

Abstract
Hearing loss can be caused by a variety of insults, including acoustic trauma and exposure to ototoxins, that principally effect the viability of sensory hair cells via the MAP kinase (MAPK) cell death signaling pathway that incorporates c-Jun N-terminal kinase (JNK).
We evaluated the otoprotective efficacy of D-JNKI-1, a cell permeable peptide that blocks the MAPK–JNK signal pathway. The experimental studies included organ cultures of neonatal mouse cochlea exposed to an ototoxic drug and cochleae of adult guinea pigs that were exposed to either an ototoxic drug or acoustic trauma. Results obtained from the organ of Corti explants demonstrated that the MAPK–JNK signal pathway is associated with injury and that blocking of this signal pathway prevented apoptosis in areas of aminoglycoside damage. Treatment of the neomycin-exposed organ of Corti explants with D-JNKI-1 completely prevented hair cell death initiated by this ototoxin. Results from in vivo studies showed that direct application of D-JNKI-1 into the scala tympani of the guinea pig cochlea prevented nearly all hair cell death and permanent hearing loss induced by neomycin ototoxicity. Local delivery of D-JNKI-1 also prevented acoustic trauma-induced permanent hearing loss in a dose-dependent manner. These results indicate that the MAPK–JNK signal pathway is involved in both ototoxicity and acoustic trauma-induced hair cell loss and permanent hearing loss. Blocking this signal pathway with D-JNKI-1 is of potential therapeutic value for long-term protection of both the morphological integrity and physiological function of the organ of Corti during times of oxidative stress.
Keywords: neomycin, ototoxicity, acoustic trauma, noise-induced hearing loss, apoptosis of hair cells, c-Jun N-terminal kinase (JNK), JNK inhibition, organ of Corti
Introduction
Hearing impairment affects ∼10% of the population and increases in frequency fourfold with aging. Of all the sensory organs, the organ of Corti functions with the smallest number of sensory receptor cells, the auditory hair cells. The human cochlea contains only 20,000 sensory receptor cells [i.e., 5,000 inner hair cells (IHCs) and 15,000 outer hair cells (OHCs)] (Bredberg, 1968), as compared with the retina that contains 137,000,000 visual sensory receptor cells. Hearing deficits are often caused by loss of sensory hair cells because of a variety of factors that generate oxidative stress including noise, infection, and ototoxic drugs (e.g., aminoglycoside antibiotics and cisplatin). In mammals, auditory hair cells are produced only during embryonic development (Ruben, 1967) and do not regenerate if lost during postnatal life, therefore, a loss of hair cells will result in profound and irreversible deafness. Unfortunately, at present, there are no effective therapies to treat the cochlea and reverse this condition. Thus, an effective therapy to prevent oxidative stress-induced cell death of auditory hair cells would be of great therapeutic value.
Previous studies have shown that ototoxic drug exposure leads to the apoptotic cell death of hair cells based on the morphological characteristics of nuclear fragmentation that occur during apoptosis (Forge, 1985; Vago et al., 1998; Pirvola et al., 2000). The c-Jun N-terminal kinase (JNK) group of MAP kinases (MAPKs), also known as stress-activated protein kinases, phosphorylate the transcription factor c-Jun (Kyriakis et al., 1994). The MAPK–JNK signal transduction pathway is activated in response to exposure of cells to environmental stress and contributes to an apoptotic response by stressed cells (for review, see Davis, 2000) that has been damaged by reactive oxygen species (ROS). The MAPK–JNK signal transduction pathway, therefore, contributes to pathophysiological responses of neurons to stress.
A cellular regulator of the MAPK–JNK signal transduction pathway has been identified and termed c-Jun-interacting protein-1 (JIP1)/islet brain 1 (IB1) (Dickens et al., 1997; Bonny et al., 2001). JIP1/IB1 is a scaffold protein that retains JNK and other members of MAPK–JNK signal cascade in the cytoplasm (Gupta et al., 1995). A deficiency of activated JNK in the cell will prevent c-Jun phosphorylation, thereby preventing the formation of transcription complexes, and in that manner protects against stress-induced apoptosis (Tournier et al., 2000).
In the present study, we used a chemically synthesized cell permeable JNK ligand named D-JNKI-1, which blocks JNK-mediated activation of c-Jun (Bonny et al., 2001). D-JNKI-1 is an efficient inhibitor of the action of all three JNK isoforms produced by linking the 20 amino acid terminal JNK-inhibitory sequence (JNK binding domain) of JIP1/IB1 to a 10 amino acid HIV–TAT transporter sequence (Vives et al., 1997). The ability of D-JNKI-1 to protect auditory hair cells from cell death induced by an ototoxic drug and acoustic trauma was tested in 3-d-old [postnatal day 3 (P3)] mouse organ of Corti explants exposed to an ototoxin and in an in vivo adult guinea pig model of ototoxicity and acoustic trauma.
Materials and Methods
The care and use of animals followed the animal welfare guidelines of the Institut National de la Santé et de la Recherche Médicale and under the approval of the French Ministèredel'Agriculture et de la Forêt. All efforts were made to minimize the number of animals used and their suffering.
In vitro studies
Organ of Corti explants. Organ of Corti explant cultures were prepared from P3 mice as described previously (Zine and de Ribaupierre, 1998). Because of the known partial resistance of apical turn hair cells to damage by aminoglycosides, only basal and mid-cochlear turns were used in this study. Briefly, explants that were composed of the basal and middle turns of the organ of Corti were placed individually on transparent membranes with 0.4 μm pores (Millipore, Bedford, MA) and kept in a 6-well culture plate containing 800 μl/well of serum-free medium. The serum-free medium consisted of DMEM and Ham's F-12 supplemented with insulin (25 μg/ml), transferrin (100 μg/ml), progesterone (2 × 10 -8m), selenium (3 × 10 -8m), and putrescine (10 -4m). The glucose concentration was increased from 3.15 to 6 gm/l. The organ of Corti explants were placed in a humidified incubator for 24 hr at 37°C and 5% CO2. On the second day, the medium was replaced with new medium that contained 1 mm neomycin sulfate (Sigma, St. Louis, MO) or the combination of 1 mm neomycin and 2 μm D-JNKI-1 for an additional 24–48 hr in vitro. Parallel untreated, control explants were maintained in medium for 3 d without either neomycin or D-JNKI-1 and were run concurrently with the experimental cultures.
Confocal microscopy. At the end of the culture period, organ of Corti explants were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer for 1 hr and prepared for immunocytochemical study. Explants were immunolabeled with a polyclonal myosin VIIa antibody (a generous gift from M. Mooseker, Yale University, New Haven, CT) to identify hair cells. A few specimens were labeled with phalloidin-tetramethyl rhodamine (TRIC) conjugates (Sigma) to visualize the f-actin present in the cuticular plates and stereocilia of the hair cells. Observations were performed on a Leica confocal laser scanning microscope equipped with epifluorescence optics (Leica Lasertechnik GmbH, Nussloch, Germany). Images were assembled by stacking digital images acquired within a 15–20 μm depth beginning from the apical surface of the explant.
Immunolabeling and a terminal deoxynucleotidyl tranferase-mediated biotinylated UTP nick end labeling. DNA fragmentation in the cells of the organ of Corti explants was detected by a terminal deoxynucleotidyl tranferase-mediated biotinylated UTP nick end labeling (TUNEL) (Fluorescein In Situ Cell Detection Kit; Intergen, Purchase, NY), according to the manufacturer's instructions. In brief, specimens were digested with 20 μg/ml proteinase K in PBS for 5 min and incubated with terminal deoxynucleotidyl transferase (TdT) and fluorescein nucleotide in a humid atmosphere at 37°C for 1 hr. For myosin VIIa antibody and TUNEL double labeling, explants were first processed for myosin VIIa immunolabeling (TRIC-tag) and then for TUNEL staining (FITC-tag). Negative controls for TUNEL staining omitted TdT. Labeled preparations were washed in PBS, mounted in Fluor-Save (Calbiochem, La Jolla, CA), and viewed with a confocal microscope. Fragmented nuclei were verified from sections of paraffin-embedded cochlear explants stained with a standard hematoxylin and eosin protocol. Adjacent sections were immunolabeled with a polyclonal phospho-c-Jun antibody (Santa Cruz Biotechnology, Santa Cruz, CA). For immunohistochemical detection, ABC complex-HRP (Vector Laboratories, Burlingame, CA) and DAB were used, and sections were lightly counterstained with 1% toluidine blue and analyzed with an Olympus bright-field microscope.
