

Abstract
Phenyl-N-tert-butylnitrone (PBN) protects rat retinas against light damage. Because the degenerative process involved in light damage and inherited retinal degeneration both lead to a common final cell death, apoptosis, we used transgenic rats with a P23H or S334ter rhodopsin mutation to test the effects of PBN on retinal degeneration and light damage and the susceptibility of the transgenic rats to light damage. In the first study, 3-week-old mutant and wild-type rats were given no drug, 0.25% PBN in drinking water, or 0.25% PBN in drinking water plus three daily intraperitoneal injections of PBN (100 mg/kg, i.p., every 8 hr). Electroretinograms were recorded at postnatal day 49, after which the rats were killed for morphometric analysis. There was no photoreceptor rescue by PBN in P23H or S334ter rats, as evidenced by equivalent loss of function and photoreceptor cells in the three treatment groups. In the second study, P23H, S334ter, and wild-type rats were exposed for 24 hr to 2700 lux light. The rats were untreated or treated with PBN (50 mg/kg per injection, every 6 hr, starting before exposure). ERGs were recorded before and 1 d after exposure. Animals were killed 6 d later for morphometric analysis. PBN protected wild-type and P23H but not S334ter retinas from light damage. S334ter retinas were relatively less susceptible to light damage than P23H and wild-type rats. The results suggest that the initiating event(s) that causes photoreceptor cell death in the mutated rats is different from that which occurs in light damage, although both ultimately undergo an apoptotic cell death.
Keywords: neuroprotection, inherited retinal degeneration, free radical, phenyl-N-tert-butylnitrone, electroretinography, light damage
Introduction
Retinitis pigmentosa (RP) is a genetically and clinically heterogeneous group of retinal degenerative diseases, affecting ∼1 in 3500 people (Pagon, 1988). Symptoms include night blindness, progressive loss of peripheral visual field, and eventual loss of central vision caused by degeneration of photoreceptor cells. A number of genes responsible for RP have now been identified (Dryja and Li, 1995; Sullivan and Daiger, 1996), most of which are expressed specifically in photoreceptor cells, and the degeneration primarily affects photoreceptor cells. There are now >100 different mutations in the rod photoreceptor visual pigment rhodopsin that are associated with RP (RetNet; http://sph.uth.tmc.edu/Ret-Net/). The rhodopsin molecule has distinct regions that are specialized for light capture, initiation of the phototransduction cascade, and rapid deactivation after light absorption. Other regions ensure its proper folding and sorting within the photoreceptor membranes. Therefore, it is not surprising that the phenotype depends on the location of the mutation and may be expressed as either dominant or recessive RP. Transgenic rat lines have been developed that express one of two mutated rhodopsin genes, as well as the wild-type gene (Steinberg et al., 1996). In S334ter rats, the opsin transgene contains a termination codon at residue 334, resulting in the expression of a rhodopsin protein lacking the 15 C-terminal amino acids. In P23H rats, the opsin transgene contains a histidine substitution at the proline 23 position. The P23H transgenic rats have a slow rod degeneration with normal cone function initially, consistent with the clinical findings of P23H patients (Machida et al., 2000).
Although transgenic rats and other animal models have been developed to study the cellular etiologies of inherited retinal degenerations and to test potential therapies, there is still little known about the molecular mechanisms leading to photoreceptor cell death, and there are no adequate therapies for RP at present. Because mutations in integral, peripheral, and soluble proteins can all lead eventually to cell death, there must be a number of initiating events. However, most studies to date have found that the photoreceptor cells undergo apoptosis as the final common death pathway converging the primary defects (Remé et al., 1998). Light-induced damage represents a suitable model system to study retinal degeneration, because apoptotic cell death underlies both light-induced damage (Li et al., 1995; Organisciak et al., 1995) and inherited retinal degeneration (Chang et al., 1993; Lolley et al., 1994; Portera-Caillau et al., 1994; Tso et al., 1994) in various animal models. This suggested that strategies used in light-damage studies could be applied to animals with inherited retinal degenerations. Using this approach, LaVail and coworkers showed that the intravitreal administration of basic FGF (bFGF) (Faktorovich et al., 1992) and other neurotrophic factors (LaVail et al., 1992) rescued photoreceptors of albino rats from light damage. bFGF also prevented inherited retinal degeneration in the Royal College of Surgeons (RCS) rat (Faktorovich et al., 1990). Other classes of survival factors that protect photoreceptors from light damage or mutations have also been discovered, including α-2-adrenergic agonists (Wen et al., 1996), a melatonin receptor antagonist (Sugawara et al., 1998), a caspase-3 inhibitor (Liu et al., 1999), pigment epithelium-derived factor (Cayouette et al., 1999; Cao et al., 2001), lens epithelium-derived growth factor (Machida et al., 2001), heat shock protein (Yu et al., 2001), and additional neurotrophic factors such as glial cell line-derived neurotrophic factor (Frasson et al., 1999) and ciliary neurotrophic factor (LaVail et al., 1998). The antioxidants vitamin C (Organisciak et al., 1991) and dimethylthiourea (Organisciak et al., 1992) have been shown to protect against light-induced damage. Recently, Organisciak et al. (2002) found that dimethylthiourea protected P23H and S334ter transgenic rats from light damage; its effect on the inherited retinal degeneration was not studied.
Phenyl-N-tert-butylnitrone (PBN) is a free radical scavenger that is frequently used as a spin trap agent in free radical research. PBN given intravenously to gerbils was found to be neuroprotective in an ischemia–reperfusion experimental paradigm (Novelli et al., 1986a). More recently, PBN has been shown to have a variety of pharmacological effects, including reduction in the mortality associated with endotoxin shock (McKechnie et al., 1986; Novelli et al., 1986b; Hamburger and McCay, 1989; Miyajima and Kotake, 1995), neuroprotection in ischemia-reperfusion and aging models (Phillis and Clough-Helfman, 1990; Carney et al., 1991), amelioration of the CNS damage associated with the human immunodeficiency virus envelope protein glycoprotein 120 (Tabatabaie et al., 1996), and prevention of streptozotocin-induced diabetes in mice (Tabatabaie et al., 1997). Other reports have indicated that the action of PBN is related to the expression of various cytokine genes and to the activation of the transcription factor nuclear factor κB (Kotake et al., 1998) and activator protein-1 (AP-1) (Sang et al., 1999).
