

Abstract
The specific mechanisms underlying general anesthesia are primarily unknown. The intravenous general anesthetic etomidate acts by potentiating GABAA receptors, with selectivity for β2 and β3 subunit-containing receptors determined by a single asparagine residue. We generated a genetically modified mouse containing an etomidate-insensitive β2 subunit (β2 N265S) to determine the role of β2 and β3 subunits in etomidate-induced anesthesia. Loss of pedal withdrawal reflex and burst suppression in the electroencephalogram were still observed in the mutant mouse, indicating that loss of consciousness can be mediated purely through β3-containing receptors. The sedation produced by subanesthetic doses of etomidate and during recovery from anesthesia was present only in wild-type mice, indicating that the β2 subunit mediates the sedative properties of anesthetics. These findings show that anesthesia and sedation are mediated by distinct GABAA receptor subtypes.
Keywords: GABA, transgenic, etomidate, general anesthesia, sedation, loreclezole, GABAA β2 subunit
Introduction
The way in which general anesthetics produce their CNS effects is still not clear. Historically, general anesthetics were thought to interfere with membrane fluidity, affecting the functioning of cellular processes. However, growing evidence indicates that general anesthetics behave in a more specific manner by interacting with proteins located within the plasma membrane (Franks and Lieb, 1994). A number of ion channels that control neuronal excitability have now been shown to be targets for clinically relevant concentrations of many anesthetic agents (Krasowski and Harrison, 1999; Thompson and Wafford, 2001). GABAA receptors are likely targets for anesthetic agents because they constitute the major inhibitory neurotransmitter system in the CNS. In this respect, the intravenous anesthetic etomidate is of particular interest. At clinically relevant concentrations, this anesthetic has little or no effect on a range of transmitter-gated ion channels, including the glycine (α1), neuronal nicotinic (α4β2 and α7), 5-HT3 (5-HT3A), AMPA (GluR1/GluR2), and NMDA (NR1a/NR2A) receptors, but greatly enhances the actions of GABA acting at the GABAA receptor (Belelli et al., 2003). Furthermore, the GABA-enhancing actions of etomidate are enantioselective and has selectivity in vitro for β2- and β3-containing receptors over β1 subunit-containing receptors (Hill-Venning et al., 1997; Tomlin et al., 1998; Belelli et al., 2003). This selectivity is entirely dependent on an asparagine residue at position 265 in the transmembrane 2 domain of the β2 subunit that is not shared by other anesthetics such as pentobarbital (Belelli et al., 1997). The activity of the anticonvulsant loreclezole is also governed by this same residue (Wingrove et al., 1994) and provides another useful tool in this study. Here we used a gene-targeting approach, combined with the unique selectivity profile of etomidate to investigate mechanisms underlying anesthesia. β2-containing receptors make up ∼50% of GABAA receptors in the CNS (McKernan and Whiting, 1996) and therefore have a major contribution to inhibitory neuronal transmission. We therefore used gene targeting to generate a mouse containing a mutation in the β2 subunit (β2 N265S) in which etomidate will selectively modulate β3-containing receptors. Here we demonstrate that sedative and anesthetic effects are mediated by different GABAA receptor subtypes. We also demonstrate that recovery of function in the β2 N265S mouse is considerably improved after etomidate anesthesia. This indicates that GABAA β3-selective agents could be used clinically to produce surgical anesthesia with a significantly improved recovery profile.
Materials and Methods
All animal experiments were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and associated guidelines.
Generation of GABAA receptor β2 N265S mutant mice
A murine 129/SvEv λ fixII library (Stratagene, LaJolla, CA) was screened using a cDNA probe containing part of the GABAA receptor β2 gene as a probe. Two positive hybridizing λ clones were subcloned into pBluescript and the full 17.5-kb-long DNA sequence, which contained exons 6, 7, and 8 of the β2 subunit gene, was determined. The Asp 265 to Ser 265 codon change was labeled by the novel restriction endonuclease restriction site ScaI, which was introduced by site-directed mutagenesis using oligonucleotide 5′-ACCACAATCAGTACTCACCTCCGGG-3′. An 8-kb-long DNA fragment containing the β2 N265S mutation (long arm) and a 2 kb DNA fragment (short arm) were cloned into the pBS246-neo-tk-1 multiple utility targeting vector (Collinson et al., 2002). After linearization with NotI, the targeting vector was introduced into AB2.2 embryonic stem (ES) cells (Lexicon Genetics, The Woodlands, TX) as described previously (Rosahl et al., 1993). Homologous recombinants were identified by PCR using the oligonucleotides P1 5′-CTATGATGCCTCTGCTGCACGGGTTGC-3′ and P2 5′-GGATGCGGTGGGCTCTATGGCTTCTGA-3′ and were further confirmed by genomic Southern blotting. Presence of the β2 N265S mutation was confirmed by cutting the 800-bp-long PCR product amplified by primers P3 5′-AACCTTTCATCTTTAGTCCCCTGG-3′ and P4 5′-TGGAAATTTGAGCAGCCCATTGTG-3′ with the restriction endonuclease ScaI. Correctly targeted ES cell clones were injected into C57BL/6 blastocysts, and one clone gave rise to highly chimeric males, which transmitted the targeted allele into the germ line. β2N265S +/- (heterozygous; F1 generation) mice were further bred with a cre-transgenic mouse (Schwenk et al., 1995) to remove the neomycin resistance gene in their offspring. cre-mediated recombination was confirmed by PCR using oligonucleotides P5 5′-AGATCTAGGTGATGACTGTC-3′ and P6 5′-AGACAGACGCCACATCACAC-3′. By additional breeding, wild-type and homozygousβ2 N265S mice were generated using a randomized breeding strategy and were kept in a mixed 75% C57BL/6–25% 129SvEv genetic background. Mice were genotyped using oligonucleotides P5 and P6 (see above), which amplified a 1.1 kb (wild-type) and 1.2 kb (targeted allele including the loxP site), respectively.
Quantitative immunoblotting
Whole brains from heterozygous, homozygous β2 N265S, and wild-type mice were collected and frozen on dry ice. Brains were homogenized for the preparation of P2 membrane fractions in 50 mm Tris-HCl, pH 7.4, and 320 mm sucrose buffer containing appropriate protease inhibitors. The resulting samples were equalized for protein concentration, and 25–100 μg were analyzed by reducing SDS-PAGE using 7% Tris-acetate minigels (Novex, San Diego, CA). After Western transfer, nitrocellulose membranes were immunoblotted using 1 μg/ml of either rabbit anti-β1 (334–360), anti-β2 (375–429), or anti-β3 (370–433) specific antibodies and a 1:5000 dilution of mouse anti-actin monoclonal antibody (Abcam, Cambridge, UK). IRDye800 anti-rabbit (Rockland Immunochemicals, Gilbertsville, PA) and AlexaFluor 680 (Molecular Probes, Eugene, OR) anti-mouse infrared labeled secondary antibodies were applied simultaneously at a 1:5000 dilution. Immunoreactive intensities were scanned and quantified using an Odyssey Infrared Imager (LI-COR Biosciences, Cambridge, UK). Independent immunoblotting experiments were repeated at least twice.