Quantification of hair cells. To quantify hair cell loss in organ of Corti explants after various treatments, surviving IHCs and OHCs were counted over a 0.1 mm distance from three or four randomly selected fields from a middle turn segment of each culture. Hair cells were counted on confocal images using NIH-Image program on myosin VIIa- or phalloidin-stained whole-mount preparations. Only intact hair cells that were traceable throughout the optical sections of the organ of Corti were counted as hair cells. The average of hair cell counts from three or four randomly selected fields was considered as a single sample. Data were collected from five or more cultures for each experimental group and expressed as a mean ± SD. A Student's t test was used for statistical analysis of comparison of hair cell counts from the different culture conditions. A p value of <0.01 as considered significant.
Real-time reverse transcription-PCR. We performed real-time reverse transcription (RT)-PCR by using specific primers on a light cycler apparatus (Roche). The mRNA of a house keeping gene (i.e., tubulin) was used to normalize for the amount and quality of the mRNAs that were extracted by the Chomczynski method (Chomczynski and Sacchi, 1987). Sequences of the primer pairs were: c-fos—F5′-GCTGACAGATACACTCCAAG-3′, R 5′-CCTAGATGATGCCGGAAACA-3′; tubulin—F 5′-GGAGGATGCTGCCAATAACT-3′, R 5′-GGTGGTGAGGATGGAATTGT-3′.
In vivo studies
Experimental protocol. The experiments were designed to determine whether local application of D-JNKI-1 into the scala tympani could rescue the cochlea from aminoglycoside, or noise-induced damage in vivo. Pigmented guinea pigs (250–300 gm; Charles River, St. Aubain, France) were used in both the ototoxin and sound trauma studies. D-JNKI-1 was perfused into the scala tympani of the cochlea via an osmotic minipump over a period of 7 d. To ensure that the cochlea was perfused with D-JNKI-1 at the time of the aminoglycoside or noise exposure, the right cochlea of each animal was implanted with the minipump 2 d before the beginning of either the aminoglycoside or noise exposure studies. Nonperfused left cochleae served as controls for the effectiveness of the aminoglycoside ototoxicity or acoustic trauma on causing auditory hair cell loss and loss of hearing function. At the end of the electrophysiological testing, the animals were sacrificed, and cochleae were prepared for a quantitative evaluation of hair cell losses using scanning electron microscopy to examine the surface of the organ of Corti and count all hair cells for the entire length of the cochlear duct.
Two days after minipump implantation (i.e., day 0), animals were given daily intraperitoneal injections of neomycin sulfate (300 mg/kg/d during 5 d; cumulative dose, 1500 mg/kg) or were exposed to acoustic trauma [6 kHz, 120 dB sound pressure level (SPL), 30 min]. Audiograms of both ears were derived from the compound action potentials (CAPs) recorded from the auditory nerves via chronic round window membrane electrodes. The CAP audiograms from both ears were measured daily in awake animals. Five days after the administration of the final neomycin injection (day 10) or 30 d after sound exposure (day 30), animals were sacrificed, and their cochleae were prepared for quantitative evaluation of hair cell loss using SEM.
Drug preparation. An Alzet osmotic minipump (model 2001; Alza Corp., Palo Alto, CA) was used to introduce D-JNKI-1 into the scala tympani of the cochlea. The drug was delivered in an artificial perilymph solution of the following composition (in mm): 137 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 1 NaHCO3, and 11 glucose, pH 7.4; osmolarity, 304 ± 4.1 mOsm/kg). One millimolar stock solution of D-JNKI-1, the D form of TAT peptide (i.e., “TAT-empty”) and the inactive mutant form of JNKI-1 (i.e., JNKI-1-mut) were prepared in 0.1 m PBS at pH 7.2.
In the neomycin exposure experiment, the D-JNKI-1 was freshly diluted in artificial perilymph to a final concentration of 10 μm on the day it was loaded into the minipump. Before surgery, the minipump was filled under sterile conditions with 200 μl of artificial perilymph containing 10 μm D-JNKI-1. In the acoustic trauma experiment, the D-JNKI-1 was freshly diluted in artificial perilymph to a final concentration of 0, 0.1, 1, 3, 10, 33, or 100 μm on the day of use. Both TAT-empty and JNKI-1-mut were freshly diluted in artificial perilymph to a final concentration of 10 μm on the day of use. Before surgery, minipumps were filled with 200 μl of artificial perilymph containing 0–100 μm D-JNKI-1 or 10 μm TAT-empty or 10 μm JNKI-1-mut under sterile conditions. A glass pipette with a ring of glue placed near the tip, to give a leak-proof seal against the cochlea, was connected to PE50 polyethylene tubing. The pipette and the tubing were then filled and connected to the flow moderator on the pumps. The minipump flow rate was 1 μl/hr, and the entire content of the pump as delivered into the scala tympani over a period of 7 d. In the neomycin toxicity experiment, neomycin sulfate was dissolved in a physiological saline solution to a final concentration of 200 mg/ml.
Surgical procedure. Guinea pigs free of middle ear infection (determined by otoscopic examination) were anesthetized with an intraperitoneal injection of a 6% solution of sodium pentobarbital (Sanofi, Montpellier, France; dose, 0.3 ml/kg). A supplementary dose (quarter dose, 0.07 ml/kg) of pentobarbital was administered after 2 hr, or more often if the animal withdrew its paw in response to deep pressure applied to the paw during the course of the surgery. Each surgical procedure lasted for ∼3 hr.
Each bulla was exposed under sterile conditions via a postauricular approach. A recording electrode of insulated platinum wire was placed on the round window membranes of both the left and right cochleae, and a reference electrode of noninsulated platinum was placed in a neck muscle. Using a micromanipulator, the tip of a glass perfusion pipette (attached by polyethylene tubing to the minipump) was inserted into a 0.2 mm hole (cochleostomy) that had been drilled close to the round window in the basal turn of the right cochlea. Dental cement was used to secure both the electrode and the tubing connected to the pump, and to close the defect created in the bulla to prevent formation of secondary otitis media. The Alzet 2001 osmotic minipump was inserted into a pocket under the skin of the animal's back between the two scapulae. Finally, both round window electrodes and the reference electrode were soldered to an electric plug and fixed with dental cement onto the skull.
CAP recordings. To elicit CAPs, we used tone bursts with a 1 msec rise/fall time and an 8 msec total duration generated by an arbitrary function generator (Instrument type 9100R; LeCroy Corp., Chestnut Ridge, NY). The signals were passed through a programmable attenuator and presented to the ear in free field via a JBL (Northridge, CA) 075 earphone. Tone burst levels of alternating polarity were applied from 0 to 100 dB SPL, in 5 dB steps. The rate of tone burst presentation was 10 per second. Cochlear responses were amplified (gain, 2000) by a differential amplifier (Grass P511K; Astro-Med, Inc., West Warwick, RI), averaged (128 samples), and stored on a Pentium PC computer. The sampling rate of the analog-to-digital converter was 50 kHz, with a dynamic range of 12 bits and 1024 samples per record. CAPs were measured peak-to-peak, between negative depression N1 and the subsequent positive wave P1. The threshold of the CAP was defined as the intensity in dB SPL needed to elicit a measurable response (≥5 μV).
Acoustic trauma. Acoustic trauma was induced by a continuous pure tone of 6 kHz generated by a waveform synthesizer (Hewlett-Packard 8904A). The animals were anesthetized and exposed to 120 dB SPL for 30 min, which was routed through a programmable attenuator and presented to the ears in free field via a JBL O75 earphone positioned 10 cm in front of the animal's head. Sound level was measured using a calibrated Bruel and Kjaer microphone (4314) and a Bruel and Kjaer calibrating amplifier (2606).
Scanning electron microscopy assessment. Five days after the final administration of neomycin (day 10) or 30 d after sound exposure (day 30), the animals were killed by an overdose of anesthetic. Both cochleae were immediately removed, fixed with a solution of 0.1 m phosphate buffer, pH 7.4, containing 1% osmic acid, and immersed in the same fixative for 1 hr, washed in phosphate buffer (0.2 m), and dehydrated in a graded series of ethanol baths (30–100%). During the 70% ethanol stage, the bony capsules of each cochlea were dissected, and the stria vascularis and Reissner's membrane were removed to expose the organ of Corti. The cochleae were then processed to 100% ethanol, critical-point dried in CO2, coated with gold palladium, and observed with a Hitachi S4000 scanning electron microscope.
