

Abstract
It is impossible to obtain and amplify live neurons from Alzheimer's disease (AD) patients. To establish the neurons harboring AD abnormality, we constructed mouse embryonic stem (ES) cells, in which the AD-causative V642I mutation was introduced to the endogenous amyloid precursor protein (APP) gene, in combination with a protocol to efficiently differentiate ES cells into postmitotic neurons without using a cell sorter. By this protocol, ES cells differentiated into >90% of the central type of adult postmitotic neurons. Neurons derived from V642I–APP knock-in ES cells were indistinguishable from wild-type ES-derived neurons, as determined by the expression of various markers for neuronal differentiation. Notably, V642I–APP knock-in ES cell-derived neurons exhibited significantly increased secretion of Aβ42 without AD-related hyperphosphorylation of tau, indicating that the direct output of the AD-causative mutation is increased Aβ42 secretion. In this study, we analyze created neurons with wild-type and AD genotypes and propose a new strategy for generating neurons for any dominantly inherited neurodegenerative diseases. The strategy can be applied to create human neurons with AD or any other neurodegenerative disease by using human ES cells.
Keywords: embryonic stem cell, postmitotic neuron, differentiation, Alzheimer's disease, gene knock-in, sharable neuron with disease genotype, amyloid precursor protein, amyloid-β protein, tau
Introduction
Alzheimer's disease (AD), the most prevalent neurodegenerative disease, is characterized pathologically by neuronal loss, intracellular neurofibrillary tangles (NFT), and extracellular neuritic plaques. The major constituent of the plaque is amyloid-β (Aβ), which is cleaved off from a single transmembrane precursor, termed amyloid precursor protein (APP). After the identification of APP695 (consisting of 695 amino acid residues) by Kang et al. (1987), at least 10 isoforms of APP have been identified, all resulting from alternative splicing of a single gene (Sandbrink et al., 1994). APP751 and APP770, expressed predominantly in non-neuronal tissues, contain a Kunitz-type protease inhibitor (KPI) domain in their extracellular region. APP695, which lacks the KPI domain, is preferentially expressed in the brain (Neve et al., 1988) and is, thus, considered to be a neuronal version of APP.
Certain cases with early-onset AD occur in a hereditary manner. Genetic studies have demonstrated that three forms of known mutant genes cause some of the familial AD (FAD): APP mutants, presenilin (PS) 1 mutants, and PS2 mutants (Shastry and Giblin, 1999). The most frequent known causes of FAD are the mutations of PS1, numbering more than 100, whereas only a few mutations, such as N141I and M239V, are in PS2 as established causes. Among FAD-linked mutations of APP, V642 mutations to I, F, and G are the most common cause (Hardy, 1992). In fewer FAD families, A617G, E618G, L648P, and K595N/M596L in APP have been identified as the causes of FAD (Mullan et al., 1992; Hendriks and van Broeckhoven, 1996; Kwok et al., 2000; Nilsberth et al., 2001).
One difficulty in advancing AD research, in view of the AD pathophysiology analysis and anti-AD therapeutic development, is that it is hard to obtain and examine live postmitotic neurons from AD patients. Even if such neurons are obtained, it is impossible to amplify them for reproduction of the obtained data or for sharing those neurons with other laboratories. In addition, as long as there is a possibility that different mechanisms may underlie different AD cases with different causes (Hashimoto et al., 2000a, 2002a,b), it is considerably disadvantageous to investigate neurons from AD patients with unidentified causes. The present study was conducted to overcome these difficulties. We noticed that if combined with recent progress in the technique for differentiation of embryonic stem (ES) cells into postmitotic neurons in vitro (Bain et al., 1995; Fraichard et al., 1995; Strübing et al., 1995; Okabe et al., 1996; Li et al., 1998; Renoncourt et al., 1998; Mujtaba et al., 1999), construction of FAD gene knock-in ES cells would allow for the creation of postmitotic neurons bearing AD abnormality. In this study, we find a new strategy for the creation of AD neurons by combining two elemental steps: the construction of ES cells with an endogenous APP gene harboring the FAD-causative V642I mutation, termed V642I–APP knock-in ES cells, and an efficient protocol for differentiation of ES cells into postmitotic neurons.
Materials and Methods
Cloning of the mouse genomic DNA of APP exon 17 region. A 3 kb mouse APP gene probe was obtained by PCR amplification of C57BL/6 mouse genomic DNA, using primers 5′-GATTCAGGATTTGAAGTCCGC-3′ and 5′-ATGATGGATGGATGTGTACTG-3′. The primers were designed to amplify intron 16 and part of the flanking exons. A mouse genomic DNA library (Invitrogen, Carlsbad, CA) of TT2 ES cells was screened with the probe, and a clone with an ∼14 kb insert containing exons 16, 17, and 18 was isolated. After characterization by restriction site mapping and nucleotide sequencing, a 4.4 kb SalI-Hind III fragment (SH1) and a 3.6 kb Hind III-XhoI fragment (HX1) were chosen as 5′ and 3′ homology, respectively, for subsequent vector construction.
V642I mutation knock-in vector construction. A 371 bp SmaI-SmaI fragment (SM1) within SH1 contained the entire mouse APP exon 17 that includes the codon for valine at position 642 (by APP695 numbering). This fragment was submitted to PCR-based mutagenesis to introduce a point mutation to alter Val 642 ( GTC) to isoleucine ( ATC), and the mutated SM1 fragment (termed mSM1) was sequenced to confirm its integrity. Subsequently, the SM1 region of the parental SH1 fragment was replaced by the mSM1 to derive SH2. SH2 was ligated to floxP-TK-Neo, then to HX1. Finally, the assembled 11.6 kb fragment was inserted into pBluescript-SK(+) (Stratagene, La Jolla, CA) to serve as a V642I–APP mutation knock-in vector.
Establishment of homologously recombinant ES clones. TT2 ES cells (derived from C57BL/6 female and CBA male) (Yagi et al., 1993) were maintained in ES medium [Knock-out DMEM (Invitrogen) plus 15% Knock-out Serum Replacement (Invitrogen), 100 μm nonessential amino acids (Invitrogen), 100 μm β-mercaptoethanol (Sigma, St. Louis, MO), 30 μm nucleosides mixture (A/G/C/T/U; Sigma), 1000 U/ml leukemia inhibitory factor (Chemicon, Temecula, CA), 2 mm l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin]. Mitomycin C mitotically inactivated mouse embryonic fibroblasts (Invitrogen) were used as a feeder layer. V642I–APP knock-in vector DNA (22 μg) was introduced by electroporation into ES cells passaged nine times in our laboratory. Electroporated cells were seeded onto a neomycin-resistant mouse embryonic fibroblast (Invitrogen) feeder layer at 2 × 10 6 cells per 100 mm dish and selected with 250 μg/ml G418. Genomic DNA of recombinant ES clones was submitted to Southern analysis using a Renaissance Random Primer Fluorescein Labeling kit (NEN Life Science Products, Boston, MA). Cointroduction of the intended V642I point mutation was confirmed by PCR amplification of the region, including the V642 locus by the primers 5′-GCTTACCTGTTAAAGGGCTTC-3′and 5′-GCACATTAAATTCATGGCACCC-3′, and nucleotide sequencing by the primer 5′-AGGTGTTCTTTGCTGAAGATGTGG-3′.
Excision of floxP-TK-Neo insert by transient expression of Cre recombinase. Homologously recombinant ES clones were transiently transfected with pIC-Cre (30 μg) that expresses Cre recombinase (Gu et al., 1993) by electroporation, seeded at 1 × 10 4 cells per 60 mm dish, and maintained in the G418-free medium. Genomic DNA samples prepared from each clone were submitted to PCR screening by the primers 5′-TCTAGAACCCAAAGAACGCCGAGTCC-3′ and 5′-CCAATGAATGTGAGATCTCTGGCTTTC-3′. Detection of the 363 bp product (mutant allele derived) in addition to the 266 bp product (wild-type allele derived) suggested a successful Cre-loxP recombination event. Putative floxP-cleaved clones were further examined for correct recombination by additional PCR, sequencing, and Southern analysis. To eliminate any possible contamination of floxP-TK-Neo remaining cells, the established clones were recloned in the ES medium containing gancyclovir (2 μm), and all the resultant clones showed the expected results for various genetic analyses. The V642I mutation was maintained throughout this step in all clones examined. Among these final mutant recombinants, the clone R34C(30)-9G was used for this study. The V642I–APP knock-in ES clone was passaged three more times to make a large frozen cell stock, kept under strict administration. In each differentiation experiment, cells from the same frozen stock were newly thawed and differentiated into neurons within two passages. We, thus, strictly controlled and maintained the quality of our ES cells. The frequency of diploid cells was 84% just before differentiation into neurons.
ES cell differentiation. We differentiated ES cells into neurons, as shown in Figure 2 A, by modifying previously described methods (Bain et al., 1995; Okabe et al., 1996). ES cells were seeded onto gelatin-coated 60 mm dishes at a density of 2 × 10 6 cells/dish and cultured for 2–3d in ES medium for amplification. Cells were then trypsinized and seeded onto bacteriological dishes as a suspension for 5 d to form embryoid bodies (EBs) in DMEM supplemented with 10% FBS, 10% newborn calf serum, nucleoside mixture, penicillin, and streptomycin. In the last 24 hr of EB formation, 5 μm retinoic acid (RA; Sigma) was added; then EBs were seeded onto fibronectin-coated 60 mm dishes (Becton-Dickinson, Franklin Lakes, NJ) in insulin–transferrin–selenium (ITS) medium [DMEM/F12 (1:1) supplemented with ITS-G supplement (Invitrogen), 50 U/ml penicillin, and 50 μg/ml streptomycin]. Two days after culturing on fibronectin-coated dishes in ITS medium, cells were dissociated with trypsin and reseeded onto fibronectin-coated 60 mm dishes in ITS medium in the presence of basic FGF (bFGF; 10 ng/ml; R & D Systems, Minneapolis, MN). After 2 d, cells were dissociated again and seeded onto poly-L-ornithine/laminin (polyO/L)-coated 6-well plates (Becton-Dickinson) at a density of 10 6 cells/well and cultured in ITS medium with bFGF for 2 d. Subsequently, the medium was changed to DMEM/F12 (1:1) plus N2 supplement (Invitrogen), penicillin, and streptomycin without bFGF. Cytosine-β-d-arabinofuranoside (AraC; 10 μg/ml; Sigma) was added to the medium to obtain postmitotic neurons efficiently and cultured for 2 d. Then, medium was replaced with Neurobasal medium (Invitrogen) supplemented with B27 supplement (Invitrogen), l-glutamine, penicillin, and streptomycin. A half volume of the medium was then replaced every 3 d.