Autoradiography
Brains from five male wild-type and four male β2 N265S mice were collected and immediately dipped in ice-cold isopentane. Cryostat-cut sections (14 μm) were then processed for t-[35S]butylbicyclophosphorothionate ([35S]TBPS) autoradiography. Slide-mounted coronal sections were washed for 10 min in 50 mm Tris/citrate and 200 mm NaBr, pH 7.4, buffer and then incubated in the same buffer containing 8 nm [35S]TBPS. The modulation of [35S]TBPS binding was studied in the presence of 5 μm loreclezole, and nonspecific binding was determined by the addition of 10 μm picrotoxin. After 90–120 min incubation at room temperature, slides were washed twice for 5 min in cold buffer, rinsed in distilled water, and exposed to film (Hyperfilm; Amersham Biosciences, Arlington Heights, IL). Autoradiograms were analyzed with MCID M2 imaging system (Imaging Research, St. Catharines, Ontario, Canada), and optical densities from various brain areas were computed. Optical densities from three to four sequential slices from each animal were combined to give a mean value for each brain region per animal.
Electrophysiology
Dissociated Purkinje neuronal recordings. Cerebellum was removed from wild-type and β2 N265S mutant mice at postnatal days 11–17, and the vermal layer was isolated and placed into ice-cold oxygenated dissociation media containing (in mm): 82 Na2SO4, 30 K2SO4, 5 MgCl2, 10 HEPES buffer, and 10 glucose, pH 7.4. Tissue was then prepared as described previously (Sur et al., 2001), and cells were plated onto a glass coverslip and left to settle for at least 30 min before use. Their characteristic size and morphology identified Purkinje cell bodies. Cells could be used for up to 5 hr after preparation. Glass coverslips containing the dissociated cells were placed in a Perspex recording chamber on the stage of a Nikon (Tokyo, Japan) Diaphot inverted microscope. Cells were perfused continuously with artificial CSF (aCSF) containing (in mm): 149 NaCl, 3.25 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 11 d-glucose, and 22 d(+)-sucrose, pH 7.4, and were observed with phase-contrast optics. Fire-polished patch pipettes were pulled on a WZ, DMZ universal puller (Zeitz Instrumente, Munich, Germany) using conventional 120TF-10 electrode glass. Pipette tip diameter was ∼1.5–2.5 μm, with resistances ∼4 MΩ. The intracellular solution contained the following: 130 mm CsCl, 10 mm HEPES, 10 mm BAPTA-Cs, 5 mm ATP-Mg, 0.1 mm leupeptin, 1 mm MgCl2, and 100 μm NaCO3, pH adjusted to 7.3 with CsOH and 320–340 mOsm. Cells were voltage clamped at -60mV using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Drug solutions were applied to the cells through a multi-barrel drug delivery system, which pivot the barrels into place using a stepping motor. This ensured rapid application and washout of the drug within 12–15 msec. GABA was applied to the cell for 5 sec with a 30 sec washout period between applications. Allosteric potentiation of GABAA receptors was measured relative to a GABA EC20 determined for each cell to account for differences in GABA potency. Modulators were preequilibrated for 30 sec before coapplication of GABA.
Cerebellar slice recordings. Cerebellar slices were prepared from wild-type or β2 N265S mice of either sex (postnatal days 16–25) according to standard protocols. Animals were killed by cervical dislocation, and the brain was then rapidly removed and placed in ice-cold aCSF containing the following (in mm): 225 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 0.5 CaCl2, 10 d-glucose, 10 MgSO4, 1 ascorbic acid, and 3 pyruvic acid (osmolarity of 328–335 mOsm). Parasagittal cerebellar slices (250 μm thick) were cut using a Leica (Nussloch, Germany) vibratome at 0–4°C and subsequently incubated at 30–32°C in a holding chamber with extracellular solution (containing 126 mm NaCl, 2.95 mm KCl, 26 mm NaHCO3, 1.25 mm NaH2PO4, 2 mm CaCl2, 10 mmd-glucose, and 2 mm MgCl2 2, pH 7.4 when bubbled with 95% O2–5% CO2, and osmolarity of 300–310 mOsm, supplemented with 10 μm AP-5) for 1 hr before recording. Slices were then kept at room temperature in extracellular solution with a continuously refreshed atmosphere of 95% O2–5% CO2 for up to 8 hr. Whole-cell patch-clamp recordings were made at 35°C from submerged cerebellar Purkinje neurons in extracellular recording solution (see above), additionally containing 2 mm kynurenic acid and 0.5 μm tetrotodoxin to block ionotropic glutamate receptors and action potentials, respectively. Purkinje cells were visually identified with an Olympus Optical (Tokyo, Japan) BX50WI microscope equipped with differential interference contrast–infrared optics. Patch pipettes were prepared from thick-walled borosilicate glass (1.55 mm outer diameter; Garner Glass, Claremont, CA) using a PP830 Pipette Puller (Narishige, Tokyo, Japan) with a resistance of ∼3–4MΩ. Electrodes were filled with intracellular solution containing (in mm): 135 CsCl, 10 HEPES, 10 EGTA, 2 MgCl2, 2 Mg 2+-ATP, and 5 QX-314, pH 7.2–7.3 with CsOH, and osmolarity of ∼300 mOsm). Miniature IPSCs (mIPSCs) were recorded at a holding potential of -60 mV using an Axopatch 1D amplifier (Axon Instruments) and filtered at 2 kHz with an eight-pole low-pass Bessel filter. Series resistance was between 7 and 20 MΩ and was compensated up to 80%. Only cells with stable access resistance were used. mIPSCs were recorded on a DTR 1204 digital tape recorder (Bio-Logic Science Instruments, Claix, France) and at a later stage digitized using a DigiData 1200A/B Series Interface (Axon Instruments) at a sampling rate of 10 kHz.
Drugs were applied via the perfusion system (2–4 ml/min) and were allowed to infiltrate the slice for a minimum of 5 min before recordings were acquired. Vehicle (ethanol and DMSO; maximum of 0.3%) had no effect on any of the mIPSC parameters.
Data was analyzed using the Strathclyde Electrophysiology Software (electrophysiology data recorder; whole cell analysis program; J. Dempster, Department of Physiology and Pharmacology, University of Strathclyde, Glasgow, UK). Individual mIPSCs were detected (offline) using a software trigger (-5 pA threshold, duration of 3 msec). For each experiment, the detected events were examined manually, and any noise that falsely met the trigger specifications was rejected. For analysis of the decay phase of mIPSCs, events were selected only if they met the following criteria: (1) traces containing overlapping events were discarded; (2) events had to have a stable baseline before and after the event; and (3) events had to return completely to baseline after the event. To average mIPSCs for each cell, the histogram distribution of the rise time was plotted, and those events that fell within a Gaussian distribution were used for additional analysis. Rise time histograms generally displayed a normal distribution with a clear peak <0.6 msec. Peak amplitudes, rise times, time for events to decay to 90% (T90), and decay time constants were calculated for a minimum of 50 averaged mIPSCs per cell. Decay time constants were calculated by fitting either of the following: a one-exponential equation in which f(t) = Aexp(-t/τ); or a two-exponential equation in which f(t) = Asexp(-t/τf) + Afexp(-t/τs), where τf and τs represent the fast and slow time constants, and Af and As represent the current contribution of the fast and slow components, respectively, to the resulting averaged mIPSC. An improvement of the fit by two exponentials compared with one resulted in a reduction in the SD of the residuals and was confirmed by use of the F test.