Quantitative SEM observations of the surface morphology of the organ of Corti were performed by determining the number of hair cells from the apex to the base of each cochlea. The results of these hair cell counts were expressed as the percentage of remaining hair cells in each row of IHCs and OHCs over the entire length of the cochlea. In control conditions, the guinea pig cochlea contained ∼2000 IHCs and 7500 OHCs. For hair cell counts, the whole cochlea was divided into 20 segments (1 mm lengths of basilar membrane each); each segment contained ∼100 IHCs and ∼375 OHCs. A hair cell was counted as missing if the stereociliary bundle was absent or the stereocilia of the bundle were completely fused. All the results are presented as means ± SEM. Differences were assessed using an unpaired Student's t test. Values of p < 0.05 were considered significant.
Results
The JNK signal transduction pathway is activated in response to the exposure of cells to enviromental stress and contributes to the apoptotic response (for review, see Davis, 2000). Components of the MAPK–JNK signaling pathway interact with the JIP1 scaffold protein (Fig. 1A). In vitro biochemical assays and transfection assays have shown that JNK, MMK7, and MLK all bind to separate sites on JIP1 (Whitmarsh and Davis, 1998). These assays show that JIP scaffold protein is a critical component of a MAPK signal tansduction pathway that contributes to JNK activation in response to the exposure of cells to enviromental stress. Here, we have used a cell-penetrating peptide to block selectively the access of JNK to c-Jun and other substrates by a competitive mechanism (Bonny et al., 2001; Barr et al., 2002). This blocking peptide (Fig. 1B) was constructed by linking the 20 amino acid JNK-binding motif of JIP1 to the 10 amino acid HIV–TAT transporter sequence and by synthesizing a highly protease-resistant D-retroinverso form of this peptide (i.e., D-JNKI-1) that doubles its half-life within the intracellular compartment.
Figure 1.

A, Schematic illustration of JIP1 and its association with components of the MAPK–JNK signaling module. The JIP1 group of scaffold proteins selectively facilitates the sequential phosphorylation cascade by a mixed-lineage kinase (MLK) to MAP kinase kinase 7 (MKK7) to c-Jun NH2-terminal kinase (JNK). The location of the JNK binding domain (JBD) of JIP1 is indicated by hatched lines. This scaffold assembly is thought to organize the MAPK–JNK signal module to respond to an oxidative stress insult to a cell. B, A schematic representation of the structural organization of the D-JNKI-1 peptide. D-JNKI-1 (30 aa) is a chimeric peptide starting with a 10 aa TAT transporter sequence, followed by a 20 aa minimal JBD sequence from the JIP1 molecule. The D-JNKI-1 peptide functions by strongly interacting with JNK, thereby preventing its interaction with and subsequent phosphorylation of c-Jun. This prevents c-Jun from interacting with itself (homodimerization) or other immediate early proteins such as c-Fos (heterodimerization) to form activator protein-1 complexes.
Organ of Corti explants
D-JNKI-1 protects P3 mouse organ of Corti hair cells against neomycin ototoxicity
We tested the ability of the D-JNKI-1 peptide to protect auditory hair cells against neomycin ototoxicity in organotypic explants. We initially explored whether D-JNKI-1 peptides could effectively translocate to inside the cell types present within the organ of Corti explants, including the hair cells. FITC-conjugated D-JNKI-1 peptide was added to the culture medium at a concentration of (1 μmol/l). Explants were then fixed and stained with rhodamine-conjugated phalloidin to identify hair cells. The images of Figure 2 shows that the FITC-labeled peptides in the culture medium rapidly entered the cells of the explants, including the sensory hair cells. A temporal study of D-JNKI-1 incorporation indicated initial uptake of the labeled peptides by explant cells as early as 1 hr and mainly confined to few scattered supporting cells (Fig. 2A). After 4 hr (Fig. 2B), other cells within the explants incorporated the D-JNKI-1 peptide including auditory hair cells and cells with the greater epithelial ridge area, with maximal incorporation occurring by 24 hr in vitro (Fig. 2C).
Figure 2.
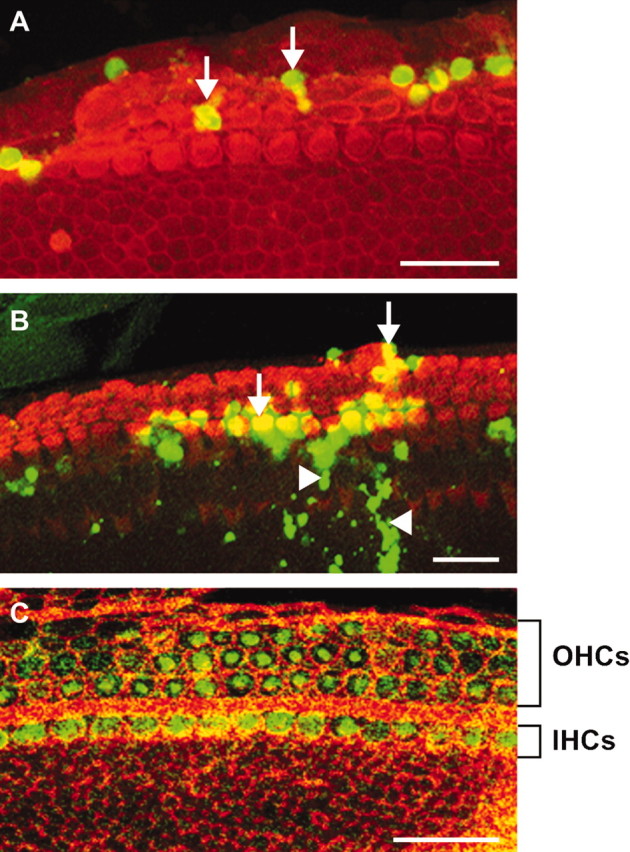
D-JNKI-1 is rapidly taken up by the hair cells in the organ of Corti explants. A–C, Three-day-old (P3) mouse organ of Corti explants incubated with FITC-conjugated D-JNKI-1 (1 μmol/l; FITC, green label) and then labeled with phalloidin-TRIC (red label) to identify the hair cells. Images were taken at 1 hr (A), 4 hr (B), and 24 hr (C) after the addition of FITC-labeled D-JNKI-1 to the medium. Green fluorescent-labeled D-JNKI-1 was initially visible at the level of a few supporting cell nuclei at 1 hr (A, arrows), was present in some hair cells (B, arrow) and cells in the greater epithelial ridge area (B, arrowheads) by 4 hr, and was present in almost all IHCs and OHCs by 24 hr (C). Scale bars, 20 μm.
To characterize the pattern of hair cell loss induced by neomycin exposure in P3 cochlear explants, we used immunostaining for myosin VIIa to identify surviving hair cells. Cultures were also double-labeled with TUNEL staining to detect apoptosis of hair cells. In control explants maintained in serum-free medium for 3 d, all hair cells including one row of IHCs and three rows of OHCs were present (Fig. 3A). Exposure of the cultures to a high dose of neomycin (1 mm) for 48 hr (Zine and de Ribaupierre, 1998, 1999) destroyed all hair cells in the basal turn and >95% of them in the middle cochlear turn. The remnant of dying hair cells within the damaged area of the sensory epithelium include many cells with TUNEL-positive nuclei and some scattered myosin VIIa-labeled, irregular-shaped dying hair cells (Fig. 3B). In contrast, cotreatment of explants with neomycin and D-JNKI-1 for 48 hr prevented almost all neomycin-induced apoptotic cell death of both IHCs and OHCs, as determined by double-labeling with anti-myosin VII a and TUNEL (Fig. 3C) and by quantification of hair cells present in the explants (Fig. 3D). Treatment with a combination of D-JNKI-1 and neomycin also preserved the four-row organization of hair cells that is characteristic of a normal, undamaged organ of Corti (Fig. 3C). Dose-response experiments (data not shown) were performed by examining the otoprotective efficacy of concentrations of D-JNKI-1 ranging from 1 μm to 1 mm in the neomycin-treated explants. The maximum level of protection of hair cells from neomycin ototoxicity was achieved at 2 μm D-JNKI-1.
Figure 3.

