

Abstract
Gonadotropin-releasing hormone (GnRH) neurons form the final common pathway for the central regulation of reproduction and are inhibited by negative energy balance. In normal adults, these neurons maintain elevated intracellular chloride so that GABAA receptor activation is excitatory. We hypothesized that fasting alters homeostatic mechanisms to eliminate excitatory responses to GABA but rejected this hypothesis when brief, local GABA application elicited action currents in GnRH neurons from fed and fasted mice. This response was specific to GABAA receptors, because glycine elicited no response. We next found that fasting reduced the frequency of spontaneous GABAergic postsynaptic currents (PSCs) and that this was reversed by in vivo treatment with leptin during the fast. In the presence of tetrodotoxin to minimize presynaptic actions, leptin also potentiated the postsynaptic response of these cells to GABAA receptor activation. Postsynaptic effects of leptin on GABAergic miniature PSCs were eliminated by inhibiting JAK2/3 (Janus kinase), the tyrosine kinase through which leptin receptors signal. In all experiments, elimination of PSCs at ECl or by treatment with the GABAA receptor antagonist bicuculline confirmed that PSCs were specifically mediated by GABAA receptor chloride channels. These data demonstrate that fasting and leptin act presynaptically and postsynaptically to alter GABAergic drive to GnRH neurons, providing evidence for GABAergic communication of metabolic cues to GnRH neurons, and suggest the possibility for functional leptin receptors on GnRH neurons. They further demonstrate cytokine modulation of the postsynaptic response to GABA in mammals, which may be important to central neural regulation in both healthy and diseased states.
Keywords: GnRH, GABA, PSC, leptin, cytokine, fertility
Introduction
Darwin linked the fate of species to the outcome of the search for food and mates over one century ago (Darwin, 1859). Subsequent experiments have shown that negative energy balance is a potent inhibitor of fertility, as evidenced by numerous animal models (Dyer et al., 1985; Ahima et al., 1996; Schneider and Zhou, 1999; Ohkura et al., 2000), and clinical syndromes such as anorexia nervosa (Stoving et al., 1999). Information regarding energy homeostasis is relayed to the gonadotropin-releasing hormone (GnRH) neurons of the hypothalamus to regulate reproductive function. These cells are the final common pathway controlling fertility and produce a frequency-modulated endocrine signal that is conveyed to the anterior pituitary gland. Metabolic signals affect reproduction at least in part by altering the frequency and amplitude of GnRH secretion (I'Anson et al., 2000). The nature of these metabolic signals is only beginning to be understood, although, among potential candidates, the adipocyte hormone leptin has received considerable attention because it both inhibits food intake (Rentsch et al., 1995; Weigle, 1995) and reverses fasting-induced reproductive suppression (Ahima et al., 1996; Nagatani et al., 1998; Sullivan et al., 2002). No conclusive evidence exists to date for leptin receptor expression in GnRH neurons (Finn et al., 1998; Cunningham et al., 1999), suggesting that presynaptic effects of this hormone may be important, although direct action on GnRH neurons cannot be discounted. In the following report, we attempt to elucidate some of the neurobiological mechanisms by which metabolic information is communicated to the GnRH neuronal axis to regulate fertility.
The neurotransmitter GABA is one candidate mediator of metabolic signals to GnRH neurons. GnRH neurons receive functional synaptic input via GABAA receptors (Sim et al., 2001). Furthermore, GABAergic neurons express leptin receptors, indicating that their activity may be under direct metabolic control (Ovesjo et al., 2001). The A subtype of the GABA receptor gates an intrinsic ion channel that passes mainly chloride; thus, GABAA receptor activation can either inhibit or excite a cell depending on chloride levels (Kaila, 1994; Rivera et al., 1999; Wagner et al., 2001). Unlike most mature neurons, adult GnRH cells are depolarized by GABAA receptor activation sufficiently to induce action potential firing in ad libitum fed animals (DeFazio et al., 2002).
We examined effects of energy balance on three facets of GABAA receptor-mediated inputs to GnRH neurons. First, we asked whether negative energy balance sufficient to suppress reproduction alters GnRH neuron chloride homeostasis so that the response to GABAA receptor activation becomes inhibitory. Such a change in response has been proposed to underlie circadian changes in the firing rate of neurons of the suprachiasmatic nucleus of the hypothalamus (Wagner et al., 1997, 2001; De Jeu and Pennartz, 2002), as well as changes in neuronal excitability after injury (Nabekura et al., 2002). Second, we examined the effects of negative energy balance and leptin on GABAergic drive to GnRH neurons, a presynaptic effect. Finally, we tested for functional evidence of direct, postsynaptic leptin action on these cells via leptin receptors.
