

Abstract
Direct immunization with amyloidβ protein (Aβ) and passive transfer of anti-Aβ antibodies reduce Aβ accumulation and attenuate cognitive deficits in transgenic models of Alzheimer's disease (AD). The reduction in Aβ deposition has been proposed to involve microglial phagocytosis of Aβ immune complexes via Fc receptors (FcRs). We have examined the efficacy of Aβ immunization in amyloid precursor protein (APP) transgenic mice crossed into FcR-γ chain knock-out mice (FcRγ-/-). As might be expected from previous studies on macrophages, phagocytosis of Aβ immune complexes via FcR was completely impaired in microglia cells isolated from FcRγ-/- mice. Thus, we immunized APP Tg2576 transgenic mice that were crossed in the FcRγ-/- background with Aβ1–42 and then analyzed the effect on Aβ accumulation. In APP Tg2576 transgenic mice crossed to FcRγ-/-, Aβ1–42 immunization significantly attenuated Aβ deposition, as assessed by both biochemical and immunohistological methods. The reduction in Aβ accumulation was equivalent to the reduction in deposition seen in Aβ1–42 immunized, age-matched, FcR-sufficient Tg2576 mice. We conclude that after Aβ immunization, the effects of anti-Aβ antibodies on Aβ deposition in APP Tg2576 transgenic mice are not dependent on FcR-mediated phagocytic events.
Keywords: Alzheimer's disease, β-amyloid protein, Fc receptor, scavenger receptor, microglia, vaccination
Introduction
Multiple strategies targeting the accumulation of amyloid β (Aβ) peptides, the primary constituent of senile plaques in Alzheimer's disease (AD) (Selkoe, 1997; Golde et al., 2000), have been actively pursued as a potential therapeutic target for the treatment of AD. Recent studies have shown that immunization with fibrillar Aβ1–42 or passive transfer of anti-Aβ antibodies can lead to the attenuation of Aβ deposition and associated pathologies (Schenk et al., 1999; Bard et al., 2000, 2003; Janus et al., 2000; Lemere et al., 2000; Bacskai et al., 2001, 2002; Das et al., 2001; DeMattos et al., 2001; Dodart et al., 2002; McLaurin et al., 2002) as well as prevent cognitive deficits in mice (Janus et al., 2000; Morgan et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002). Several potentially nonexclusive hypotheses have been proposed regarding how Aβ immunization might alter Aβ deposition in the brain (Das and Golde, 2002). Efficacy of immunization has been proposed to involve increased microglial uptake via Fc receptors (FcRs) of antibody-bound Aβ immune complexes (Schenk et al., 1999; Bard et al., 2000; Bard et al., 2003). Anti-Aβ antibodies could interfere with the process of amyloid deposition by disrupting preexisting fibrils or preventing new fibril formation (Solomon et al., 1996, 1997). Alternatively, Aβ immunization might result in selective activation of microglia, leading to internalization of Aβ by non-FcR receptors, such as scavenger receptors (Brazil et al., 2000). Another possible mechanism, termed the “peripheral sink” hypothesis (DeMattos et al., 2001), suggests that binding of Aβ by anti-Aβ IgG in the plasma creates a sink, which leads to enhanced efflux of Aβ from the brain into the plasma, resulting in decreased levels of soluble Aβ in the brain.
To optimize the immunization protocol for the clinical setting, one key issue that must be resolved is the actual mechanism or mechanisms by which Aβ immunizations works. In this regard the aforementioned mechanisms might be divided into two categories: those reliant on intact antibodies and those that simply require a high-affinity Aβ binding agent that mimics the interaction of the anti-Aβ antibody with Aβ. Importantly, only one of the possible mechanisms is likely to have an absolute requirement for intact antibodies, namely microglial FcR-mediated phagocytosis of Aβ immune complexes. In this study, we have used the well characterized FcR-γ chain knock-out mice (FcRγ -/-) (Takai et al., 1994) to study the precise contribution of FcR in the alteration of Aβ deposition after immunization with Aβ42. We first verified that microglia isolated from FcRγ -/- mice are defective in mediating phagocytosis of Aβ immune complexes in vitro. We then actively immunized amyloid precursor protein (APP) transgenic Tg2576 mice in an FcRγ -/- background with Aβ1–42. Aβ1–42 immunization attenuated Aβ deposition to an equivalent extent in Tg2576 mice crossed to FcRγ -/- mice as in Tg2576 mice with functional FcR. These results indicate that FcR-mediated phagocytosis of Aβ immune complexes is not likely to play a key role in Aβ immunotherapies.
Materials and Methods
Mice. FcRγ -/- mice (C3H/129S) were produced as described previously (Takai et al., 1994) and were obtained from The Jackson Laboratory (Bar Harbor, ME). To generate Tg2576 mice (B6/SJL, hAPP +/-) (Hsiao et al., 1996) in the FcRγ -/- background, male Tg2576 mice (hAPP +/-) were mated with female FcRγ -/- mice to generate Tg2576 (hAPP +/-) x FcRγ +/- mice. The Tg2576 (hAPP +/-) x FcRγ +/- males were then backcrossed to FcRγ -/- female mice to generate the Tg2576 (hAPP +/-) x FcRγ -/- mice. Genotyping of Tg2576 mice and FcRγ -/- mice from various crosses were performed by PCR as described previously (Takai et al., 1994; Hsiao et al., 1996).