D-JNKI-1 treatment prevents apoptosis and loss of neomycin-exposed hair cells. A–C, Confocal images of anti-myosin VIIa (red) and TUNEL (green) double-labeled P3 organ of Corti explants. A, Untreated, control explant. B, Explant exposed to 1 mm neomycin for 48 hr. C, Explant exposed to1 mm neomycin in the presence of 2 μm D-JNKI-1 for 48 hr. Neomycin exposure resulted in a severe loss of hair cells and the presence of many TUNEL-labeled nuclei within the region of the damaged auditory sensory epithelium (B). Most hair cells were already missing, but a few remaining damaged hair cells that were in the process of apoptosis are indicated by TUNEL labeling of their nuclei (arrows). D-JNKI-1 completely prevented neomycin-induced apoptotic cell death of both IHCs and OHCs; no TUNEL-positive cells were present (C). Arrowhead, IHC; rows 1–3, OHCs. Scale bars, 20 μm. D, Results of quantitative analysis of hair cell counts to determine the protective effect of D-JNKI-1 on IHCs, OHCs, and total hair cells (Total HCs) against neomycin induced loss in organ of Corti explants obtained from the middle turns of P3 mouse cochleae. Hair cell counts are presented as means ± SD (bars) from five separate experiments (n = 3–5 explants/condition). D-JNKI-1 treatment of explants provided protection against neomycin-induced hair cell loss that was highly significant (p < 0.001).
Activation of the JNK pathway and apoptosis of auditory hair cells
We initially examined the changes in the normal morphology of hair cells in both neomycin-exposed cochlear explants and neomycin-exposed, D-JNKI-1-treated cochlear explants from histological sections of these cochlear explants. The unexposed control cultures showed a characteristic morphology of the organ of Corti that was well preserved with one row of IHCs and three rows of OHCs (Fig. 4A). In contrast, explants exposed to neomycin showed evidence of a degenerative process in the organ of Corti characterized by dying hair cells that were irregularly shaped and had condensed pyknotic nuclei, a hallmark of apoptotic cell death (Fig. 4B), whereas in neomycin-exposed (1 mm), D-JNKI-1-treated (2 μm) cochlear explants hair cells stained uniformly with normal morphology (Fig. 4C) and exhibited the same characteristic pattern of IHC and OHC organization present in untreated, control explants (Fig. 4A).
Figure 4.
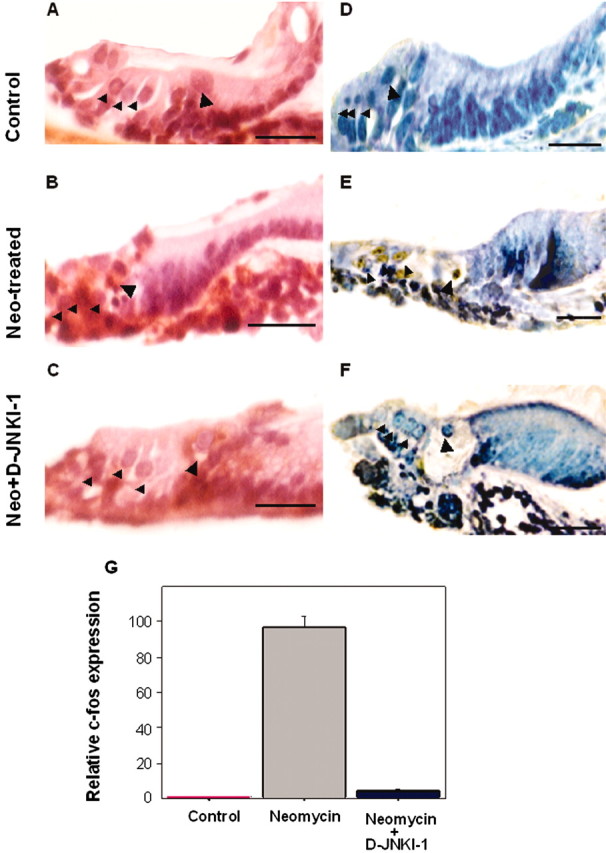
Neomycin exposure caused both hair cell loss and c-Jun phosphorylation in sensory cell nuclei in organ of Corti explants. Transverse plane tissue sections of P3 mouse organ of Corti explants. A, D, Untreated, control explants. B, E, Explants exposed to 1 mm neomycin for 24 hr. C, F, Explants exposed to 1 mm neomycin in the presence of D-JNKI-1 (2 μm) for 24 hr. A–C, Sections stained with hematoxylin and eosin. Nuclear condensations were present in some of the remaining hair cells in the neomycin-exposed explant (B) but were not present in either control explant hair cells (A) or in the hair cells of neomycin-exposed cultures that were protected by D-JNKI-1 (C). D–F, Sections immunostained with an anti-phospho-c-Jun antibody. Phospho-c-Jun antibody-immunolabeled hair cells were present in the neomycin-exposed explant (E) but were not detected in either control explants (D) or in the explants that had been coincubated with neomycin and D-JNKI-1 peptide for 24 hr. (F). Large arrowheads indicate the area of the IHCs, and the small arrowheads indicate the area of the OHCs. Scale bars, 5 μm. G, Results from real-time RT-PCR analysis of c-fos expression in cochlear explants. Bar graphs show relative levels of c-fos expression normalized to tubulin mRNA (n = 3). RNAs were extracted from untreated control, neomycin-exposed (24 hr), and neomycin-exposed, D-JNKI-1-treated (24hr) P3 cochlear explants, as described in Materials and Methods. Note that c-fos expression was significantly upregulated in cochlear explants after neomycin exposure and was at a near untreated, control explant level of expression in the neomycin-exposed, D-JNKI-1-treated explants.
Immunohistochemistry was performed to determine whether the c-Jun transcription factor, a major target of JNK, was activated (i.e., phosphorylated) in neomycin-exposed mouse cochlear explants. Immunostaining of paraffin sections of explants with anti-phospho-c-Jun antibody revealed that JNK activation of c-Jun had occurred within the neomycin-exposed cochlear explants (Fig. 4E). Phosphorylation of c-Jun was evident in the nuclei of hair cells within the regions of aminoglycoside damage in the lower and mid-cochlear turns of the explants. In contrast, in the D-JNKI-1-treated, neomycin-exposed explants (Fig. 4F), all of the hair cells were negative from activated c-Jun immunostaining, and only few immunopositive cells were detected at the border of these cochlear explants. In unexposed, control explants, none of the hair cells immunolabeled with the phospho-c-Jun antibody (Fig. 4D).
Inhibition of c-fos expression
To confirm that D-JNKI-1 peptide could interfere with a downstream target of JNK signaling, we measured its effect on the level of c-fos gene expression (Fig. 4G). The transcriptional activity of the c-fos promoter is positively modulated by a JNK target, the ELK-1 transcription factor. Exposure of cochlear explants to 1 mm neomycin for 24 hr resulted in a highly significant increase of c-fos transcription (a 96 ± 6.51 times increase over control; p < 0.001) above the level of c-fos expression in unexposed, control explants. The addition of D-JNKI-1 to the medium at the time of neomycin exposure prevented the increase in c-fos expression that occurred in response to neomycin ototoxic damage, bringing the level of its expression near to that observed in for unexposed, control explants.
Quantification of these mRNA results and normalization to tubulin mRNA real-time PCR results indicated that c-fos transcription in response to neomycin exposure was greatly reduced (i.e., a 21 ± 1.5 times increase; representing a 78% decrease when compared with the response evoked by neomycin alone) by cotreatment of the neomycin-exposed explants with D-JNKI-1 peptide. These results strongly suggest that the transcriptional regulation of c-fos is at least partially under the control of activated JNK in the neomycin damaged organ of Corti explants.
In vivo: inner ear delivery of D-JNKI-1
D-JNKI-1 protects the cochlea against neomycin ototoxicity
To extend the in vitro studies, we tested the ability of D-JNKI-1 to protect hearing function in vivo from neomycin ototoxicity. Neomycin exposure consisted of a series of daily intraperitoneal injections of 300 mg/kg/d neomycin sulfate for 5 d. Within the group of the 20 animals that were tested, 5 animals were excluded because they died during the course of the treatment (likely from effects of neomycin toxicity), and 8 animals were excluded because they did not show a clear, definable hearing loss. Therefore, we reported functional results from seven animals with hearing losses ≥20 dB at 20 kHz after 5 d of neomycin treatment.