Frequency of mIPSCs in each cell was determined by counting the number of events in a 30–60 sec period.
Because all resultant mIPSCs were best fit by a two-exponential equation, the weighted mean τ (τw) was also determined using the following equation: τw = [Af/(Af + As)] * τf + [As/(Af + As)] * τs, where parameters τf, τs, Af, and As are the same as above.
Neuroscreen of the β2 N265S mouse
A neurological screen was performed on the β2 N265S mice to investigate whether the point mutation has any behavioral phenotype. β2 N265S mice did not differ from wild-type mice in body weight, rectal temperature, grip strength, beam balancing, or swimming ability.
Behavioral studies
F4 generation mice, aged 2–5 months, were used for the intraperitoneal loss of righting reflex (LORR) and low-dose locomotor and rotarod experiments. The intravenous LORR, loss of pedal withdrawal reflex (LOPWR), and recovery rotarod experiments used F7 mice at 7 months of age. The EEG studies used 7-month-old F4 mice.
Loss of righting reflex. Wild-type and β2 N265S mice received intraperitoneal injections of either etomidate (20–40 mg/kg; n = 10–11) or pentobarbital (50 mg/kg; n = 11) or intravenous tail vein injections of etomidate (5–15 mg/kg; n = 6–8) or propofol (30 mg/kg; n = 5) and were then placed into a clean cage. The time to LORR was recorded. The mice were then laid on their backs, and the time until they could right themselves was recorded. To reduce heat loss, a heat lamp was suspended above the supine mice to maintain the air temperature at 27 ± 2°C.
Loss of pedal withdrawal reflex. Wild-type and β2 N265S mice received intravenous injections (flow rate, 300 μl/min) of etomidate (5–15 mg/kg; n = 7–8) or propofol (30 mg/kg; n = 7–8). The mice were then laid on their back. Again, a heat lamp was suspended above the supine mice to maintain air temperature at 27 ± 2°C. The hindlimb of each subject was slightly extended, and the interdigital webbing of the foot was pinched firmly by atraumatic forceps. The forceps were fitted with a block to prevent full closure, thus allowing uniform application of pressure. A clear attempt to withdraw the limb was recorded as regaining the pedal withdrawal reflex. This was tested, in alternative limbs, every 1 min in the case of mice treated with 5 mg/kg etomidate and propofol and every 3 min in the other treatment groups, until withdrawal reflex was regained.
Spontaneous locomotor activity. Naive wild-type and β2 N265S mice were dosed with etomidate (0.3–12.5 mg/kg, i.p.) and immediately placed in activity chambers as described previously (Reynolds et al., 2003). Cage crossing activity (i.e., consecutive breaks of beams on either side of the cage) was recorded for 20 min. This experiment was run on separate groups of mice over three dose ranges (0.3–3, 3–7.5, and 7.5– 12.5 mg/kg) for the β2 N265S mice with a vehicle group in each experiment. There were no differences in the vehicle responses or the overlap doses between experiments, and so the three data sets have been combined. The final group sizes were as follows: n = 31 for vehicle; n = 12 for 0.3, 1, and 5 mg/kg; n = 24 for 3 mg/kg; n = 18 for 7.5 mg/kg; and n = 6 for 10 and 12.5 mg/kg.
Rotarod. Mice were trained to remain on a rod (constant speed model 7600; Ugo Basile, Comerio, Italy) revolving at 16 rpm until they could perform three consecutive 120 sec trials. Mice were then dosed with vehicle (n = 5 for wild type and n = 6 for β2 N265S) or etomidate (5 or 7.5 mg/kg, i.p.; n = 10 for wild type and n = 11 for β2 N265S) and, 5 min later, were given a single rotarod trial. The time that the mouse could remain on the rotarod was recorded up to a maximum of 120 sec. For the experiments examining recovery of motor coordination after anesthesia, trained mice were intravenously dosed and handled as described in the LORR experiments. For each dose of etomidate (n = 6–8), rotarod testing began when the mouse recovered its righting reflex. Mice were tested every 3 min for a total of 30 min. Between trials, the mice were placed back in their home cages.
EEG studies
Male β2 N265S mice and wild-type controls (n = 4 each genotype) were implanted with radiotelemetry transmitters (Data Sciences International, St. Paul, MN) under isoflurane anesthesia for the recording of EEG activity (unilateral electrode coordinates were -2 mm anteroposterior and -2.2 mediolateral from lambda and bregma). Mice were allowed to recover for at least 14 d before the experiment.
Anesthesia experiment. Mice were restrained in a tube, and etomidate was administered as a single bolus via the lateral tail vein at doses of 10 and 12.5 mg/kg. Mice were returned to their home cage for collection of EEG (sampling rate, 250 Hz) data. EEG data were analyzed (Somnologica 3.1; Flaga, Reykjavik, Iceland) in 100 msec epochs for percentage suppression versus burst activity per minute. Isoelectric “suppression” activity was defined as waveform data <5μV in amplitude (typical peak-to-peak amplitude of “burst” activity, 250 μV).
Assessment of sedation experiment. Amount of slow-wave sleep (SWS) was assessed under baseline conditions on experimental day 1 by scoring 8 sec epochs of EEG and activity data for “wake” or “slow-wave sleep” according to standard electrophysiological criteria (van Gool and Mirmiran, 1986). On experimental day 2, after recovery from etomidate anesthesia, the amount of sleep per hour was compared with the same circadian hour the previous day.
Statistics
In vitro electrophysiology. All results are reported as the arithmetic mean ± SEM. All normalized data are expressed as percentage of control, and statistical significance was assessed using the Student's t test or repeated-measures ANOVA as appropriate.
Autoradiography. Optical densities were analyzed with unpaired t tests to determine the effect of genotype and addition of loreclezole.
Quantitative Western blotting. All β subunit optical densities are arithmetic mean ± SD. Values are normalized to actin optical densities and are expressed as percentage of the maximum intensity. Statistical differences between genotype were assessed using unpaired t tests.
Behavioral experiments. LORR, LOPWR, locomotor activity, and low-dose rotarod experiments were analyzed using two-way ANOVA, with genotype and drug dose as the two factors. When a significant effect was found, a Newman–Keuls post hoc test was then used to compare between genotypes at a given drug dose. The recovery rotarod experiment was analyzed using two-way repeated-measures ANOVA for the three doses of etomidate (factors of genotype and drug dose) and a one-way repeated-measures ANOVA for the propofol-treated mice. For the EEG experiments, the Mann–Whitney test was used to test for statistical significance between genotypes.
Drug preparation
In vitro electrophysiology. All drugs were made initially as concentrated stock solutions (10 -2m) and subsequently diluted in extracellular solution. R(+)Etomidate (Organon, West Orange, NJ), bicuculline methobromide (Sigma, St. Louis, MO), and pentobarbitone (Aldrich, St. Louis, MO) were prepared in H2O and diluted in extracellular solution to the desired concentration. Loreclezole (Janssen Pharmaceutica, Titusville, NJ) was prepared in DMSO.
Behavioral experiments. Commercially available etomidate (Hypnomidate; Janssen Pharmaceutica) was administered as a 2 mg/ml solution in 35% propylene glycol–water for injection. Commercially available propofol (Rapinovet; Schering-Plough Research Institute, Union, NJ) was administered as a 10 mg/ml solution in an aqueous isotonic emulsion.