Microglial isolation. Microglial cells were obtained from cerebral cortices of neonate (1–3 d old) FcRγ -/- mice (C3H/129S) and FcRγ +/+ wild-type (wt) mice (C3H/129S or C57BL/6) as described previously (Bard et al., 2000). Isolated cortices were minced and triturated in HBSS containing 50 μg/ml DNase I (Sigma, St. Louis, MO). Microglia were then resuspended in media and plated in a chamber slide system for analysis (Lab-Tek-II slide system, Fisher Scientific, Pittsburgh, PA). We used uptake of acetylated low-density lipoprotein (Dil-Ac-LDL) (Molecular Probes, Eugene, OR) to assess the purity of isolated microglia. All studies were conducted on cultures in which >90% of cells stained with Dil-Ac-LDL.
Preparation of Aβ microaggregates. Aβ1–42 peptide was purchased from American Peptide Company (Sunnyvale, CA). Aβ1–42 (1 mg/ml) was fluorescently labeled by derivatizing with Cy3 (Molecular Probes) according to manufacturer's instructions. The labeled Aβ1–42 was then allowed to aggregate by incubating the diluted stock solution at 37°C for 48 hr. The aggregated Aβ1–42 was then sonicated (3 × 10 sec burst) to generate smaller fibrillar structures (microaggregates) for use in microglial phagocytosis assay.
Preparation of Aβ: anti-Aβ monoclonal antibody IgG complexes. Cy3-labeled Aβ microaggregates (5 μg/ml) were incubated with increasing concentrations (0, 5, 10, and 20 μg/ml) of anti-Aβ monoclonal antibody (mAb) Ab9 (human Aβ1–16 specific, IgG2a) or Ab42–5 (human Aβ1–16 specific, IgG2b) for 1 hr at 37°C, centrifuged at 100,000 × g for 30 min, and resuspended to original volume. Binding of antibody to Aβ was confirmed by Western blotting.
Microglial Aβ phagocytosis assay. The microglial phagocytosis assay was performed as described previously (Brazil et al., 2000). Briefly, isolated microglia were first rinsed and incubated with Cy3-labeled Aβ1–42 microaggregates (5 μg/ml) coated with anti-Aβ mAbs for 10 min at 37°C. For scavenger receptor blocking studies, fucoidan (500 μg/ml) was added to microglial cultures 10 min before incubation as well as during incubation with Aβ. For Fc receptor blocking studies, 100 μg/ml of normal IgG was added to microglial cultures during incubation with antibody-coated Cy3–Aβ. After incubation, cells were washed in PBS, fixed in 2% paraformaldehyde, and mounted for confocal microscopy. Cy3–Aβ uptake was assessed by fluorescence microscopy and digital image collection using an Olympus confocal microscope with Fluoview 2 software. For quantification of fluorescence, images of at least five randomly selected fields of cells were obtained using the 20× objective. Fluorescence intensity levels on individual cells were measured using Analytical Imaging System (AIS, 4.0) (Imaging Research, Ontario, Canada). The average fluorescent intensity level per cell was determined by summing the fluorescent intensity of all cells (>100 cells) divided by the total number of cells counted.
Aβ1–42 immunizations. For immunizations, freshly prepared aggregated Aβ1–42 (100 μg per immunization) was emulsified in 1:1 (v/v) of complete Freund's adjuvant/incomplete Freund's adjuvant (CFA/IFA) (Sigma) as described previously (Das et al., 2001). Groups of mice were immunized intraperitoneally with 100 μg of Aβ1–42 peptide in CFA (first injection) and IFA (2 weeks and monthly thereafter) for 3 months.
Anti-Aβ IgG ELISA. Anti-Aβ serum titers in immunized mice were determined by ELISA techniques as described previously (Das et al., 2001). Anti-Aβ-specific antibody titers were quantified by using serial dilutions of monoclonal anti-Aβ antibody (4G8, human Aβ17–14 epitope; Signet, Dedham, MA) as the standard. IgG isotypes were measured by ELISA according to manufacturer's protocols (Southern Biotechnology, Birmingham, AL). Serum IgG isotypes were quantified by comparison with purified isotype standard added to each plate.
Biochemical analysis of Aβ. Frozen hemibrains were sequentially extracted in a two-step extraction involving sonication in (1) 2% SDS and (2) 70% formic acid (FA) as described previously (Kawarabayashi et al., 2001). Extracted Aβ was then measured using a sandwich ELISA system with the following newly developed monoclonal antibodies: Aβ42-capture with mAb 2.1.3 (human Aβ42 specific) and detection with HRP-conjugated mAb Ab9 or Ab33.1.1 (human Aβ1–16 specific); Aβ40-capture with mAb Ab9 and detection with HRP-conjugated mAb 13.1.1 (human Aβ40 specific) (see supplemental data, Fig. 1, available at www.jneurosci.org).
Figure 1.