Averaged audiograms from unperfused, contralateral cochleae revealed that the earliest effect of the intraperitoneal injection of neomycin on hearing acuity was observed 3 d after the beginning of this treatment (i.e., a cumulative dose of 900 mg/kg) (Fig. 5A). Hearing loss continued to develop over the time course of the neomycin treatment. By day 6 (cumulative dose, 1500 mg/kg), most hearing losses in the higher frequencies (12–26 kHz) exceeded 20 dB (Fig. 5A). In contrast, no significant hearing losses were observed in the cochlea of neomycin-injected animals that were perfused for 7 d with a 10 μm solution of D-JNKI-1 (Fig. 5B). At day 6, in the contralateral control ears, a clear dissociation was seen between the responses to sound stimulation (i.e., at 8 kHz). The CAPs evoked by low sound intensity level stimulation were more affected than at the high-intensity levels, a typical feature of OHC damage (see Fig. 5C). In the D-JNKI-1-perfused ears, the CAP amplitudes remained unchanged for all of the frequencies and levels of sound stimulation tested (Fig. 5D).
Figure 5.

Local delivery of D-JNKI-1 into the cochlea strongly protected against neomycin-induced hearing and hair cell losses. A, B, A comparison of hearing threshold shifts of contralateral unperfused left cochleae (A) to those of the D-JNKI-1-perfused right cochleae (B) from the same neomycin-treated animals (n=7). Hearing loss was calculated as the difference in decibels between auditory thresholds before neomycin treatment and after 1 d (black circles), 3 d (white circles), and 6 d (white triangle). Changes in hearing thresholds are expressed as mean values ± SEM. Note that the neomycin treatment (300 mg/kg/d during 5 d) induced dose-dependent cumulative hearing losses in the contralateral unperfused left cochleae (A) but there were no significant hearing losses in the neomycin-exposed, D-JNKI-1-perfused inner ears (B). C, D, Protective effect of 10 μm solution of D-JNKI-1 against neomycin ototoxicity on amplitude–intensity function of the CAP evoked by stimulation with 8 kHz tone bursts. Shown are the results obtained before neomycin treatment (white circles) and at 6 d after neomycin (black circles). In the contralateral, unperfused cochleae, neomycin treatment caused a reduction in the CAP amplitudes predominantly in the low portion of amplitude–intensity function (C). In contrast, there was no reduction in CAP amplitude seen in the cochleae perfused with a 10μm solution of D-JNKI-1 (D). A–D, All points represent mean ± SEM values calculated from seven animals. E, F, Scanning electron micrographs from the basal turns of cochleae from the same neomycin-exposed animal. Note the extensive loss of OHCs from the organ of Corti of the basal turn of the contralateral unperfused, neomycin-exposed left cochlea. Only four OHCs remain in the area viewed in E as apposed to the 29 OHCs present in the image presented in F, in which perfusion of a 10 μm solution of D-JNKI-1 prevented loss of OHCs, O, Area of all three rows of OHCs. I, Single row of IHCs. Scale bar, 15μm. G, H, Cytocochleograms obtained from contralateral unperfused left cochleae (G; n = 3) and D-JNKI-1-perfused right cochleae (H; n = 3) of the same neomycin-treated animals 10 d after the start of neomycin injections. Cytocochleograms show the percentage of surviving IHCs (white circles) and OHCs from the first (black circles), second (dark gray circles), and third (light gray circles) rows as a function of the distance from the cochlear apex (millimeter). Note the extensive loss of OHCs in the basal turns and loss of a few IHCs in both the basal and apical turns of the contralateral unperfused left cochleae. An average of only 58.2% of the OHCs remained intact in the damaged area of the basal turns of contralateral unperfused cochleae (G). In contrast, the D-JNKI-1-perfused cochleae show only minimal losses of OHCs (i.e., 4%), which occur predominantly in the basal turns (H).
Scanning electron microscopic observations of the surface morphology of each organ of Corti was performed at the end of the physiological survey of hearing function [i.e., 5 d after the final injection of neomycin (day 10)]. In the contralateral unperfused cochlea subjected to a cumulative dose of 1500 mg/kg of systemic neomycin, most of the OHCs were missing from the basal turn (Fig. 5E). A protective effect of locally administered D-JNKI-1 against neomycin-induced hair cell loss was confirmed by morphological evaluation (Fig. 5F). Hair cells were then quantified in: control cochleae (n = 3); neomycin-exposed, unperfused cochleae (n = 3); and neomycin-exposed, D-JNKI-1-perfused cochleae (n = 3). Drastic losses of OHCs in the basal, medial, and apical cochlear turns, with a typical base to apex pattern of hair cell loss and first to the third row gradient of OHC loss, were observed in the cochleae, subject to systemic neomycin (Fig. 5G). A few damaged IHCs were also detected in the apical and basal turns (Fig. 5G). Cochleae that were perfused with 10 μm D-JNKI-1 for 2 d before and during neomycin exposure showed a very small loss of OHCs from the basal and apical turns, with no apparent damage to the OHCs of the middle cochlear turns (i.e., less than a 5% loss of OHCs; Fig. 5H). There was no damage to the IHCs in any turn of the neomycin-exposed, D-JNKI-1-perfused cochleae at day 10 (Fig. 5H).
D-JNKI-1 protects the cochlea against acoustic trauma
We next investigated the protective effect of D-JNKI-1 against acoustic trauma-induced hair cell and hearing losses. Forty-two animals were divided into seven different groups of six animals for each concentration of D-JNKI-1 tested (i.e., 0, 0.1, 1, 3, 10, 33, or 100 μm). Noise-exposed, unperfused left cochleae served as controls for the effectiveness of acoustic trauma-induced hair cell losses and hearing loss. In contralateral noise-exposed, unperfused cochleae, the average hearing loss measured 20 min after noise exposure reached 60 dB in the higher frequencies of the CAP audiogram, with a maximum temporary hearing loss achieved between the frequencies of 12 and 16 kHz (Fig. 6A). During the first 5 d after the noise exposure, there was a partial recovery in the CAP thresholds, to reach a permanent hearing loss from 30 to 40 dB with no further improvement in hearing threshold levels over the remaining month of the experiment (Fig. 6A). This impairment observed at 30 d after exposure represents a definitive hearing loss that is permanent [i.e., a permanent threshold shift (PTS)]. When the cochlea was perfused with 10 μm D-JNKI-1, the immediate elevation of hearing threshold caused by acoustic trauma [i.e., temporary threshold shift (TTS)] was significant and was >50 dB in the 8–20 kHz range of the high frequencies. However, a clear improvement in the recovery of CAP thresholds, with a significant reduction in the initial (i.e., 20 min) TTS, was observed 1 month after acoustic trauma (Fig. 6B) without development of a significant PTS for any of the frequencies tested over the next 30 d (p < 0.05 between 4 and 6 kHz and p < 0.001 between 8 and 26 kHz).
Figure 6.

Perfusion of D-JNKI-1 into the scala tympani protected against acoustic trauma-induced permanent hearing loss. A, B, Hearing thresholds from contralateral noise-exposed, unperfused left cochleae (A; n = 6) and the noise-exposed right cochleae perfused with a 10 μm solution of D-JNKI-1 (B; n = 6) from the same animals. Hearing loss was calculated as the difference in decibels between auditory thresholds before acoustic trauma, 20 min (black circles) and 30 d (white circles) after noise exposure. Acoustic trauma (6 kHz, 120 dB SPL, 30 min) induced a maximum hearing loss of 60 dB when measured 20 min after exposure. Note the spontaneous, but incomplete, recovery of thresholds in the contralateral unperfused cochleae (A). Protection against a permanent hearing loss was clearly observed for the 10μm D-JNKI-1-treated cochleae, with an initial hearing loss (TTS) that was similar to the contralateral unperfused cochleae at 20 min but with a near complete recovery of hearing function by 30 d after exposure (B). C, D, Protective effect of 10 μm D-JNKI-1 against acoustic trauma on amplitude–intensity function of the CAP evoked by stimulation with 8 kHz tone bursts. Shown are the results obtained before (white circles) and 6 d after exposure to the acoustic trauma paradigm (black circles). Acoustic trauma induced a drastic decrease in the CAP amplitude for all intensity levels of sound stimulation (8 kHz) in the contralateral noise-exposed, unperfused left cochleae by day 6 (C). Note a near complete recovery of the amplitude–intensity function by 6 d after exposure in cochleae perfused with 10 μm D-JNKI-1 (D). A–D, All points represent mean ± SEM values calculated from six animals.
Amplitude-intensity functions recorded at day 6 from unperfused, noise-exposed cochleae showed a dramatic reduction in the CAP evoked by both low- and high-intensity stimulation of the 8 kHz frequency (Fig. 6C), suggesting that, in addition to the OHCs, IHCs were also being affected by the sound trauma paradigm. In contrast, no change was observed in the amplitude of the CAP of the D-JNKI-1-perfused right cochleae of noise exposed guinea pigs (Fig. 6D).