Results
The β2 N265S mutant mice were generated in a similar way to that described previously for the α1H101R mutant mice (McKernan et al., 2000). The site-directed codon change from N265 to S265 was introduced into exon 8 of the murine β2 subunit gene of the GABAA receptor by homologous recombination and confirmed at the DNA level (Figs. 1, 2A). Northern blot data confirmed normal expression of the β2 subunit gene in the β2 N265S mice (data not shown). Quantitative immunoblot analysis of whole-brain membranes using β subtype-specific antibodies (Fig. 2B–G) showed there was no change in the expression of any β subunit in heterozygous and homozygous β2 N265S mice compared with wild type. The quantitative analysis of autoradiograms (Fig. 2) revealed no changes in the total number of [35S]TBPS binding sites in the cortex, hippocampus, striatum, and thalamus of β2 N265S mice compared with wild-type mice, indicating that the introduced point mutation is without effect on the expression of β2-containing receptors. However, striking differences in the [35S]TBPS binding were observed between wild-type and β2 N265S animals in the presence of loreclezole, a selective modulator of β2 and β3 subunit-containing GABAA receptors. Indeed, this anticonvulsant greatly reduced [35S]TBPS binding in wild-type (in which binding to both β2- and β3-containing receptors will be allosterically inhibited) (Fig. 2J) compared with β2 N265S (in which only β3-containing receptors will be modulated) (Fig. 2K) mice. For example, reduction of >50% of [35S]TBPS binding was found in cortex, hippocampus, and striatum of wild-type mice, whereas [35S]TPBS signal was decreased only by 16–35% in these regions in β2 N265S mice (Table 1). This was particularly striking in thalamus, a region expressing high levels of β2 in which 47% was displaced in wild-type mice compared with only 17% in the β2 N265S mutant mice. Altogether, the data confirmed that the N265S mutation has been successfully introduced in the mutant mice without changing the expression level of the β2 subunit of the GABAA receptor.
Figure 1.

Generation of β2 N265S mutant mice. Targeting strategy for the GABAA receptor β2 N265S mice. Schematic representation of the wild-typeβ2 allele of the GABAA receptor (a) and targeting vector including the site-specificβ2N265S mutation indicated by an asterisk (b). E, EcoRI; K, KpnI restriction sites; 6, 7, 8, exons 6, 7, and 8, respectively; black arrowhead, loxP site; neo, neomycin resistance gene; TK, thymidine kinase gene; P1–P6, oligonucleotides used for PCR (see Materials and Methods). Targeted β2 allele after homologous recombination before (c) and after (d) cre-mediated excision of the loxP flanked neomycin cassette.
Figure 2.
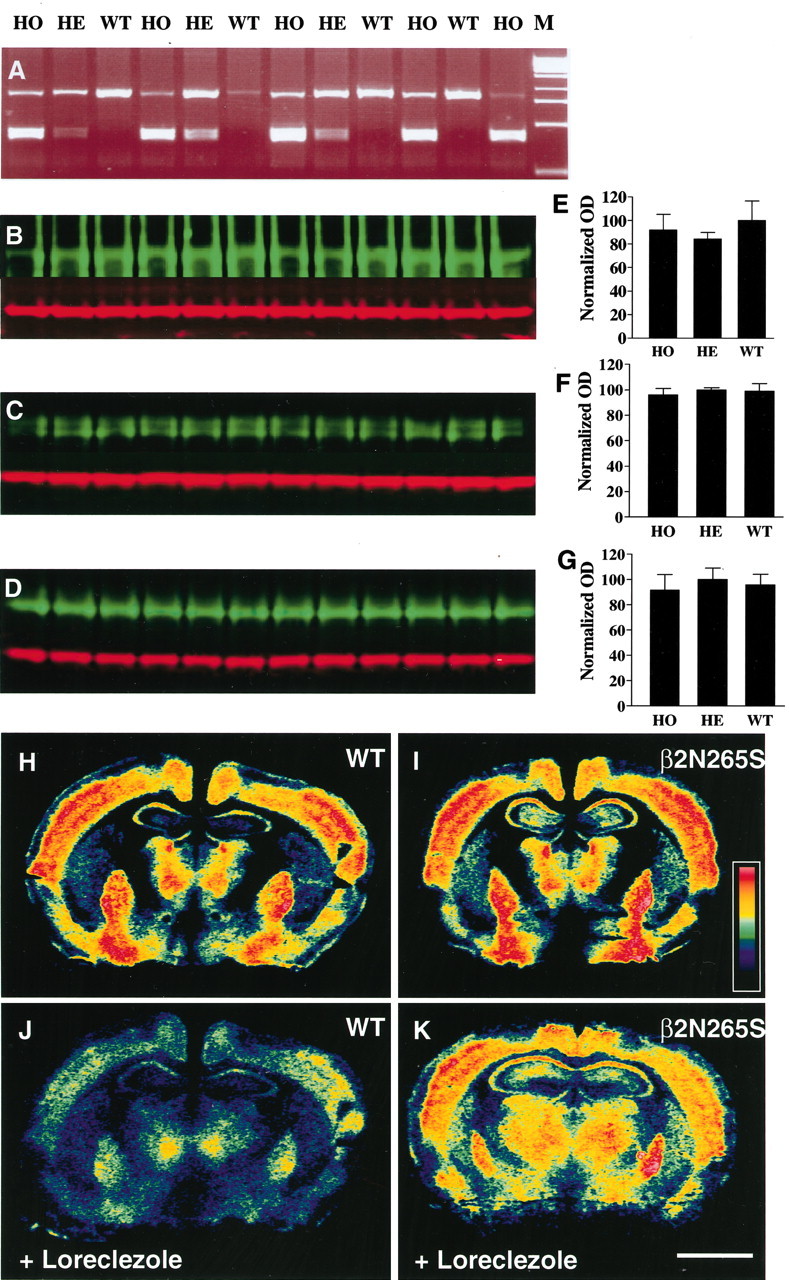
In vitro validation ofβ2 N265S mice. Genotyping examples of tail biopsy samples from wild-type (WT), heterozygous (HE), and homozygous (HO) mice using primers P3 and P4 (see Material and Methods) and digestion of the resulting PCR product with the restriction endonuclease ScaI (A). The 800 bp PCR product is nearly fully cut into two 400 bp fragments for homozygous, less cut for heterozygous, and not cut at all for wild-type samples. M, Marker. Quantitative Western blots showing analysis of P2 membranes from wild-type (WT), heterozygous (HE), and homozygous (HO) mice using anti-β1 (B), anti-β2 (C), and anti-β3 (D) antibodies (green). Immunoreactive intensities were quantified and normalized against anti-actin controls (red). The comparison of β subunit expression across genotypes is represented graphically in E–G. OD, Optical density. Example autoradiographs of brain sections from wild-type (H, J) and β2 N265S (I, K) mice showing [35S]TBPS binding sites. In J and K, [35S]TBPS binding was performed in the presence of 5 μm loreclezole to allosterically inhibit binding toβ2 andβ3 subunit-containing receptors. Scale bar, 2 mm.
Table 1.