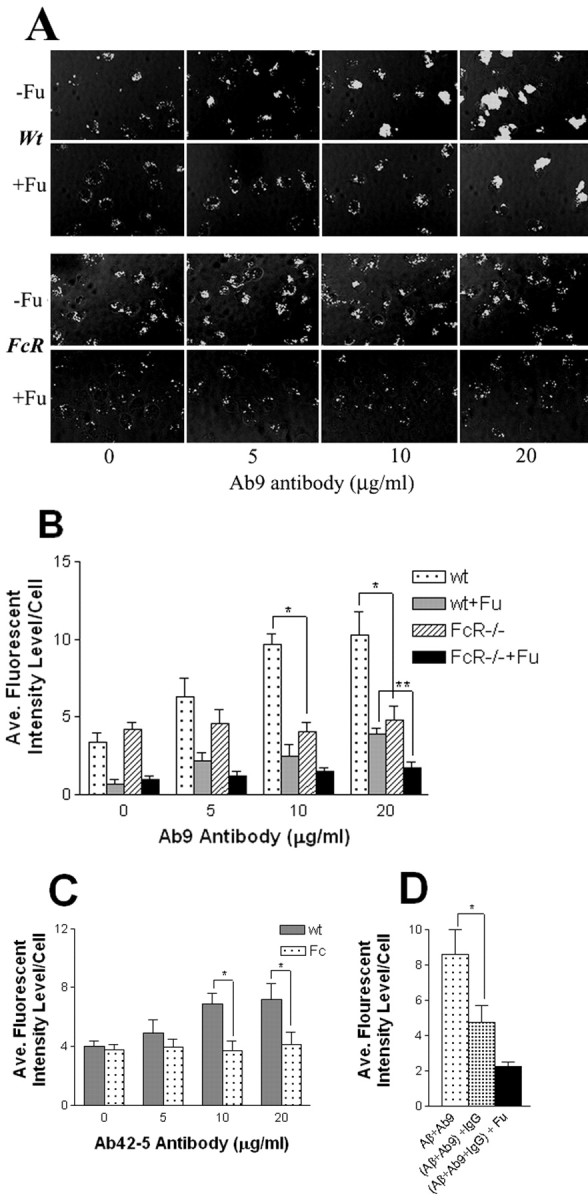
Microglia from FcRγ-/- mice exhibit defective uptake of anti-Aβ immune complexes. A, Microglia isolated from wild-type (Wt, top panels) mice and FcRγ-/- mice (bottom panels) were incubated with Cy3–Aβ in the presence of increasing concentrations of Ab9 antibody for 10 min without fucoidan (-Fu) or in the presence of 500μg/ml fucoidan (+Fu). Magnification, 40×. Data represented are from one of three independent experiments. B, Quantitation of Cy3–Aβ internalization in the presence of increasing concentrations of Ab9 and fucoidan. *p < 0.01; **p < 0.01. C, Quantitation of Cy3–Aβ internalization in the presence of increasing concentrations of Ab42–5. *p < 0.01. D, Quantitation of Cy3–Aβ internalization by wt microglia in the presence of competing IgG. Microglia were incubated with Cy3–Aβ complexed with 20 μg/ml of Ab9 antibody (Aβ + Ab9), in the presence of 100 μg of purified mouse IgG (Aβ + Ab9 + IgG), and in the presence of 500 μg/ml of fucoidan (Aβ + Ab9 + IgG) + Fu. *p < 0.01. Data represented are from one of two independent experiments.
Immunohistology. Tissue samples fixed in 4% paraformaldehyde were stained for Aβ as follows. Uniformly spaced sections spanning the neocortex and hippocampus were cut coronally at 25 μm. Five sections per brain were probed for the presence of amyloid deposits as follows: sections were pre-treated with 80% formic acid for 5 min, washed in PBS, and then incubated with Ab9 antibody (1:1000), followed by detection with anti-mouse-HRP using a MOMS kit (Vector Labs, Burlingame, CA). Free-floating tissue sections were stained for the presence of activated microglia with rat anti-mouse CD45 (1:3000; Serotec, Oxford, UK), followed by detection with anti-rat-HRP (ABC system, Vector Labs), and then counterstained with Congo red as described previously (Wilcock et al., 2001). Quantitation of plaque burden and CD45 staining was performed using Sigma Scanpro program (Jandel Scientific, San Rafael, CA). Serial coronal sections stained as above were captured, and the threshold for plaque staining and CD45 staining was determined and kept constant throughout the analysis. All of the above analyses were performed in a blinded manner.
Statistics. Statistical analysis between treatment groups was performed using the Student's t test. A Bonferroni correction was incorporated to correct for the number of all possible pairwise comparisons.
Results
Microglia from FcRγ-/- mice exhibit defective phagocytosis of anti-Aβ immune complexes
To test whether microglia from FcRγ -/- mice are defective in their ability to phagocytose Aβ immune complexes, we performed an in vitro phagocytosis assay using isolated microglia from primary cultures of mixed glial cells of newborn FcRγ +/+ (wt) mice and FcRγ -/- mice. To determine the effects of anti-Aβ antibodies on microglial uptake of Cy3–Aβ microaggregates, we used two different anti-Aβ monoclonal antibodies: Ab9, an IgG2a, and Ab42–5, an IgG2b. Both recognize an epitope in the amino terminus of Aβ (1–16) and have high affinity for monomeric and fibrillar Aβ. They also recognize native plaques on unfixed frozen sections (see supplemental data, Fig. 2), a feature reportedly predictive of an anti-Aβ antibodies efficacy in passive immunization (Schenk et al., 1999; Bard et al., 2000). In wild-type microglia, anti-Aβ immune complexes were rapidly internalized into intracellular vesicles (see supplemental data, Fig. 3). In the presence of increasing concentrations of Ab9, Cy3–Aβ uptake was significantly increased in the wt microglia (Fig. 1A, top panels) but not in FcRγ -/- microglia (Fig. 1A, bottom panels). Quantitative assessment of the uptake showed that in wt microglia, 5 μg/ml of Ab9 increased uptake by >200% and 10 or 20 μg/ml Ab9 increased Aβ uptake by >300% (Fig. 1B). Increased Cy3–Aβ uptake was also observed in FcRγ +/+ but not FcRγ -/- microglia, with increasing concentrations of the Ab42–5 (Fig. 1C). These data demonstrate that FcRγ -/- microglia exhibit a deficit in uptake of anti-Aβ immune complexes.
Figure 2.