We have shown that PBN protects the retina against light-induced apoptosis (Ranchon et al., 2001, 2002). Because of the common apoptotic degenerative pathway in both inherited and light-induced retinal degenerations, and because some of the agents described above were neuroprotective in both types of degeneration, we tested the effect of chronic administration of PBN on the degeneration induced by the S334ter and P23H transgenes in the rat retina. In addition, because P23H rats (Nir et al., 2001; Organisciak et al., 2002) and mice (Naash et al., 1996a,b; Wang et al., 1997) are more susceptible to light damage than wild-type animals, and the onset of degeneration in P23H-3 and S334ter-4 rats occurs between postnatal day 10 (P10) and P15, approximately coinciding with eye opening (P12), we considered that light might be a contributing factor in the degenerative mechanism. Therefore, we tested the effect of PBN on light-induced damage in P23H and S334ter rat retinas.
Materials and Methods
Materials. PBN was synthesized by Dr. Yashige Kotake (Oklahoma Medical Research Foundation, Oklahoma City, OK). All other chemicals were reagent grade and purchased from Sigma (St. Louis, MO), except as indicated.
Animals. Wild-type control rats used in this study were Sprague Dawley rats (Harlan, Indianapolis, IN). Heterozygous P23H-3 and S334ter-4 rats were generated by mating homozygous P23H (line 3) or S334ter (line 4) rats with wild-type Sprague Dawley rats. (The transgenic line designations are omitted hereafter for brevity.) All animals were born and kept in dim cyclic light (12 hr light/dark cycle; 5–10 lux) and had ad libitum access to lab chow and water. These two particular lines were chosen because they show a significant degree of retinal degeneration by 7 weeks of age, which would allow the determination of any rescue by the various experimental treatments. The animal care strictly conformed to the Association for Research in Vision and Ophthalmology statement for the Use of Animals in Vision and Ophthalmic Research and the University of Oklahoma Health Sciences Center Guidelines for Animals in Research. All protocols were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Oklahoma Health Sciences Center and Dean A. McGee Eye Institute.
Electroretinography. Rats were dark-adapted overnight and prepared under dim red light for the ERG study. They were anesthetized with intramuscular injections of ketamine (120 mg/kg body weight) and xylazine (6 mg/kg body weight). One drop of 1% tropicamide was applied to the cornea to dilate the pupil. The white light stimulus used to evoke ERGs was delivered in 10 msec pulses by an integrating sphere (Lab-sphere, North Sutton, NH) at an interval between flashes of 60 sec. ERGs were recorded with gold electrodes at six intensities presented in ascending order, starting below threshold, to obtain the b-wave sensitivity curves. The software program Origin 6.0 (Microcal Software, Northampton, MA) was used to fit the data of each rat, giving the saturated a-wave (Amax) and b-wave (Bmax) amplitudes.
Histology. Rats were killed by carbon dioxide asphyxiation for light microscopic evaluation of retinal structure. Immediately after death, eyes were excised, placed in fixative (4% paraformaldehyde, 2% trichloroacetic acid, 20% isopropyl alcohol, 2% zinc chloride, and 72% distilled water), and embedded in paraffin. Sections of 5 μm were cut along the vertical meridian through the optic nerve. Illustrations of photoreceptor degeneration in the P23H line 3 and S334ter line 4 mutant rhodopsin transgenic rats (Lewin et al., 1998; Liu et al., 1999; Machida el al., 2000; Green et al., 2001; Nir et al., 2001; Organisciak et al., 2002) and the protection from light damage by PBN (Ranchon et al., 2001, 2002) have been presented previously and will not be repeated here. Instead, a quantitative, morphometric assessment of photoreceptor cell loss was performed. The thickness of the outer nuclear layer (ONL) was measured at 0.5 mm distances from the optic nerve to the inferior and superior ora serrata and plotted as shown in Figure 2. The area under the curves was integrated using the Origin 6.0 program (Microcal Software), and the results were used for quantitative comparisons. The ONL area is proportional to ONL thickness and is a measure of the number of viable photoreceptor cells (Williams and Howell, 1983; Michon et al., 1991).
Figure 2.
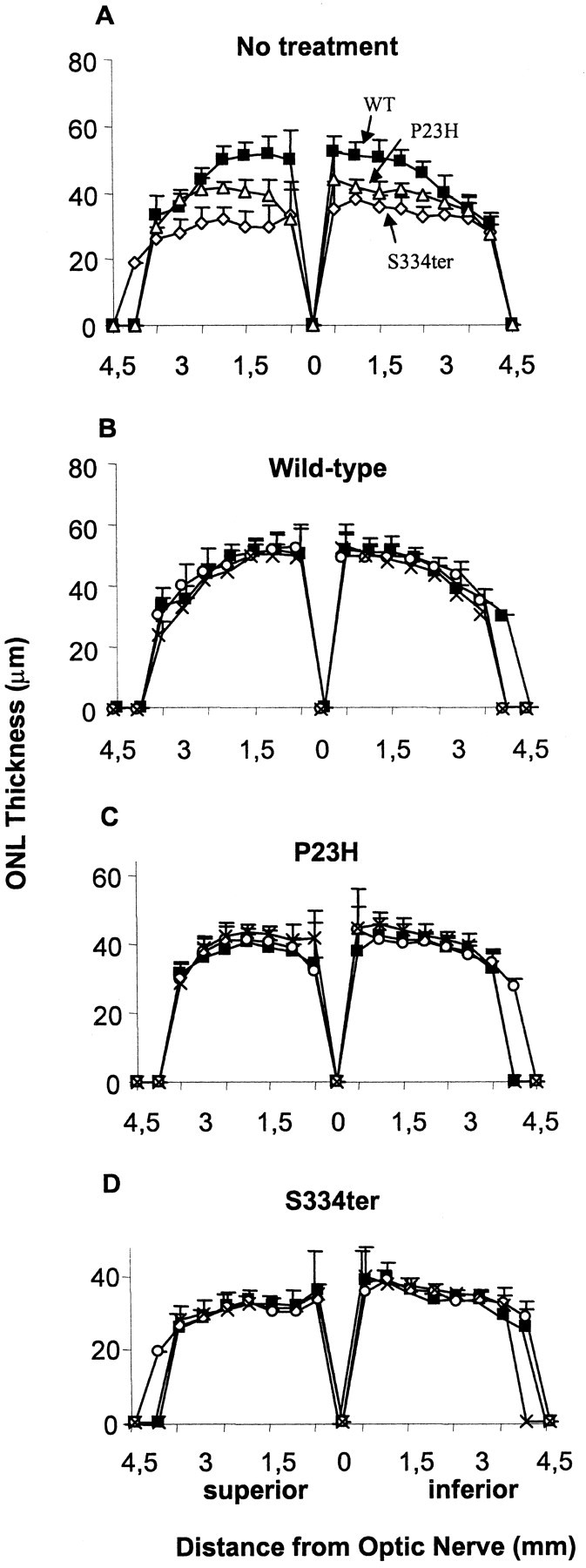
ONL thickness (in micrometers) at P49 plotted as a function of the retinal location (in millimeters) from the optic nerve head. A, S334ter (⋄), P23H (▵), and wild-type (WT; □) values from untreated rats. Wild-type (B), P23H (C), and S334ter (D) rats were given water (▪), 0.25% PBN in drinking water (○), or 0.25% PBN in drinking water plus three intraperitoneal injections of PBN (100 mg/kg per injection) (×), starting at P21.