Figure 2.

Differentiation of ES cells into postmitotic neurons. A, The protocol for differentiation of ES cells into postmitotic neurons is schematically illustrated. See Materials and Methods for details. B, Time course of the expression of markers in cells differentiating into neurons from wtES cells. Small numbers represent the days after being plated on polyO/L-coated dishes. B represents adult mouse whole brain in all figures. C, Northern blot analysis of neuronal markers in neurons derived from wild-type or knock-in ES cells after 23 d of differentiation. W, K, and N represent wtES cells, knock-in ES cells, and mouse cortical neurons, respectively, in all panels. D, Neurotransmitter phenotypes of neurons derived from wild-type or knock-in ES cells after 23 d of differentiation. S represents spinal cord. E, Expression of BACE1 in neurons derived from wild-type (WT) or knock-in (KI) ES cells after being plated onto polyO/L-coated dishes. Syn, Synapsin I; Syp, synaptophysin. Each of the results was reproduced at least three times.
Probes. Each probe was cloned by reverse transcription (RT)-PCR using avian myeloblastosis virus reverse transcriptase (Life Sciences Inc.), followed by Taq DNA polymerase (Takara, Shiga, Japan) from total RNA of mouse brain, spinal cord, or P19 mouse embryonal carcinoma cells. Primers used, and the sizes of the products are as follows: Oct3 (463 bp), 5′-GTGGAGGAAGCCGACAACAATG-3′ and 5′-GATATCAGTTTGAATGCATGGGAGAG-3′; nestin (407 bp), 5′-CTGGAACAGAGATTGGAAGGCCGCT-3′ and 5′-GGATCCTGTGTCTTCAGAAGGCTGTCAC-3′; MAP2 (455 bp), 5′-GGAGAGGCAGAATTTCCACTCCTGA-3′ and 5′-TCCTGCTCTGCGAATTGGTTCTGAC-3′; neurofilament-M (NF-M; 528 bp), 5′-TTCGCCGGCTACATCGAGAAAGTG-3′ and 5′-CTGGTGCATGTTCTGGTCTGAGTG-3′;GFAP(592bp),5′-CTCGAGATGATGGAGCTCAATGACCGCT-3′ and 5′-CAGCGTCTGTGAGGTCTGCAAACTT-3′; MAG (401 bp), 5′-TGAGAACCAGTATGGCCAGAGAGCC-3′ and 5′-CAATCAGGATGGCAAAGGCGACCAC-3′; synaptophysin (529 bp), 5′-ACATCTTCCTGCAGAACAAGTACCG-3′ and 5′CCGTAGCCTTGCTGCCCATAGTCGC-3′; synapsin I (493 bp), 5′-GGTGGATTCTCTGTGGACATGGAAG-3′ and 5′-AGTTCTGCCCAATCTTCTGGACACG-3′; Rab3A (513 bp), 5′-ATGGCTTCCGCCACAGACTCTCGCT-3′ and 5′-ATTAATGTTGTCCTTGGCGCTGGCCT -3′; β-site APP-cleaving enzyme 1 (BACE1; 926 bp), 5′-CACAAGGCCCGGGCTCACTATG-3′ and 5′-CAAATACTTTCTTGGGCAAGCGAAGGTTGG-3′; tau (1033 bp), 5′-CTTTGAACCAGTATGGCTGACCCT-3′ and 5′-TCACAAACCCTGCTTGGCCAAGGAAGC-3′; ChAT (493 bp), 5′-AGCAGTTCAGGAAGAGCCAGGCCAT-3′ and 5′-AAATTAATGACAACATCCAAGAC-3′; GAD 1 (528 bp), 5′-GCCCATGGATGCACCAGAAAACTGG-3′ and 5′-GGTATTGGCCGTTGATGTCAGCCAT-3′; GAD 2 (578 bp), 5′-TTGGGAATTGGCAGACCAACCGCAA-3′ and 5′-TGGTTCCAGCTGTGGCACTCACCAG-3′; TH (478 bp), 5′-AGAGTCTCATCGAGGATGCCCGCAA-3′ and 5′-TGGTCAGAGAAGCCCGGATGGTCCA-3′; tryptophan hydroxylase (TPH; 566 bp), 5′-GACCACCCTGGCTTCAAAGACAATG-3′ and 5′-CCACAGTGAAAAAGTAGCACGTTGCC-3′; vesicular glutamate transporter (VGluT) 1 (591 bp), 5′-CGCCTACTTTGAAGAAGTGTTCGG-3′ and 5′-TCACTTTCGTCACTGCCAGCCAGC-3′; VGluT2 (720 bp), 5′-GAGCCCTGCAAAGCATCCTACCAT-3′ and 5′-GCCCAAGGTTGTTTCTCTCCTGAG-3′. The PCR products were purified and subcloned into pCRII-topoisomerase I (TOPO) deoxythymidine deoxyadenosine cloning vector (Invitrogen) and sequenced using an automated sequencer (ABI-PRISM310 Genetic Analyzer; Perkin-Elmer Applied Biosystems, Foster City, CA). Other cDNAs used for probes as templates were: mouse APP695 cDNA, XhoI fragment (1.2 kb); mouse amyloid precursor-like protein 1 (APLP1) cDNA, EcoR I-EcoR V fragment (1.3 kb); and mouse APLP2 cDNA, Hind III fragment (1.3 kb). Each probe was labeled with [α-32P] dCTP (Amersham Pharmacia Biotech, Uppsala, Sweden) by the random priming method.
Northern blot analysis. Cellular total RNA was extracted with ISOGEN (Nippon Gene, Tokyo, Japan). Total RNA (10 μg) was separated by formaldehyde/agarose gel electrophoresis, transferred to a Hybond-N nylon membrane (Amersham Pharmacia Biotech), and hybridized with each probe.
RT-PCR analysis. To examine the neurotransmitter phenotype of neurons derived from ES cells, expression of ChAT, GAD1, GAD2, TH, TPH, VGluT1, and VGluT2 were determined by one-step RT-PCR using rTth DNA polymerase (RT-PCR high Plus; TOYOBO, Osaka, Japan) using the same primer sets for cloning of each probe. Total RNA (1 μg) was submitted to RT at 60°C for 30 min, followed by PCR (40 cycles) at 94°C for 1 min and 60°C for 1.5 min. For GAD2, PCR (25 cycles) was performed at 94°C for 1 min and 65°C for 1.5 min. For VGluT1 and VGluT2, PCR (40 cycles) was performed at 94°C for 1 min and 65°C for 1.5 min. PCR products were separated by agarose gel electrophoresis and transferred to Hybond-N + nylon membrane (Amersham Pharmacia Biotech), followed by Southern hybridization using the corresponding probe for each marker.
Expression of APP770, APP751, and APP695 was determined by one-step RT-PCR described above using the following primers: a common sense primer, 5′-GAGGATGACTCTGATGTCTGGTG-3′; an APP770 antisense primer, 5′-GTTTATCAGGATCTTGGGGAAGAGG-3′; an APP751 antisense primer, 5′-TGCTGGCTGCTGTCGTGGGAGACAC-3′; an APP695 antisense primer, 5′-TGCTGTCGTGGGAACTCGGACCAC-3′; a glyceraldehyde-3-phosphate dehydrogenase (G3PDH) sense primer, 5′-TCCACCACCCTGTTGCTGTA-3′; and a G3PDH antisense primer, 5′-ACCACAGTCCATGCCATCAC-3′. PCR (25 cycles) was performed at 94°C for 1 min and either 60°C (for APP770, APP751, and G3PDH) or 65°C (for APP695) for 1.5 min. The PCR product (273 bp) amplified using the common sense primer and the APP695 antisense primer from mouse APP695 cDNA was subcloned into pCRII-TOPO as described above and used for Southern hybridization. To determine whether splice variants lacking exon 15 [leukocyte-derived (L)-APPs] exist, we also performed one-step RT-PCR described above, using primers 5′-CCGTGGAGCTCCTTCCCGTGAATG-3′ and 5′-CATATCCGTTCTGCTGCATCTTGG-3′. PCR (35 cycles) was performed at 94°C for 1 min and 60°C for 1.5 min.
To examine whether tau isoforms expressed in ES cell-derived neurons are of the CNS type or the PNS type, one-step RT-PCR was performed using a sense primer for amplifying between exons 4 and 9, 5′-GAAGAAGCAGGCATCGGAGACACC-3′; a sense primer for amplifying between exons 5 and 9, 5′-GCCAGCAAAGACAGGACAGGAAATG-3′; and a common antisense primer, 5′-TGTAGCCGCTTCGTTCTCCGGATT-3′. PCR (40 cycles) was performed at 94°C for 1 min and 60°C for 1.5 min.
Alternative splicing of tau involving exons 2 and 3 was examined by one-step RT-PCR using the following primers and PCR condition: a sense primer for the splice variant lacking both exons 2 and 3, 5′-GACCATGGCTTAAAAGTGGAAG-3′ (94°C for 1 min and 60°C for 1.5 min, 20 cycles); a sense primer for the splice variant lacking exon 3, 5′-ACTCCAACTGCTGAACTCGAAGAA-3′ (94°C for 1 min and 65°C for 1.5 min, 25 cycles); a sense primer for the splice variant containing both exons 2 and 3, 5′-ACTCCAACTGCTGATCACGTGACT-3′ (94°C for 1 min and 60°C for 1.5 min, 20 cycles); a common antisense primer, 5′-TGTAGCCGCTTCGTTCTCCGGATT-3′. The PCR product (301 bp) amplified using the sense primer for the splice variant lacking both exons 2 and 3 and the common antisense primer from mouse primary cultured cortical neurons was subcloned into pCRII-TOPO and used for Southern hybridization. We also performed the one-step RT-PCR, which amplifies between exons 1 and 4 to detect all three types of CNS-type isoforms caused by alternative splicing of exons 2 and 3 using primers 5′-CTTTGAACCAGTATGGCTGACCCT-3′ and 5′-GGTGTCTCCGATGCCTGCTTCTTC-3′. PCR (30 cycles) was performed at 94°C for 1 min and 65°C for 1.5 min.
