Cav1.2α1 and Cav1.3α1 LTCCs possess distinct functional properties that thus allow them to serve distinct neuronal functions. Cav1.3α1 channels activate at more negative voltages, inactivate slower during depolarizing pulses, and exhibit lower dihydropyridine (DHP) antagonist sensitivity than Cav1.2α1. Their biophysical properties make them highly suitable to mediate tonic neurotransmitter release in sensory cells (such as in cochlear inner hair cells) (Platzer et al., 2000) to support plateau potentials in spiking neurons (Carlin et al., 2000; Alaburda et al., 2002; Morisset and Nagy, 2000) and contribute to diastolic depolarization and pacemaking in the sinoatrial node (Mangoni et al., 2003) (for review, see Lipscombe, 2002). In the mammalian retina, Cav1.3α1 subunits are expressed in photoreceptor nerve terminals and selected bipolar cell synapses (Morgans et al., 1998; Taylor and Morgans, 1998; Morgans, 1999). ICa,L in photoreceptors and bipolar cells shares most features of heterologously expressed Cav1.3α1 currents. It was therefore proposed that this channel underlies retinal ICa,L (Wilkinson and Barnes, 1996).
Recently, Cav1.4α1 subunits were discovered as a putative neuronal LTCC subunit (Bech-Hansen et al., 1998; Strom et al., 1998). Cav1.4α1 is expressed predominantly in the retina but also in other neurons such as dorsal root ganglia (Murakami et al., 2001). In the mammalian retina, its expression pattern resembles Cav1.3α1. Cav1.4α1 immunoreactivity has also been localized in the synapses of the outer and inner plexiform layer as well as on photoreceptor cell bodies (Firth et al., 2001; Morgans, 2001; Morgans et al., 2001; Ball et al., 2002; Berntson et al., 2003). Its physiological relevance for normal retinal function is evident from Cav1.4 α1 mutations causing incomplete X-linked congenital stationary night blindness (iCSNB2) in humans (Bech-Hansen et al., 1998; Strom et al., 1998). Cav1.4α1 could therefore also represent a synaptically localized Ca2+ channel in the retina. However, this interpretation is complicated by the fact that Cav1.4α1 is the only cloned mammalian Ca2+ channel α1 subunit that has not yet been functionally characterized. Its characterization in isolated neurons is hampered by the lack of Cav1.4α1-deficient mouse models. These would allow the identification of neurons, such as photoreceptors, with predominantly Cav1.4α1-mediated currents and isolate them from the residual Ca2+ current components. In the absence of such models, the biophysical and pharmacological characterization of recombinant Cav1.4α1 channels would allow us to address many open questions: can Cav1.4α1 form DHP-sensitive LTCCs and ICa,L as described in retinal neurons? Do its biophysical properties resemble Cav1.2α1 or rather the lower voltage-activated Cav1.3α1? Do Cav1.4α1 currents exhibit Ca2+-dependent inactivation? Cav1.4α1 subunits lack a tyrosine in transmembrane segment IVS6, which was found previously to be part of the DHP-binding pocket of other LTCCs (Peterson et al., 1996; Striessnig et al., 1998). This raises the question of whether Cav1.4α1 exhibits the typical DHP sensitivity by which retinal ICa,L has been defined.
Here we describe for the first time the successful functional expression of a human retinal Cav1.4α1 in mammalian cells. We show that Cav1.4α1 channels share many of the properties of Cav1.3α1, including intermediate DHP sensitivity, but lack Ca2+-dependent inactivation under identical experimental conditions.
Materials and Methods
Cloning of human CaV1.4α1 subunits. The Cav1.4α1 cDNA (Strom et al., 1998) (GenBank AJ224874; open reading frame length, 5898 bp) was cloned from five subfragments (F1–F5) using different native or artificial restriction enzyme (RE) sites [nucleotide numbers (nt) are given in parentheses; asterisks indicate artificial RE sites introduced by PCR]: F1,SalI*-BamHI (nt, -5–812), F2, BamHI-SphI (nt, 812–1993), F3 SphI-ClaI (nt, 1993–3255), F4, ClaI-EcoRI (nt, 3255–4349), F5, EcoRI-XbaI* (nt, 4349–5907). Fragments were generated by reverse transcriptase (RT)-PCR using proofreading pfu DNA polymerase (Stratagene, La Jolla, CA). First strand cDNA as a PCR template was synthesized from 1–1.5 μg of human retinal poly A+ RNA (Clontech, Cambridge, UK) with the Ready-To-Go T-primed first-strand reaction kit (Amersham Biosciences, Arlington Heights, IL). PCR fragments were subcloned into vectors pBluescript SK+ (Stratagene) or pSport-1 (Invitrogen, San Diego, CA). Sequence integrity of the subclones was determined by DNA sequencing (MWG Biotech, Ebersberg, Germany). The construction of the complete Cav1.4α1 was performed as follows: fragment F1 + 2 was generated by ligating the BamHI-SphI fragment (F2) into the corresponding RE sites of pSport-1-containing fragment F1. Fragment F4 + 5 was generated by ligating the ClaI-EcoRI fragment (F4) into the corresponding RE sites of pBluescript SK+-containing fragment F5. These steps were followed by a three fragment ligation of the SalI*–SphI fragment (F1 + 2) and the SphI-ClaI fragment (F3) into the SalI and ClaI sites of the F4 + 5-containing pBluescript SK+. For subsequent expression studies, the Cav1.4α1 construct was either inserted into plasmid pGFP+ (Grabner et al., 1998; Koschak et al., 2001) (yielding Cav1.4α1 with GFP fused to its N terminus GFP-Cav1.4α1) or into the corresponding vector pGFP-, which lacks the GFP sequence.
Transient expression of LTCCs in tsA-201 cells. tsA-201 cells were maintained at 37°C and 5% CO2 in DMEM—Coon's F12 medium (Invitrogen) supplemented with 10% (v/v) FCS (Sebak, Aidenbach, Germany), 2 mml-glutamine, and 100 U/ml of penicillin streptomycin. For transient Ca2+ channel expression, cells were plated onto 10 cm tissue culture dishes 12 hr before transfection with Ca2+ phosphate precipitation using standard protocols. Human Cav1.4α1, human Cav1.3α1 (Koschak et al., 2001), human Cav2.1α1 (Wappl et al., 2002), or rabbit Cav1.2α1-a (Mikami et al., 1989) subunits were expressed together with α2δ1 (Ellis et al., 1988), rat β3 subunits (Castellano et al., 1993), or rat β2a (Perez-Reyes et al., 1992). Transfection protocols for Cav2.1α1, Cav1.2α1, and Cav1.3α1 subunits were as described previously (Koschak et al., 2001). Cav1.4 α1-transfected cells were incubated at 30°C and 5% CO2 6 – 8 hr after transfection for 2 – 3 d before recording. One day before recording, cells were transferred to 3 cm culture dishes containing glass coverslips for drug application experiments. Transfected cells were visualized as GFPCav1.4α1 or by cotransfected GFP fluorescence.
Membrane preparation and immunoblotting with affinity-purified sequence-directed antibodies. Immunoblotting was performed as described previously (Safayhi et al., 1997; Platzer et al., 2000) using a generic anti-α1 sequence-directed antibody (anti-α11382-1400; raised against residues 1382–1400 of Cav1.1α1) (Safayhi et al., 1997). Membranes from tsA-201 cells transfected with 3 μg of α1, 2 μg of β, 2.5 μg of α2δ1 subunit cDNA, and 2.5 μg of pUC18 carrier DNA in a 10 cm culture dish were prepared as described previously (Huber et al., 2000).