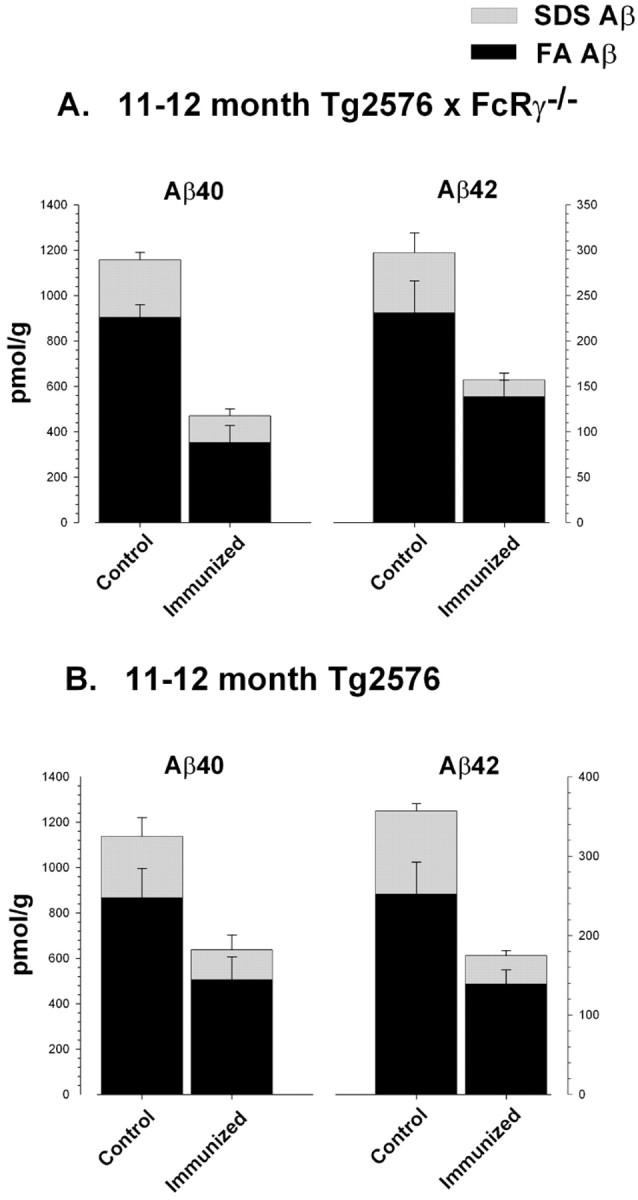
Aβ levels were significantly reduced in the Aβ1–42-immunized 11- to 12-month-old Tg2576 × FcRγ-/- mice. Mice were killed after immunization with Aβ1–42 for 3 months, and both SDS-soluble (SDS) and SDS-insoluble formic acid extractable (FA) fractions were analyzed by capture ELISA. A, Tg2576 × FcRγ-/- mice, 11–12 month old (n = 5 per group); B, wt Tg2576 mice, 11–12 month old (n = 6 per group). Statistical analyses are provided in Results.
Figure 3.
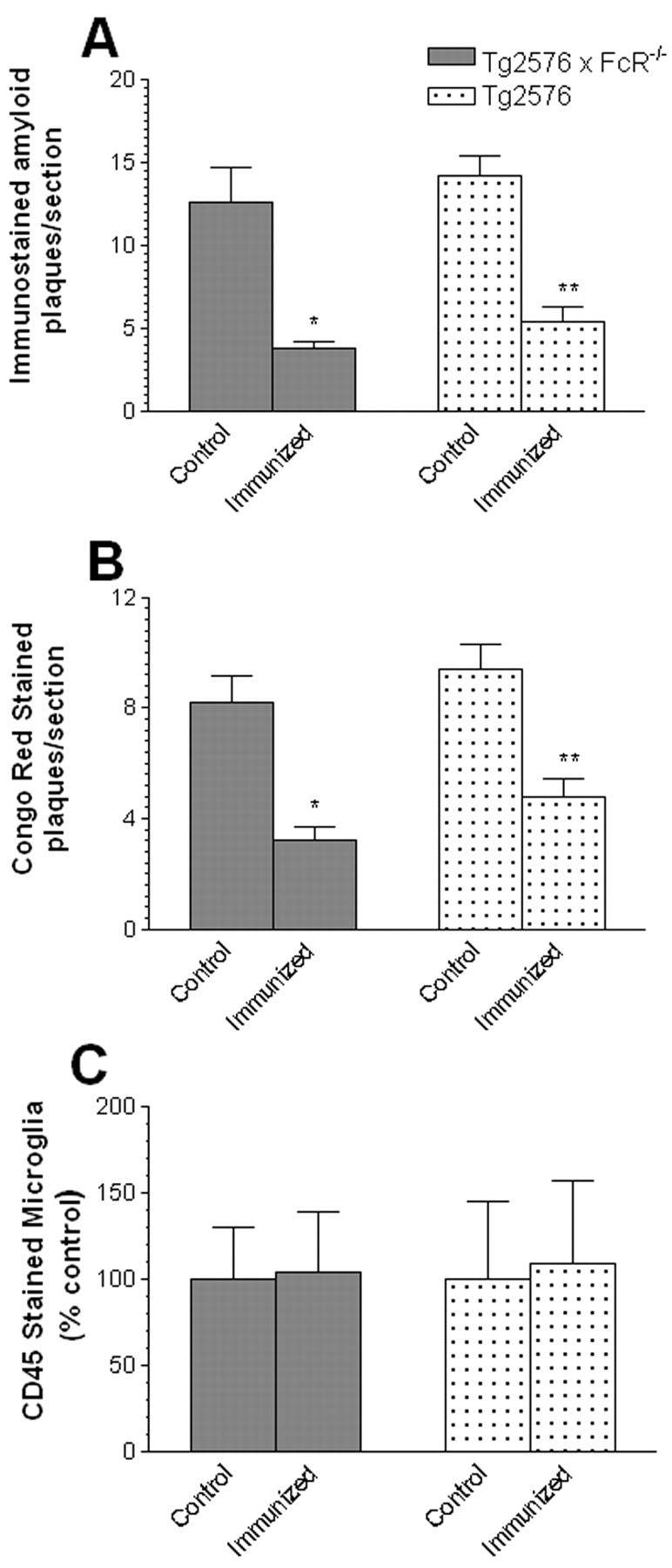
Quantitative image analysis of amyloid plaque burden in the 11- to 12-month-old Tg2576 × FcRγ-/- mice. A, Immunostained amyloid plaque burden is reduced in the 11- to 12-month-old Tg2576 × FcRγ-/- mice immunized with Aβ1–42 (*p < 0.004) and in the 11- to 12-month-old Tg2576 mice (**p < 0.005). B, Congo red-stained plaque burden is significantly reduced in the 11- to 12-month-old Tg2576 × FcRγ-/- mice immunized with Aβ1–42 (*p < 0.005) and in the 11- to 12-month-old Tg2576 mice (**p < 0.05). C, Quantitation of CD45-stained activated microglia revealed no statistically significant differences between immunized and control groups of the 11- to 12-month-old Tg2576 × FcRγ-/- and the 11- to 12-month-old Tg2576 mice.