Alternative splicing of tau involving exon 10 was examined by one-step RT-PCR using primers 5′-GTGCCCATGCCAGACCTAAAGAAT-3′ and 5′-CCTGGCTTGTGATGGATGTTCCCT-3′. PCR (25 cycles) was performed at 94°C for 1 min and 65°C for 1.5 min. The PCR product (164 bp) amplified using the above primers, which corresponds to the fragment that lacks exon 10, from mouse primary cultured cortical neurons was subcloned into pCRII-TOPO and used for Southern hybridization.
Primary culture of CNS-derived neurons. Primary cultured cortical neurons were prepared from embryonic day (E) 14 mouse embryo brain, as described previously (Sudo et al., 2000; Hashimoto et al., 2001a,b). On the seventh day in vitro (DIV), cells were used for extraction of total RNA for Northern blot and RT-PCR, fixed with 4% paraformaldehyde in PBS for immunostaining, or subjected to cell survival assay. Primary cultured hippocampal neurons were prepared from hippocampi of E17 mouse embryos with the same protocol as cortical neurons and subjected to cell survival assay. For this assay, CNS-derived neurons were seeded onto 24-well plates at a density of 1.25 × 10 5 cells/well. On 4DIV, medium was replaced with Neurobasal medium supplemented with B27 supplement. On 7DIV, CNS-derived neurons were cocultured with ES-derived neurons that had been cultured for 2–3 weeks on a poly-O/L-coated filter membrane of culture inserts (Becton-Dickinson; 1.6 × 10 6 pores/cm 2, pore size, 1 μm), cultured in medium conditioned from ES-derived neurons, or treated with Aβ1–42 (Peptide Institute, Osaka, Japan) for 3 d, followed by WST-8 assay (Hashimoto et al., 2000b) using Cell Counting Kit-8 (Wako Pure Chemicals, Tokyo, Japan).
Antibodies. Monoclonal anti-nestin (clone Rat401) antibody and anti-embryonic (E)-neuronal cell adhesion molecule (NCAM) (clone 5A5) antibody were from Developmental Studies Hybridoma Bank. Monoclonal antibodies, anti-MAP2 (clone HM-2), and Cy3-conjugated anti-GFAP (clone G-A-5) were purchased from Sigma. Polyclonal rabbit anti-NF-M (145 kDa) C-terminal antibody, rabbit anti-galactocerebroside, and monoclonal anti-O4 antibodies were obtained from Chemicon. Monoclonal anti-phosphorylated tau antibody (clone AT8) and polyclonal anti-phosphorylated tau antibody AP422 were obtained from Innogenetics (Gent, Belgium) and Wako Pure Chemicals, respectively. FITC-conjugated goat anti-mouse IgG, TexasRed-conjugated goat anti-mouse IgG, and Cy3-conjugated goat anti-mouse IgM antibodies were from Jackson Immunoresearch Laboratories (West Grove, PA). Rhodamine-conjugated porcine anti-rabbit IgG was from DAKO (Glostrup, Denmark).
Immunofluorescence analysis. Cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS, and blocked with 0.1% BSA in PBS. These cells were incubated with each primary antibody at room temperature for 1 hr, followed by incubation with corresponding secondary antibody at room temperature for 1 hr. Cells were observed with an LSM510 laser-scanning confocal microscope (Carl Zeiss, Oberkochen, Germany). For counting, nuclei of the cells were also stained with Hoechst 33258 (Sigma).
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay was performed using a DeadEnd fluorometric TUNEL system (Promega, Madison, WI). Briefly, cells were fixed with 4% paraformardehyde and permeabilized with 0.2% Triton X-100. Fragmented DNA was labeled with fluorescein-12-dUTP at 37°C for 1 hr. The reaction was terminated by the addition of 2× SSC, followed by washing twice with PBS. Total nuclei were labeled with Hoechst 33258. Both TUNEL-positive and Hoechst-positive nuclei were detected with a confocal microscope and counted.
Quantitation of Aβ by two-site ELISA. Culture media were collected every 3 d between days 7 and 28 after being plated on polyO/L-coated dishes in Neurobasal medium with B27 supplement. Two-site ELISA (BNT77/BA27 or BC05) for quantitation of secreted Aβ species ending at 40 or 42/43 residues was performed, as described previously (Yamatsuji et al., 1996; Tomita et al., 1997).
Results
Establishment of V642I–APP knock-in ES cells
For the purpose of producing neurons affected by a genetic mutation found in FAD patients, we took advantage of the Cre-loxP recombination system in combination with the conventional gene targeting method. A point mutation in a codon coding Val642 of APP to isoleucine was the first genetic alteration identified in FAD (Hardy, 1992). To introduce this point mutation of the APP gene in TT2 mouse ES cells, a gene-targeting vector was constructed based on the mutagenesis scheme depicted in Figure 1A. This vector is composed of three elements: (1) a 4.4 kb 5′ homology, which contains exon 17 with Val642 (GTC) to isoleucine (ATC) mutation; (2) a 3.6 kb floxP-TK-Neo cassette, which includes expression cassettes for herpes simplex virus TK and Neo (neomycin phosphotransferase) as negative and positive drug selection markers, respectively, flanked by two loxP sequences; and (3) a 3.6 kb 3′ homology. The separation between Val642 codon in exon 17 and the upstream loxP site was ∼1.0 kb in this targeting vector. Southern analysis screening (Fig. 1B) of the 144 G418-resistant clones incorporated with this mutation knock-in vector identified four homologous recombinant clones: R34, R37, R58, and R60. Subsequent sequencing of these clones, though, revealed that only R34 cointroduced the V642I mutation together with floxP-TK-Neo cassette in intron 17 in the expected manner. The Val642 codon remained unaltered in the other three clones, most likely because the 5′-side crossover had occurred in the 1.0-kb homology between the Val642 point mutation and the first loxP sequence, and not in the 5′ upstream region of the Val642 codon in the targeting vector. The R34 clone was further analyzed by PCR and sequencing, and the genetic integrity of the exon 17 region was confirmed (data not shown).
Figure 1.

Establishment of the mutant mouse ES cell clone with FAD-related V642I mutation in the APP gene. A, Two-step modification of the mouse APP gene. By homologous recombination, the targeting vector introduces V642I mutation in exon 17 and a TK-Neo cassette flanked by two loxP sequences (floxP-TK-Neo) in the downstream intron (Intermediate mutant). Transient expression of Cre recombinase excises the drug selection cassette in intron 17, leaving a single loxP sequence (Final mutant). Probes for Southern analysis and the strategy to identify homologous recombinants are shown. The restriction sites shown are BamHI(B), Hind III (H), and XhoI(X). B, Southern analysis to identify the Intermediate mutant by different probes. Genomic DNA samples prepared from the Intermediate mutant clone [Mut(I); R34 in the text] were digested by BamH I and Hind III and hybridized to each probe. The expected sizes of the fragments are shown in kilobase. WT, Wild-type control. C, Identification of the Final mutant. A 97 bp exogenous sequence composed of a loxP (triangle) and the accompanying restriction sites remain after the successful Cre-mediated recombination event. Thus, PCR reaction depicted in the scheme was used to identify the Final mutant clone [Mut(F); R34C(30)-9 in the text]. Size markers: 100 bp DNA ladder.
Insertion of the long selection marker cassette sequence in intron 17 may alter the expression of APP from the mutated allele. Therefore, drug selection cassettes flanked by two loxP sequences located in intron 17 were excised by transient expression of Cre recombinase. The Cre expression construct pIC-Cre (Gu et al., 1993) was introduced in R34 cells by electroporation, and a total of 128 clones were isolated without drug selection. Among these, three clones showed the PCR amplification pattern of floxP-excised allele (Fig. 1C), and in these clones, additional PCR, sequencing, and Southern analysis confirmed that the excision had occurred as expected (data not shown). To eliminate possible contamination of parental (nonexcised) ES cells, one of the excised clones, R34C(30)-9, was further recloned in the ES medium containing gancyclovir (2 μm). All the resultant clones were free of floxP-TK-Neo sequence when examined by PCR and nucleotide sequencing (data not shown), and one established clone, R34C(30)-9G, was submitted for the following analysis.
Differentiation of ES cells into postmitotic neurons
The differentiation protocol of ES cells into neurons was performed as described in Figure 2A and Materials and Methods. To accelerate the differentiation process, EBs were stimulated with 5 μm RA during the last 24 hr of 5 d EB formation. When these RA-stimulated EBs were subsequently attached to fibronectin-coated tissue culture surface and cultured in the serum-free medium, expression of the undifferentiated ES cell marker Oct3 mRNA unidirectionally decreased, whereas expression of the neural stem cell marker nestin, which was originally negative, began to be significantly detected on day 7 (Fig. 2B). The nestin-positive cells grew out from the EBs as determined by immunostaining (Fig. 3A). The small population of cells had already expressed the mature neuronal marker MAP2 and NF-M at this time point (Fig. 3B). The nestin-positive cells were concentrated during the next 2 d of culture on fibronectin-coated dishes in the presence of bFGF (Fig. 3C). In addition, mature neurons positive with MAP2 and NF-M increased during this period (Fig. 3D). This finding was consistent with the Northern blot data using these markers (Fig. 2B). On day 9, these cells were detached and seeded on polyO/L-coated dishes and cultured in the presence of bFGF.
Figure 3.