Loreclezole inhibition of [35S]TBPS binding
|
Brain |
Wild-type percentage |
β2 N2655 percentage inhibition |
|---|---|---|
| regiona
|
inhibition by loreclezole
|
by loreclezole
|
| Cortex | 54.3 ± 0.75 (5) | 35.1 ± 2.7 (4)** |
| Hippocampus | 54.1 ± 4.2 (5) | 35.3 ± 8.7 (4) |
| Striatum | 55.6 ± 3.2 (5) | 28.5 ± 1.9 (4)** |
| Thalamus
|
46.6 ± 4.6 (5)
|
16.5 ± 5.7 (4)*
|
[35S]TBPS autoradiograms were analyzed with an MCID M2 system, and optical densities from various brain regions were determined under different pharmacological conditions in wild-type and β2 N2655 mice. Data represent the percentage inhibition of [35S]TBPS-specific binding by loreclezole in different brain regions and are the mean ± SD. Brackets indicate the number of individual animals used. *p < 0.01 compared with the inhibition in wild type; **p < 0.001 compared with the inhibition in wild type.
[35S]TBPS binding in the absence of loreclezole was not significantly different between genotypes
In vitro modulation of β2-containing receptors
Cerebellar Purkinje neurons express predominantly α1β2γ2 GABAA receptors (Laurie et al., 1992a) and are consequently useful tools for evaluating the effects of genetic manipulation of any of these subunits. Cerebellar neurons from 11- to 17-d-old mice were dissociated, and visually identified Purkinje cell bodies were used to study the pharmacology of GABAA receptors expressed therein. Application of GABA elicited currents that were equivalent amplitude in both wild-type and β2 N265S mice (wild type, 2963 ± 276 pA, n = 11; β2 N265S, 3067 ± 296, n = 14). The EC20 concentration for GABA used in subsequent experiments was also unchanged (wild type, 3.1 ± 0.3 μM, n = 11; β2N265S, 3.9 ± 0.4 μM, n = 14), indicating no difference in GABA potency. Submaximal GABA responses in wild-type Purkinje neurons were potentiated by coapplication of 3 μM etomidate or 3 μM loreclezole; however, in neurons isolated from β2 N265S mice, this potentiation was absent or severely reduced (Fig. 3a,b). In contrast, potentiation of GABA responses by the barbiturate pentobarbital was unaffected (Fig. 3c). Analysis of mIPSCs from Purkinje neurons in cerebellar slices revealed no difference in the amplitude and kinetic properties of these currents (Fig. 3d) or frequency of occurrence (wild type, 16.5 ± 3.6 Hz, n = 15; β2 N265S, 15.8 ± 2.9 Hz, n = 15). On application of 10 μM etomidate to slices from wild-type mice, the currents showed an increase in peak amplitude (133 ± 21 to 168 ± 28 pA; p < 0.05) and a marked prolongation of the decay phase of the current (τw of 2.8 ± 0.2 msec compared with 29.2 ± 5.8 msec; p < 0.01). In β2 N265S mice, there was no significant increase in peak amplitude (147 ± 13 to 163 ± 13 pA) and a reduction in the prolongation of the decay in the presence of etomidate (τw of 2.4 ± 0.1 msec compared with 7.3 ± 0.3 msec; p < 0.01) (Fig. 3e). Cumulative probability plots of time taken to reach 90% of decay (Fig. 3f) illustrate the prolongation of the synaptic current by etomidate and the reduced effect in the β2 N265S mice.
Figure 3.

Electrophysiological properties of cerebellar Purkinje neurons in wild-type andβ2 N265S mice. Representative recordings from dissociated cerebellar Purkinje neurons isolated from wild-type or β2 N265S mice in response to a predetermined EC20 concentration of GABA, in the absence and presence of 3 μm loreclezole (a), 3 μm etomidate (b), and 100 μm pentobarbital (c). Associated histograms illustrate mean potentiation ± SEM from four or more cells in each case. d, Overlayed averaged mIPSCs recorded from Purkinje cells from wild-type (dark) and β2 N265S (light) mice. e, Representative traces illustrating the effect of 10 μm etomidate on wild-type (i) and β2 N265S (ii) mIPSCs from Purkinje cells. f, Cumulative probability plots for T90 values in the presence and absence of 10μm etomidate obtained from an exemplar neuron of wild-type (i) and β2 N265S (ii) mice show that etomidate preferentially prolonged mIPSC decay of wild-type mice [from 6.32 ± 0.47 to 61.9 ± 10.55 msec (n = 5) in wild-type mice, p < 0.01, paired Student's t test; from 5.56 ± 0.21 to 18.52 ± 0.62 msec (n = 5) inβ2 N265S mice, p < 0.001, paired Student's t test] Mean percentage increase of T90 values by 10 μm etomidate from five neurons was 927 ± 220 and 225 ± 14 for wild-type andβ2 N265S mice, respectively (p < 0.01 by repeated-measures ANOVA).
In vivo response to anesthetics
The anesthetic effects of etomidate in β2 N265S mice were first investigated using the LORR test (Fig. 4a). The time taken for mice to lose their righting reflex after intraperitoneal injection of etomidate did not show dose dependence and was not different between the two genotypes (wild type, 125 ± 16 sec; β2 N265S, 125 ± 6.0 sec at 30 mg/kg). The duration of LORR induced by intraperitoneal etomidate was highly dose dependent, and wild-type and β2 N265S mice displayed very similar LORR times when compared across the three doses of etomidate tested. Likewise, another class of anesthetic (pentobarbital, a barbiturate) produced similar LORR durations in both wild-type and β2 N265S mice. None of the intraperitoneal doses of etomidate produced a robust LOPWR (data not shown). Intravenous administration of etomidate also produced a dose-dependent LORR in both genotypes, although the duration of LORR was significantly longer (p < 0.05 for 10 and 15 mg/kg, i.v.) in the wild-type mice at the higher doses (Fig. 4a). A 5 mg/kg intravenous dose of etomidate was not sufficient to induce LOPWR (Fig. 4b), but higher doses of 10 and 15 mg/kg intravenously produced a dose-dependent increase in LOPWR duration. β2 N265S mice showed a shorter LOPWR duration than wild-type mice (genotype, F(1,41) = 13.4; p = 0.0007), and this effect was dose dependent (genotype × drug dose interaction, F(2,41) = 5.25; p = 0.009). An intravenous anesthetic that is unaffected by the point mutation propofol did not show any differences between wild-type and β2 N265S mice in either LORR (Fig. 4a) or LOPWR (Fig. 4b).
Figure 4.