To more closely examine this phenomenon and to demonstrate whether the defective uptake can be attributable to loss of FcR function in the FcRγ -/- microglia, we performed additional experiments. First, we determined the effect of scavenger receptor-A (SRA)-mediated uptake of Cy3–Aβ ligand by using the SRA competitive ligand, fucoidan (Fig. 1B). In the absence of antibody, fucoidan decreased Cy3–Aβ uptake by FcRγ +/+ and FcRγ -/- microglia by >75% (Fig. 1B). In the presence of increasing amounts of Ab9 and fucoidan, a dose-dependent increase in uptake of Cy3–Aβ is observed in FcRγ +/+ microglia. This increase was >500% at 20 μg/ml of Ab9 (Fig. 1B). In the presence of fucoidan and 20 μg/ml of Ab9, a slight increase (<70%) in the level of anti-Aβ immune complexes was detected at the cell surface of the FcRγ -/- microglia (Fig 1B) (see also supplemental data, Fig. 3). This binding could be attributable to interaction of the complex with residual FcR or other cell surface receptors. To ensure that FcR-mediated the increased uptake of Cy3–Aβ in the presence of Ab9, Cy3–Aβ uptake was evaluated in the presence of 20 μg/ml of Ab9 with and without competing mouse IgG (100 μg/ml). These studies show that IgG partially blocks uptake of Ab9 Cy3–Aβ uptake by wt microglial cells (Fig. 1D). Collectively, these data show that FcRγ -/- microglia exhibit normal scavenging of Aβ in the absence of antibody but exhibit a pronounced deficit in FcR-mediated phagocytosis of Cy3–Aβ microaggregates in the presence of anti-Aβ antibody or mouse IgG.
Biochemical extractable Aβ levels and plaque loads were significantly reduced in the brains of Aβ1–42 immunized 11- to 12-month-old Tg2576 × FcRγ-/- mice
To investigate the effects of FcRγ -/- knock-out on Aβ clearance at various stages of Aβ deposition in vivo, we immunized two age groups of Tg2576 × FcRγ -/- mice (female 11- to 12-month-old and 14- to 15-month-old mice at the time of immunization). Age and sex-matched FcR-sufficient Tg2576 mice were immunized as controls. All groups of mice generated qualitatively similar anti-Aβ antibody titers with the main IgG subtype produced being IgG2b (Table 1). Except for a small decline in the titers apparent in the 14- to 15-month-old group, absolute titers of anti-Aβ among the various groups were also quite similar. At the end of 3 months of immunization, mice were killed, and the levels of both SDS-soluble (SDS) and SDS-insoluble FA-extractable fractions of Aβ40 and Aβ42 were analyzed by ELISA. In the 11- to 12-month-old Tg2576 × FcRγ -/- mice immunized with Aβ1–42, there was a 42% reduction (p < 0.03) in Aβ42 levels in the SDS-insoluble FA fraction and a 66% reduction of Aβ42 in the SDS fractions (p < 0.01) (Fig. 2A). Aβ40 levels were also reduced by 64% (p < 0.01) in the FA fractions and by 55% (p < 0.03) in the SDS fractions. The 11- to 12-month-old FcR-sufficient Tg2576 mice immunized with Aβ1–42 (Fig. 2B) showed similar reductions in Aβ40 and Aβ42; Aβ42 was reduced by 44% (p < 0.03) in the FA fraction and by 65% in SDS fractions (p < 0.01), whereas Aβ40 was reduced by 42% (p < 0.05) in the FA fraction and by 49% in SDS fractions (p < 0.02). In both groups of 14- to 15-month-old immunized mice, there were only slight decreases in the levels of either SDS or FA extractable Aβ40 and Aβ42 (see supplemental data, Fig. 4).
Table 1.