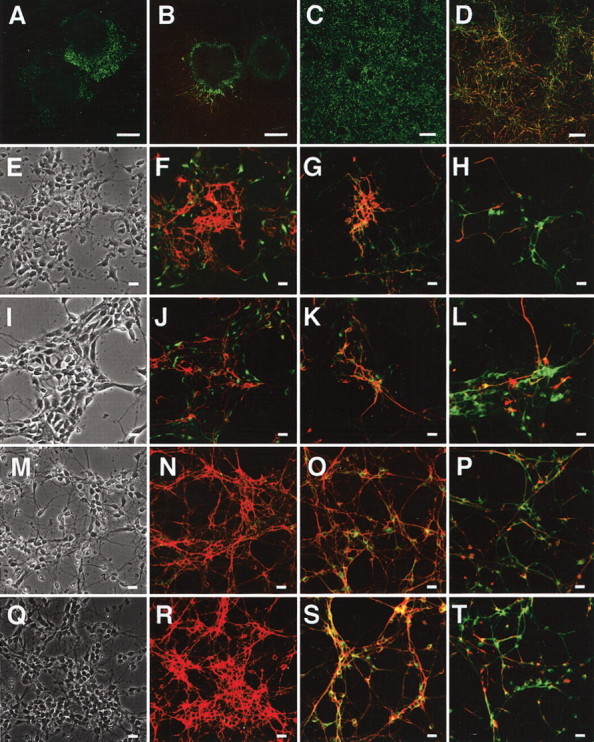
Analysis of morphology and marker expression in ES-derived cells. A, B, Day 7 of wild-type EBs attached on fibronectin-coated dishes and stained with anti-nestin antibody (A, green) or both anti-MAP2 (green) and anti-NF-M (red) antibodies (B). C,D, Day 9 of cells derived from wtES cells cultured on fibronectin-coated dishes and stained with anti-nestin antibody (C, green) or both anti-MAP2 (green) and anti-NF-M (red) antibodies (D). E–H, Day 11 (postpolyO/L day 2) of wtES-derived cells stained with anti-nestin (green) plus anti-E-NCAM (red) antibodies (F), anti-MAP2 (green) plusanti-E-NCAM antibodies (red) (G), or anti-MAP2 (green) plus anti-NF-M (red) antibodies (H). E is a phase-contrast image of F. Representative fields are indicated. I–L, Day 11 of knock-in ES-derived cells stained with anti-nestin (green) plus anti-E-NCAM (red) antibodies (J), anti-MAP2 (green) plus anti-E-NCAM (red) antibodies (K), or anti-MAP2 (green) plus anti-NF-M (red) antibodies (L). I is a phase-contrast image of J. M–P, Day 13 (post-polyO/L day 4) of wtES-derived cells stained with anti-nestin (green) plus anti-E-NCAM (red) antibodies (N), anti-MAP2 (green) plus anti-E-NCAM (red) antibodies (O), or anti-MAP2 (green) plus anti-NF-M (red) antibodies (P). M is a phase-contrast image of N. Q–T, Day 13 of knock-in ES-derived cells stained with anti-nestin (green) plus anti-E-NCAM (red) antibodies (R), anti-MAP2 (green) plus anti-E-NCAM (red) antibodies (S), or anti-MAP2 (green) plus anti-NF-M (red) antibodies (T). Q is a phase-contrast image of R. Scale bars: A, B, 200 μm; C, D, 100 μm; E–T, 25 μm.
Two days after the onset of culturing on polyO/L-coated dishes (day 11), ∼70% of the cells differentiated from both wild-type ES (wtES) and V642I–APP knock-in ES cells were MAP2 positive, whereas around 30% of the cells were nestin positive (Table 1). Approximately 40% of the cells expressed polysialylated NCAM (PSA-NCAM or E-NCAM) (Table 1). E-NCAM-positive cells did not express nestin but simultaneously expressed MAP2 as determined by immunostaining (Fig. 3F,G), as was also the case with the cells derived from V642I–APP knock-in ES cells (Fig. 3J,K). Expression of nestin began to decrease after plating on the poly-O/L-coated surface, and AraC treatment abolished the expression of nestin and instead augmented that of MAP2 (Fig. 2B). Counting the immunostained cells derived from both wtES and V642I–APP knock-in ES cells on day 13 revealed ∼90% of cells were both MAP2 and E-NCAM positive (Fig. 3O,S; Table 1). In clear contrast, <5% of cells were nestin positive, and nestin-positive cells did not overlap with E-NCAM-positive cells (Fig. 3N,R; Table 1). These results indicate that proliferating nestin-positive neural stem cells were almost eliminated by treatment with AraC. Expression of NF-M was observed in some, but not all, MAP2-positive cells (Fig. 3H,L,P,T). Neither GFAP-positive cells nor O4/galactocerebroside-positive cells were observed by day 13 (data not shown). The expression of MAP2 and the synapse markers Rab3A and synaptophysin continued to increase and stayed at levels as high as in the adult brain during the next 2 weeks of culture (Fig. 2B). In contrast, expression of tau transiently increased after AraC treatment and drastically decreased over 3 weeks of culturing on polyO/L-coated dishes (Fig. 2B). Two weeks after the onset of culturing on polyO/L-coated dishes (day 23), expressions of synaptic markers including Rab3A, synaptophysin, and synapsin I as well as MAP2 and tau in the cells derived from V642I–APP knock-in ES cells were similar to those in wtES-derived cells (Fig. 2C). Three weeks after the onset of culturing on polyO/L-coated dishes (day 30) in the Neurobasal medium with B27 supplement, relatively short MAP2-positive processes and long NF-M-positive fibers were observed in wtES-derived cells (Fig. 4A,B). Similar results were obtained from the cells derived from V642I-APP knock-in ES cells (Fig. 4C,D). At this time point, nestin-positive cells were still observed, and some of them also expressed GFAP (Fig. 4E,F; data not shown). Although nestin was not detectable by Northern blot analysis, transcript of GFAP was weakly detected after being plated on polyO/L-coated dishes and increased in 3 weeks of culture (Fig. 2B). The expression of MAG, a marker for oligodendrocytes, was not detected throughout the process of this differentiation protocol (Fig. 2B). Likewise, cells positive with O4 and galactocerebroside were not induced by this protocol (data not shown). The expression of BACE1 was constant during the culture on poly-O/L for 3 weeks, and then slightly decreased (Fig. 2E). These results were in agreement with the notion that the V642I mutation does not affect the neuronal differentiation of ES cells.
Table 1.
The efficiency of the differentiation of ES cells into neuronal lineages
|
|
Day 2 |
Day 4 |
||||
|---|---|---|---|---|---|---|
| Markers
|
WT (%)
|
V642I-APP KI (%)
|
WT (%)
|
V642I-APP KI (%)
|
||
| Nestin | 29.9 ± 3.7 (1710) | 37.1 ± 11.3 (1197) | 3.4 ± 1.0 (1534) | 4.8 ± 0.3 (1377) | ||
| E-NCAM | 39.5 ± 13.2 (1399) | 39.8 ± 10.0 (1061) | 91.2 ± 0.5 (1118) | 90.4 ± 1.7 (1215) | ||
| MAP2
|
70.0 ± 2.4 (1417)
|
67.1 ± 2.9 (1141)
|
87.7 ± 0.4 (1571)
|
88.6 ± 1.5 (1669)
|
||
Wild-type (WT) and V642I-APP knock-in (V642I-APP KI) ES cells were differentiated in accordance with the protocol described in Materials and Methods. Cells were counted on day 2 and day 4 after being plated on polyO/L-coated dishes. The percentages of neural precursors (nestin positive) and postmitotic neurons (E-NCAM and MAP2 positive) in total cells (Hoechst 33258 positive) were estimated. Approximately 1100-1700 Hoechst-positive nuclei in several randomly chosen fields are counted in each experiment, and the averages of total cell numbers are shown in parentheses. Values are mean ± SE of three independent differentiation experiments.
Figure 4.

Analysis of morphology and marker expression in ES-derived cells cultured on polyO/L-coated dishes for 3 weeks. A, B, Cells derived from wtES cells cultured on polyO/L-coated dishes for 3 weeks and stained with anti-MAP2 (green) and anti-NF-M (red) antibodies. C, D, Cells derived from knock-in ES cells cultured on polyO/L-coated dishes for 3 weeks and stained with anti-MAP2 (green) and anti-NF-M (red) antibodies. E, F, Cells derived from wtES cells cultured on polyO/L-coated dishes for 3 weeks and stained with anti-nestin (green) and Cy3-conjugated anti-GFAP (red) antibodies. B, D, and F are phase-contrast images of A, C, and E, respectively. Scale bar, 100 μm. Each of the results was reproduced at least three times.
Transmitter phenotypes of neurons derived from ES cells
To determine what kind of neurons are differentiated from ES cells, we investigated the transmitter phenotypes using RT-PCR methods. GAD1, GAD2, and VGluT2 were detected 2 weeks after the onset of culturing on polyO/L-coated dishes in the neurons derived from ES cells, suggesting the existence of GABAergic and glutamatergic neurons (Fig. 2D). In contrast, little ChAT, TH, or TPH was expressed in these neurons (Fig. 2D). There was no obvious difference between neurons from wtES and V642I–APP knock-in ES cells (Fig. 2D). Although VGluT1 was detected in primary cultured cortical neurons as well as adult whole brain and spinal cord, we did not observe its expression during 4 weeks of culturing on polyO/L in neurons derived from ES cells (Fig. 2D; data not shown).
Expression of APP isoforms
We next focused on the expression of APP and its family. As shown in Figure 5A, we observed a single band of APP, ∼3.5 kb, in the total RNA from differentiating cells of all time points, suggesting that nonamyloidogenic forms such as APRP 563 (de Sauvage and Octave, 1989) and APRP 365 (Jacobsen et al., 1991) are not expressed in these cells. The total amount of the transcripts of the APP gene drastically increased during the differentiation process. This was also the case with expression of APLP1, whereas expression of APLP2 only slightly increased during the differentiation process (Fig. 5A). The level of expression of each APP family member was indistinguishable between wtES-derived and V642I–APP knock-in ES-derived neurons (Fig. 5B).
Figure 5.

Expression of APP isoforms. A, Northern blot analysis for APP, APLP1, and APLP2. Small numbers represent the days after being plated on polyO/L-coated dishes. B, Northern blot analysis for APP, APLP1, and APLP2 in neurons derived from wild-type or knock-in ES cells after 23 d of differentiation. C, RT-PCR analysis of the splicing at exon 15 in differentiating cells derived from wild-type or knock-in ES cells after 4, 9, and 23 d of differentiation. M represents markers in all figures. D, RT-PCR analysis of the expression of APP770, APP751, and APP695. PCR products were submitted to Southern blot analysis using a 273 bp fragment of APP. E, RT-PCR analysis of the expression of APP770, APP751, and APP695 in differentiating cells derived from wild-type or knock-in ES cells after 4, 9, and 23 d of differentiation. PCR products were submitted to Southern blot analysis using a 273 bp fragment of APP. Each of the results was reproduced at least three times.
We then examined one of the splice sites of the APP gene involving exon 15. We performed RT-PCR using primers flanking the sequence of exons 14–18; the corresponding PCR products were 465 and 411 bp for cDNAs with and without exon 15, respectively, in accordance with the method described by König et al. (1992), with slight modification. As shown in Figure 5C, we were able to detect only 465 bp species in cells derived from ES cells and in ES cells themselves, regardless of genotype, as well as primary cultured cortical neurons and adult whole brain. The result is consistent with the fact that L-APPs that lack exon 15 are not expressed in neurons (Sandbrink et al., 1994).