Electrophysiological recordings. Whole-cell patch-clamp experiments were performed at room temperature (Axopatch 200B amplifier; Axon Instruments, Foster City, CA) and linked to a personal computer equipped with pClamp version 7.0. Currents were recorded at sampling rates of 5 or 25 kHz and low-pass filtered at 2 or 5 kHz with a Digidata 1322A analog-to-digital board (Axon Instruments). Borosilicate glass pipettes were pulled using a Sutter P-97 (Linton Instruments, Palgrave, UK), microelectrode puller and fire polished, showing typical resistances of 2–3 MΩ when filled with internal solution. Capacitance compensation and series resistance compensations of 60% were used. The solutions for whole-cell measurements were as follows (in mm): (internal solution) 135 CsCl, 10 Cs-EGTA, and 1 MgCl2, adjusted to pH 7.4, with CsOH; (recording solution) 15 BaCl2 or 15 CaCl2, 10 HEPES, 150 Choline-Cl, and 1 MgCl2, adjusted to pH 7.4, with CsOH. The holding potential (HP) was -80 mV, unless stated otherwise. The presence of ATP in the pipette solutions did not affect run down of heterologously expressed L-type channels (see below) and was therefore omitted. All voltages were corrected for a liquid junction potential of -9 mV for Ba2+ and -8 mV for Ca2+-containing solutions. Leak and capacitative currents were measured using hyperpolarizing pulses. Raw currents were corrected for linear leak currents. The voltage dependence of activation was determined from current—voltage (I—V) curves obtained by step depolarizations from the holding potential to various test potentials. I—V curves were fitted according to the following:
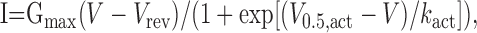 |
1 |
where Vrev is the extrapolated reversal potential of IBa, V is the membrane potential, I is the peak current, Gmax is the maximum conductance of the cell, V0.5, act is the voltage for half-maximal activation, and kact is the slope factor of the Boltzmann term. The time course of current activation was fitted to the following exponential functions:
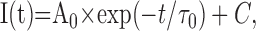 |
2 |
where I(t) is the current at time t after the depolarization, A0 the steady state current amplitude with the respective time constant of activation, τ0, and C the remaining steady state current or to the following:
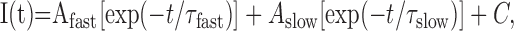 |
3 |
yielding time constants for a fast (τfast) and a slow (τslow) component.
Effects of DHPs were monitored continuously using 0.1 Hz depolarizing pulses (40 msec) to Vmax. DHPs were dissolved in the recording solution from a 10 mm stock solution in dimethyl sulfoxide and perfused through a microcapillary onto cells using a gravity driven perfusion system. Only cells exhibiting stable currents (run down <5% during the first 60 sec) were used for analysis of DHP effects. The DHPs isradipine and BayK 8644 (kindly provided by Novartis, Basel, Switzerland, and Bayer, Wuppertal, Germany) were used as their racemic mixtures.
Activation of G-proteins was achieved by intracellular perfusion with guanosine 5′-[γ-thio]triphoshate (GTPγS; Sigma, St. Louis, MO) for >3 min under whole-cell conditions. The degree of voltage-dependent current facilitation was determined as the ratio (facilitation ratio) of absolute peak current amplitudes before [-PP (prepulse)] and after (+PP) a conditioning prepulse (5–200 msec to voltages between 80 and 140 mV).
Statistics. Data were analyzed using Clampfit 8.0 (Axon Instruments) and Origin 5.0 (Microcal Software, Northampton, MA). All data are presented as mean ± SE for the indicated number of experiments. Statistical significance was determined by unpaired student's t test except when stated otherwise (Kruskal—Wallis test followed by Dunn's multiple comparison procedure, or one-way ANOVA followed by Bonferroni test as indicated).
Results
Although the DNA sequences of the human and mouse Cav1.4α1 subunits are known (Bech-Hansen et al., 1998; Strom et al., 1998; Naylor et al., 2000), their successful functional expression has not been reported so far. We therefore constructed a full-length Cav1.4α1 cDNA derived from human retina for functional expression in tsA-201 cells. The Cav1.4α1 cDNA contains exons 1, 2, and 9a (Strom et al., 1998; Boycott et al., 2001).
We first confirmed the efficient expression of full-length Cav1.4α1 subunits (calculated molecular mass, 220 kDa) on the protein level by immunoblot analysis of transfected tsA-201 cell membranes (Fig. 1). As expected for the full-length form of Cav1.4α1, the immunostained band comigrated with the prestained myosin molecular mass standard (217 kDa), slightly faster than Cav1.3α1 (calculated molecular mass, 242.5 kDa) and Cav1.2α1 (Fig. 1). Its expression density was slightly lower than that of Cav1.2α1 and Cav1.3α1 (Fig. 1) (n = 4).
Figure 1.
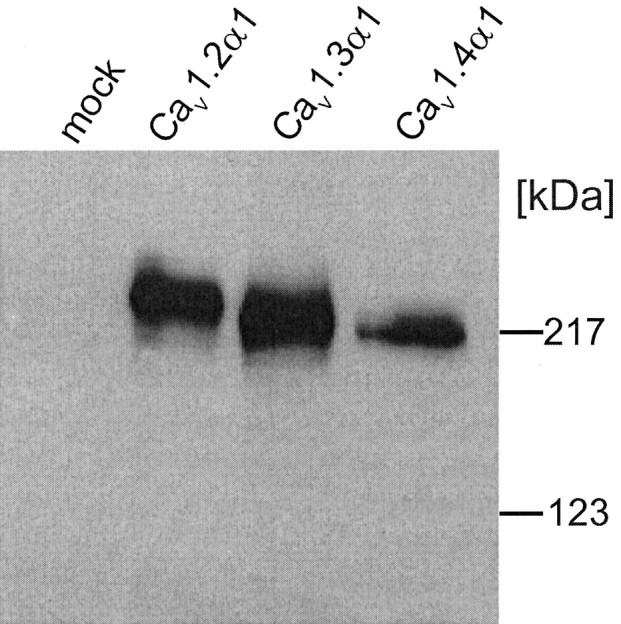
Heterologous expression of different LTCC subunits in tsA-201 cells. Cells were transfected with Cav1.2α1, Cav1.3α1, or Cav1.4α1 together with β3 and α2δ1 subunit cDNA as described in Materials and Methods. Expression of α1 subunit proteins was analyzed in immunoblots of membranes prepared from lyzed cells after separation on 8% SDS-PAGE gels (10 μg of membrane protein per lane) using a generic anti-α1 sequence directed antibody (anti-CP1382–1400). No α1 immunoreactivity was present in mock-transfected cells used as a control. One of four experiments yielding similar results is shown.
Next, we investigated whether the heterologously expressed Cav1.4α1 subunits can also form functional channels after expression in tsA-201 cells using the whole-cell patch-clamp technique. Using a standard transfection protocol and cotransfection with α2δ1 and β3 subunits, significant IBa was measurable during depolarization from an HP of -90 mV for both Cav1.4α1 and the GFP-Cav1.4α1 fusion protein (Fig. 2). Compared with Cav1.3α1, the expression efficiency was lower. Only 50 of 227 (22%) Cav1.4α1 + β3 + α2δ1 and 19 of 71 (28%) GFP-Cav1.4α1 + β3 + α2δ1 transfected (i.e., GFP-positive) cells yielded IBa (15 mm Ba2+ as charge carrier), exceeding endogenous currents (Fig. 2, legend). In contrast, 66% of GFP-positive cells transfected with Cav1.3α1 + β3 + α2δ1 (and >90% with Cav1.2α1 + β3 + α2δ1; data not shown) expressed L-type currents. When Cav1.4α1 was coexpressed with β3 subunits in the absence of α2δ1, current densities did not exceed those of endogenous currents measured in untransfected tsA-201 cells (Fig. 2A) (p > 0.05). This shows that Cav1.4α1 can associate with α2δ subunits. Because β subunits exert modulatory effects on Cav1.4α1-meditated currents (see below), the smallest functional complex is Cav1.4α1 + β + α2δ. Coexpression of Cav1.4α1 + α2δ1 with β2a subunits, which are important for normal retinal function (Ball et al., 2002), also yielded significant IBa above endogenous currents (p < 0.01) (Fig. 2A).
Figure 2.