Anti-Aβ antibody titers and IgG isotypes from Aβ1–42 immunized micea
|
Mice |
Total titer (μg/ml) |
IgG1 |
IgG2a |
IgG2b |
IgG3 |
|---|---|---|---|---|---|
| 11-12 month Tg × FcRγ-/- | 36.4 ± 8.2 | 2.3 ± 0.2 | 6.1 ± 1.3 | 52.7 ± 11.7 | 1.7 ± 0.6 |
| 11-12 month Tg2576 | 32.1 ± 5.1 | 1.8 ± 0.3 | 7.5 ± 1.8 | 44.5 ± 7.2 | 0.9 ± 0.2 |
| 14-15 month Tg × FcRγ-/- | 33.7 ± 6.2 | 1.1 ± 0.3 | 3.2 ± 0.6 | 37.2 ± 4.1 | 1.3 ± 0.7 |
| 14-15 month Tg2576
|
34.6 ± 7.8
|
0.8 ± 0.3
|
2.9 ± 1.1
|
35.9 ± 10.7
|
0.9 ± 0.2
|
Titers are reported with values from unimmunized mice control substrate. Anti-Aβ antibody titers in unimmunized control mice were negligible
Figure 4.

Representative pictures of immunostained Aβ plaques (stained with Ab9 antibody) in the neo cortex of 11- to 12-month-old Tg2576 × FcRγ-/- mice (A) and control group (B) immunized with Aβ1–42 (A, B, magnification 40×). Representative pictures of Congo red-stained plaques (red) decorated with microglia immunostained with anti-mouse CD45 (black) in the neo cortex of 11- to 12-month-old Tg2576 × FcRγ-/- mice (C) and control group (D) immunized with Aβ1–42 (C, D, magnification 100×).
To determine whether there were alterations in Aβ plaque loads in the immunized mice, coronal sections of each mouse hemibrain were analyzed for changes in immunostained Aβ plaque loads as well as Congo red-stained plaques using quantitative image analysis. In the 11- to 12-month-old Tg2576 × FcRγ -/- mice, there were significant reductions in both the immunostained Aβ plaque burdens (Fig. 3A) as well as Congo redstained plaque loads (Fig. 3B). Representative immunostained sections and Congo red-stained plaques are shown from immunized and nonimmunized Tg2576 × FcRγ -/- mice in Figure 4. Similar levels of reductions in Aβ plaque loads were also seen in the 11- to 12-month-old Tg2576 (FcR sufficient) mice immunized with Aβ42 (Fig. 3A,B). In the older 14- to 15-month-old mouse groups, a trend toward reduction of immunohistochemical amyloid load was seen in the immunized Tg2576 (48% compared with control) and Tg2576 × FcRγ -/- mice (42% compared with control); however, no change was seen in the number of Congo red plaques. These data are consistent with previous studies in which we observe reduced efficacy of immunization in Tg2576 animals that had high preexisting plaque loads (Das et al., 2001).
We also examined whether there were any differences in global microglial activation in either the immunized or nonimmunized Tg2576 × FcRγ -/- mice. Coronal sections of each mouse hemibrain were immunostained with anti-mouse CD45, a marker for activated microglia (Irie-Sasaki et al., 2003). As shown in Figure 4, C and D, there were abundant numbers of CD45 immunoreactive microglia present, surrounding congophilic plaques both in the Aβ1–42-immunized as well as control Tg2576 × FcRγ -/- mice. Quantitative image analysis of the CD45 staining between the immunized and control groups revealed no significant differences in the density of activated microglial surrounding congophilic plaques in the Tg2576 × FcRγ -/- mice as well as FcR-sufficient Tg2576 mice (Fig. 3C).
Discussion
Despite the setbacks in the phase II AN-1792 Aβ vaccination trial, which was halted because of a meningioencephalitic presentation in ∼5% of the patients (Check, 2002), Aβ immunotherapy or derivative strategies remain a novel and promising approach for the treatment or prevention of AD. Two recent reports involving a subset of patients that were enrolled in this trial have shed some light on the potential therapeutic value of this strategy. Results from one study showed evidence in one patient for a reduced number of Aβ plaques in the cortex after immunization with AN-1792 (Nicoll et al., 2003). In a more recent study, in 20 patients who generated significant anti-Aβ antibody titers, there were slower rates of decline of cognitive functions and activities of daily living as determined by a battery of tests compared with 9 patients without such high titer anti-Aβ antibodies (Hock et al., 2003). Ultimately, the success and tolerability of future studies may depend on the mechanism or mechanisms through which Aβ immunization works. One possible mechanism supported by some experimental observations is that Aβ immunization triggers phagocytosis of antibody-bound Aβ immune complexes via microglial FcR. After immunization, an increased number of microglial cells stained with anti-Aβ antibodies have been observed (Schenk et al., 1999). Using an ex vivo strategy, it was shown that anti-Aβ antibodies induced phagocytosis of Aβ plaques. Importantly, Fab fragments of these antibodies failed to induce Aβ phagocytosis, suggesting that the enhanced uptake was attributable to FcR (Bard et al., 2000). More recently, passive immunization with anti-Aβ IgG2a monoclonal antibodies, which exhibit higher affinity for the phagocytic FcγRI receptor, was shown to more effectively attenuate Aβ deposition compared with anti-Aβ mAbs of the IgG2b and IgG1 isotypes, again suggesting a potential role for FcR-mediated mechanisms in Aβ immunotherapies (Bard et al., 2003).