When compared with the unperfused cochleae, perfusion of artificial perilymph alone (i.e., 0 μm) did not modify either the acute effect of the acoustic trauma or the time course of functional recovery (Fig. 7A). When cochleae were perfused with a range of dosages of D-JNKI-1, there was an immediate elevation of CAP thresholds (TTS) because of acoustic trauma similar to those observed in the unperfused, control ears. In contrast, a clear improvement in the recovery of CAP thresholds, with a significant reduction in the development of a PTS was observed in D-JNKI-1-treated cochleae over a range of dosages (Fig. 7A). This improvement was evident as early as 2 d after the initial exposure to the noise exposure paradigm, and this sparing effect was maintained for at least 25 d after the pump had stopped delivering the D-JNKI-1 treatment solution into the scala tympani (i.e., 5 d after the acoustic trauma) (Fig. 7A). Statistical analysis of the data obtained at 8 kHz 6 d after acoustic trauma showed a significant improvement in CAP thresholds for all concentrations of D-JNK above 1 μm (Fig. 7B), and there was complete recovery to preexposure thresholds in response to treatment with 100 μm D-JNKI-1 (Fig. 7A, B). The concentration of D-JNKI-1 in the scala tympani perfusion solution required to prevent 50% of the permanent threshold shift caused by exposure to the sound trauma paradigm was 2.31 μm (Fig. 7B).
Figure 7.

Intracochlear perfusion with D-JNKI-1, but not with D-TAT-empty or inactive JNKI-1-mut peptides, prevented the hearing loss induced by exposure to the acoustic trauma paradigm. A, Shown are the functional recoveries over time of CAP threshold shifts, for a 8 kHz pure tone from contralateral control cochleae (black circles) and cochleae perfused with artificial perilymph alone (i.e., 0 μm; white circles) or with artificial perilymph containing either 10 μm (white squares) or 100 μm (white triangles) D-JNKI-1 peptide. Note the initial rapid phase of recovery within the first 2 d, followed by a slower recovery phase in all animals. D-JNKI-1 both accelerated and improved the final functional recovery of the CAP. Time-response data have been fitted to exponential curves. B, The dose-dependent effects of D-JNKI-1 at 6 d after acoustic trauma with hearing loss at 8 kHz expressed as the percentage of recovery is shown. Dose–response data were then fitted to a curve using a nonlinear least-square logistic equation. The Boltzman equation was used for fitting sigmoid curves. The EC50 was calculated as 2.31 μm. C, A comparison a 10 μm D-TAT-empty-perfused right cochleae (white circles) with contralateral unperfused left cochleae (black circles) in the same animals exposed to acoustic trauma (n = 3) showed no significant differences (i.e., both ears showed both the same level and pattern of hearing loss). D, A comparison of a 10 μm JNKI-1-mut-perfused right cochleae (black circles) with contralateral unperfused left cochleae (white circles) in the same animals exposed to acoustic trauma (n = 3) showed similar patterns of hearing losses.
In contrast to these results, animals that were implanted with minipumps containing either D-TAT-empty (n = 3) or JNKI-1-mut (n = 3) perfusate showed no significant differences between the treated right ears (i.e., subjected to acoustic trauma and perfused with either of these two control perfusion solutions) and their unperfused left ears, which were subjected to acoustic trauma alone (Fig. 7C, D).
SEM observation of the surface morphology of each organ of Corti was performed at the end of the physiological survey of hearing thresholds (i.e., 30 d after acoustic trauma). In the noise-exposed unperfused, contralateral cochleae, a massive loss of both IHCs and OHCs was found in the area located 14–16 mm from the apex (Fig. 8A). This area corresponds to the position in the cochlear duct that codes for 8–14 kHz (Cody and Johnstone, 1980; frequency map of the guinea pig cochlea). In contrast to the site of extensive focal damage documented in the unprotected cochleae, scala tympani perfusion of 10 μm D-JNKI-1 for 2 d before and for 5 d after noise exposure provided a significant level of protection to all of the cochlear hair cells present in the area that was maximally stimulated by the noise exposure paradigm (Fig. 8B). Counting all of the hair cells present over the entire length of the cochlear duct allowed us to construct cochleograms for noise-exposed, contralateral unperfused cochleae (n = 3) and noise-exposed cochleae perfused with 10 μm D-JNKI-1 (n = 3). In the contralateral unperfused, noise-exposed cochleae, most of the hair cell losses were found in the single row of IHCs and in the first row of OHCs, and to a lesser extent in the second and third rows of the OHCs (Fig. 8C). Scala tympani perfusion of 10 μm D-JNKI-1 for 2 d before, during, and for 5 d after noise exposure resulted in a clear protective effect of both the IHCs and OHCs from the permanent effects of sound trauma (i.e., PTS). In this group, only 12% of IHCs and 6% of the first row of OHCs were missing in the area of the noise-induced lesion (Fig. 8D), which is consistent with the physiological data that showed almost no PTS.
Figure 8.

Local delivery of D-JNKI-1 into the scala tympani protected against acoustic trauma-induced hair cell loss. A, B, Scanning electron micrographs of areas of acoustic trauma damage in cochleae from the same noise exposed animal. In the damaged area of the contralateral unperfused cochleae, the most severe damage was observed in the row of IHCs (I) and the first row of OHCs (O), with a gradation of damage in the second and the third rows of OHCs (A). Note that direct delivery of 10 μm D-JNKI-1 into the scala tympani of the cochlea effectively prevented acoustic trauma-induced hair cell loss (B). Scale bar, 15μm. C, D, Quantitative analysis of hair cell damage consisted of counting all hair cells along the entire length of the cochlear ducts. Cochleograms represent the mean survival of hair cells as the function of the distance from the apex (in millimeters) in contralateral unperfused cochleae (C; n = 3) and in the 10μm D-JNKI-1-perfused cochleae (D; n = 3) of the same animals. Noise exposure caused a narrow band of hair cell trauma in the cochlea located 14–16 mm from the apex of the cochlea. Ninety-one percent of the IHCs (white circles) and 43% of the OHCs were lost from this area by 30 d after the initial acoustic trauma in the unprotected cochleae (C). Note the typical gradient of loss from the first row (black circles) to the second (dark gray circles) and third (light gray circles) rows of OHCs. In contrast, only 6% OHCs and 11.9% IHCs were lost as a consequence of acoustic trauma if cochleae that were treated with local application of a 10 μm solution of D-JNKI-1 (D).
Discussion
Hair cell counts and TUNEL staining of organ of Corti explants exposed to neomycin reveal that many of the ototoxin-damaged hair cells show features characteristic of apoptosis. This result supports earlier studies that report apoptosis of aminoglycoside-damaged hair cells (Vago et al., 1998; Forge and Li, 2000; Ylikoski et al., 2002) and noise trauma-damaged hair cells (Pirvola et al., 2000; Hu et al., 2000, 2002; Wang et al., 2002). Another observation from neomycin-exposed explants is that many nuclei of damaged hair cells immunostained for phosphorylated c-Jun, indicating involvement of the MAPK–JNK signal pathway in apoptosis of damaged sensory cells. The MAPK–JNK signal pathway can be activated by a high level of cellular stress and contributes to apoptosis of ROS-damaged cells (Gupta et al., 1995; Ip and Davis, 1998). c-Jun is an important target of the MAPK–JNK pathway (Kyriakis et al., 1994; Davis, 2000), and phosphorylation of regulatory amino-terminal serine residues increases its transcriptional activity (Derijard et al., 1994; Gupta et al., 1995).
D-JNKI-1 treatment can effectively block apoptosis of insulin-secreting cells by inhibiting the MAPK–JNK signal pathway (Bonny et al., 2001). Direct evidence that MAPK–JNK signaling participates in apoptosis of ototoxin-injured hair cells comes from experiments in which D-JNKI-1 and neomycin were introduced simultaneously into organ of Corti explants. Treatment of aminoglycoside-exposed explants with D-JNKI-1 prevents: (1) phosphorylation c-Jun within hair cell nuclei; (2) TUNEL labeling of hair cells; and (3) loss of hair cells. This in vitro result and results of aminoglycoside-challenged/CEP-1347-protected cochlear explants (Pirvola et al., 2000) strongly implicate MAPK–JNK signaling as a major pathway involved in aminoglycoside-induced death of hair cells.