Etomidate-induced LORR is preserved but sedation is decreased in β2 N265S mice. The duration of LORR (a; mean ± SEM duration) induced by intraperitoneal etomidate was very similar for both wild type (WT) and β2 N265S at all three doses of etomidate examined (F(1,57) < 0.005; p > 0.1). Likewise, 50 mg/kg intraperitoneal pentobarbital produced similar LORR durations in both wild-type andβ2 N265S mice (F(1,20) = 2.24; p > 0.1). n = 10–11 for etomidate and pentobarbital groups. Intravenous etomidate produced a dose-dependent increase in LORR duration, although this was significantly shorter for the β2 N265S mice at the higher doses (n = 6–8). Propofol, an intravenous anesthetic not effected by the point mutation, gave a similar LORR in both genotypes (n = 5). Intravenous etomidate produced a dose-dependent increase in LOPWR (b; mean ± SEM duration), although this was shorter in theβ2 N265S mice (n = 7–8). Low doses of etomidate (0.3–7.5 mg/kg, i.p.) dose-dependently decreased spontaneous cage crossings (c; exploration–locomotion) in wild-type mice. In contrast, these doses of etomidate did not reduce activity inβ2N265Smice. At higher doses of etomidate (10 and 12.5 mg/kg), both wild-type andβ2 N265S mice behaved similarly (n = 31 for vehicle; n = 12 for 0.3, 1, 3, and 5 mg/kg; n = 18 for 7.5 mg/kg; n = 6–7 for 10 and 12.5 mg/kg). In the rotarod test of motor coordination (d), wild-type mice showed a marked deficit compared with vehicle-treated mice at 5 and 7.5 mg/kg intraperitoneal etomidate, whereas the performance ofβ2 N265S mice was not significantly affected. Data shown are mean ± SEM time on rotarod; n = 5–6 for vehicle and n = 10–11 for etomidate; +p < 0.05 compared with vehicle-treated mice; *p < 0.05 compared with wild-type mice receiving the same dose of etomidate.
The effects of low intraperitoneal doses of etomidate on spontaneous activity and forced motor activity were examined in locomotor activity chambers and the rotarod, respectively. Vehicle-treated wild-type and β2 N265S mice showed the same level of locomotor activity (Fig. 4c) and total activity (data not shown) as they explored the unfamiliar environment of the activity chambers. Etomidate (0.3–7.5 mg/kg, i.p.) dose dependently decreased activity in the wild-type mice, with significantly less activity being seen at 5 and 7.5 mg/kg etomidate (p < 0.05 compared with β2 N265S). In contrast, etomidate-treated β2 N265S mice did not show any reduction in activity over this dose range and were significantly more active than wild-type controls at 3–7.5 mg/kg etomidate. At the higher doses of 10 and 12.5 mg/kg etomidate, both genotypes showed very low levels of activity. From this experiment and others, it was observed that 10 mg/kg intraperitoneal etomidate produced LORR in some mice, and, if the dose was increased to 15 mg/kg intraperitoneally, all mice displayed LORR (data not shown). Thus, the low level of activity seen at the high end of the dose range in this activity experiment indicated that the LORR threshold for etomidate had been reached. Forced motor activity (Fig. 4d) produced similar results. The wild-type mice displayed significant deficits compared with vehicle-treated control mice after 5 or 7.5 mg/kg etomidate, whereas β2 N265S mice did not.
The lack of sedation–ataxia inβ2 N265S mice receiving low doses of etomidate suggested that recovery from anesthesia may be more rapid. We examined the rate of recovery of motor function using the rotarod after varying durations of LORR induced by intravenous administration of etomidate (Fig. 5). The results clearly show thatβ2 N265S mice recover performance on this motor task more quickly than wild-type mice (genotype, F(1,38) = 63.1; p < 0.00005), especially as the dose of etomidate is increased (drug dose, F(2,38) = 4.52; p < 0.02). Indeed, at 15 mg/kg intravenous etomidate, wild-type mice hardly improved above baseline performance during the 30 min test period after recovery of LORR, and yet β2 N265S mice reach maximum performance by 21 min (Fig. 5c). Wild-type and β2 N265S mice did not differ in their recovery rate after propofol-induced LORR (genotype, F(1,8) = 1.35; p > 0.1) (Fig. 5d).
Figure 5.

β2 N265S mice recover more quickly from etomidate-induced LORR. Mice were tested for recovery of motor coordination after administration of etomidate (5–15 mg/kg, i.v.) or propofol (30 mg/kg, i.v.). Each group of mice were repeatedly tested on the rotarod starting from the recovery of righting reflex. a–c,β2 N265S mice recovered maximal rotarod performance much more quickly than wild-type mice (WT) (genotype, F(1,38) = 63.1; p < 0.00005), and this effect was more pronounced at higher doses of etomidate (drug dose, F(2,38) = 4.52; p < 0.02). d, Both genotypes recovered at a similar rate after propofol-induced LORR (F(1,8) = 1.35; p > 0.1). n = 6–8 for all etomidate groups; n = 5 for propofol.
Encephalographic activity after etomidate anesthesia
Etomidate anesthesia results in a characteristic EEG pattern called “burst suppression” (Vijn and Sneyd, 1998). Such activity is characterized by large-amplitude, fast spikes interspersed by relatively isoelectric periods in the EEG. Such waveforms are distinctly different to the waking and sleeping EEG (Fig. 6a). Burst suppression activity is evident in the cortical EEG of both the wild-type and β2 N265S mice, and quantification of such activity (percentage of each minute EEG in suppression) reveals no significant difference in the EEG pattern of both genotypes (Fig. 6bi). Furthermore, the level of EEG suppression is dose dependent (Fig. 6bii).
Figure 6.

EEG analysis of anesthesia and recovery inβ2 N265S mice. a, Data from wild-type (WT) andβ2 N265S mice showing differences in EEG across different stages of vigilance. i, Wake EEG is characterized by a low-amplitude signal, containing higher frequencies. ii, During slow-wave sleep, signal amplitude is increased, with slower frequencies evident. iii, Etomidate anesthesia produces a very different type of EEG waveform that is defined as burst suppression. Large-amplitude, fast bursts are interspersed by periods of relatively isoelectric EEG. iv, Magnification of a portion of EEG trace shown in iii. Suppressed EEG was defined as a waveform of <5μV amplitude, of not <100 msec duration, and is defined by dotted window. b, Depth of anesthesia as defined by EEG burst suppression activity is similar in both wild-type and β2 N265S mice. i, Percentage of suppression in EEG per minute after etomidate anesthesia (12.5 mg/kg, i.v.) is not significantly different inβ2 N265S mice compared with wild type. ii, Percentage EEG suppression is dose dependent. Higher levels of suppression are evident at 12.5 mg/kg intravenously in both genotypes (wild type, filled squares;β2 N265S, open triangles) compared with 10 mg/kg intravenously (wild type, open squares;β2 N265S, filled triangles), although there is no effect of genotype at both doses. c, There is increased sedation in wild-type compared with β2N265S mice after recovery from etomidate anesthesia.i, After recovery from 10mg/kg intravenous etomidate, increased levels of slow-wave sleep (compared with baseline circadian matched control) are evident in the first 2 hr after recovery. Sleep in β2 N265S mice is not significantly different from baseline. ii, After recovery from 12.5 mg/kg intravenous etomidate, there is enhanced SWS in the wild-type mice in the following 3 hr. Amount of SWS is not significantly enhanced in theβ2 N265S mouse.
After recovery from etomidate anesthesia, the amount of SWS was expressed as a percentage of the circadian timed matched baseline (see Materials and Methods). After recovery, wild-type mice have dose-dependent enhanced levels of SWS (Fig. 6c), whereas β2 N265S mice exhibit normal levels of sleep. Wild-type mice therefore display a “hypnotic hangover” effect after recovery from etomidate anesthesia, which prolongs the anesthetic recovery phase.