To definitively ascertain the role of microglial FcR in Aβ immunotherapies, we used a direct genetic approach using APP Tg2576 mice bred into FcRγ -/- mice for these studies. The FcR-γ chain is required for surface expression of FcγRIII (Kurosaki and Ravetch, 1989) and for signaling effector functions such as phagocytosis of immune complexes by FcγRI (Ernst et al., 1993). Therefore, knock-out of the FcR-γ chain results in mice that lack expression of FcγRIII and are defective in effector functions mediated by FcγRI (Takai et al., 1994). Although the FcR-γ chain is not directly involved in the expression and maturation of FcγRII (Ravetch and Bolland, 2001), the complete absence of phagocytosis activity in the FcRγ -/- mice indicated an unexpected role of this subunit in the effector functions of FcγRII as well. Of note, although expression of the FcγRI receptor was previously presumed to be completely abolished in FcRγ -/- mice, a recently published study using newly generated antibodies against FcγRI showed that the FcRγ -/- mice express low levels of FcRI on the surface in bone marrow-derived macrophages, approximately one-fifth the level compared with wt macrophages (Barnes et al., 2002). These residual FcRI receptors were shown to bind and internalize small amounts of monomeric IgG2a; however, the absence of FcR-γ chain subunit still renders the FcRγ -/- mice unable to perform FcR-mediated phagocytosis. Because of these later reports and the lack of information on the phagocytic phenotype of microglia from FcRγ -/- mice, we first performed a series of in vitro experiments to directly assess microglial phagocytosis of Aβ in microglial isolated from FcRγ -/- mice. These data show that the microglia isolated from FcRγ -/- mice exhibit almost no uptake of anti-Aβ immune complexes via FcR. Aggregated Aβ was readily scavenged by both FcRγ +/+ and FcRγ -/- microglia in the absence of anti-Aβ. Thus, there did not appear to be any defects in the non-FcR-mediated Aβ uptake by microglia in the FcRγ -/- mice.
Having demonstrated that knock-out of the FcR-γ chain significantly impairs microglial FcR-mediated phagocytosis of Aβ immune complexes, we analyzed the effectiveness of Aβ1–42 immunizations in Tg2576 × FcRγ -/- crossed mice. Aβ deposition was unaltered in these mice, and when immunized with Aβ1–42, they developed qualitatively and quantitatively similar anti-Aβ titers, as assessed by direct comparison with the Tg2576 strain. In the 11- to 12-month-old Tg2576 × FcRγ -/- mice and Tg2576 mice, which have moderate amounts of Aβ deposition at the time of immunization, there were significant reductions in Aβ deposition after immunization. As shown previously, Aβ immunization was less effective in mice with higher initial plaque loads (Das et al., 2001). In this case, Aβ1–42 immunization of 14- to 15-month-old Tg2576 × FcRγ -/- mice and Tg2576 mice had minimal impact on Aβ deposition. Although an increase in activated microglia surrounding congophilic plaques after Aβ immunizations was shown in one report (Wilcock et al., 2001), after immunization we found no significant differences in the density of activated microglial surrounding congophilic plaques in the Tg2576 × FcRγ -/- mice as well as FcR-sufficient Tg2576 mice. These data are consistent with the notion that Aβ deposition itself and not immunization is driving the microglial activation.
Our results indicate that FcR-mediated uptake of anti-Aβ immune complexes is not required for the attenuation of Aβ deposition after Aβ1–42 immunization in Tg2576 mice. These data contrast with the aforementioned studies suggesting a potential role for FcR-mediated mechanisms in Aβ immunotherapies (Bard et al., 2000, 2003). Using an ex vivo phagocytosis assay, the authors (Bard et al., 2000) demonstrate that wt microglia are capable of phagocytosing anti-Aβ immune complexes in an FcR-dependent manner. A subsequent study (Bard et al., 2003) showing that IgG2a anti-Aβ antibodies are more effective than other IgG isotypes in reducing Aβ deposition in PDAPP mice is more difficult to reconcile with our findings. Because IgG2a antibodies have a higher affinity for FcγRI than other antibodies, this result was interpreted as providing evidence for the role of FcR in mediating the efficacy of immunization. Although care was taken to show that affinity for aggregated or a soluble Aβ did not correlate with efficacy, we would suggest that some other property of the IgG2a antibodies used in that study, independent of interaction with FcR, must account for the enhanced efficacy observed. Certainly, genetic differences between the PDAPP mice and Tg2576 mice could account for differences in Aβ immune responses as well as efficacy in these studies.
Our results do not rule out a possible role for enhanced non-FcR-mediated cellular scavenging of anti-Aβ immune complexes after immunization. Anti-Aβ antibodies in the CNS could bind to and alter deposited Aβ so that it could be more readily internalized by SRA or other receptors present on microglia cells (Brazil et al., 2000; Webster et al., 2001; Bamberger et al., 2003). Indeed, it has been shown that high concentrations of either intact anti-Aβ or Fab fragments of the anti-Aβ applied directly to the brains of transgenic mice results in rapid clearance of Aβ and is associated with a local microglial infiltration (Bacskai et al., 2002). Although the high concentrations of intact anti-Aβ and anti-Aβ Fab achieved after direct application to the brain are not likely to be present after either active immunization with Aβ1–42 or passive immunization, these studies are consistent with non-FcR-mediated uptake. Further study will be needed to determine the possible role of non–FcR-mediated microglial or CNS cell scavenging of Aβ after Aβ immunotherapy. Of course, one area of intense debate regarding the mechanism of Aβ immunotherapy is whether access of the antibody to the CNS is required. Our data do not address this issue but do demonstrate that if a peripheral mechanism is at work, it again is likely to be independent of FcR-mediated phagocytosis.