Biophysical properties of IBa and ICa through Cav1.4α1 subunits. Cav1.4α1 subunits were expressed together with β3 + α2δ1 (A—C) or β2a + α2δ1 (A), as described in Materials and Methods, using 15 mm Ba2+ (A—C) or 15 mM Ca2+ (D) as charge carrier. A, Expression density was determined by depolarizing pulses to Vmax.IBa was measured 48 – 82 hr after transfection for the indicated number of cells. Small currents measured in untransfected cells were attributable to endogenous non-L-type currents that were <1.72 pA/pF. Asterisks indicate statistically significant difference to untransfected cells (p < 0.01; Kruskal—Wallis test, followed by Dunn's multiple comparison test). B, Superimposed currents were activated by depolarizing Cav1.4α1-transfected cells during 50 msec pulses from an HP of -90 mV to between -60 and 40 mV in 10 mV steps. C, Normalized I—V curves for Cav1.4α1 coexpressed with β3 and α2δ1 subunits using 15 mm Ba2+ (black squares) or Ca2+ (black circles) as charge carriers. Biophysical parameters are given in Table 1. D, Protocol as in B but with 15 mm Ca2+ in the bath solution.
We analyzed the biophysical properties of Cav1.4α1-mediated currents in comparison with Cav1.2α1 and Cav1.3α1, which, like Cav1.4α1, are also expressed in sensory cells including the retina (Morgans et al., 1998; Taylor and Morgans, 1998; Morgans, 1999; Berntson et al., 2003). Cav1.4α1 IBa typically activated at more negative voltages (-38.8 ± 0.7 mV; n = 36; β3 + α2δ1 coexpression) than Cav1.2α1 but slightly more positive than Cav1.3α1 (p < 0.001) (Table 1) (Koschak et al., 2001). Representative currents activated by 50 msec step depolarizations to different test potentials (HP, -90 mV) are illustrated in Figure 2B. The I—V relationship (V0.5,act, Vmax) was shifted to more positive potentials with 15 mm Ca2+ as the charge carrier (p < 0.01) (Table 1, Fig. 2C).
Table 1.
Biophysical properties of Ba2+and Ca2+currents through heterologously expressed Cav1.4α1 subunits
|
|
|
|
|
|
|
|
+ BayK 8644 |
||
|---|---|---|---|---|---|---|---|---|---|
| Subunits coexpressed with α2δ1
|
Charge carrier (mM)
|
V0.5,act (mV)
|
Kact (mV)
|
Vmax (mV)
|
Activation threshold (mV)
|
Current density (pA/pF)
|
V0.5,act (mV)
|
Kact (mV)
|
|
| Cav1.4α1 + β3 | 15Ba2+ | −8.9 ± 0.8 | −8.0 ± 0.4 | 4.2 ± 0.7 | −38.8 ± 0.7 | 15.5 ± 2.0 | −21.5 ± 1.3xxx | −5.7 ± 0.3xx | |
| n = 36 | n = 36 | n = 36 | n = 36 | n = 36 | n = 9 | n = 9 | |||
| GFP-Cav1.4α1 + β3 | 15Ba2+ | −11.0 ± 1.4 | −9.1 ± 0.4 | 2.7 ± 1.1 | −42.0 ± 1.1 | 6.0 ± 1.8 | −24.9 ± 1.5xxx | −4.7 ± 0.5xxx | |
| n = 18 | n = 18 | n = 18 | n = 18 | n = 18 | n = 8 | n = 8 | |||
| Cav1.4α1 + β2a | 15Ba2+ | −10.7 ± 1.4 | −9.5 ± 0.3 | 2.6 ± 1.3 | −45.6 ± 1.2** | 4.6 ± 1.3 | |||
| n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | ND | ND | |||
| Cav1.3α1 + β3 | 15Ba2 | −16.8 ± 0.7*** | −9.2 ± 0.2 | −1.7 ± 0.7*** | −44.5 ± 0.5*** | 22.4 ± 4.2 | |||
| n = 52 | n = 52 | n = 52 | n = 52 | n = 52 | ND | ND | |||
| Cav1.2α1 + β3 | 15Ba2 | −3.0 ± 1.1** | −8.0 ± 0.2 | 13.4 ± 1.9*** | −31.5 ± 0.5*** | 20.4 ± 5.6 | |||
| n = 20 | n = 20 | n = 20 | n = 20 | n = 20 | ND | ND | |||
| Cav1.4α1 + β3 | 15Ca2+ | 0.6 ± 2.2** | −9.4 ± 0.5 | 15.1 ± 2.2*** | −33.6 ± 1.8 | 12.7 ± 3.7 | |||
| n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | ND | ND | |||
| Cav1.3α1 + β3 | 15Ca2 | −7.8 ± 1.1 | −9.4 ± 0.4 | 7.1 ± 1.6* | −37.3 ± 0.9 | 13.5 ± 2.2 | |||
| n = 13 | n = 13 | n = 13 | n = 13 | n = 13 | ND | ND | |||
| Cav1.2α1 + β3 | 15Ca2 | 9.2 ± 1.5+ | −10.5 ± 0.3*** | 21.4 ± 1.2 | −27.6 ± 1.3+ | 10.4 ± 2.1 | |||
|
|
|
n = 22
|
n = 22
|
n = 22
|
n = 22
|
n = 22
|
ND
|
ND
|
|
GFP-Cav1.4α1 and Cav1.4α1 Ca2+ channels were coexpressed with rat β3 or rat β2a and rabbit α2δ1 subunits in tsA-201 cells, and the biophysical properties were determined using 15 mm Ba2+ or Ca2+ as a charge carrier. V0.5,act, Kact, and Vmax were obtained by fitting the data as described in Materials and Methods. The activation threshold was determined as the test potential at which 5% of the maximal current was activated. Data are given as means ± SE; ND, not determined. Statistical differences for V0.5,act, Kact, Vmax, and activation thresholds (calculated by one-way ANOVA, followed by Bonferroni test) are given in comparison with Cav1.4α1 + β3 + α2δ1 with Ba2+ (*p<0.05; **p<0.01; ***p<0.001) or Ca2+ (+p<0.05; ++p<0.01; +++p<0.001) as charge carrier. For cells used for the calculation of biophysical parameters, current densities (no BayK 8644 present) are also given. For statistical comparisons of current densities, see Figure 2A. Statistically significant differences for BayK 8644 effects (xxp<0.01; xxxp<0.001; Student's t tests) are also indicated.
The time course of activation determined during 50 msec depolarizations to Vmax revealed similar activation time constants for IBa through GFP-tagged or non-GFP-tagged Cav1.4α1 subunits. When coexpressed with β3 + α2δ1, activation could be described by a monoexponential time course in the majority of cells (Cav1.4α1; 0.43 ± 0.08 msec; 9 of 14 cells). In the remaining cells, a biexponential onset of activation was measured (τfast = 0.46 ± 0.08 msec; τslow = 16.4 ± 3.9 msec; relative contribution of slow component, 6.03 ± 1.93%). Coexpression of β2a subunits primarily resulted in activation by a biexponential time course (three of four cells; τfast = 0.70 ± 0.13 msec; τslow = 6.6 ± 0.1 msec; relative contribution of slow component, 3.9 ± 1.1%). Therefore, with respect to the activation properties, IBa through Cav1.4α1 Ca2+ channels closely resembled Cav1.3α1 currents, which also activate with faster time courses and at lower voltages than Cav1.2 (Koschak et al., 2001; Scholze et al., 2001; Xu and Lipscombe, 2001). No changes in the (monoexponential) activation time course were detected for Cav1.3α1 currents in the presence of β2a subunits (monophasic activation; α2δ1 + β3, τact = 0.77 ± 0.06; n = 24; α2δ1 + β2a, τact = 0.73 ± 0.1; n = 4; p > 0.05).
One property that distinguishes Cav1.3α1 from Cav1.2α1 currents is its slower inactivation during prolonged depolarizations (Koschak et al., 2001). The experiments in Figure 3 illustrate that inactivation of Cav1.4α1 was even slower than for Cav1.3α1 (Fig. 3). Only 50.2 ± 2.9% (n = 17) of IBa inactivated after 5 sec of depolarization to Vmax (Fig. 3A,B). After 10 sec, 84.2 ± 6.4% (n = 4) of Cav1.3α1 but only 68.1 ± 2.7% (n = 17) of Cav1.4α1IBa inactivated (p < 0.05) (Fig. 3A,B). Substitution of β2a for β3 subunits also significantly slowed inactivation (Fig. 3E,F). This also demonstrates that β subunits participate in fine tuning the Cav1.4α1 channel complex.
Figure 3.