To determine which isoform of APP is expressed in differentiating cells, we used the RT-PCR method using specific primers recognizing either a sequence in an exon (common sense primer recognizing exon 5 and antisense primer for APP770 recognizing exon 8) or the junction of exons (antisense primers for APP 751 recognizing the junction of exons 7 and 9 and APP695 recognizing the junction of exons 6 and 9). Primer sets for APP770, APP751, and APP695 can amplify 484 bp, 448 bp, and 273 bp of fragments, respectively, and each primer set could amplify specifically the corresponding fragment from adult mouse brain total RNA (Fig. 5D). As shown in Figure 5D, expression of both APP770 and APP751 increased transiently as the nestin-positive cells were concentrated. However, after treatment with AraC, these transcripts decreased. In contrast, expression of APP695 increased accordingly with expression of the markers for mature neurons (Fig. 5D). Our primer set for APP770 was also expected to amplify APP714, one of the other isoforms with exon 8. The predicted size for PCR product of APP714 was estimated to be 316 bp, and we also detected it by RT-PCR, followed by Southern hybridization in total RNA from the differentiating cells, especially on day 13, as well as from mouse whole brain, although the level of its expression was much lower than APP770 (Fig. 5D). As far as we have investigated, there was no difference between neurons derived from wtES and V642I–APP knock-in ES cells (Fig. 5E). These results add additional lines of evidence that ES cell-derived neurons have the same characteristics as neurons in vivo. In addition, the V642I mutation of APP affects neither the expression level of APP family nor alternative splicing of APP.
Expression of tau isoforms
CNS neurons express six types of the tau isoforms, all of which lack exons 4a, 6, and 8 (Lee et al., 2001), whereas the isoforms of tau expressed in PNS neurons have exon 4a and occasionally exon 6 (Couchie et al., 1992; Goedert et al., 1992). To clarify the type of the neurons derived from ES cells by our protocol, we performed RT-PCR, which amplifies either between exons 5 and 9 (primer set 1) or between exons 4 and 9 (primer set 2). Primer set 1 amplifies a 409 bp fragment when the tau is PNS type (exons 6 and 8 are present and absent, respectively), whereas the same primer set amplifies a 211 bp fragment when the tau is of the CNS type (both exons 6 and 8 are absent). As shown in Figure 6A, we detected only the 211 bp fragment in total RNA of ES cell-derived neurons, as well as mouse primary cultured cortical neurons and the adult whole brain. This result indicates that the tau isoforms expressed in ES-derived neurons possess neither exon 6 nor exon 8. We also performed similar experiments using primer set 2. This primer set amplifies a 994 bp fragment when the exon 4a exists, but both exons 6 and 8 are absent; and it amplifies a 283 bp fragment when neither exons 4a, 6, nor 8 exists. In ES cell-derived neurons, only the 283 bp fragment was amplified (Fig. 6B), indicating the absence not only of exons 6 and 8 but also of exon 4a in the tau isoforms expressed in the ES cell-derived neurons. Taken together, these data indicate that neurons derived from ES cells by the present protocol were of the CNS type.
Figure 6.

Expression of tau isoforms. A, RT-PCR analysis of the splicing at exons 6 and 8 using the primer set 1 in neurons derived from wild-type or knock-in ES cells after 23 d of differentiation. B, RT-PCR analysis of the splicing at exons 4a, 6, and 8 using the primer set 2 in neurons derived from wild-type or knock-in ES cells after 23 d of differentiation. C, RT-PCR analysis of the splicing at exons 2 and 3 in neurons derived from wild-type (WT) or knock-in (KI) ES cells during the period from 1 to 5 weeks after plating on polyO/L dishes. -/-,2/- or -/3, and 2/3 represent tau isoforms lacking both exons 2 and 3, containing either exon 2 or exon 3, and containing both exons 2 and 3, respectively. D, RT-PCR analysis of the splicing at exons 2 and 3 in neurons derived from wild-type (WT) or knock-in (KI) ES cells during the period from 1 to 5 weeks after plating on polyO/L dishes. -/-,2/-, and 2/3 represent tau isoforms lacking both exons 2 and 3, containing exon 2 but not exon 3, and containing both exons 2 and 3, respectively. PCR products were submitted to Southern blot analysis using 301 bp fragment of tau. E, RT-PCR analysis of the splicing at exon 10 in neurons derived from wild-type (WT) or knock-in (KI) ES cells during the period from 1 to 5 weeks after plating on polyO/L dishes. +10 and -10, tau isoforms containing and lacking exon 10, respectively. PCR products were submitted to Southern blot analysis using 164 bp fragment of tau. Each of the results was reproduced at least three times.
We next analyzed the CNS-type tau isoforms expressed in neurons derived from ES cells. CNS-type tau consists of six isoforms caused by alternative splicing at exons 2, 3, and 10, although the smallest isoform lacking exons 2, 3, and 10 is predominantly expressed in fetal brain. To examine whether exons 2 and 3 exist in tau expressed in neurons derived from ES cells, we primarily performed RT-PCR using primers flanking the sequence of exons 1–4; the corresponding PCR products were 136, 225, and 312 bp for cDNAs without exon 2 or exon 3, with either only exon 2 or only exon 3, and with both exons 2 and 3, respectively. We detected only the smallest fragment in neurons derived from ES cells in 2 weeks of culturing on polyO/L-coated dishes like primary cultured cortical neurons derived from E14 embryos (Fig. 6C). In contrast, similar to adult whole brain, we detected all three fragments in ES-derived neurons after 3 weeks of culturing on poly O/L-coated dishes. The level of the expression of the smallest fragment was higher than those of others (Fig. 6C). To examine the expression of each tau isoform in detail, we further performed RT-PCR using primer sets recognizing the junction of exons 1 and 4 corresponding to the isoform(s) lacking both exons 2 and 3 (Fig. 6D, -/-), the junction of exons 2 and 4 corresponding to the isoform(s) containing exon 2 but not exon 3 (Fig. 6D, 2/-), or the junction of exons 2 and 3 corresponding to the isoform(s) containing both exons 2 and 3 (Fig. 6D, 2/3). Predicted sizes for their products are 301 bp, 301 bp, or 388 bp, respectively. Primary cultured cortical neurons derived from E14 embryos dominantly expressed the isoform(s) lacking both exons 2 and 3, whereas adult whole brain expressed all three types of isoform (Fig. 6D). Neurons derived from ES cells originally expressed only the isoform(s) lacking exons 2 and 3 (Fig. 6D). However, its expression gradually decreased (Fig. 6D). Instead, they began to express the other two types of isoform, which correspond to the adult forms, 2 weeks after culturing on polyO/L-coated dishes (Fig. 6D). We could not detect any fragment at all by the same RT-PCR using a primer set that recognized the junction of exons 1 and 3, confirming the absence of an isoform(s) containing exon 3 but not exon 2 (data not shown).
As for exon 10, we performed RT-PCR using primers flanking the sequence of exons 9–12; the corresponding PCR products are expected to be 257 bp and 164 bp for cDNA with and without exon 10, respectively. In primary cultured cortical neurons, the species lacking exon 10 was predominantly amplified, whereas the species containing exon 10, which reflects the adult forms, was predominantly amplified in adult whole brain (Fig. 6E). Consistent with the results of exons 2 and 3, neurons derived from ES cells on 1 week after plating on polyO/L dishes predominantly expressed the isoform(s) lacking exon 10 (Fig. 6E). Then, expression of the isoform(s) lacking exon 10 gradually decreased, and, instead, the isoform(s) containing exon 10 increased during the 5-week culture on polyO/L-coated dishes (Fig. 6E). Taken together, tau isoforms expressed in neurons derived from ES cells alter from the fetal to the adult type during the period from 2 to 4 weeks after the last plating. Also, a similar result was obtained from both wild-type and V642I–APP knock-in.
Cell death in neurons derived from ES cells
Neuronal cell death is one of the pathological characteristics of AD. To examine whether we can observe the effect of the V642I mutation of APP on neuronal cell death, we performed TUNEL assay using neurons derived from ES cells. In our protocol, a considerable number of ES-derived cells died because of treatment with AraC, and some of them did not detach from the dishes. In fact, many shrunken and sometimes fragmented nuclei of dead cells were labeled with both fluorescein-12-dUTP and Hoechst 33258 in both cells derived from wild-type and V642I–APP knock-in ES cells (Fig. 7, A and B, for wild-type; data not shown for knock-in). Therefore, we investigated the time course of the ratio of TUNEL-positive nuclei versus Hoechst-positive ones to detect newly occurred cell death caused by the mutation. If the mutation enhances cell death, the increment of TUNEL-positive/Hoechst-positive nuclei in V642I–APP knock-in ES cells would be larger than that in wild-type cells. As shown in Figure 7C, the ratios of TUNEL-positive/Hoechst-positive nuclei in the neurons derived from V642I–APP knock-in ES cells were similar at each time point to those in wild-type neurons, suggesting that the V642I mutation of APP does not enhance apoptotic neuronal death as long as the 3 weeks of the culture on polyO/L-coated dishes.
Figure 7.