The main question that we have addressed in this study is whether FcR-mediated phagocytosis of anti-Aβ plays a role in determining the efficacy of Aβ immunotherapy. Given that microglial cells from FcRγ -/- mice are deficient in phagocytosis of anti-Aβ immune complexes and that there is no evidence for compensatory mechanisms enabling phagocytosis of immune complexes in FcRγ -/- mice, these studies indicate that FcR-mediated mechanisms play little or no role in the effectiveness of Aβ immunotherapy in APP Tg2576 mice. Thus, it appears that the Fc portion of the anti-Aβ antibody required for interaction with FcR may not be necessary for Aβ immunotherapy to work. This hypothesis is supported by a recent study showing that peripheral administration of two Aβ binding agents, gelsolin and GM-1 ganglioside, had a modest effect on Aβ deposition in transgenic mice (Matsuoka et al., 2003). Future studies with these or other Aβ binding agents (i.e., anti-Aβ single chain variable fragments) will be needed to definitively test this hypothesis in mice. If intact antibodies are not needed in mice, then it is likely that therapies using high-affinity Aβ binding agents that lack the immunologic effector functions of antibodies can then be tested in humans.
Footnotes
This work was supported by a Smith fellowship to P.D. and by the National Institutes of Health (AG18454) (T.E.G). We thank Dr. S. G. Younkin for providing the Tg2576 mice.
Correspondence should be addressed to Dr. Todd. E. Golde, Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224. E-mail: golde.todd@mayo.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238532-07$15.00/0
References
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT ( 2001) Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7: 369-372. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT ( 2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci 22: 7873-7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE ( 2003) A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci 23: 2665-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T ( 2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916-919. [DOI] [PubMed] [Google Scholar]
- Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, Khan K, Kholodenko D, Lee C, Lee M, Motter R, Nguyen M, Reed A, Schenk D, Tang P, Vasquez N, Seubert P, Yednock T ( 2003) Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci USA 100: 2023-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes N, Gavin AL, Tan PS, Mottram P, Koentgen F, Hogarth PM ( 2002) FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity 16: 379-389. [DOI] [PubMed] [Google Scholar]
- Brazil MI, Chung H, Maxfield FR ( 2000) Effects of incorporation of immunoglobulin G and complement component C1q on uptake and degradation of Alzheimer's disease amyloid fibrils by microglia. J Biol Chem 275: 16941-16947. [DOI] [PubMed] [Google Scholar]
- Check E ( 2002) Nerve inflammation halts trial for Alzheimer's drug. Nature 415: 462. [DOI] [PubMed] [Google Scholar]
- Das P, Golde TE ( 2002) Open peer commentary regarding Abeta immunization and CNS inflammation by Pasinetti et al. Neurobiol Aging 23: 671-674. [DOI] [PubMed] [Google Scholar]
- Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE ( 2001) Reduced effectiveness of Abeta1–42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging 22: 721-727. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM ( 2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 98: 8850-8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM ( 2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci 5: 452-457. [DOI] [PubMed] [Google Scholar]
- Ernst LK, Duchemin AM, Anderson CL ( 1993) Association of the high-affinity receptor for IgG (Fc gamma RI) with the gamma subunit of the IgE receptor. Proc Natl Acad Sci USA 90: 6023-6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Eckman CB, Younkin SG ( 2000) Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta 1502: 172-187. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, deq Uervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM ( 2003) Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron 38: 547-554. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G ( 1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Irie-Sasaki J, Sasaki T, Penninger JM ( 2003) CD45 regulated signaling pathways. Curr Top Med Chem 3: 783-796. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St. George-Hyslop P, Westaway D ( 2000) A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408: 979-982. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KS, Younkin SG ( 2001) Age-dependent changes in brain, CSF, and plasma amyloid protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: 372-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH ( 2002) Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci 22: 6331-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Ravetch JV ( 1989) A single amino acid in the glycosyl phosphatidylinositol attachment domain determines the membrane topology of Fc gamma RIII. Nature 342: 805-807. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, Desai R, Hancock WW, Weiner HL, Selkoe DJ ( 2000) Nasal A beta treatment induces anti-A beta antibody production and decreases cerebral amyloid burden in PD-APP mice. Ann NY Acad Sci 920: 328-331. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Saito M, LaFrancois J, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K ( 2003) Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to β-amyloid. J Neurosci 23: 29-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Lambermon MH, Darabie AA, Brown ME, Janus C, Chishti MA, Horne P, Westaway D, Fraser PE, Mount HT, Przybylski M, St. George-Hyslop P ( 2002) Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med 8: 1263-1269. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW ( 2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982-985. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO ( 2003) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 9: 448-452. [DOI] [PubMed] [Google Scholar]
- Ravetch JV, Bolland S ( 2001) IgG Fc receptors. Annu Rev Immunol 19: 275-290. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P ( 1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173-177. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ ( 1997) Alzheimer's disease: genotypes, phenotypes, and treatments. Science 275: 630-631. [DOI] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Hanan E, Katzav T ( 1996) Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc Natl Acad Sci USA 93: 452-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frankel D, Hanan-Aharon E ( 1997) Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci USA 94: 4109-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV ( 1994) FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 76: 519-529. [DOI] [PubMed] [Google Scholar]
- Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ ( 2001) Antibody-mediated phagocytosis of the amyloid beta-peptide in microglia is differentially modulated by C1q. J Immunol 166: 7496-7503. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Ugen KE, Gottschall PE, DiCarlo G, Dickey C, Boyett KW, Jantzen PT, Connor KE, Melachrino J, Hardy J, Morgan D ( 2001) Number of Abeta inoculations in APP+PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA Cell Biol 20: 731-736. [DOI] [PubMed] [Google Scholar]