Inactivation properties of Cav1.4α1 Ca2+ channels. A, IBa (black traces) through Cav1.3α1 and Cav1.4α1 subunits coexpressed with β3 and α2δ1 subunits were elicited by 10 sec depolarizing pulses from an HP of -90 mV to Vmax. Representative current traces for Cav1.3α1 (n = 4) and Cav1.4α1 (n = 17) channels are shown. Traces were normalized to the peak current amplitudes. For the experiments shown, inactivation measured during 5 and 10 sec depolarizing pulses was as follows: Cav1.4α1, 41 and 56%; Cav1.3α1, 89 and 97%. A representative trace for current through Cav1.4α1 recorded with 15 mm Ca2+ as charge carrier is illustrated in gray superimposed on the IBa trace indicated in black. B, Percent current inactivation measured after 0.25, 5, and 10 sec depolarizations to Vmax in Cav1.4α1-transfected cells using either 15 mm Ba2+ or 15 mM Ca2+ as the charge carrier. Inactivation of currents during pulses was not significantly different for Ba2+ (black bars) and Ca2+ (gray bars) (n = 7; p > 0.05). C, D, Inactivation for Cav1.2α1 (C) and Cav1.3α1 (D) during 2 sec depolarizing pulses toVmax with 15 mm Ba2+ (black trace) or 15 mM Ca2+ (gray trace) as charge carriers. For Cav1.3α1, a variable noninactivating ICa component was found (9 – 40%; n = 4), whereas remaining Cav1.2α1 currents were always <3.5% (n = 7). E, Inactivation of IBa through Cav1.4α1 cotransfected with β3 (black) or β2a (gray) and α2δ1. Currents were normalized to peak IBa. Currents were elicited by depolarization from an HP of -90 mV to Vmax. F, Percentage of inactivation of IBa through Cav1.4α1 cotransfected with β3 (black; n = 17) or β2a (gray;n = 7), and α2δ1 was determined after 5 and 10 sec during a depolarization from an HP of -90 mV toVmax. Currents were normalized to peakIBa. Inactivation with β2a coexpression was 27.2±4.6% (after 5 sec) and 44.6 ± 6.4% (after 10 sec; n=7), respectively. Asterisks indicate a statistically significant difference to β2a coexpression (p < 0.01).
In addition to voltage, Ca2+ is also an important determinant of LTCC inactivation. Figure 3, C and D, illustrates that not only inactivation of Cav1.2α1 (for review, see Budde et al., 2002) but also of heterologously expressed Cav1.3α1 occurred in a Ca2+-dependent manner (percentage of current inactivation after 250 msec; Cav1.2α1, IBa, 60.7 ± 8%; n = 4; ICa, 84.5 ± 3.1%; n = 6;p < 0.05; Cav1.3α1, IBa, 37.5 ± 2.9%; n = 13; ICa, 68.8 ± 4.7%;n = 12; p < 0.001). In contrast, under the same experimental conditions (10 mm EGTA in the pipette solution), Cav1.4α1 did not exhibit accelerated inactivation with Ca2+ as charge carrier throughout its slow inactivation time course (Fig. 3A,B). As a consequence, the majority of ICa through Cav1.2α1 and Cav1.3α1, but hardly any Cav1.4α1 current, inactivated during 200 – 400 msec.
LTCCs are defined by their high sensitivity to DHP antagonists and their activation by DHP Ca2+ channel activators (Peterson et al., 1996), such as BayK 8644. In Cav1.4α1 subunits, a IVS6 tyrosine (position 1414 in the human Cav1.4α1 sequence) (Fig. 8), previously shown to contribute to the formation of the binding pocket (Peterson et al., 1996), is replaced by a phenylalanine. The corresponding mutation in Cav1.1α1 subunits reduces DHP antagonist sensitivity ∼3- to 5-fold (Peterson et al., 1996). Its role for agonist action has not yet been studied. Therefore, we tested the DHP sensitivity of heterologously expressed Cav1.4α1 channels (+ β3 + α2δ1). At -90 mV HP, the DHP antagonist isradipine (1 μm) blocked 82.7 ± 2.9% (n = 7) of IBa elicited by 0.1 Hz depolarizing pulses to Vmax (Fig. 4A). The same concentration completely inhibited Cav1.3α1 currents under identical experimental conditions, as reported previously (Koschak et al., 2001) (Fig. 4B). Current inhibition by 300 nm concentrations was also slightly less pronounced for Cav1.4α1, compared with Cav1.3α1 (Koschak et al., 2001) (Fig. 4B). Changing the HP from -90 to -50 mV dramatically increased isradipine sensitivity of Cav1.4α1 (Fig. 4B, open triangle), indicating a voltage-dependent mechanism of DHP block, which is also typical for both Cav1.2α1 and Cav1.3α1 (Welling et al., 1997; Koschak et al., 2001). Activation of IBa through Cav1.4α1 by the Ca2+ channel activator BayK 8644 occurred in an LTCC-typical manner (Fig. 5). Perfusion of Cav1.4α1-transfected cells (yielding significant IBa already in the absence of drug) with 5 μm BayK 8644 resulted in a robust (9.6 ± 1.9-fold; n = 7) increase of the maximal IBa (Fig. 5A), similar to Cav1.3α1 stimulation (Koschak et al., 2001). Furthermore, BayK 8644 produced a typical ∼10 mV hyperpolarizing shift of the I—V curve (Table 1, Fig. 5B). Interestingly, in some GFP-positive cells (four of four tested) with no significant current under basal conditions, the presence of Cav1.4α1 currents was unmasked by application of the Ca2+ channel activator BayK 8644 (5 μm; 10.5 ± 1.3 pA/pF; n = 4). This suggested that an even >10-fold stimulation of Cav1.4α1 currents occurred in some cells. Interestingly, a similarly strong BayK 8644 dependence of L-type current components has also been described in retinal cone bipolar cells (Pan, 2000). Our data demonstrate that phenylalanine in position 1414 of Cav1.4α1 still supports full agonist sensitivity. Therefore, a tyrosine in this position is unlikely to be required for DHP agonist action in LTCCs.
Figure 8.

Sequence alignment of L-type Cav1.4α1 with Cav1.2α1 and Cav1.3α1. An amino acid exchange within the proposed DHP-binding domain of LTCCs is indicated by an arrow. A tyrosine residue conserved in all L-type α1 subunits is replaced by phenylalanine in position 1414 of Cav1.4α1. The sequence alignment also illustrates amino acid differences between Cav1.4α1 and Cav1.2α1 or Cav1.3α1, which might explain the differences in Ca2+-dependent inactivation described under our experimental conditions. Sequence stretches previously identified as critical determinants for calmodulin-binding and Ca2+-dependent inactivation (de Leon et al., 1995; Zuhlke et al., 1999; Mouton et al., 2001; Pitt et al., 2001) are indicated.
Figure 4.

DHP antagonist sensitivity of Cav1.4α1 Ca2+ channels. Cav1.4α1 was coexpressed with β3 and α2δ1 subunits in tsA-201 cells and recorded in bath solution containing 15 mm Ba2+ as charge carrier. A, IBa was elicited from depolarizations to Vmax (filled circles) in the absence or presence (gray bar) of the DHP antagonist isradipine. For the experiment shown,IBa block by 1 μm isradipine was 80%. Corresponding peak current traces elicited by 40 msec depolarizations in the absence (control) and presence of isradipine are shown in the inset. B, Concentration-dependent inhibition was measured from a holding potential of -90 mV for Cav1.4α1 (black triangles) during superfusion of the cell with bath solution containing the indicated concentrations of isradipine. The dose—response relationship was compared with Cav1.3α1 (squares) (Koschak et al., 2001). Pronounced voltage dependence of isradipine block of IBa through Cav1.4α1 subunits was observed by changing the HP from -90 to -50 mV (open triangle). Asterisks indicate statistically significant difference (p < 0.05; data are means ± SE).
Figure 5.