Rhodopsin measurement. Thirty-day-old rats were dark-adapted overnight, killed the next morning, and their retinas removed under dim red light. Two retinas from the same rat (n = 6) were homogenized in 500 μl of 67 mm phosphate buffer containing 2% N-octyl-β-d-glucopyranoside (Calbiochem, La Jolla, CA) and shaken at 4°C for 1 hr. Hydroxylamine (25 μl, 1 m) was added, and the samples were centrifuged at 4°C for 30 min at 27,000 × g. The clear supernatant was scanned from 400 to 800 nm (Ultrospec 3000; Amersham Biosciences, Cambridge, UK), after which the samples were bleached by exposure to the room light for at least 15 min and scanned again. The difference in absorption at 500 nm was used to determine the concentration of rhodopsin using a molar extinction coefficient of 42,000 (Shichi et al., 1969).
Effect of PBN on inherited retinal degeneration. In each strain (wild-type, P23H, and S334ter), rats were divided into three groups of 10 rats each (90 rats total). The first group was given 0.25% PBN in drinking water, the second group was given 0.25% PBN in drinking water plus three intraperitoneal injections (800, 1400, and 2000) of PBN (100 mg/kg body weight, in an aqueous solution of 25 mg/ml PBN), and the third group was given only drinking water. The treatments were begun at P21 and continued until P49. ERGs were recorded at P49, after which the animals were killed and retinas taken for morphologic examination.
Light damage paradigm. Control ERGs were recorded on 8 wild-type, 20 S334ter, and 20 P23H rats (P38–P41). At P45–P52, rats from each strain were divided into two subgroups that were dark-adapted overnight before being exposed for 24 hr to constant illumination of 2700 lux intensity. One subgroup was treated with PBN (aqueous solution, 50 mg/kg, i.p.) every 6 hr over the 24 hr of light exposure, the first treatment starting 0.5 hr before exposure (Ranchon et al., 2001). The other subgroup was not treated. No sham injections were done, because they have been shown to have no effect (Ranchon et al., 1999, 2001). After light exposure, the animals were placed in the dark for 24 hr and ERGs were recorded the next day (day 1). Thereafter, the animals returned to dim cyclic light until they were killed 5 d later for morphometric analysis.
Statistical analysis. ANOVA was performed on the derived parameters and ONL areas to determine the effects of treatments and/or light exposure on the ERG values and the ONL areas. A significance level of p = 0.05 was set. If ANOVA was significant, multiple comparisons were performed to decide which pairs of mean values were different. Significant differences across groups were assessed using the post hoc Scheffé test with the significance level set at p = 0.05 (Scheffé, 1959)
Results
Effect of PBN on inherited retinal degeneration
Animals were untreated or treated with 0.25% PBN in drinking water with or without PBN injections from P21 to P49, at which time retinal structure and function were evaluated. In the untreated groups, retinal function was reduced in the P23H and S334ter rats compared with that of wild-type rats (Fig. 1A). The maximal b-wave amplitude (Bmax) was reduced significantly to 54% in P23H animals and 51% in S334ter animals (p < 0.0001), with no significant difference found between the two transgenic lines. PBN treatment did not alter the b-wave amplitudes in wild-type rats (Fig. 1B). PBN treatment also had no significant protective effect on retinal function in the mutant rhodopsin transgenic rats (Fig. 1C,D), indicating that the drug did not protect their retinal photoreceptors from degeneration.
Figure 1.
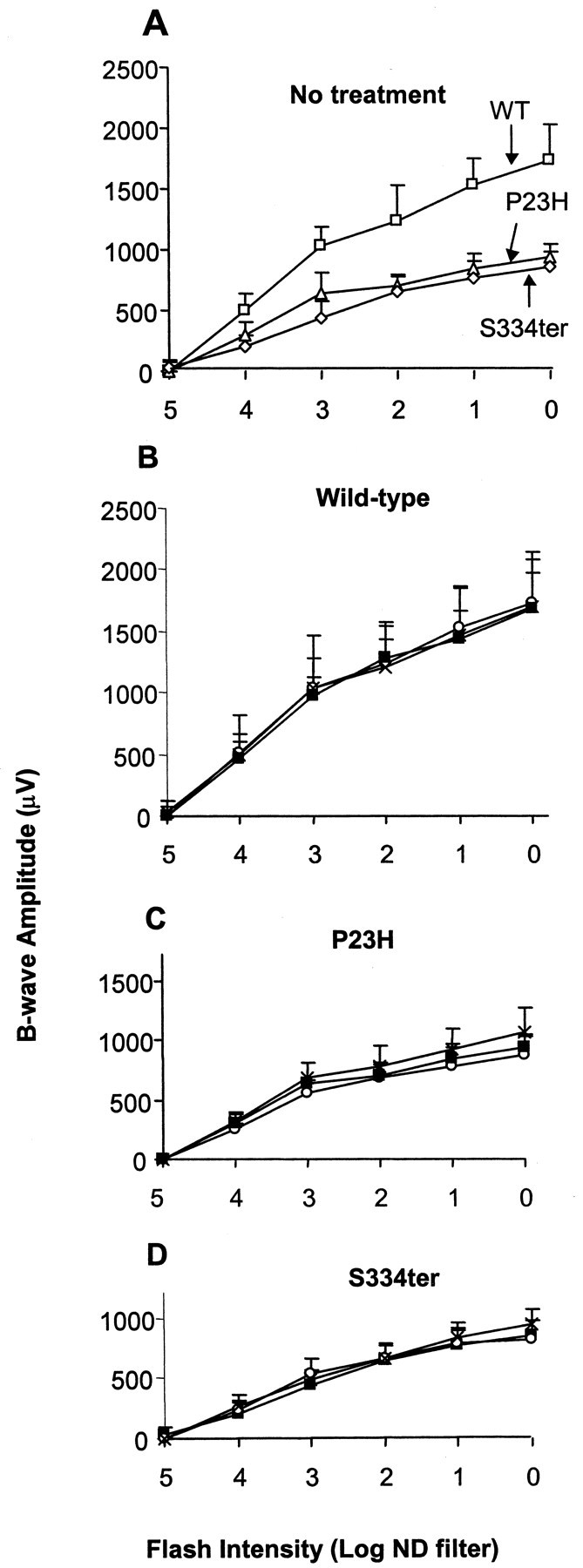
Retinal function at P49 in cyclic light. The b-wave amplitude (in microvolts) of the ERG response is plotted as a function of the stimulus intensity [log neutral density (ND) filter]. A, S334ter (⋄), P23H (▵), and wild-type (WT; □,) values from untreated animals. Wild-type (B), P23H (C), and S334ter (D) rats were given water (▪), 0.25% PBN in drinking water (○), or 0.25% PBN in drinking water plus three intraperitoneal injections daily of PBN (100 mg/kg per injection) (×), starting at P21.