Discussion
Our understanding of what happens during clinical anesthesia is still rudimentary; however, it is clear that the major requirements are unconsciousness as determined by lack of sensitivity to noxious stimuli, amnesia, and preferably lack of movement. The low potency and lack of selectivity of many anesthetic compounds has hampered research into defining the mechanisms of hypnotic and anesthetic activity. Etomidate is unusual in that it is a GABAergic modulator that has selectivity for receptors containing β2 and β3 subunits. Etomidate was also without effect in a general screen of other types of receptor and enzymes. In addition, the separated (±)enantiomers of etomidate differ in their in vivo anesthetic potency in a manner consistent with their individual potency at GABAA receptors (Dunwiddie et al., 1986; Tomlin et al., 1998; Belelli et al., 2003). Estimated effective plasma concentrations of etomidate also correlate well with the concentration range over which this drug potentiates GABAA receptors (Krasowski and Harrison, 1999), particularly those containing β2 or β3 subunits. The in vitro potentiation of GABAA receptors by etomidate is determined by N265; however, it is currently unclear whether this residue determines part of a binding site for the drug or is essential for transducing the effects of the anesthetic. In β1, the equivalent residue is serine, and in vitro the β2 N265S mutation does not confer any other apparent difference in the pharmacological or biophysical properties of the channel (Wingrove et al., 1994). We hypothesized that generating this mutation in vivo would not produce any phenotype by itself that would compromise additional evaluation, i.e., the phenotype would only be manifest during pharmacological challenge. This proved to be the case because naive β2 N265S mice were phenotypically normal in a general behavioral screen. In addition, the levels of expression and physiological properties of GABAA receptors that contained β2 and corresponding synaptic inhibitory currents were, as far as could be determined, identical to those in wild-type animals.
Lack of allosteric inhibition of [35S]TBPS binding by loreclezole in brain sections from β2 N265S mice demonstrated first that the β2 subunit was appropriately expressed and correlated well with reported distribution and quantification of this subunit (Pirker et al., 2000, Sur et al., 2001). Similarly, using β subunit-selective antibodies, levels of β1, β2, and β3 subunit protein were equivalent in wild-type heterozygote and homozygote mice. Electrophysiological investigation of receptors expressed in cerebellar Purkinje neurons demonstrates that these cells express primarily β2-containing receptors and that the sensitivity of these receptors to etomidate and loreclezole was lost in cells from β2 N265S mice compared with the anesthetic pentobarbital, which was unaffected. In addition, inhibitory synaptic currents that are prolonged by application of etomidate were considerably less sensitive compared with wild type.
The data presented here complement those published recently by Jurd et al. (2003), who showed that β3 is absolutely required for surgical anesthesia induced by etomidate. The current results significantly extend the findings of Jurd et al. by defining the role of β2. Clearly, activity of etomidate at the β2 subunit is not required to obtain an LORR. However, β2 does contribute to the duration of LORR, depending on the route of administration used. These differences are most likely to be accounted for by the very different pharmacodynamics of etomidate when dosed either intraperitoneally or intravenously. Intraperitoneal administration results in slow absorption of the drug (LORR onset, ∼2 min) and produces prolonged LORR but no LOPWR, presumably because insufficient β3-containing GABAA receptors are occupied. Intravenous administration, in contrast, rapidly (LORR onset, <5 sec) produced both LORR and LOPWR. Detailed pharmacokinetic and pharmacodynamic studies would be required to clarify this point, which is beyond the scope of this investigation. A second measure of the depth of anesthesia is EEG burst suppression, which is commonly used in the clinic. We observed that burst suppression was identical in wild-type and β2 N265S mice, demonstrating a β3-specific mechanism of anesthesia in the brain. The significantly shorter duration of LOPWR in the β2 N265S mice reflects that using spinal reflexes as the anesthesia endpoint can yield different results from using physiological or central measures, which is well documented in the literature (Arras et al., 2001; Antognini and Carstens, 2003). We observed that, after intravenous etomidate, the β2 N265S mice recovered their motor abilities quicker than wild-type mice and spent less time asleep. This demonstrated that β2-containing receptors mediate the sedative–hypnotic properties of etomidate. Recovery from propofol-induced anesthesia was the same in both genotypes, suggesting that the point mutation has not caused a nonspecific change in excitability of these mice.
The data presented here clearly demonstrate that the sedative and anesthetic properties of etomidate can be separated. These findings are consistent with the recent study of β3 N265M mice (Jurd et al., 2003) and a number of other studies that have potentiated β2- and/or β3-containing receptors by other means. GABAA receptors containing β2 subunits (in combination withα1 andγ2 subunits) account for ∼50% of GABAA receptors in the brain (McKernan and Whiting, 1996). This receptor combination is sensitive to benzodiazepines. Several studies using both α1 H101R mutated mice (Rudolph et al., 1999; Crestani et al., 2000; McKernan et al., 2000) and subunit-selective compounds (McKernan et al., 2000) have clearly shown thatα1-containing receptors mediate the sedative and ataxic effects of benzodiazepines. This is consistent with our finding that the selective potentiation of β2-containing receptors produces sedation.
Previous studies have looked at the effect of anesthetics in β3 -/- mice and found that the LORR duration was reduced for the benzodiazepine midazolam and for etomidate (Quinlan et al., 1998). These data are consistent with the present findings that β3-containing receptors mediate the anesthetic effects of etomidate. They do not explain why an anesthetic effect (LORR) is observed in β3 -/- mice. Although there is no definitive answer, this discrepancy may well occur through compensatory changes that occur in the β3 -/- mice (Ramadan et al., 2003). β3 is normally expressed in the embryo (Laurie et al., 1992b), and β3 -/- mice show severe developmental abnormalities similar to those seen in Angelman's syndrome (Homanics et al., 1997; DeLorey et al., 1998). Developmental compensation, perhaps by other GABA subunits, in brain regions critical for anesthesia may underlie the reduced effect of etomidate in these mice. The use of a β2 N265S mouse in our current study avoids these problems because the normal complement of functional GABAA receptors is expressed in the brains of these mice, and the function of these receptors in the absence of anesthetic is completely unaffected by the mutation. Our data also support the hypothesis that at least the majority of β2 and β3 subunits are not assembled together in a single receptor complex because they appear to serve different functions and, in many regions, are differentially localized (Pirker et al., 2000).
A recent study has demonstrated that anesthetics can produce sedative effects via a specific sleep pathway in the hypothalamus. By direct injection into the tuberomammillary nucleus, Nelson et al. (2002) discovered that propofol could induce sedation that was reduced by the GABA antagonist gabazine in a very precise manner. This study demonstrated that individual pathways in the brain are involved in determining the properties of anesthetics that until now have been viewed as being produced by general dampening down of synaptic signaling. Clearly, considered in combination with our study, it is apparent that anesthetic hypnosis is generated by a much more specific mechanism, involving discrete pathways and receptor subtypes. The reticular thalamic nucleus has been reported to play a major role in generation of dynamic changes in thalamo-cortical oscillations across the sleep–wake cycle and has been shown to express primarily GABAA receptors that contain β3 subunits (Pirker et al., 2000). The bursting properties of neurons in this region are critically dependent on GABAergic interactions (Kim et al., 1997), and genetic disruption of β3 results in abolition of GABAA-mediated inhibition in this region and enhanced synchrony of thalamic oscillations accounting for the seizure activity in these animals (Huntsman et al., 1999). The clear demarcation of β3 and β2 in this region suggests very defined functional roles consistent with our findings here.