DHP agonist sensitivity of Cav1.4α1 Ca2+ channels. For all experiments, Cav1.4α1 subunits were coexpressed with β3 and α2δ1 subunits in tsA-201 cells. Charge carrier was 15 mm Ba2+. One representative experiment (of 9) is shown. A, Stimulation of IBa through Cav1.4α1 Ca2+ channels by the Ca2+ channel activator BayK 8644 (BayK). IBa was elicited by depolarizations to Vmax before and after application of BayK 8644-containing solution (5 μm). Maximal IBa is plotted against time. The inset shows representative traces in the absence (control) and presence of BayK 8644. B, Current—voltage relationship for Cav1.4α1 in the absence (black circles) and presence of 5 μm BayK 8644 (BayK; gray circles). V0.5, act was -8 and -21.1 mV for the control and BayK 8644-modulated current, respectively. One representative experiment (of 7) is shown.
We also tested the modulation of Cav1.4α1 currents by G-protein activation and/or strong depolarizing pulses. Figure 6A shows the effect of 200 msec depolarizing prepulses to 80 mV on IBa through Cav1.4α1, elicited by a subsequent test pulse toVmax. Prepulses facilitated IBa in 59% (10 of 17) of Cav1.4α1 + β3+ α2δ1-transfected cells. The absence of facilitation in some cells has also been reported previously for Cav1.2α1 after expression in mammalian cells (Kamp et al., 2000). In cells showing facilitation, the extent of facilitation was slightly (but not significantly) smaller for Cav1.4α1 (facilitation ratio, 1.13 ± 0.02) than for Cav1.2 α1 (1.27 ± 0.07) and Cav1.3 α1 (1.26 ± 0.09) (Fig. 6B) when measured under identical experimental conditions. As expected (Bourinet et al., 1994), no facilitation was observed for the P/Q-type channel Cav2.1α1 subunit (Fig. 7B,C, lower panel) (n = 7) under these experimental conditions. The application of 200 msec prepulses not only increased the peak current but also accelerated the activation and inactivation kinetics of the facilitated Cav1.4α1 current (Fig. 6A). Whereas control IBa hardly inactivated (>97% of current remained at the end of the 400 msec test pulse) (Fig. 6A), slightly accelerated inactivation was detected for the facilitated current (92.8 ± 0.8% residual current;p < 0.001). Similar kinetic changes have also been observed for other facilitated L-type currents (Dai et al., 1999; Kamp et al., 2000). Coexpression with β2a + α2δ1 subunits also supported facilitation, which was observed in 85% of the experiments, and the average facilitation ratio was similar, as measured for β3 +α2δ1 (1.18 ± 0.03; n = 6). Similar facilitation was observed with Ca2+ as a charge carrier. Prepulses facilitated ICa in five of five Cav1.4α1 + β3 + α2δ1-transfected cells (facilitation ratio, 1.16 ± 0.06).
Figure 6.

Voltage-dependent facilitation of Cav1.4α1 subunits. A, Schematic representation of the voltage protocol used to elicit facilitation. Channel activity was recorded in tsA-201 cells transfected with Cav1.4α1, β3, and α2δ1 subunits in 15 mm Ba2+ solution. Test pulses (TPs) of 400 msec were applied with or without a 200 msec PP. A representative current trace (facilitation ratio, 1.11) is shown. Note the differences in activation and inactivation kinetics time courses. B, Comparison of voltage-dependent facilitation of different LTCCs. Facilitation of Cav1.2α1, Cav1.3α1, and Cav1.4α1 IBa is expressed as the amplitude ratio of currents recorded with a prepulse (+PP) over the respective control currents without prepulse (-PP). A similar extent of facilitation was observed for all LTCC subtypes tested (p > 0.05). Facilitation was observed in seven of seven Cav1.2α1-transfected cells, eight of 20 Cav1.3α1-transfected cells, and 10 of 17 Cav1.4α1 (+ β3 + α2δ1)-transfected cells.
Figure 7.
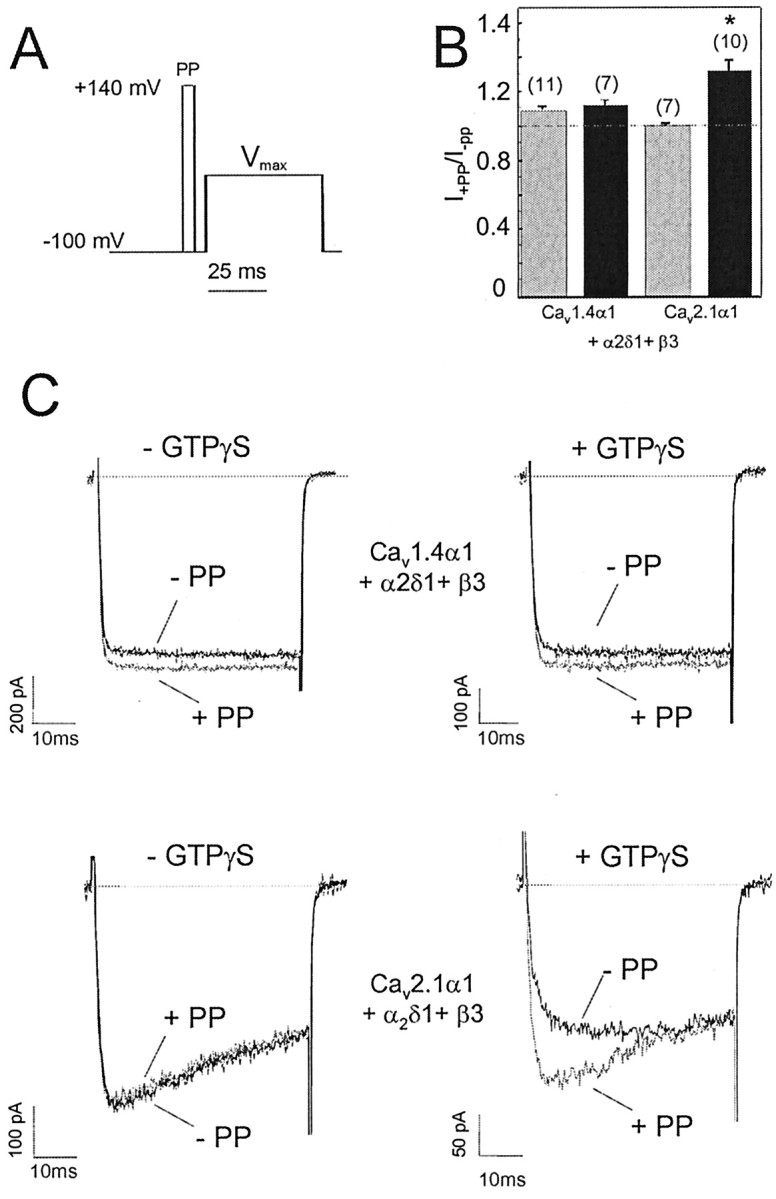
Depolarization-induced facilitation of Cav1.4α1 subunits. A, Pulse protocol used to determine G-protein modulation of Cav1.4α1- and Cav2.1α1-mediated IBa. Facilitation was measured during 50 msec test pulses (TP) to Vmax either with or without a 5 msec PP to 140 mV. B, Facilitation ratios for Cav1.4α1 and Cav2.1α1 Ca2+ channels (coexpressed with β3 and α2δ1) in the absence (control; gray bars) and presence of 300 μm intracellular GTPγS (black bars) using the pulse protocol described in A. Recordings were started after ≥3 min of dialysis with GTPγS. The following facilitation ratios were obtained in the absence and presence of GTPγS, respectively:Cav1.4α1, 1.1±0.02 (n=11 of 17 cells), 1.12±0.02 (n=7 of 14 cells); Cav2.1α1, 0.99 ± 0.01 (n = 7 of 7 cells), 1.3 ± 0.07 (n = 10 of 10 cells). Asterisk indicates statistically significant difference (p < 0.01). C, Representative current traces for the experiments described in B.