Measurement of ONL areas in untreated wild-type, P23H, and S334ter rat retinas showed a significant reduction in P23H (16%) and S334ter (31%) compared with wild type (p < 0.008 and p < 0.00001, respectively), with photoreceptor loss in both superior and inferior hemispheres (Fig. 2A). There was no significant effect of PBN treatment on ONL area in any of the test groups (Fig. 2B–D).
Light damage susceptibility
Because PBN had no protective effect on the photoreceptor degeneration because of the genetic mutations, we tested whether it could provide protection against light damage in the mutant animals. Wild-type, P23H, and S334ter rats were divided into two subgroups: one group was not treated, and the other was given five intraperitoneal injections of aqueous PBN (50 mg/kg per injection, the first given 0.5 hr before light exposure) (Ranchon et al., 2001). Both subgroups were exposed to continuous illumination (2700 lux) for 24 hr. Retinal function was determined by ERG analysis before and 1 d after light treatment. In wild-type (Fig. 3A,B) and P23H (Fig. 3C,D) untreated rats, 24 hr of light exposure resulted in complete loss of the a-wave and b-wave responses of the ERG. However, in the untreated S334ter animals (Fig. 3E,F), retinal function was only partially reduced by the same light treatment [Amax was 20 ± 14% (p < 0.0001) and Bmax was 40 ± 14% (p < 0.0001) compared with their respective values before exposure to the damaging light]. Treatment with PBN had a significant effect on the preservation of function in the wild-type and P23H rats. Amax and Bmax values were preserved (p < 0.00001) to some extent in wild-type (42 ± 9 and 62 ± 16%, respectively) and P23H (16 ± 9 and 49 ± 19%, respectively) animals. However, PBN treatment was without effect in the S334ter rats; Amax and Bmax values were 29 ± 15 and 55 ± 16%, respectively, which although slightly higher than the values for untreated animals, were not significantly different.
Figure 3.
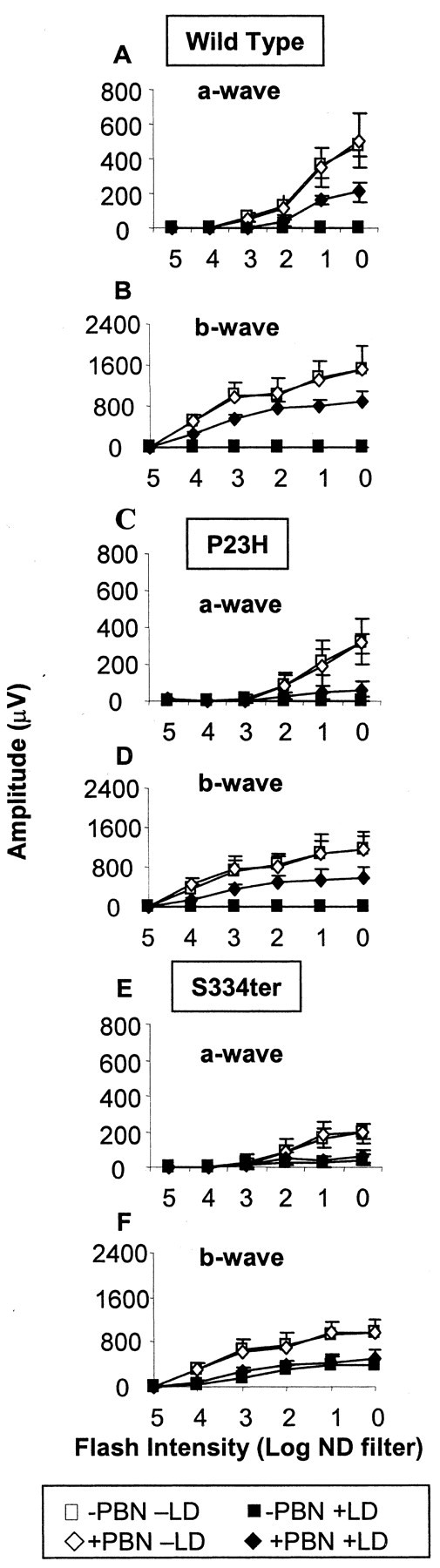
a-wave (A, C, and E) and b-wave (B, D, and F) sensitivity curves at P38–P41. The a-wave and b-wave amplitudes (in microvolts) are plotted as a function of the stimulus intensity [log neutral density (ND) filter] before (open symbols) and after (filled symbols) damaging light. Rats were untreated (squares) or treated with PBN (diamonds). LD, Light damage.
Exposure of untreated rats to 2700 lux of continuous illumination caused loss of photoreceptor cells in all three groups, with wild-type (Fig. 4A) and P23H (Fig. 4B) animals being more affected than S334ter animals (Fig. 4C). Values for the 7-week-old control animals used in the first study (Fig. 2) are presented here for comparison. In wild-type rats, light stress induced a significant reduction of the ONL area to 3.6% of unexposed wild-type retinas in the superior hemispheres and to 14% of unexposed wild-type retinas in the inferior hemispheres (Fig. 4A). In P23H and S334ter animals, the ONL thickness was affected to the same extent in superior and inferior hemispheres of the retina, and the integrated area was reduced to 10% of unexposed wild-type in P23H rats (Fig. 3B) and to 40% of unexposed wild-type in S334ter rats (Fig. 3C).
Figure 4.

ONL thickness after exposure to damaging light. A, Wild type; B, P23H; C, S334ter. Control values (filled circle; data from Fig. 2) before light damage are shown. Rats were either untreated (open triangles) or treated with five intraperitoneal injections of PBN (50 mg/kg per injection, filled triangles, as described in Materials and Methods).