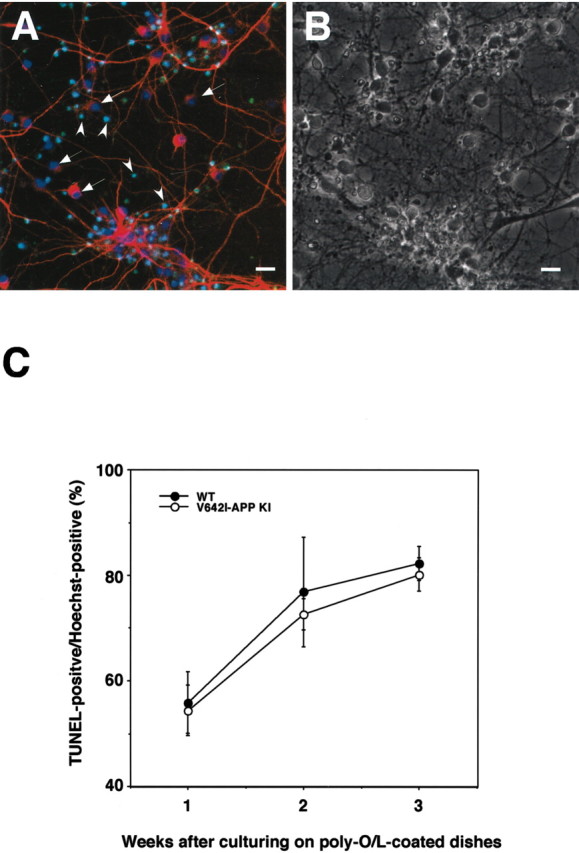
TUNEL assay of cells derived from ES cells. A, wtES-derived cells cultured on polyO/L-coated dishes for 3 weeks, and labeled with Hoechst 33258 (blue), fluorescein-12-dUTP (green), and anti-MAP2 antibody (red). Relatively large nuclei of MAP2-expressing living cells were labeled with Hoechst 33258 only (arrow), whereas the other small nuclei were both Hoechst and TUNEL positive (arrowheads). B is a phase-contrast image of A. Scale bar, 25 μm. C, Time course of TUNEL-positive/Hoechst-positive nuclei. Cells derived from either wtES (closed circles) or V642I–APP knock-in ES (open circles) cells were cultured for either 1, 2, or 3 weeks on polyO/L-coated dishes and were subjected to TUNEL assay, followed by staining of whole nuclei with Hoechst 33258. Each signal was detected with a confocal microscope, and the percentages of TUNEL-positive nuclei in Hoechst-positive (total) nuclei (at least 1000 nuclei were counted) were estimated. Values are means ± SE of three independent differentiation experiments.
V642I–APP knock-in ES cell-derived neurons increase the secretion of Aβ42
We next focused on the production of Aβ, a major component of senile plaques, from ES-derived neurons. We collected the media of culturing neurons derived from ES cells and measured the concentration of both Aβ40 and Aβ42 during the period from post-polyO/L days 7–28. Whereas the Aβ40 secretion was quantitatively similar from wtES- or V642I–APP knock-in ES-derived neurons, the amounts of Aβ42 secretion were specifically augmented during this period (Fig. 8A). As a result, the ratio of secreted Aβ42 versus total Aβ (Aβ40+Aβ42) in the culture medium of V642I–APP knock-in ES-derived neurons reproducibly and constantly increased to the ∼1.5-fold level of that in the cultured medium of wtES-derived neurons (Fig. 8B). Although the observed initial increase and late-phase decrease in the secreted Aβ amounts might be attributable to increased expression of APP (Fig. 5A) and decreased expression of BACE1 (Fig. 2E), respectively, the real mechanisms must be elucidated in detail in the next study. These data demonstrate that V642I–APP knock-in ES cell-derived neurons exhibit one of the central phenotypes of AD.
Figure 8.
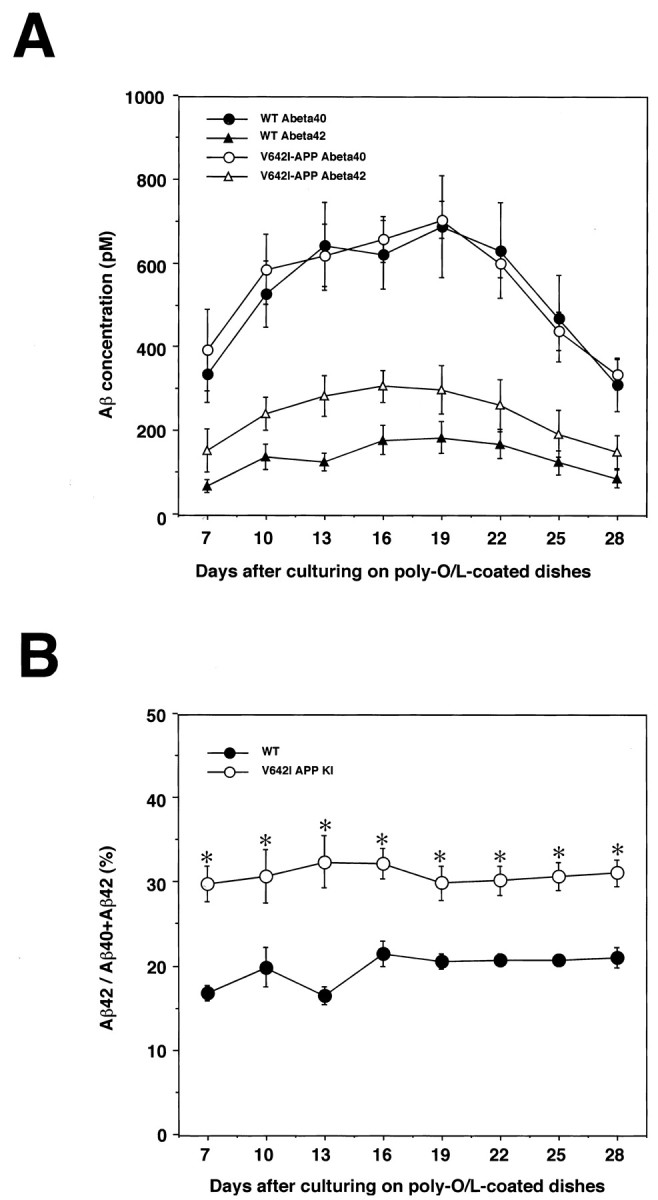
Secretion of Aβ in the culture medium of ES-derived neurons. wtES and knock-in ES cells were similarly differentiated by our protocol, and the cultured media were collected every 3 d during the period between 7 and 28 d after being plated on polyO/L-coated dishes. The concentrations of Aβ40 (circles) and Aβ42 (triangles) secreted from wtES-derived neurons (closed symbols) or knock-in ES-derived neurons (open symbols) were determined by two-site ELISA (A). Values are means ± SE of six of seven independent series of similar experiments. The result from one series of the experiment, in which each obtained value deviated remarkably from the values of the other six experiments, was excluded because values remeasured in this experiment were not reproducible. Aβ42/total Aβ (Aβ40+Aβ42) ratios of wtES-derived neurons (closed circles) or knock-in ES-derived neurons (open circles) were calculated from the Aβ amounts (B). Statistical analysis was performed with one-way ANOVA, followed by a post hoc test, in which p < 0.05 was assessed as significant. Asterisks indicate a significant difference between wtES- and knock-in ES-derived neurons.
It should be noted here that we were unable to observe massive neuronal death induced by the V642I mutation of APP and by increased secretion of Aβ42 in neurons derived from V642I–APP knock-in ES cells (Fig. 7). However, we cannot rule out the possibility that our ES-derived neurons are not a vulnerable population in the AD brain, and, therefore, that the increased Aβ42 did not enhance cell death in our system. To examine whether the increased secretion of Aβ42 from V642I–APP knock-in neurons enhances cell death in the CNS-derived vulnerable population, we cocultured ES-derived neurons with primary cultured hippocampal neurons obtained from E17 mouse embryos. We considered that the hippocampal neurons from E17 mouse embryos are, so far, the best model for the vulnerable population. Primary cultured cortical neurons derived from E14 mouse embryos were also applied to confirm the results with the hippocampal neurons. For this experiment, ES cells were differentiated by our protocol, and on day 9, they were seeded onto a filter membrane of culture inserts (1.6 × 106 pores/cm2; pore size, 1 μm), followed by final neuronal differentiation as described in Materials and Methods. They were cultured for 2–3 weeks on the filter membrane until the secretion of Aβ reached the maximum (Fig. 8A). Three days after the last medium change, the ES-derived neurons on the filter membrane were transferred to the 24-well plates in which the 7DIV of CNS-derived neurons were cultured, and then they were cocultured for 3 d, followed by determination of viability of the CNS-derived neurons by WST-8 assay. As shown in Figure 9A, the viability of both hippocampal and cortical neurons was not influenced by coculturing with neurons derived from either wild-type cells (closed columns) or V642I–APP knock-in ES cells (open columns), whereas 25 μm synthetic Aβ1–42 significantly reduced the viability of both hippocampal and cortical neurons through the filter membrane of the culture insert (Fig. 9C,D). These results suggest that although Aβ secretion was increased from V642I–APP knock-in ES-derived neurons, the level of the increment was below the threshold with which we can observe neuronal cell death by Aβ. We also examined whether the viability of CNS-derived neurons is influenced by culturing in medium conditioned from either wild-type or V642I-APP knock-in ES-derived neurons. No difference in cell viability was detected (Fig. 9B). As shown in Figure 8A, the maximal concentration of secreted total Aβ was ∼1 nm. The same concentration of synthetic Aβ1–42 was not neurotoxic (Fig. 9C,D); therefore, it is possible that the concentration of Aβ secreted from V642I–APP knock-in neurons is too low to induce neuronal cell death of CNS-derived neurons acutely and directly.
Figure 9.

Effect of the increased secretion of Aβ42 from neurons derived from V642I–APP knock-in ES cells on primary cultured CNS-derived neurons. A, Cocultures of neurons derived from ES cells with primary cultured CNS neurons. The 7DIV of hippocampal neurons derived from E17 mouse embryos and cortical neurons derived from E14 mouse embryos were cocultured with either wtES-derived neurons (closed columns) or V642I–APP knock-in ES-derived neurons (open columns) on filter membranes of culture inserts for 3 d, followed by determination of their viability with WST-8 assay. Cell viability was estimated as percentage of control (cultured with culture insert in the absence of ES-derived neurons). Values were means ± SD of three independent experiments. B, Culture of CNS neurons in conditioned medium from ES-derived neurons. The 7DIV of hippocampal neurons derived from E17 mouse embryos and cortical neurons derived from E14 mouse embryos were cultured in medium conditioned from either wtES-derived neurons (closed columns) or V642I–APP knock-in ES-derived neurons (open columns) for 3 d, followed by determination of their viability with WST-8 assay. Cell viability was estimated as the percentage of control (cultured in fresh medium). Values were means ± SD of five and three independent experiments for hippocampal and cortical neurons, respectively. C, D, The effect of synthetic Aβ 1–42 on primary cultured CNS-derived neurons. The 7DIV of hippocampal (C) and cortical (D) neurons from E17 and E14 mouse embryos, respectively, were treated with (open columns) or without (closed columns) Aβ1–42 in the absence (-) or presence (+) of culture insert for 3 d, followed by determination of their viability with WST-8 assay. Cell viability was estimated as the percentage of control (without Aβ). Values were means±SD of three determinations. Asterisks indicate a significant difference between control and Aβ treatment, as determined by Student's t test (p < 0.01).