We found no evidence for a G-protein dependence of this prepulse facilitation of Cav1.4α1 currents. When the nonhydrolyzable GTP analog GTPγS was included in the pipette solution to activate expressed G-proteins in tsA-201 cells (Herlitze et al., 1997; Meza and Adams, 1998), facilitation induced by 5 msec depolarizing prepulses to 140 mV remained unaffected (Fig. 7). To prove that G-protein activation is feasible under our experimental conditions, the modulation of Cav2.1α1 channels was determined using the same experimental protocol. At least 3 min after establishing the whole-cell configuration, prepulse application caused the typical relief of G-protein modulation of Cav2.1α1 currents characterized by faster activation and the reduction of peak current amplitude (Fig. 7B,C). All Cav2.1α1 +β3 + α2δ1-expressing cells (seven of seven) dialyzed with GTPγS exhibited this typical G-protein-mediated inhibition (Fig. 7B,C), consistent with previous studies (Bourinet et al., 1996; Zhang et al., 1996; Herlitze et al., 1997; Meza and Adams, 1998; Canti et al., 1999). These experiments clearly demonstrated that voltage-dependent G-protein modulation can be measured under our experimental conditions, but that facilitation of Cav1.4α1 is unlikely to result from relief of G-protein-induced channel inhibition.
Discussion
Biophysical properties of Cav1.4α1
Here we report the first successful functional characterization of Ca2+ currents through Cav1.4α1 subunits. Experiments were performed under the same conditions previously used to compare the biophysical and pharmacological properties of Cav1.3α1 and Cav1.2α1 LTCCs using the whole-cell patch-clamp technique. Cav1.4α1 currents resemble more closely Cav1.3α1 than Cav1.2α1. Like Cav1.3α1, Cav1.4α1 activated more rapidly and at more negative voltages than heterologously expressed Cav1.2α1 and also inactivated more slowly. Cav1.4α1 exhibited no Ca2+-induced inactivation, which was found to exist in Cav1.3α1 (Xu and Lipscombe, 2001; our observations) and is well studied for Cav1.2α1 (for review, see Budde et al., 2002). Therefore, the slower inactivation of Cav1.4α1 became especially prominent when Ca2+ was the permeating cation (Fig. 3).
Ca2+-dependent inactivation is mediated through Ca2+ and calmodulin interaction with the C-terminal tail and is a typical property of LTCCs (for review, see Budde et al., 2002). However, the absence of Ca2+-dependent inactivation is not a specific property of Cav1.4α1. A putative neuronal Cav1.2α1 C-terminal splice variant, Cav1.2α186 (α1C,86), also lacks Ca2+-induced inactivation (Soldatov et al., 1997). In this splice variant, 80 amino acid residues of the C-terminal tail are replaced by 81 essentially nonidentical amino acid residues. This eliminates important motifs essential for calmodulin-mediated Ca2+-dependent inactivation and also causes a profound acceleration of voltage-dependent inactivation of IBa. In contrast, inspection of the Cav1.4α1 sequence (Fig. 8) revealed that the regions recently identified as determinants for Ca2+-dependent inactivation in Cav1.2α1 (Qin et al., 1999; Zuhlke et al., 1999; Pate et al., 2000; Peterson et al., 2000; Romanin et al., 2000; Mouton et al., 2001; Pitt et al., 2001) are highly conserved in this subunit. Only a few amino acid differences, as compared with Cav1.2α1 and Cav1.3α1, exist in the F-helix of the EF hand (Peterson et al., 2000), peptide A (Pitt et al., 2001), the CB peptide (Pate et al., 2000), and peptide C (Pitt et al., 2001). Therefore, Cav1.4α1 subunits represent an ideal model to further study the role of these amino acid changes for the molecular mechanisms of Ca2+-dependent inactivation. Additional studies must also address the question whether Cav1.4α1 undergoes N-lobe calmodulin-mediated calcium-dependent inactivation revealed at low intracellular Ca2+ buffering (Liang et al., 2003).
DHP-sensitivity of Cav1.4α1
As for Cav1.3α1, the apparent DHP antagonist sensitivity of Cav1.4α1 was significantly lower (∼15-fold) than for Cav1.2α1 at negative holding potentials. This intermediate DHP antagonist sensitivity of Cav1.4α1 and Cav1.3α1 is in good accordance with data obtained on L-type currents in retinal cells, in which relatively high concentrations of DHPs are required to block ICa,L (Wilkinson and Barnes, 1996; Protti and Llano, 1998; Taylor and Morgans, 1998). Cav1.4α1 current inhibition by DHP antagonist was highly voltage dependent. At a more positive membrane potential, 100 nm isradipine inhibited >80% of IBa (Fig. 4B), indistinguishable from the block of Cav1.3α1 under the same experimental conditions (Koschak et al., 2001). Therefore, the low apparent affinity of Cav1.4α1 in comparison with Cav1.2α1 is likely to be attributable to differences in the voltage-dependent interaction of the DHP antagonist, as recently demonstrated also for Cav1.3α1 (Koschak et al., 2001). We could exploit a “natural mutation,” an amino acid exchange from tyrosine to phenylalanine in position 1414 (Fig. 8), of the DHP-binding domain to investigate the role of this residue for agonist action. In Cav1.1α1 (data not shown), Cav1.2α1, and Cav1.3α1, a tyrosine is found in this position (Tyr 1463; α1C-a numbering) (Striessnig et al., 1998). Mutation of this residue to phenylalanine was found to decrease DHP antagonist-binding affinity at least in Cav1.1α1 (Peterson et al., 1996). We show that the tyrosine to phenylalanine exchange in Cav1.4α1 does not cause a major change in DHP antagonist sensitivity, as compared with Cav1.3α1. The role of this tyrosine for DHP agonist sensitivity has not been investigated thus far. Because we found a robust stimulation of Cav1.4α1-mediated currents by BayK 8644, we can clearly demonstrate that the tyrosine hydroxyl is not required for BayK 8644 stimulation of LTCCs.
Functional implications
Our data provide a first answer to the important question whether Cav1.4α1 LTCCs can contribute to the L-type currents in retinal photoreceptors and bipolar cells, which are tightly coupled to neurosecretion. In photoreceptors (Wilkinson and Barnes, 1996; Taylor and Morgans, 1998; Kourennyi and Barnes, 2000; Stella et al., 2002) and bipolar cells (von Gersdorff and Matthews, 1996; Protti and Llano, 1998) of different species, DHP-sensitive ICa was found to posses properties not typically found for L-type currents in cardiac myocytes or neurons. These were described as faster activation, slower inactivation, negative activation thresholds, and intermediate DHP sensitivity. Such properties were described previously both in the current study and by others (for review, see Lipscombe, 2002) for Cav1.3α1. Cav1.3α1 is expressed in photoreceptor terminals in the outer plexiform layer (OPL) and, most likely, also bipolar cells synapses in the inner plexiform layer (IPL) (Morgans et al., 1998; Taylor and Morgans, 1998; Morgans, 1999). Therefore, these channels could account for the retinal ICa,L in these cells. However, we can now demonstrate that Cav1.4α1 can also mediate currents with similar properties. Because Cav1.4α1 is also expressed in the synapses of the OPL and IPL (Firth et al., 2001; Morgans, 2001; Morgans et al., 2001; Ball et al., 2002; Berntson et al., 2003), it may also participate in the formation of photoreceptor and bipolar cell ICa,L (Berntson et al., 2003). In humans, Cav1.4α1 mutations cause iCSNB2. Most of these mutations result in truncated subunits and should cause a complete loss of function. The most promising animal model to directly quantitate the contribution of Cav1.4α1 to retinal ICa,L are Cav1.4α1-deficient mice. In these animals, one should be able to correlate visual defects with the relative contribution of Cav1.4α1 to retinal ICa,L. Cav1.3α1-deficient mice do not seem to represent such a useful model because they do not exhibit electroretinogram changes (Platzer et al., 2000) (M.W. Seeliger, E. Schmid, J. Platzer, and J. Striessnig, unpublished observations).