In PBN-treated groups, the ONL area of wild-type (Fig. 4A) and P23H (Fig. 4B) rat retinas was significantly preserved to 54% (wild type) (p < 0.00001) and 50% (P23H) (p < 0.00001), respectively, in the superior retina and to 92% (wild type) (p < 0.03) and 72% (P23H) (p < 0.004), respectively, in the inferior retina of comparable hemispheres in unexposed rats. In the S334ter rats, the ONL area (Fig. 4C) was significantly preserved to 74% (p < 0.005) in the inferior retina by PBN treatment, but not in the superior hemisphere.
Comparison of rhodopsin content and ONL area in wild-type, P23H, and S334ter rats
Before exposure to the damaging light, the status of the retinas in each strain was determined in P30 animals. The ONL area was significantly reduced to 85% of wild-type controls in P23H (p < 0.014) and to 70% of wild-type in S334ter (p < 0.0001) retinas (Table 1), and there was a significant difference (p < 0.01) between P23H and S334ter retinas. The rhodopsin content was reduced to 61% in P23H (p < 0.0001) and 36% in S334ter (p < 0.00001) retinas (Table 1). Retinal function was reduced in the two transgenic strains; Amax was 55 ± 11% in the P23H (p < 0.002) and 36 ± 5% in the S334ter (p < 0.0001) rats, compared with that in wild-type rats. Amax was also significantly different between P23H and S334ter rats (p < 0.005). Bmax was significantly (p < 0.0001) reduced to 55 ± 4% in P23H rats and to 54 ± 5% in S334ter rats compared with the wild-type (1620 ± 155 μV) rats (Table 1). Bmax was not significantly different between S334ter and P23H rats.
Table 1.
Wild-type, P23H, and S334ter rats at 30 d of age
|
|
Wild-type |
P23H |
S334ter |
|---|---|---|---|
| ONL (area) | 298 ± 11 | 254 ± 22 | 210 ± 10 |
| Rhodopsin (nmol/retina) | 1.87 ± 0.27 | 1.14 ± 0.20 | 0.68 ± 0.24 |
| Amax (μV) | 507 ± 45 | 280 ± 57 | 185 ± 23 |
|
Bmax (μV)
|
1620 ± 155
|
897 ± 69
|
872 ± 79
|
Area under the ONL is expressed as μm2 × 10−3. Rhodopsin content per retina was measured by spectrophotometry. Maximal a-wave amplitude (Amax) and b-wave amplitude (Bmax) are expressed in microvolts.
Discussion
Light damage to photoreceptors is triggered by the excessive absorption of photons by the visual pigment rhodopsin (Grimm et al., 2000), and the vulnerability of these cells is positively correlated to the rhodopsin content (Noell et al., 1966; Noell and Albrecht, 1971; Remé et al., 1998). In this study, we found that the critical determinant for light-damage sensitivity is not solely the rhodopsin content, because P23H rats had a lower retinal rhodopsin content than the wild-type rats, yet they were as susceptible to light damage as the wild types. This is consistent with the finding by Wenzel et al. (2001a) that dark-adapted rhodopsin level does not necessarily correlate with photons caught in the light, and that the steady-state bleach level and the regeneration rate are also important determinants.
The S334ter rats were relatively more resistant to light damage than the P23H rats. The resistance to light damage may be attributable in part to the significantly lower levels of rhodopsin compared with the levels seen in P23H rats, but other factors may be involved. For example, at least some of the rhodopsin is mis-sorted in S334ter photoreceptors (Green et al., 2000). At present, however, we do not know whether other factors in photon capture or rhodopsin regeneration (Wenzel et al., 2001a) or other genetic factors (Danciger et al., 2000; Hao et al., 2002) that influence the susceptibility to light damage are different in the two mutant lines. Thus, the exact cause of the reduced susceptibility of S334ter rats to light damage remains to be determined.
In this study, we found that acute administration of PBN protects wild-type and P23H but not S334ter rat retinas from the damaging effect of light. This suggests that the protection by PBN is dependent on the normal activation–deactivation of the rhodopsin molecule. In wild-type and P23H rats, PBN protection might involve mechanisms other than free radical scavenging, such as a downregulation of the rhodopsin regeneration rate or effects on cellular metabolism. PBN has also been shown to reduce basal protein phosphorylation in primary rat glia (Robinson et al., 1999), and phosphorylation is a major step in rhodopsin regeneration (Bennett and Sitaramayya, 1988; McDowell et al., 2001). This may explain why PBN was less effective in the S334ter rats, because S334ter-rhodopsin is missing the phosphorylation sites required for the inactivation process.
Because PBN protected wild-type and P23H rat retinas from light damage but did not protect P23H (or S334ter) from inherited degeneration, the underlying mechanisms that cause these two types of degeneration and the action of PBN on them must be different. Although relatively little is known about the molecular and cellular mechanisms of the inherited retinal degenerations compared with those of light damage, a few points can be suggested from the present findings. First, apoptosis is known to be the “final cell death pathway” in both light-induced and hereditary retinal degenerations. The fact that PBN protects against light-induced but not against P23H-induced or S334ter-induced degeneration suggests that PBN does not interact directly with the apoptotic pathway or that the apoptotic pathway in the mutation-induced and light-induced degeneration are different. This latter possibility must be considered until shown otherwise in view of the recent demonstration of two apoptotic pathways in light-induced retinal degeneration (Hao et al., 2002).
The second point that can be made from the present data on differential effects of PBN on inherited and light-induced degenerations concerns the role of free radicals in the degenerative process. Chronic administration of PBN for 4 weeks did not protect against inherited retinal degeneration induced by the expression of the P23H or S334ter mutant rhodopsin transgenes. Because PBN was injected at ∼60 mg/day per rat and/or 0.25% PBN was present in the drinking water, the lack of protection cannot be attributed to a too small amount of the drug, but rather suggests that: (1) free radicals do not play an early role in the hereditary degenerative process, (2) radicals are involved too far down in the cascade of degenerative events for the cells to be rescued, or (3) other cellular pathways affected by PBN are not involved in inherited retinal degenerations.
The fact that S334ter rats are less susceptible to light damage than P23H rats and are the least rescued by PBN and the fact that P23H rats are not rescued to the same extent as wild type are consistent with PBN as a protective agent against the primary events in light-induced cell death but not on the mutation-induced pathways. PBN may act as an antioxidant/free radical scavenger in the early stages of light damage. There is probably a complex interaction between the secondary processes of light damage and inherited degeneration. The effects of light on cells already stressed by photoreceptor mutations probably differ according to the interactions of light damage with the pathways of degeneration in each type of mutation. Although these suggestions would explain our current findings, they remain to be shown experimentally.