This study demonstrates that a defined subtype of GABAA receptor can completely confer the hypnotic properties of an anesthetic and that the associated sedation is controlled primarily independently via a different subtype and a separate mechanism. The previous hypothesis of anesthesia producing “general global depression” should be revised in light of these data. Using a combination of genetically modified mice and receptor subtype-specific compounds, we will be able to more closely define the mechanism of action of anesthetics and our understanding of which neuronal pathways are involved. It may also enable a closer understanding of what differentiates sleep and unconsciousness.
Footnotes
D.B. is a Medical Research Council Senior Fellow. A.H. is supported by a Biotechnology and Biological Sciences Research Council-funded CASE award with Merck Sharp & Dohme. We thank J. Dempster for providing software tools to analyze synaptic currents and David Osborne for expert technical assistance. We also acknowledge the kind gift of β1, β2, and β3 subunit antibodies from Prof. Werner Sieghart.
Correspondence should be addressed to K. Wafford, Merck Sharp & Dohme Research Laboratories, The science Research Centre, Terlings Park, Harlow, Essex CM20 2QR, UK. E-mail: keith_wafford@merck.com.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238608-10$15.00/0
D.S.R., T.W.R., and J.C. contributed equally to this work.
References
- Antognini JF, Carstens E ( 2003) Anesthesia, the spinal cord and motor responses to noxious stimulation. In: Neural mechanisms of anesthesia (Antognini JF, Carstens EE, Raines DE, eds), pp 183-204. Totowa, NJ: Humana.
- Arras M, Autenried P, Rettich A, Spaeni D, Rulicke T ( 2001) Optimization of the intraperitoneal anesthesia in mice: drugs, doseages, adverse effects, and anesthesia depth. Comp Med 51: 443-456. [PubMed] [Google Scholar]
- Belelli D, Lambert JJ, Peters JA, Wafford KA, Whiting PJ ( 1997) The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci USA 94: 11031-11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Muntoni A-L, Merrywest SD, Gentet LJ, Casula A, Callachan H, Madau P, Gemmell DK, Hamilton NM, Lambert JJ, Sillar KT, Peters JA ( 2003) The in vitro and in vivo enantioselectivity of etomidate implicates the GABAA receptor in general anaesthesia. Neuropharmacology 45: 57-71. [DOI] [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, McKernan RM, Seabrook GR, Dawson GR, Whiting PJ, Rosahl TW ( 2002) Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α5 subunit of the GABAA receptor. J Neurosci 22: 5572-5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Martin JR, Mohler H, Rudolph U ( 2000) Mechanism of action of the hypnotic zolpidem in-vivo. Br J Pharmacol 131: 1251-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW ( 1998) Mice lacking the β3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioural characteristics of Angelman syndrome. J Neurosci 18: 8505-8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Worth TS, Olsen R ( 1986) Potentiation of recurrent inhibition in rat hippocampus by barbiturate and related nonbarbiturate depressant drugs. J Pharmacol Exp Ther 238: 564-575. [PubMed] [Google Scholar]
- Franks NP, Lieb WR ( 1994) Molecular and cellular mechanisms of general anesthesia. Nature 367: 607-614. [DOI] [PubMed] [Google Scholar]
- Hill-Venning C, Belelli D, Peters JA, Lambert JJ ( 1997) Subunit-dependent interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br J Pharmacol 120: 749-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CE, Korpi ER, Makela R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW ( 1997) Mice devoid of γ-aminobutyrate type A receptor β3 subunit have epilepsy, cleft palate and hypersensitive behaviour. Proc Natl Acad Sci USA 94: 4143-4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR ( 1999) Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science 283: 541-543. [DOI] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U ( 2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABAA receptor beta3 subunit. FASEB J 17: 250-252. [DOI] [PubMed] [Google Scholar]
- Kim U, Sanchez-Vives MV, McCormick DA ( 1997) Functional dynamics of GABAergic inhibition in the thalamus. Science 278: 130-134. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Harrison NL ( 1999) General anesthetic actions on ligandgated ion channels. Cell Mol Life Sci 55: 1278-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH, Wisden W ( 1992a) The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J Neurosci 12: 1063-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH, Wisden W ( 1992b) The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci 12: 4151-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan RM, Whiting PJ ( 1996) Which GABA-A receptor subtypes really occur in the brain? Trends Neurosci 19: 139-143. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ, Castro JL, Ragan CI, Dawson GR, Whiting PJ ( 2000) Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat Neurosci 3: 587-592. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M ( 2002) The sedative component of anesthesia is mediated by GABAA receptors in an endogenous sleep pathway. Nat Neurosci 5: 979-984. [DOI] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G ( 2000) GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101: 815-850. [DOI] [PubMed] [Google Scholar]
- Quinlan JJ, Homanics GE, Firestone LL ( 1998) Anesthesia sensitivity in mice that lack the beta3 subunit of the gamma-aminobutyric acid type A receptor. Anesthesiology 88: 775-780. [DOI] [PubMed] [Google Scholar]
- Ramadan E, Fu Z, Losi G, Homanics GE, Neale JH, Vicini S ( 2003) GABAA receptor beta3 subunit deletion decreases alpha2/3 subunits and IPSC duration. J Neurophysiol 89: 128-134. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, O'Meara GF, Newman RJ, Bromidge FA, Atack JR, Whiting PJ, Rosahl TW, Dawson GR ( 2003) GABA-A alpha1 subunit knockout mice do not show a hyperlocomotor response following amphetamine or cocaine treatment. Neuropharmacology 44: 190-198. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Sudhof TC ( 1993) Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell 75: 661-670. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Mohler H ( 1999) Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature 401: 796-800. [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky KA ( 1995) cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23: 5080-5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur C, Wafford KA, Reynolds DS, Hadingham KL, Bromidge F, Macaulay A, Collinson N, O'Meara G, Howell O, Newman R, Myers J, Atack JR, Dawson GR, McKernan RM, Whiting PJ, Rosahl TW ( 2001) Loss of the major GABAA receptor subtype in the brain is not lethal in mice. J Neurosci 21: 3409-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S-A, Wafford KA ( 2001) Mechanism of action of general-anesthetics—new information from molecular pharmacology. Curr Opin Pharmacol 1: 78-83. [DOI] [PubMed] [Google Scholar]
- Tomlin SL, Jenkins A, Lieb WR, Franks NP ( 1998) Stereoselective effects of etomidate optical isomers on gamma-aminobutyric acid type A receptors and animals. Anesthesiology 88: 708-717. [DOI] [PubMed] [Google Scholar]
- van Gool WA, Mirmiran M ( 1986) Effects of aging and housing in an enriched environment on sleep-wake patterns in rats. Sleep 9: 335-347. [DOI] [PubMed] [Google Scholar]
- Vijn PCM, Sneyd JR ( 1998) I.v. anesthesia and EEG burst suppression in rats: bolus injections and closed-loop infusions. Br J Anaesth 81: 415-421. [DOI] [PubMed] [Google Scholar]
- Wingrove PB, Wafford KA, Bain C, Whiting PJ ( 1994) The modulatory action of loreclezole at the gamma-aminobutyric acid type A receptor is determined by a single amino acid in the beta 2 and beta 3 subunit. Proc Natl Acad Sci USA 91: 4569-4573. [DOI] [PMC free article] [PubMed] [Google Scholar]