The inhibitory action of D-JNKI-1 is fundamentally different from the action of small chemical inhibitors (e.g., CEP-1347) (Maroney et al., 1998; Saporito, 1999; Maroney et al., 2001; Harris et al., 2002). D-JNKI-1 does not interfere with intrinsic JNK activity that might be involved in such physiological activities as differentiation (Ip and Davis, 1998) and neuritic outgrowth (Bodner et al., 2002b), but rather interrupts access of MAPK– JNK to substrates within the nucleus of a cell (Bonny et al., 2001; Barr et al., 2002).
Both c-fos and c-jun belong to a group of transcription factors called immediate early genes, which can promote apoptosis of neurons in response to cellular stress. MAPK–JNK signaling can activate ELK-1, resulting in increased c-fos transcription (Meloche et al., 1992; Traverse et al., 1994; Marshall, 1995; Halazonetis et al., 1998). The present study indicates that c-fos is involved in MAPK–JNK-mediated apoptosis and that treatment of neomycin-exposed explants with D-JNKI-1 blocks the MAPK–JNK-evoked increase in c-fos mRNA. The ability of MAPK–JNK signaling to affect both c-fos and c-jun transcription factors may, in part, explain why D-JNKI-1 is so highly effective in protecting auditory hair cells against neomycin. D-JNKI-1 provides excellent otoprotection against aminoglycoside-induced hair cell loss within the explants, however, these in vitro experiments cannot determine whether D-JNKI-1-protected hair cells retain their ability to function as receptor cells.
The results of in vivo studies with D-JNKI-1 in guinea pigs provides physiological evidence that D-JNKI-1-protected hair cells are functionally intact after both aminoglycoside exposure and sound trauma. Unilateral, local scala tympani delivery of D-JNKI-1 provides an internal control (i.e., the contralateral unperfused cochlea) to control for intra-animal variability in response to either noise exposure or neomycin ototoxicity.
In neomycin-exposed guinea pigs, the pattern of OHC losses correlates with the observed hearing loss. These patterns of neomycin-induced hair cell and hearing losses are in sharp contrast to the almost complete protection against both hair cell losses and hearing loss provided by local perfusion of D-JNKI-1.
Until now, MAPK–JNK signal pathway activation has been evident only after neomycin or gentamicin treatment or after noise exposure (Pirvola et al., 2000; Ylikoski et al., 2002). However, most aminoglycosides (e.g., kanamycin and amikacin) are known to induce ROS generation (Garetz et al., 1994; Priuska and Schacht, 1995; Clerici et al., 1996; Hirose et al., 1997; Conlon et al., 1999) and apoptotic cell death of aminoglycoside-damaged hair cells (Nakagawa et al., 1998a,b; Pirvola et al., 2000; Ylikoski et al., 2002). Because ROS can activate the JNK pathway (for review, see Finkel, 1998), it can be assumed that aminoglycosides capable of generating ROS can also activate JNK signaling. On the basis of this hypothesis, we suggest that intracochlear perfusion of D-JNKI-1 will also be effective in protecting auditory hair cells against other aminoglycosides.
It has long been known that administration of aminoglycosides early in life has a more profound effect on the viability of the auditory hair cells than administration of an equal dose in an adult. The reason(s) for this difference in aminoglycoside sensitivity between adult and young cochlear hair cells is not known. This critical period of aminoglycoside hypersensitivity occurs when hair cells are already well differentiated and functional (Pujol, 1986). The level of hair cell maturity for both young and adult animal hair cells suggests that molecular mechanisms of ototoxin-induced hair cell death are similar for both the critical period and the adult stage. Therefore, it may be expected that D-JNKI-1 treatment will also protect the cochlear hair cells from aminoglycosides during this hypersensitive period in young animals.
All noise-exposed guinea pigs have a similar initial TTS with no significant difference between the TTS of the protected (D-JNKI-1 perfused) and unprotected noise-exposed inner ears within the same animal. In contrast, significant differences in hearing thresholds are evident in noise-exposed animals tested at 1 month after exposure when the TTS converts into a PTS. The D-JNKI-1-perfused cochleae (protected) showed significantly less PTS than the contralateral unprotected cochleae. Cytocochleograms prepared 1 month after noise exposure showed that treatment with D-JNKI-1 preserves >88% of the IHCs and OHCs that were shown to be lost from noise-exposed, unprotected cochleae within the same animals.
The otoprotective effects of D-JNKI-1 are in agreement with previous studies in which systemic delivery of CEP-1347 lessened the effects of both aminoglycoside-induced (Ylikoski et al., 2002) and sound trauma-induced (Pirvola et al., 2000) hair cell and hearing losses in guinea pigs.
Both the D-JNKI-1 and the CEP-1347 studies (Pirvola et al., 2000; Ylikoski et al., 2002) target MAPK–JNK signaling, but there are major differences between these studies that include both route of delivery (i.e., systemic vs site specific) and mode of action of these compounds (i.e., D-JNKI-1 works by selectively blocking access of MAPK–JNK to their substrates within the nucleus) (Bonny et al., 2001), whereas CEP-1347 inhibits MAPK–JNK signaling at the level of the mixed lineage kinases (Harris et al., 2002). Both of these compounds (i.e., CEP-1347 and D-JNKI-1) are highly effective in preventing aminoglycoside-induced hair cell losses in vitro, and in their prevention of both hair cell and hearing losses within in vivo models of ototoxicity and sound trauma (Pirvola et al., 2000; Ylikoski et al., 2002; the present study). However, because there were many variables between the CEP-1347 studies (Pirvola et al., 2000; Ylikoski et al., 2002) and the current study, it is not possible to directly compare the otoprotective efficacy of these two MAPK–JNK signal pathway-blocking compounds.
The present study and the CEP-1347 studies both support activation of MAPK–JNK signaling as a major stress-activated cell death pathway in the mammalian cochlea. Therefore, this signaling pathway offers opportunities toward identifying other potential targets and designing other specific compounds to inhibit activation of the MAPK–JNK signal pathway. One such molecule of interest may be POSH (plenty of SH3 sec), because it can act as a scaffold protein for MAPK–JNK signaling and it appears to link activation of this cell death pathway to GTP-Rac (Xu et al., 2003). In vitro inhibitor experiments have implicated small GTPases in aminoglycoside-induced loss of auditory hair cells (Bodner et al., 2002a).
Systemic delivery of an otoprotective molecule can compromise the effectiveness of an otoprotective therapy by producing unwanted side effects (Reser et al., 1999; Li et al., 2001). Therefore, long-term intracochlear perfusion of a compound such as D-JNKI-1 may avoid unwanted side effects that may result from systemic application of this or any similar MAPK–JNK signal pathway inhibitor. The present study prevents the apoptotic cell death of auditory hair cells by pretreatment (2 d) and treatment during and after exposure by either acoustic trauma or exposure to an ototoxic aminoglycoside. However, most clinical interventions are delivered after trauma (e.g., sound trauma). In some models, including cerebral-induced and myocardial ischemia-induced apoptosis (Markgraf et al., 1998; Sandmann et al., 2002), investigators have found that treatment with calpain inhibitors can be effective when delivered for up to several hours after an initial insult. We are currently assessing whether D-JNKI-1 can be of therapeutic value when applied after either noise or aminoglycoside exposure. Preliminary results indicate that local delivery of D-JNKI-1 can rescue both hearing function and hair cells when the treatment is begun within several hours after the initial sound trauma insult.
Footnotes
This work was supported by Grant 31-56897.99 (A.Z. and F.d.R.) from the Swiss National Science Foundation. We thank two anonymous reviewers for critical reading and helpful comments on the first version of this manuscript.