Recent studies provide some insight into the differences in the mechanisms of cell death in light damage and inherited degenerations with molecules other than PBN. Mice lacking the protooncogene c-fos were protected from light damage (Hafezi et al., 1997), whereas the transcription factor AP-1 was increased during bright-light stress (Hafezi et al., 1999). Suppression of AP-1 expression with steroids protected retinas from light damage (Wenzel et al., 2001b). In retinal degeneration mutants, however, the absence of c-fos resulted in no protective effect in rd/rd (Hafezi et al., 1998) and rhodopsin knock-out mice (Hobson et al., 2000). Similarly, overexpression of the survival-promoting growth factor FGF-2 reduces hyperoxia-induced photoreceptor cell death in mice but does not reduce cell death in rd/rd or Q344ter mutant rhodopsin transgenic mice (Yamada et al., 2001). These studies, along with our present findings, provide substantial evidence that the mechanisms of cell death initiation in the mutant mice are different from those in light damage, although apoptosis is the ultimate cause of cell demise in each case.
Substantial effort in retinal degeneration research has focused on a protective strategy to slow the rate of retinal degeneration. There exists a significant genetic heterogeneity in RP phenotypes, even in patients with the same gene mutation, and a large number of mutations exist in multiple retinal genes leading to the common pathway of photoreceptor cell death. Specific gene therapies, such as antisense or ribozymes (Lewin et al., 1998), may not readily treat a significant fraction of patients. As a result, it is important to develop a generalized survival factor therapy that does not target the mutant gene product, but rather alters the photoreceptor or its environment in a manner that promotes cell survival. The use of light damage and S334ter and P23H rats in combination allows the design of therapies for animal models that mimic human disorders, yet provides different degeneration and gene defects with which to test such generalized therapies.
Footnotes
This work was supported by grants from the National Institutes of Health (EY00871, EY01919, EY02162, EY06842, EY04149, EY12190, and RR17703), Research to Prevent Blindness (RPB), The Foundation Fighting Blindness, Oklahoma Center for the Advancement of Science and Technology–Oklahoma Applied Research Support, Samuel Roberts Nobel Foundation, The IPSEN Foundation, and Presbyterian Health Foundation. M.M.L. is an RPB senior scientist investigator. We gratefully acknowledge the assistance of Mark Dittmar in the breeding and husbandry of the animals and in the ERG measurements.
Correspondence should be addressed to Dr. Isabelle Ranchon, Laboratoire de Biophysiques Sensorielles, Facultés de Médecine et de Pharmacie, 28 Place Henri Dunant, 63000 Clermont-Ferrand, France. E-mail: isabelle.ranchon@uclermont1.fr.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236050-08$15.00/0
References
- Bennett N, Sitaramayya A ( 1988) Inactivation of photoexcited rhodopsin in retinal rods: the roles of rhodopsin kinase and 48-kDa protein (arrestin). Biochemistry 27: 1710–1715. [DOI] [PubMed] [Google Scholar]
- Cao W, Tombran-Tink J, Elias R, Sezate S, Mrazek D, McGinnis JF ( 2001) In vivo protection of photoreceptors from light damage by pigment epithelium-derived factor. Invest Ophthalmol Vis Sci 42: 1646–1652. [PubMed] [Google Scholar]
- Carney JM, Starke-Reed PE, Oliver CN, Landum RW, Cheng MS, Wu JF, Floyd RA ( 1991) Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound N-tert-butyl-alpha-phenylnitrone. Proc Natl Acad Sci USA 88: 3633–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayouette M, Smith SB, Becerra SP, Gravel C ( 1999) Pigment epithelium-derived factor delays the death of photoreceptors in mouse models of inherited retinal degenerations. Neurobiol Dis 6: 523–532. [DOI] [PubMed] [Google Scholar]
- Chang GQ, Hao Y, Wong F ( 1993) Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron 11: 595–605. [DOI] [PubMed] [Google Scholar]
- Danciger M, Matthes MT, Yasumura D, Akhmedov NB, Rickabaugh T, Gentleman S, Redmond TM, La Vail MM, Farber DB ( 2000) A QTL on distal chromosome 3 that influences the severity of light-induced damage to mouse photoreceptors. Mamm Genome 11: 422–427. [DOI] [PubMed] [Google Scholar]
- Dryja TP, Li T ( 1995) Molecular genetics of retinitis pigmentosa. Hum Mol Genet 4: 1739–1743. [DOI] [PubMed] [Google Scholar]
- Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM ( 1990) Photoreceptor degeneration in inherited retinal dystrophy by basic fibroblast growth factor. Nature 347: 83–86. [DOI] [PubMed] [Google Scholar]
- Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM ( 1992) Basic fibroblast growth factor and local injury protect photoreceptors from light damage in the rat. J Neurosci 12: 3554–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasson M, Picaud S, Leveillard T, Simonutti M, Mohand-Said S, Dreyfus H, Hicks D, Sabel J ( 1999) Glial cell line-derived neurotrophic factor induces histologic and functional protection of rod photoreceptors in the rd/rd mouse. Invest Ophthalmol Vis Sci 40: 2724–2734. [PubMed] [Google Scholar]
- Green ES, Menz MD, LaVail MM, Flannery JG ( 2000) Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 41: 1546–1553. [PubMed] [Google Scholar]
- Green ES, Rendahl KG, Zhou S, Ladner M, Coyne M, Srivastava R, Manning WC, Flannery JG ( 2001) Two animal models of retinal degeneration are rescued by recombinant adeno-associated virus-mediated production of FGF-5 and FGF-18. Mol Ther 3: 507–515. [DOI] [PubMed] [Google Scholar]
- Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Remé CE ( 2000) Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration? Nat Genet 25: 63–66. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Steinbach JP, Marti A, Munz K, Wang ZQ, Wagner EF, Aguzzi A, Remé CE ( 1997) The absence of c-fos prevents light-induced apoptotic cell death of retinal photoreceptors in retinal degeneration in vivo Nat Med 3: 346–349. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Abegg M, Grimm C, Wenzel A, Munz K, Sturmer J, Farber DB, Remé CE ( 1998) Retinal degeneration in the rd mouse in the absence of c-fos. Invest Ophthalmol Vis Sci 39: 2239–2244. [PubMed] [Google Scholar]
- Hafezi F, Marti A, Grimm C, Wenzel A, Remé CE ( 1999) Differential DNA binding activities of the transcription factors AP-1 and Oct-1 during light-induced apoptosis of photoreceptors. Vision Res 39: 2511–2518. [DOI] [PubMed] [Google Scholar]
- Hamburger SA, McCay PB ( 1989) Endotoxin-induced mortality in rats is reduced by nitrones. Circ Shock 29: 329–334. [PubMed] [Google Scholar]
- Hao W, Wenzel A, Obin MS, Chen CK, Brill E, Krasnoperova NV, Eversole-Cire P, Kleyner Y, Taylor A, Simon MI, Grimm C, Remé CE, Lem J ( 2002) Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet 32: 254–260. [DOI] [PubMed] [Google Scholar]
- Hobson AH, Donovan M, Humphries MM, Tuohy G, McNally N, Carmody R, Cotter T, Farrar GJ, Kenna PF, Humphries P ( 2000) Apoptotic photoreceptor death in the rhodopsin knockout mouse in the presence and absence of c-fos. Exp Eye Res 71: 247–254. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Sang H, Miyajima T, Wallis GL ( 1998) Inhibition of NFκB, iNOS mRNA, COX2 mRNA, and COX catalytic activity by phenyl N-tertbutylnitrone. J Biol Chem 268: 11520–11527. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Unoki K, Yasumura D, Matthes MT, Yancopoulos GD, Steinberg RH ( 1992) Multiple growth factors, cytokines, and neurotrophins rescue photoreceptors from the damaging effects of constant light. Proc Natl Acad Sci USA 89: 11249–11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Lau-Villacorta C, Unoki K, Sung CH, Steinberg RH ( 1998) Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci 39: 592–602. [PubMed] [Google Scholar]
- Lewin AS, Drenser KA, Hauswirth WW, Nishikawa S, Yasumura D, Flannery JG, LaVail MM ( 1998) Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med 4: 967–971. [DOI] [PubMed] [Google Scholar]
- Li S, Chang CJ, Abler AS, Tso MOM ( 1995) Inhibitory effects of cycloheximide and flunarizidine on light-induced apoptosis of photoreceptor cells. In: Degenerative diseases of the retina (Anderson RE, LaVail MM, Hollyfield JG, eds), pp 27–38. New York: Plenum.
- Liu C, Li Y, Peng M, Laties AM, Wen R ( 1999) Activation of caspase-3 in the retina of transgenic rats with the rhodopsin mutation S334ter during photoreceptor degeneration. J Neurosci 19: 4778–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolley RN, Rong H, Craft CM ( 1994) Linkage of photoreceptor degeneration by apoptosis with inherited defect in phototransduction. Invest Ophthalmol Vis Sci 35: 358–362. [PubMed] [Google Scholar]
- Machida S, Kondo M, Jamison JA, Khan NW, Kononen LT, Sugawara T, Bush RA, Sieving PA ( 2000) P23H rhodopsin transgenic rats: correlation of retinal function with histology. Invest Ophthalmol Vis Sci 4: 3200–3209. [PubMed] [Google Scholar]
- Machida S, Chaudhry P, Shinohara T, Singh DP, Reddy VN, Chylack Jr LT, Sieving PA, Bush RA ( 2001) Lens epithelium-derived growth factor promotes photoreceptor survival in light-damaged and RCS rats. Invest Ophthalmol Vis Sci 42: 1087–1095. [PubMed] [Google Scholar]
- McDowell JH, Robinson PR, Miller RL, Brannock MT, Arendt A, Smith WC, Hargrave PA ( 2001) Activation of arrestin: requirement of phosphorylation as the negative charge on residues in synthetic peptides from the carboxyl-terminal region of rhodopsin. Invest Ophthalmol Vis Sci 42: 1439–1443. [PubMed] [Google Scholar]
- McKechnie K, Furman BL, Parratt JR ( 1986) Modification by oxygen free radical scavengers of the metabolic and cardiovascular effects of endotoxin infusion in conscious rats. Circ Shock 19: 429–439. [PubMed] [Google Scholar]
- Michon JJ, Li ZL, Shioura N, Anderson RJ, Tso MOM ( 1991) A comparative study of methods of photoreceptor morphometry. Invest Ophthalmol Vis Sci 32: 280–284. [PubMed] [Google Scholar]
- Miyajima T, Kotake Y ( 1995) Spin trapping agent, phenyl N-tert-butyl nitrone, inhibits induction of nitric oxide synthase in endotoxin-induced shock in mice. Biochem Biophys Res Commun 215: 114–121. [DOI] [PubMed] [Google Scholar]
- Naash MI, Ripps H, Li S, Goto Y, Peachey NS ( 1996a) Polygenic disease and retinitis pigmentosa: albinism exacerbates photoreceptor degeneration induced by the expression of a mutant opsin in transgenic mice. J Neurosci 16: 7853–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naash ML, Peachey NS, Li ZY, Gryczan CC, Goto Y, Blanks J, Milam AH, Ripps H ( 1996b) Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci 37: 775–782. [PubMed] [Google Scholar]
- Nir I, Harrison JM, Liu C, Wen R ( 2001) Extended photoreceptor viability by light stress in RCS rats but not in the opsin P23H mutant rats. Invest Ophthalmol Vis Sci 42: 842–849. [PubMed] [Google Scholar]
- Noell WK, Albrecht R ( 1971) Irreversible effects of light on the retina: role of vitamin A. Science 172: 76–79. [DOI] [PubMed] [Google Scholar]
- Noell WK, Walker VS, Kang BS, Berman S ( 1966) Retinal damage by light in rats. Invest Ophthalmol 5: 450–473. [PubMed] [Google Scholar]
- Novelli GP, Angiolini P, Tani R, Consales G, Bordi L ( 1986a) Phenyl-T-butyl-nitrone is active against traumatic shock in rats. Free Radic Res Commun 1: 321–327. [DOI] [PubMed] [Google Scholar]
- Novelli GP, Angiolini P, Consales G, Lippi R, Tani R ( 1986b) Anti-shock action of phenyl-t-butylnitrone, a spin trapper. In: Oxygen free radicals in shock (Novelli G, Ursini F, eds), pp 119–124. Basel: Karger.
- Organisciak DT, Darrow RM, Bicknell IR, Jiang YL, Pickford M, Blank JC ( 1991) Protection against retinal light damage by natural and synthetic antioxidants. In: Retinal degenerations (Anderson RE, Hollyfield JG, La-Vail MM, eds), pp 189–201. New York: Plenum.