Phosphorylation of tau in ES cell-derived neurons
To test whether the V642I–APP knock-in ES-derived neurons are phosphorylated at the residues relevant to NFT, we stained the cells derived from ES cells with anti-phosphorylated tau antibody AT8 3 weeks after being plated on polyO/L-coated dishes. AT8 significantly stained both wtES- and V642I–APP knock-in ES-derived neurons 3 weeks after being plated on polyO/L-coated dishes (Fig. 10A,B), suggesting that tau in both wtES- and V642I–APP knock-in ES-derived neurons at this time point was similarly phosphorylated at the residues recognized by AT8. This was also the case with 7DIV primary cultured cortical neurons obtained from E14 mouse embryo brain, which were stained by AT8 (Fig. 10C).
Figure 10.
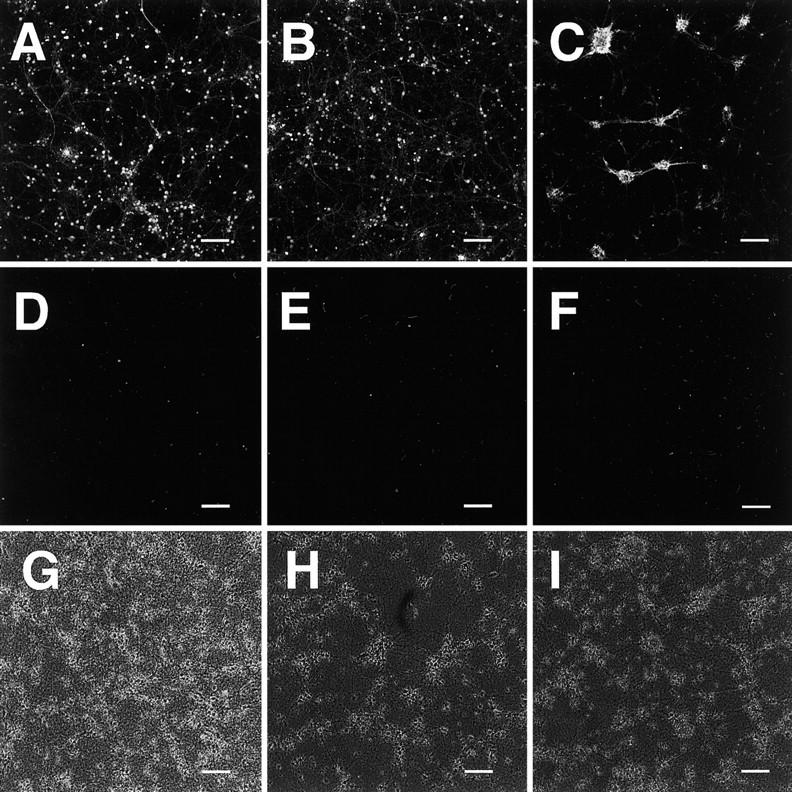
Phosphorylation of tau in neurons derived from ES cells. Cells derived from wtES (A, D, G) or knock-in ES (B, E, F, H, I) cells cultured on polyO/L-coated dishes for either 3 weeks (A, B) or 6 weeks (D–I), or 7DIV primary cultured cortical neurons (C) were stained with AT8 (A–E) or AP422 (F). G, H, and I are phase-contrast images of D, E, and F, respectively. Scale bars, 100μm. Each of the results was reproduced at least three times.
In contrast, neither wtES- nor V642I–APP knock-in ES-derived neurons 6 weeks after being plated on polyO/L-coated dishes were stained by AT8 (Fig. 10D,E). No neurons were stained by anti-pSer-422 antibody AP422, which specifically recognizes Ser422 phosphorylated tau and thereby NFT-specific phosphorylated tau (Fig. 10F for V642I–APP knock-in ES-derived neurons; data not shown for wtES-derived neurons).
Discussion
Using two-step genetic engineering, we have herein established mouse ES cells with the endogenous APP gene harboring the FAD-causative V642I mutation, termed V642I–APP knock-in ES cells. We have also established a protocol for rapidly and efficiently differentiating ES cells into the CNS-type postmitotic neurons by modifying the method developed by Okabe et al. (1996). The use of RA is based on the method of Bain et al. (1995). This protocol yielded >90% of neurons within 2 weeks without using drug-resistant genes (Li et al., 1998), the immunopanning technique (Mujtaba et al., 1999), or fluorescence-activated cell sorting (FACS) (Keyoung et al., 2001). By combining the V642I–APP knock-in ES cells with our protocol for the neuronal differentiation of ES cells, we tried to construct a novel in vitro neuronal model with the AD genotype. As a result, neurons differentiated from V642I–APP knock-in ES cells were associated with a significantly increased ratio of Aβ42 versus total Aβ secreted to the cultured medium. These data demonstrate that postmitotic neurons differentiated from V642I–APP knock-in ES cells behave as the model of live neurons harboring at least one of the central AD phenotypes. However, this mutation, although causative for AD, occurs in a small number of FAD cases, and it is not clear how relevant or important having a cell line with this mutation will be. It should be extended to other FAD genes, including PS1 and PS2.
NFT relevant hyperphosphorylation of tau was not detected in V642I–APP knock-in ES-derived neurons that overproduced Aβ42 at the same time. This suggests that Aβ42 overproduction is the direct output of the FAD-linked V642I mutation. Whereas phosphorylated tau recognized by AT8 was observed in both wtES- and V642I–APP knock-in ES-derived neurons at 3 weeks after the onset of terminal differentiation (the last plating and AraC treatment), this phenomenon was attributable to the facts that (1) ES-derived neurons express the fetal type of tau at this stage (this study) and (2) the fetal type tau is phosphorylated at the AT8-recognizable residues (Morishima-Kawashima et al., 1995). In fact, little NFT-specific phosphorylation of tau was observed in V642I–APP knock-in ES-derived neurons at 6 weeks after the onset of terminal differentiation, demonstrating that NFT-relevant hyperphosphorylation of tau was not detected in V642I–APP knock-in ES-derived neurons overproducing Aβ42. It has remained unknown whether NFT or NFT-relevant hyperphosphorylation of tau is a direct output of AD-causative mutations. Whereas neither NFT nor NFT-relevant hyperphosphorylation of tau has been detected in AD mutant gene-overexpressing mice (Irizarry et al., 1997a,b), there is a possibility that the hyperphosphorylated tau caused by AD mutant genes is secondarily suppressed in vivo or that hyperphosphorylation of tau by AD mutant requires a longer period than that required for Aβ42 overproduction.
Massive cell death in V642I–APP knock-in ES-derived cells was not observed during the period of 3 week culture on polyO/L-coated dishes. A potential reason is that neuronal death may not be a phenotype induced by the V642I mutation in young neurons or in pure neuronal cultures. Because our present protocol is with the chemical medium without serum, it is usually difficult to maintain the culture of the differentiated neurons for >2 months. A trial of their long-term culture in the medium with serum is now in progress in our laboratory. Another possibility is that a lack of extensive death in V642I–APP knock-in neurons is attributable to a lack of the certain differentiated subpopulation sensitive to AD insults, such as the entorhinal cortex, the hippocampus, and the cholinergic system of the basal forebrain. It has been speculated that these neurons are afflicted in AD brains and are vulnerable to AD insults, but little is known yet. The neurons differentiated by our present protocol contain little of the cholinergic-type neurons. At the present, there is no efficient protocol to differentiate ES cells into cholinergic neurons (Okabe et al., 1996). In theory, ES cells, which are totipotent, could be differentiated into each subpopulation of neurons with specific functionality. In fact, efficient protocols to differentiate ES cells into dopaminergic neurons (Kawasaki et al., 2000; Lee et al., 2000) and spinal motor neurons (Wichterle et al., 2002) were provided. Therefore, the combination of FAD gene–knock-in ES cells with protocols efficiently differentiating them into various types of neurons, including basal forebrain cholinergic neurons as well as neurons of the entorhinal cortex and the hippocampus, would allow us to investigate the AD sensitivity of neuronal subpopulations. It should be noted that the cholinergic deficit occurs late in the AD process (Braak and Braak, 1991).
We observed that Aβ42 overproduction in neurons derived from V642I-APP knock-in ES cells, and we examined whether the increased secretion of Aβ42 causes cell death of neurons belonging to a vulnerable population in AD by coculturing primary hippocampal neurons with neurons derived from V642I–APP knock-in ES cells as well as from wild-type ES cells. We also examined the effect of Aβ42 overproduction by culturing hippcampal neurons in conditioned medium from neurons derived from V642I-APP knock-in ES cells. Although we were not able to detect cell death induced by Aβ overproduction in V642I–APP knock-in ES cells, this suggests that the degree of Aβ overproduction would be below the thresholds with which we can detect its biological effects.
In summary, there are multiple advantages in the strategy for the creation of AD neurons by combining AD gene–knock-in ES cells created with two-step modification of the endogenous mouse APP gene with a protocol for efficiently differentiating ES cells into postmitotic neurons. First, we can obtain postmitotic neurons carrying the AD genotype without establishing animals derived from the ES cells. Postmitotic neurons are, therefore, obtainable even when the recombinant ES cell-derived mice cannot be obtained and are obtainable more rapidly than by obtaining primary cultured neurons from gene–knock-in ES cell-derived mice, when such mice are created. Hence, the effect of an embryonic lethal gene alteration on neuronal development and function can be directly investigated. Second, whereas one of the most efficient techniques to obtain ES cell-derived neurons is to gather the reporter-positive cells by FACS from differentiated ES cells expressing a reporter gene under the control of neural marker promoters, our protocol is applicable to any ES cells without reporter genes. Our protocol is not only simpler than the conventional methods but also more advantageous in obtaining an unbiased population of neurons differentiated from, theoretically, any ES cells. This implies that our strategy is immediately applicable to the establishment of in vitro model systems for a number of neurodegenerative diseases, the causative genes of which are dominantly inherited. Finally, as noted in Introduction, the greatest difficulty in investigating and treating AD pathophysiology is the fact that it is impossible to obtain sharable, live postmitotic neurons from AD patients. As human ES or germinal cells have been established (Shamblott et al., 1998; Thomson et al., 1998) and their in vitro differentiation is demonstrated (Carpenter et al., 2001; Reubinoff et al., 2001; Zhang et al., 2001), the present strategy provides the only feasible method to establish human postmitotic neurons harboring the AD genotype and potentially the AD phenotype that could be shared by researchers. The strategy of the V642I–APP knock-in ES-derived neurons, with further improvement, may allow us to overcome this difficulty.