On the basis of their biophysical characteristics and intermediate DHP sensitivity, Cav1.3α1 and Cav1.4α1 might be classified as a functional LTCC subgroup. Because of its lower activation threshold, Cav1.3α1 has indeed been shown to serve in an essential role for cardiac pacemaking in the sinoatrial node, which cannot be substituted by Cav1.2α1 expressed in the same cells (Zhang et al., 2002; Mangoni et al., 2003). Faster activation, lower activation thresholds, and slower inactivation make Cav1.3α1 and Cav1.4α1 also suited for certain neuronal functions. First, they can support neurotransmitter release from nonspiking neurons and sensory cells such as photoreceptors (Cav1.3α1 and Cav1.4α1) and cochlear inner hair cells (Cav1.3α1). In the latter, >90% of the current is carried by Cav1.3α1 (Platzer et al., 2000). In the darkness, photoreceptors are continuously depolarized by cGMP-gated channels to approximately -30 to -40 mV. During illumination, they hyperpolarize by approximately -20 to -30 mV (Witkovsky et al., 1997). To rapidly adjust tonic release to changes in illumination (i.e., changes in membrane potential),ICa,L should be rapidly gated, activated over a relatively negative voltage range, and slowly inactivated at depolarized potentials. These criteria are fulfilled by Cav1.3α1 and Cav1.4α1 but not Cav1.2α1. Second, sustained ICa,L, activating at negative voltages, are suitable to support plateau potentials in neurons elicited, for instance, by weak depolarizations to voltages just above the resting potential of a neuron. For example, such plateau potentials occur in motoneurons (Carlin et al., 2000; Alaburda et al., 2002) and second order pain neurons (Morisset and Nagy, 2000), in which they modulate motoneuron responses and pain processing, respectively. Such current components are believed to be mediated by Cav1.3α1 (Carlin et al., 2000; Alaburda et al., 2002), but systematic analysis of Cav1.4α1 expression (e.g., in motoneurons) has not yet been performed. Similarly, in bipolar cell nerve terminals, such low voltage-activated L-type Ca2+ currents also seem to account for the interesting finding that specific retinal bipolar cells, which are generally considered nonspiking cells, can respond to light-induced tonic depolarization by photoreceptors with Ca2+ action potentials and regenerative responses from a plateau potential (Burrone and Lagnado, 1997; Protti et al., 2000), mechanisms that are suitable to amplify small photoreceptor signals. Additional studies will have to determine the inactivation kinetics of Cav1.3α1 and Cav1.4α1 on even longer time scales than those shown here. Note that in mouse cochlea inner hair cells, Cav1.3α1-mediated currents inactivate to a much smaller extent than after heterologous expression (Zidanic and Fuchs, 1995; Kollmar et al., 1997; Platzer et al., 2000). It will be important to reveal the molecular substrate of this difference, which may not be only attributable to alternative splicing of α1 subunits (Koschak et al., 2001; Safa et al., 2001; Xu and Lipscombe, 2001).
Our work demonstrates for the first time that Cav1.4α1 subunits form functional LTCCs that can contribute to such currents. Therefore, this study paves the way for the analysis of Cav1.4α1 mutations responsible for iCSNB2. A detailed genotype—phenotype analysis of this disease will now be possible.
Footnotes
This work was supported by Austrian Science Fund Grants P-14541 and P-14820 (J.S.), the österreichische National Bank, and the European Community Grant HPRN-CT-2000 – 00082. We thank G. Pelster for excellent technical support.
Correspondence should be addressed to Jörg Striessnig, Institut für Pharmazie, Abteilung Pharmakologie und Toxikologie, Peter-Mayr-Strasse 1/I, A-6020 Innsbruck, Austria.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236041-09$15.00/0
References
- Alaburda A, Perrier JF, Hounsgaard J ( 2002) Mechanisms causing plateau potentials in spinal motoneurones. Adv Exp Med Biol 508: 219–226. [DOI] [PubMed] [Google Scholar]
- Ball SL, Powers PA, Shin HS, Morgans CW, Peachey NS, Gregg RG ( 2002) Role of the beta(2) subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci 43: 1595–1603. [PubMed] [Google Scholar]
- Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM ( 1998) Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet 19: 264–267. [DOI] [PubMed] [Google Scholar]
- Berntson A, Taylor WR, Morgans CW ( 2003) Molecular identity, synaptic localization, and physiology of calcium channels in retinal bipolar cells. J Neurosci Res 71: 146–151. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Charnet P, Tomlinson WJ, Stea A, Snutch TP, Nargeot J ( 1994) Voltage-dependent facilitation of a neuronal α1C L-type calcium channel. EMBO J 13: 5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Stea A, Snutch TP ( 1996) Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci USA 431: 470–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott KM, Maybaum TA, Naylor MJ, Weleber RG, Robitaille J, Miyake Y, Bergen AA, Pierpont ME, Pearce WG, Bech-Hansen NT ( 2001) A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants. Hum Genet 108: 91–97. [DOI] [PubMed] [Google Scholar]
- Budde T, Meuth S, Pape HC ( 2002) Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci 3: 873–883. [DOI] [PubMed] [Google Scholar]
- Burrone J, Lagnado L ( 1997) Electrical resonance and Ca2+ influx in the synaptic terminal of depolarizing bipolar cells from the goldfish retina. J Physiol (Lond) 505: 571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti C, Page KM, Stephens GJ, Dolphin AC ( 1999) Identification of residues in the N terminus of alpha1B critical for inhibition of the voltage-dependent calcium channel by Gβγ J Neurosci 19: 6855–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin KP, Jones KE, Jiang Z, Jordan LM, Brownstone RM ( 2000) Dendritic L-type calcium currents in mouse spinal motoneurons: implications for bistability. Eur J Neurosci 12: 1635–1646. [DOI] [PubMed] [Google Scholar]
- Castellano A, Wei X, Birnbaumer L, Perez-Reyes E ( 1993) Cloning and expression of a third calcium channel beta subunit. J Biol Chem 268: 3450–3455. [PubMed] [Google Scholar]
- Catterall WA ( 2000) Structure and regulation of voltage-gated calcium channels. Annu Rev Cell Dev Biol 16: 521–555. [DOI] [PubMed] [Google Scholar]
- Dai S, Klugbauer N, Zong X, Seisenberger C, Hofmann F ( 1999) The role of subunit composition on prepulse facilitation of the cardiac L-type calcium channel. FEBS Lett 442: 70–74. [DOI] [PubMed] [Google Scholar]
- de Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X, Soong TW, Snutch TP, Yue DT ( 1995) Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science 270: 1502–1506. [DOI] [PubMed] [Google Scholar]
- Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A, Schwartz A, Harpold MM ( 1988) Sequence and expression of mRNAs encoding the alpha1 and alpha2 subunits of a DHP-sensitive calcium channel. Science 241: 1661–1664. [DOI] [PubMed] [Google Scholar]
- Firth SI, Morgan IG, Boelen MK, Morgans CW ( 2001) Localization of voltage-sensitive L-type calcium channels in the chicken retina. Clin Experiment Ophthalmol 29: 183–187. [DOI] [PubMed] [Google Scholar]
- Grabner M, Dirksen RT, Beam KG ( 1998) Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc Natl Acad Sci USA 95: 1903–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR ( 1999) L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401: 703–708. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Hockerman GH, Scheuer T, Catterall WA ( 1997) Molecular determinant of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel alpha 1A subunit. Proc Natl Acad Sci USA 94: 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber I, Wappl E, Herzog A, Mitterdorfer J, Glossmann H, Langer T, Striessnig J ( 2000) Conserved Ca2+ antagonist binding properties and putative folding structure of a recombinant high affinity dihydropyridine binding domain. Biochem J 347: 829–836. [PMC free article] [PubMed] [Google Scholar]
- Kamp TJ, Hu H, Marban E ( 2000) Voltage-dependent facilitation of cardiac L-type Ca channels expressed in HEK-293 cells requires beta-subunit. Am J Physiol Heart Circ Physiol 278: H126–H136. [DOI] [PubMed] [Google Scholar]
- Kollmar R, Fak J, Montgomery LG, Hudspeth AJ ( 1997) Hair cell-specific splicing of mRNA for the alpha1D subunit of voltage-gated Ca2+ channels in the chicken's cochlea. Proc Natl Acad Sci USA 94: 14889–14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J ( 2001) alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem 276: 22100–22106. [DOI] [PubMed] [Google Scholar]