- Organisciak DT, Darrow RM, Jiang Y-L, Marak GE, Blank JC ( 1992) Protection by dimethylthiourea against retinal light damage in rats. Invest Ophthalmol Vis Sci 33: 1599–1609. [PubMed] [Google Scholar]
- Organisciak DT, Kutty RK, Leffack M, Wong P, Messing S, Wiggert B, Darrow RM, Chader GE ( 1995) Oxidative damage and responses of retinal nuclei arising from intense light exposure. In: Degenerative diseases of the retina (Anderson RE, LaVail MM, Hollyfield JG, eds), pp 9–18. New York: Plenum.
- Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B ( 2003) Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Invest Ophthalmol Vis Sci 44: 486–492. [DOI] [PubMed] [Google Scholar]
- Pagon RA ( 1988) Retinitis pigmentosa. Surv Ophthalmol 33: 137–177. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Clough-Helfman C ( 1990) Protection from cerebral ischemic injury in gerbils with spin trap agent N-tert-butyl-alpha-phenylnitrone (PBN). Neurosci Lett 116: 315–319. [DOI] [PubMed] [Google Scholar]
- Portera-Caillau C, Sung CH, Nathans J, Adler R ( 1994) Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci USA 91: 974–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranchon I, Gorrand JM, Cluzel J, Droy-Lefaix MT, Doly M ( 1999) Functional protection of photoreceptors from light-induced damage by dimethylthiourea and ginkgo biloba. Invest Ophthalmol Vis Sci 40: 1191–1199. [PubMed] [Google Scholar]
- Ranchon I, Chen S, Alvarez K, Anderson RE ( 2001) Systemic administration of phenyl-N-tert-butylnitrone protects the retina from light damage. Invest Ophthalmol Vis Sci 42: 1375–1379. [PubMed] [Google Scholar]
- Ranchon I, White J, Chen S, Alvarez K, Kotake Y, Anderson RE ( 2002) Chronic administration of phenyl-tert-butylnitrone protects the retina against light damage. In: New insights into retinal degenerative diseases (Anderson RE, LaVail MM, Hollyfield JG, eds), pp 95–104. New York: Kluwer–Plenum.
- Remé C, Grimm C, Hafezi F, Marti A, Wenzel A ( 1998) Apoptotic cell death in retinal degenerations. Prog Retin Eye Res 17: 443–464. [DOI] [PubMed] [Google Scholar]
- Robinson KA, Stewart CA, Pye Q, Floyd RA, Hensley K ( 1999) Basal protein phosphorylation is decreased and phosphatase activity increased by an antioxidant and a free radical trap in primary rat glia. Arch Biochem Biophys 365: 211–215. [DOI] [PubMed] [Google Scholar]
- Sang H, Wallis GL, Stewart CA, Kotake Y ( 1999) Expression of cytokines and activation of transcription factors in lipopolysaccharide-administered rats and their inhibition by phenyl N-tert-butylnitrone (PBN). Arch Biochem Biophys 363: 341–348. [DOI] [PubMed] [Google Scholar]
- Scheffé H ( 1959) The analysis of variance. New York: Wiley.
- Shichi H, Lewis MS, Irreverre F, Stone AL ( 1969) Biochemistry of visual pigments: purification and properties of bovine rhodopsin. J Biol Chem 244: 529–536. [PubMed] [Google Scholar]
- Steinberg RH, Flannery JG, Naash M, Oh P, Matthes MT, Yasumura D, Lau-Villacorta C, Chen J, LaVail MM ( 1996) Transgenic rat models of inherited retinal degeneration caused by mutant opsin genes. Invest Ophthalmol Vis Sci 37: S698. [Google Scholar]
- Sugawara T, Sieving PA, Iuvone PM, Bush RA ( 1998) The melatonin antagonist luzindole protects retinal photoreceptors from light damage in the rat. Invest Ophthalmol Vis Sci 39: 2458–2465. [PubMed] [Google Scholar]
- Sullivan LS, Daiger SP ( 1996) Inherited retinal degeneration: exceptional genetic and clinical heterogeneity. Mol Med Today 2: 380–386. [DOI] [PubMed] [Google Scholar]
- Tabatabaie T, Kotake T, Wallis G, Jacob JM, Floyd RA ( 1996) In vivo trapping of nitric oxide in the brain of neonatal rats treated with the HIV-1 envelope protein gp120: protective effects of alpha-phenyl-tertbutylnitrone. Biochem Biophys Res Commun 21: 386–390. [DOI] [PubMed] [Google Scholar]
- Tabatabaie T, Kotake Y, Wallis G, Jacob JM, Floyd RA ( 1997) Spin trapping agent phenyl N-tert-butylnitrone protects against the onset of drug-induced insulin-dependent diabetes mellitus. FEBS Lett 407: 148–152. [DOI] [PubMed] [Google Scholar]
- Tso MOM, Zhang C, Abler AS, Chang CJ, Wong F, Chang GQ, Lam TT ( 1994) Apoptosis leads to photoreceptor degeneration in inherited retinal dystrophy of RCS rats. Invest Ophthalmol Vis Sci 35: 2693–2699. [PubMed] [Google Scholar]
- Wang M, Lam TT, Tso MO, Naash MI ( 1997) Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci 14: 55–62. [DOI] [PubMed] [Google Scholar]
- Wen R, Cheng T, Li Y, Cao W, Steinberg RH ( 1996) A2-adrenergic agonists induce basic fibroblast growth factor expression in photoreceptors in vivo and ameliorate light damage. J Neurosci 16: 5986–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Remé CE, Williams TP, Hafezi F, Grimm C ( 2001a) The Rpe65 Leu450Met variation increases retina resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci 21: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Seeliger MW, Jaissle G, Hafezi F, Kretschmer R, Zrenner E, Remé CE ( 2001b) Prevention of photoreceptor apoptosis by activation of the glucocorticoid receptor. Invest Ophthalmol Vis Sci 42: 1653–1659. [PubMed] [Google Scholar]
- Williams TP, Howell WL ( 1983) Action spectrum of retinal light-damage in albino rats. Invest Ophthalmol Vis Sci 24: 285–287. [PubMed] [Google Scholar]
- Yamada H, Yamada E, Ando A, Esumi N, Bora N, Saikia J, Sung CH, Zack DJ, Campochiaro PA ( 2001) Fibroblast growth factor-2 decreases hyperoxia-induced photoreceptor cell death in mice. Am J Pathol 159: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Kent CR, Tytell M ( 2001) Retinal uptake of intravitreally injected Hsc/Hsp70 and its effect on susceptibility to light damage. Mol Vis 7: 48–56. [PubMed] [Google Scholar]