Footnotes
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The Rat401 developed by Dr. Susan Hockfield and the 5A5 developed by Drs. Thomas M. Jessel and Jane Dodd were kindly obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biological Sciences at the University of Iowa. We especially thank Dr. Shuji Matsuda, Takako Hiraki, and Yuko Ito for indispensable cooperation; Yoshiomi and Yumi Tamai for support; Dr. Dovie Wylie for expert assistance; and all members of the Departments of Pharmacology and Anatomy at KEIO University for essential cooperation.
Correspondence should be addressed to Dr. Ikuo Nishimoto, Department of Pharmacology, Medical Research Building, Sixth Floor, KEIO University School of Medicine, Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan. E-mail: nisimoto@sc.itc.keio.ac.jp.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238513-13$15.00/0
Y.A. and K.K. contributed equally to this work.
References
- Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI ( 1995) Embryonic stem cells express neuronal properties in vitro Dev Biol 168: 342-357. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E ( 1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239-259. [DOI] [PubMed] [Google Scholar]
- Carpenter MK, Inokuma MS, Denham J, Mujtaba T, Chiu C-P, Rao MS ( 2001) Enrichment of neurons and neural precursors from human embryonic stem cells. Exp Neurol 172: 383-397. [DOI] [PubMed] [Google Scholar]
- Couchie D, Mavilia C, Georgieff IS, Liem RKH, Shelanski ML, Nunez J ( 1992) Primary structure of high molecular weight tau present in the peripheral nervous system. Proc Natl Acad Sci USA 89: 4378-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sauvage F, Octave J-H ( 1989) A novel mRNA of the A4 amyloid precursor gene coding for a possibly secreted protein. Science 245: 651-653. [DOI] [PubMed] [Google Scholar]
- Fraichard A, Chassande O, Bilbaut G, Dehay C, Savatier P, Samarut J ( 1995) In vitro differentiation of embryonic stem cells into glial cells and functional neurons. J Cell Sci 108: 3181-3188. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Crowther RA ( 1992) Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci USA 89: 1983-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Zou Y-R, Rajewsky K ( 1993) Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 73: 1155-1164. [DOI] [PubMed] [Google Scholar]
- Hardy J ( 1992) Framing β-amyloid. Nat Genet 1: 233-234. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Ito Y, Nishimoto I ( 2000a) Multiple mechanisms underlie neurotoxicity by different types of Alzheimer's disease mutations of amyloid precursor protein. J Biol Chem 275: 34541-34551. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Jiang H, Niikura T, Ito Y, Hagiwara A, Umezawa K, Abe Y, Murayama Y, Nishimoto I ( 2000b) Neuronal apoptosis by apolipoprotein E4 through low-density lipoprotein receptor-related protein and heterotrimeric GTPases. J Neurosci 20: 8401-8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ito Y, Niikura T, Shao Z, Hata M, Oyama F, Nishimoto I ( 2001a) Mechanisms of neuroprotection by a novel rescue factor Humanin from Swedish mutant amyloid precursor protein. Biochem Biophys Res Commun 283: 460-468. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I ( 2001b) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Aβ. Proc Natl Acad Sci USA 98: 6336-6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ito Y, Arakawa E, Kita Y, Terashita K, Niikura T, Nishimoto I ( 2002a) Neurotoxic mechanisms triggered by Alzheimer's disease-linked mutant M146L presenilin 1: involvement of NO synthase via a novel pertussis toxin target. J Neurochem 80: 426-437. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Ito Y, Kita Y, Terashita K, Nishimoto I ( 2002b) Neurotoxic mechanisms by Alzheimer's disease-linked N141I mutant presenilin 2. J Pharmacol Exp Ther 300: 736-745. [DOI] [PubMed] [Google Scholar]
- Hendriks L, van Broeckhoven C ( 1996) Aβ A4 amyloid precursor protein gene and Alzheimer's disease. Eur J Biochem 237: 6-15. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT ( 1997a) APPsw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965-973. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT ( 1997b) Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: 7053-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Muenkel HA, Blume AJ, Vitek MP ( 1991) A novel species-specific RNA related to alternatively spliced amyloid precursor protein mRNAs. Neurobiol Aging 12: 575-583. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, Nishikawa SI, Sasai Y ( 2000) Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 28: 31-40. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire H-G, Unterback A, Salbaum JM, Masters CL, Grezeschik KH, Multhaup G, Beyreuther K, Müller-Hill B ( 1987) The precursor of Alzheimer disease amyloid A4 protein resembles cell-surface receptor. Nature 325: 733-736. [DOI] [PubMed] [Google Scholar]
- Keyoung HM, Roy NS, Benraiss A, Louissaint Jr A, Suzuki A, Hashimoto M, Rashbaum WK, Okano H, Goldman SA ( 2001) High-yield selection and extraction of two promoter-defined phenotypes of neural stem cells from the fetal human brain. Nat Biotechnol 19: 843-850. [DOI] [PubMed] [Google Scholar]
- König G, Mönning U, Czech C, Prior R, Banati R, Schreiter-Gasser U, Bauer J, Masters CL, Beyreuther K ( 1992) Identification and differential expression of a novel alternative splice isoform of the βA4 amyloid precursor protein (APP) mRNA in leukocytes and brain microglial cells. J Biol Chem 267: 10804-10809. [PubMed] [Google Scholar]
- Kwok JB, Li QX, Hallupp M, Whyte S, Ames D, Beyreuther K, Masters CL, Schofield PR ( 2000) Novel Leu723Pro amyloid precursor protein mutation increases amyloid β42(43) peptide levels and induces apoptosis. Ann Neurol 47: 249-253. [DOI] [PubMed] [Google Scholar]
- Lee S-H, Lumelsky N, Studer L, Auerbach JM, McKay RD ( 2000) Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol 18: 675-679. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ ( 2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Li M, Pevny L, Lovell-Badge R, Smith A ( 1998) Generation of purified neural precursors from embryonic stem cells by lineage selection. Curr Biol 8: 971-974. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K, Ihara Y ( 1995) Hyperphosphorylation of tau in PHF. Neurobiol Aging 16: 365-380. [DOI] [PubMed] [Google Scholar]
- Mujtaba T, Piper DR, Kalyani A, Groves AK, Lucero MT, Rao MS ( 1999) Lineage-restricted neural precursors can be isolated from both the mouse neural tube and cultured ES cells. Dev Biol 214: 113-127. [DOI] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt LA ( 1992) Pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of β-amyloid. Nat Genet 1: 345-347. [DOI] [PubMed] [Google Scholar]
- Neve RL, Finch EA, Dawes LR ( 1988) Expression of the Alzheimer amyloid precursor gene transcripts in the human brain. Neuron 1: 669-677. [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Näslund J, Lannfelt L ( 2001) The ‘Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat Neurosci 4: 887-893. [DOI] [PubMed] [Google Scholar]
- Okabe S, Forsberg-Nilsson K, Spiro AC, Segal M, McKay RDG ( 1996) Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells in vitro Mech Dev 59: 89-102. [DOI] [PubMed] [Google Scholar]
- Renoncourt Y, Carroll P, Filippi P, Arce V, Alonso S ( 1998) Neurons derived in vitro from ES cells express homeoproteins characteristic of motoneurons and interneurons. Mech Dev 79: 185-197. [DOI] [PubMed] [Google Scholar]
- Reubinoff BE, Itsykson P, Turetsky T, Pera MF, Reinhartz E, Itzik A, Ben-Hur T ( 2001) Neural progenitors from human embryonic stem cells. Nat Biotechnol 19: 1134-1140. [DOI] [PubMed] [Google Scholar]
- Sandbrink R, Masters CL, Beyreuther K ( 1994) βA4-Amyloid protein precursor mRNA isoforms without exon 15 are ubiquitously expressed in rat tissues including brain, but not in neurons. J Biol Chem 269: 1510-1517. [PubMed] [Google Scholar]
- Shamblott MJ, Axelman J, Wang S, Bugg EM, Littlefield JW, Donovan PJ, Blumenthal PD, Huggins GR, Gearhart JD ( 1998) Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc Natl Acad Sci USA 95: 13726-13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastry BS, Giblin FJ ( 1999) Genes and susceptible loci of Alzheimer's disease. Brain Res Bull 48: 121-127. [DOI] [PubMed] [Google Scholar]
- Strübing C, Ahnert-Hilger G, Shan J, Wiedenmann B, Hescheler J, Wobus AM ( 1995) Differentiation of pluripotent embryonic stem cells into the neuronal lineage in vitro give rise to mature inhibitory and excitatory neurons. Mech Dev 53: 275-287. [DOI] [PubMed] [Google Scholar]
- Sudo H, Jiang H, Yasukawa T, Hashimoto Y, Niikura T, Kawasumi M, Matsuda S, Takeuchi Y, Aiso S, Matsuoka M, Murayama Y, Nishimoto I ( 2000) Antibody-regulated neurotoxic function of cell-surface β-amyloid precursor protein. Mol Cell Neurosci 16: 708-723. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM ( 1998) Embryonic stem cell lines derived from human blastocysts. Science 282: 1145-1147. [DOI] [PubMed] [Google Scholar]
- Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grünberg J, Haass C, Iwatsubo T, Obata K ( 1997) The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increase the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci USA 94: 2025-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessel TM ( 2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110: 385-397. [DOI] [PubMed] [Google Scholar]
- Yagi T, Tokunaga T, Furuta Y, Nada S, Yoshida M, Tsukada T, Saga Y, Takeda N, Ikawa Y, Aizawa S ( 1993) A novel ES cell line, TT2, with high germline-differentiating potency. Anal Biochem 214: 70-76. [DOI] [PubMed] [Google Scholar]
- Yamatsuji T, Matsui T, Okamoto T, Komatsuzaki K, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane TB, Giambarella U, Nishimoto I ( 1996) G protein-mediated neuronal DNA fragmentation induced by familial Alzheimer's disease-associated mutants of APP. Science 272: 1349-1352. [DOI] [PubMed] [Google Scholar]
- Zhang S-C, Wernig M, Duncan ID, Brüstle O, Thomson JA ( 2001) In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol 19: 1129-1133. [DOI] [PubMed] [Google Scholar]