- Kourennyi DE, Barnes S ( 2000) Depolarization-induced calcium channel facilitation in rod photoreceptors is independent of G proteins and phosphorylation. J Neurophysiol 84: 133–138. [DOI] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori M, Alseikhan BA, Yue DT ( 2003) Toward a unified mechanism for calcium regulation of the calcium channel family. Biophys J 84: 405. [Google Scholar]
- Lipscombe D ( 2002) L-type calcium channels: highs and new lows. Circ Res 90: 933–935. [DOI] [PubMed] [Google Scholar]
- Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J ( 2003) Functional role of L-type Cav1.3 calcium channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100: 5543–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza U, Adams B ( 1998) G-Protein-dependent facilitation of neuronal alpha1A, alpha1B, and alpha1E Ca2+ channels. J Neurosci 18: 5240–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S ( 1989) Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature 340: 230–233. [DOI] [PubMed] [Google Scholar]
- Morgans CW ( 1999) Calcium channel heterogeneity among cone photoreceptors in the tree shrew retina. Eur J Neurosci 11: 2989–2993. [DOI] [PubMed] [Google Scholar]
- Morgans CW ( 2001) Localization of the alpha(1F) calcium channel subunit in the rat retina. Invest Ophthalmol Vis Sci 42: 2414–2418. [PubMed] [Google Scholar]
- Morgans CW, El Far O, Berntson A, Wassle H, Taylor WR ( 1998) Calcium extrusion from mammalian photoreceptor terminals. J Neurosci 18: 2467–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgans CW, Gaughwin P, Maleszka R ( 2001) Expression of the alpha1F calcium channel subunit by photoreceptors in the rat retina. Mol Vis 7: 202–209. [PubMed] [Google Scholar]
- Morisset V, Nagy F ( 2000) Plateau potential-dependent windup of the response to primary afferent stimuli in rat dorsal horn neurons. Eur J Neurosci 12: 3087–3095. [DOI] [PubMed] [Google Scholar]
- Mouton J, Feltz A, Maulet Y ( 2001) Interactions of calmodulin with two peptides derived from the c-terminal cytoplasmic domain of the Ca(v)1.2 Ca2+ channel provide evidence for a molecular switch involved in Ca2+-induced inactivation. J Biol Chem 276: 22359–22367. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakagawasai O, Fujii S, Kameyama K, Murakami S, Hozumi S, Esashi A, Taniguchi R, Yanagisawa T, Tan-no K, Tadano T, Kitamura K, Kisara K ( 2001) Antinociceptive action of amlodipine blocking N-type Ca2+ channels at the primary afferent neurons in mice. Eur J Pharmacol 419: 175–181. [DOI] [PubMed] [Google Scholar]
- Naylor MJ, Rancourt DE, Bech-Hansen NT ( 2000) Isolation and characterization of a calcium channel gene, cacna1f, the murine orthologue of the gene for incomplete X-linked congenital stationary night blindness. Genomics 66: 324–327. [DOI] [PubMed] [Google Scholar]
- Pan ZH ( 2000) Differential expression of high- and two types of low-voltage-activated calcium currents in rod and cone bipolar cells of the rat retina. J Neurophysiol 83: 513–527. [DOI] [PubMed] [Google Scholar]
- Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney GG, Serysheva II, Williams BY, Anderson ME, Hamilton SL ( 2000) Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem 275: 39786–39792. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei X, Birnbaumer L ( 1992) Cloning and expression of a cardiac/brainβ subunit of the L-type calcium channel. J Biol Chem 267: 1792–1797. [PubMed] [Google Scholar]
- Peterson BZ, Tanada TN, Catterall WA ( 1996) Molecular determinants of high affinity dihydropyridine binding in L-type calcium channels. J Biol Chem 271: 5293–5296. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, Lee JS, Mulle JG, Wang Y, de Leon M, Yue DT ( 2000) Critical determinants of Ca2+-dependent inactivation within an EF-hand motif of L-type Ca(2+) channels. Biophys J 78: 1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW ( 2001) Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem 276: 30794–30802. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J ( 2000) Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type calcium channels. Cell 102: 89–97. [DOI] [PubMed] [Google Scholar]
- Protti DA, Llano I ( 1998) Calcium currents and calcium signaling in rod bipolar cells of rat retinal slices. J Neurosci 18: 3715–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, von GersdorffH ( 2000) Light evokes Ca2+ spikes in the axon terminal of a retinal bipolar cell. Neuron 25: 215–227. [DOI] [PubMed] [Google Scholar]
- Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L ( 1999) Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci USA 96: 2435–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, Abernethy DR, Soldatov NM ( 2000) Ca2+ sensors of L-type Ca2+ channel. FEBS Lett 487: 301–306. [DOI] [PubMed] [Google Scholar]
- Safa P, Boulter J, Hales TG ( 2001) Functional properties of Cav1.3 (alpha1D) L-type Ca2+ channel splice variants expressed by rat brain and neuroendocrine GH3 cells. J Biol Chem 276: 38727–28737. [DOI] [PubMed] [Google Scholar]
- Safayhi H, Haase H, Kramer U, Bihlmayer A, Roenfeldt M, Ammon HP, Froschmayr M, Cassidy TN, Morano I, Ahlijanian M, Striessnig J ( 1997) L-type calcium channels in insulin-secreting cells: biochemical characterization and phosphorylation in RINm5F cells. Mol Endocrinol 11: 619–629. [DOI] [PubMed] [Google Scholar]
- Scholze A, Plant TD, Dolphin AC, Nurnberg B ( 2001) Functional expression and characterization of a voltage-gated CaV1.3 (alpha1D) calcium channel subunit from an insulin-secreting cell line. Mol Endocrinol 15: 1211–1221. [DOI] [PubMed] [Google Scholar]
- Soldatov NM, Zuhlke RD, Bouron A, Reuter H ( 1997) Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40-42 in alpha1C subunit in the kinetics and Ca2+ dependence of inactivation. J Biol Chem 272: 3560–3566. [DOI] [PubMed] [Google Scholar]
- Stella Jr SL, Bryson EJ, Thoreson WB ( 2002) A2 adenosine receptors inhibit calcium influx through L-type calcium channels in rod photoreceptors of the salamander retina. J Neurophysiol 87: 351–360. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinnegger MJ, Glossmann H ( 1998) Structural basis of drug binding to L calcium channels. Trends Pharmacol Sci 19: 108–115. [DOI] [PubMed] [Google Scholar]
- Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BH, Wutz K, Gutwillinger N, Ruther K, Drescher B, Sauer C, Zrenner E, Meitinger T, Rosenthal A, Meindl A ( 1998) An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet 19: 260–263. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Morgans C ( 1998) Localization and properties of voltage-gated calcium channels in cone photoreceptors of Tupaia belangeri. Vis Neurosci 15: 541–552. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G ( 1996) Calcium-dependent inactivation of calcium current in synaptic terminals of retinal bipolar neurons. J Neurosci 16: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wappl E, Koschak A, Poteser M, Sinnegger MJ, Walter D, Eberhart A, Groschner K, Glossmann H, Kraus RL, Grabner M, Striessnig J ( 2002) Functional consequences of P/Q-type Ca2+ channel Cav2.1 missense mutations associated with episodic ataxia type 2 and progressive ataxia. J Biol Chem 277: 6960–6966. [DOI] [PubMed] [Google Scholar]
- Welling A, Ludwig A, Zimmer S, Klugbauer N, Flockerzi V, Hofmann F ( 1997) Alternatively spliced IS6 segments of the alpha 1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle L-type Ca2+ channels. Circ Res 81: 526–532. [DOI] [PubMed] [Google Scholar]
- Wilkinson MF, Barnes S ( 1996) The dihydropyridine-sensitive calcium channel subtype in cone photoreceptors. J Gen Physiol 107: 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkovsky P, Schmitz Y, Akopian A, Krizaj D, Tranchina D ( 1997) Gain of rod to horizontal cell synaptic transfer: relation to glutamate release and a dihydropyridine-sensitive calcium current. J Neurosci 17: 7297–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Lipscombe D ( 2001) Neuronal Cav1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21: 5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW ( 1996) Multiple structural elements in voltage-dependent calcium channels support their inhibition by G proteins. Neuron 17: 991–1003. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N ( 2002) Functional roles of Cav1.3 (alpha1D) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res 90: 981–987. [DOI] [PubMed] [Google Scholar]
- Zidanic M, Fuchs PA ( 1995) Kinetic analysis of barium currents in chick cochlear hair cells. Biophys J 68: 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H ( 1999) Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399: 159–162. [DOI] [PubMed] [Google Scholar]