Correspondence should be addressed to Dr. Azel Zine, INSERM U583, 71 rue de Navacelles, 34090 Montpellier, France. E-mail: Zine@montp.inserm.fr.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238596-12$15.00/0
References
- Barr RK, Kendrick TS, Bogoyevitch MA ( 2002) Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem 277: 10987-10997. [DOI] [PubMed] [Google Scholar]
- Bodner D, Brors D, Pak K, Gloddek B, Ryan AF ( 2002a) Rescue of auditory hair cells from aminoglycoside toxicity by Clostridium difficile toxin B, an inhibitor of the small GTPases Rho/Rac/Cdc42. Hear Res 172: 81-86. [DOI] [PubMed] [Google Scholar]
- Bodner D, Gloddek B, Ryan AF, Huverstuhl J, Brors D ( 2002b) Inhibition of the c-Jun N terminal kinase signaling pathway influences neurite outgrowth of spiral ganglion neurons in vitro Laryngoscope 112: 2057-2061. [DOI] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF ( 2001) Cell-permeable peptide inhibitors of JNK: novel blockers of β-cell death. Diabetes 50: 77-82. [DOI] [PubMed] [Google Scholar]
- Bredberg G ( 1968) Cellular pattern and nerve supply of the human organ of Corti. Acta Otolaryngol Suppl 236: 1. [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N ( 1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156-159. [DOI] [PubMed] [Google Scholar]
- Clerici WJ, Hensley K, DiMartino DL, Butterfield DA ( 1996) Direct detection of ototoxicant-induced reactive oxygen species generation in cochlear explants. Hear Res 98: 116-124. [DOI] [PubMed] [Google Scholar]
- Cody AR, Johnstone BM ( 1980) Electrophysiological and morphological correlates in the guinea pig cochlea after exposure to ‘impulsive’ noise. Scand Audiol Suppl 12: 121-127. [PubMed] [Google Scholar]
- Conlon BJ, Aran JM, Erre JP, Smith DW ( 1999) Attenuation of aminoglycoside-induced cochlear damage with the metabolic antioxidant α-lipoic acid. Hear Res 128: 40-44. [DOI] [PubMed] [Google Scholar]
- Davis RJ ( 2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239-252. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ ( 1994) JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76: 1025-1037. [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ ( 1997) A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277: 693-696. [DOI] [PubMed] [Google Scholar]
- Finkel T ( 1998) Oxygen radicals and signaling. Curr Opin Cell Biol 10: 248-253. [DOI] [PubMed] [Google Scholar]
- Forge A ( 1985) Outer hair cell loss and supporting cell expansion following chronic gentamicin treatment. Hear Res 19: 171-182. [DOI] [PubMed] [Google Scholar]
- Forge A, Li L ( 2000) Apoptotic death of hair cells in mammalian vestibular sensory epithelia. Hear Res 139: 97-115. [DOI] [PubMed] [Google Scholar]
- Garetz SL, Rhee DJ, Schacht J ( 1994) Sulfhydryl compounds and antioxidants inhibit cytotoxicity to outer hair cells of a gentamicin metabolite in vitro Hear Res 77: 75-80. [DOI] [PubMed] [Google Scholar]
- Gupta S, Campbell D, Derijard B, Davis RJ ( 1995) Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267: 389-393. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Georgopoulos K, Greenberg ME, Leder P ( 1998) c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 55: 917-924. [DOI] [PubMed] [Google Scholar]
- Harris CA, Deshmukh M, Tsui-Pierchala B, Maroney A, Johnson Jr EM ( 2002) Inhibition of the c-Jun N-terminal kinase signaling pathway by the mixed lineage kinase inhibitor CEP-1347 (KT7515) preserves metabolism and growth of trophic factor-deprived neurons. J Neurosci 22: 103-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Hockenbery DM, Rubel EW ( 1997) Reactive oxygen species in chick hair cells after gentamicin exposure in vitro Hear Res 104: 1-14. [DOI] [PubMed] [Google Scholar]
- Hu BH, Guo W, Wang PY, Henderson D, Jiang SC ( 2000) Intense noise-induced apoptosis in hair cells of guinea pig cochleae. Acta Otolaryngol 120: 19-24. [PubMed] [Google Scholar]
- Hu BH, Henderson D, Nicotera TM ( 2002) Involvement of apoptosis in progression of cochlear lesion following exposure to intense noise. Hear Res 166: 62-71. [DOI] [PubMed] [Google Scholar]
- Ip YT, Davis RJ ( 1998) Signal transduction by the c-Jun N-terminal kinase (JNK) from inflammation to development. Curr Opin Cell Biol 10: 205-219. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR ( 1994) The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369: 156-160. [DOI] [PubMed] [Google Scholar]
- Li G, Frenz D, Brahmblatt S, Feghali J, Ruben RJ, Beggren D, Arezzo J ( 2001) Round window membrane delivery of l-methyionine provides protection from cisplatin ototoxicity without compromising chemotherapeutic efficacy. Neurotoxicology 22: 163-176. [DOI] [PubMed] [Google Scholar]
- Markgraf CG, Velayo NL, Johnson MP, McCarty DR, Medhi S, Koehl JR, Chmielewski PA, Linnik MD ( 1998) Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke 29: 152-158. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight Jr E, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA ( 1998) Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci 18: 104-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL ( 2001) CEP-1357 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem 276: 25302-25308. [DOI] [PubMed] [Google Scholar]
- Marshall CJ ( 1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179-185. [DOI] [PubMed] [Google Scholar]
- Meloche S, Seuwen K, Pagés G, Pouysségur J ( 1992) Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol Endocrinol 6: 845-854. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yamane H, Takayama M, Sunami K, Nakai Y ( 1998a) Apoptosis of guinea pig cochlear hair cells following chronic aminoglycoside treatment. Eur Arch Otorhinolaryngol 255: 127-131. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yamane H, Takayama M, Sunami K, Nakai Y ( 1998b) Dose-dependent response of vestibular hair cells of guinea pigs following streptomycin ototoxiation. Acta Otolaryngol 118: 530-533. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J ( 2000) Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci 20: 43-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priuska EM, Schacht J ( 1995) Formation of free radicals by gentamicin and iron and evidence for an iron/gentamicin complex. Biochem Pharmacol 50: 1749-1752. [DOI] [PubMed] [Google Scholar]
- Pujol R ( 1986) Periods of sensitivity to antibiotic treatment. Acta Otolaryngol Suppl 429: 29-33. [DOI] [PubMed] [Google Scholar]
- Reser D, Rho M, Dewan D, Herbst L, Li G, Stupak H, Zur K, Romaine J, Frenz D, Goldbloom L, Kopke RD, Arezzo J, Van De Water TR ( 1999) l- and d-methionine provide equivalent long term protection against CDDP-induced ototoxicity in vivo, with partial in vitro and in vivo retention of antineoplastic activity. Neurotoxicology 20: 731-748. [PubMed] [Google Scholar]
- Ruben RJ ( 1967) Development of the inner ear of the mouse: a radioautographic study of terminal mitosis. Acta Otolaryngol (Stockh) 220: 1-44. [PubMed] [Google Scholar]
- Sandmann S, Prenzel F, Shaw L, Schauer R, Unger T ( 2002) Activity profile of calpains I and II in chronically infarcted rat myocardium-influence of the calpain inhibitor CAL 9961. Br J Pharmacol 135: 1951-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporito MS, Brown EM, Miller MS, Carswell S ( 1999) CEP-1347/KT-7515, an inhibitor of c-Jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo J Pharmacol Exp Ther 288: 421-427. [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ ( 2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870-874. [DOI] [PubMed] [Google Scholar]
- Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P, Ullrich A ( 1994) EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr Biol 4: 694-701. [DOI] [PubMed] [Google Scholar]
- Vago P, Humbert G, Lenoir M ( 1998) Amakacin intoxication induces apoptosis and cell proliferation in rat organ of Corti. NeuroReport 9: 431-436. [DOI] [PubMed] [Google Scholar]
- Vives E, Brodin P, Lebleu B ( 1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 272: 16010-16017. [DOI] [PubMed] [Google Scholar]
- Wang J, Dib M, Lenoir M, Vago P, Eybalin M, Hameg A, Pujol R, Puel JL ( 2002) Riluzole rescues cochlear sensory cells from acoustic trauma in the guinea-pig. Neuroscience 111: 635-648. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ ( 1998) Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem Sci 23: 481-485. [DOI] [PubMed] [Google Scholar]
- Xu Z, Kukekov NV, Greene LA ( 2003) POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 22: 252-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylikoski J, Xing-Qun L, Virkkala J, Pirvola U ( 2002) Blocade of c-Jun N-terminal kinase pathway attenuates gentamicin-induced cochlear and vestibular hair cell death. Hearing Res 166: 33-43. [DOI] [PubMed] [Google Scholar]
- Zine A, de Ribaupierre F ( 1998) Replacement of mammalian auditory hair cells. NeuroReport 9: 263-268. [DOI] [PubMed] [Google Scholar]
- Zine A, de Ribaupierre F ( 1999) Tissue-specific levels and cellular distribution of epidermal growth factor receptors within control and neomycin-damaged neonatal rat organ of Corti. J Neurobiol 38: 313-322. [PubMed] [Google Scholar]