Materials and Methods
Animals. Adult transgenic female mice in which green fluorescent protein (GFP) is genetically targeted to GnRH neurons (Suter et al., 2000) were used. Animals were housed in groups of three to five and were maintained on standard rodent chow (Harlan 7012; Harlan Sprague Dawley, Bartonsville, IL) and water ad libitum. All animals were held on a 14/10 light/dark cycle with lights on at 5:00 A.M. eastern standard time. Female mice were placed in one of three treatment groups: (1) diestrous fed; (2) fasted for 48 hr during diestrus, during which only water ad libitum was given; or (3) fasted for 48 hr during diestrus and treated with leptin (1 mg/kg, i.p., two times per day) during the fast. Estrous cycle stage was determined by vaginal cytology; all animals were diestrous on the day of the experiment. Importantly, and as shown previously (Ahima et al., 1996; Sullivan et al., 2002), fasted mice remained in diestrus on the day of the experiment, indicating restraint of the 3–5 d mouse reproductive cycle by the 48 hr fast. Glucose levels were also measured on the day of the experiment (One Touch Fast Take glucose monitor; Lifescan, Milpitas, CA) to verify nutritional state. All procedures were approved by the Animal Care and Use Committee of the University of Virginia and were conducted in accordance with the National Research Council publication Guide for Care and Use of Laboratory Animals.
Slice preparation and recordings. All reagents were purchased from Sigma (St. Louis, MO). Coronal sections at 200 μm through the preoptic area and hypothalamus were prepared as described previously (Nunemaker et al., 2002) in solutions containing 5 mm rather than 10 mm glucose. For recording, individual slices were transferred to a recording chamber mounted on the stage of an Olympus Optical BX50WI upright fluorescent microscope (Opelco, Dulles, VA). Slices in the recording chamber were continuously superfused with oxygenated recording saline kept at 30–32°C with an inline heating unit (Warner Instruments, Hamden, CT), with the addition of d(-)2-amino-5-phophonovaleric acid (APV) (20 μm) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μm) to block glutamatergic currents and, in some recordings, tetrodotoxin (0.5 μm) and leptin (50 nm). Experiments were performed using Pulse Control software (Bookman Laboratory, University of Miami, Miami, FL), and currents were recorded with an EPC-8 amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany), digitized by an ITC-18 acquisition interface (Instrutech, Port Washington, NY), and stored using IGOR PRO software (Instrutech) on a G4 Macintosh computer (Apple Computers, Cupertino, CA).
On-cell response to rapid application of GABA. Local pressure application of GABA (1 mm in HEPES buffer) was accomplished using methods described previously (DeFazio et al., 2002). Recording electrodes (2–4 MΩ) were filled with either of the following (in mm): a low-chloride solution containing 139 K-gluconate, 1 KCl, 10 HEPES, 5 EGTA, 0.1 CaCl2, and 4.0 MgCl2, pH 7.2 with NaOH; or a high-chloride solution in which K-gluconate was replaced with KCl (final concentration of 140 KCl). The low-chloride solution was used to ensure that elicited action currents were not attributable to accidental rupture of the cell membrane and subsequent dialysis with pipette solution. To record the response to rapid GABA or vehicle application (5 msec pulse of 5–10 psi), gigaohm seals (1–3 GΩ) were formed in the on-cell configuration, and large-amplitude fast current spikes were detected in voltage clamp at 0 mV with signals filtered at 10 kHz. Seal resistance was periodically monitored and remained >1GΩ. In addition, the response to pressure application of glycine (1 mm in HEPES buffer) was tested (n = 2 cells fed, 2 fasted, 3 fasted with in vivo leptin) to determine whether GnRH neurons respond to another transmitter that binds a receptor with an intrinsic chloride channel.
Recording postsynaptic currents. Electrodes (2–4 MΩ) were filled with the high-chloride pipette solution with the addition of 4 mm MgATP and 0.4 mm NaGTP before adjusting to pH 7.2 with NaOH. GFP-GnRH neurons were identified, and the whole-cell recording configuration was achieved. Membrane potential was clamped at -60 mV, and signals were filtered at 7 kHz with gain set at 10 mV/pA for 180 sec recording periods. Liquid junction potential of 3 mV (Barry, 1994) was not corrected for. Postsynaptic currents (PSCs) were stored as Event Tracker files using Pulse Control and IGOR PRO software. Input resistance (Rin), series resistance (Rs), and membrane capacitance (Cm) were continually monitored as described previously (DeFazio et al., 2002). Only recordings with Rin >500 MΩ and Rs <20 MΩ were included for analysis. Mean Rin, Rs, and Cm were not different (p > 0.05) among or within cells in which comparisons were made. Bicuculline (20 μm), a GABAA receptor antagonist, was bath applied during a subset of recordings to determine that detected PSC events were GABAA receptor mediated. Individual cells were further examined to determine that changes in the PSC properties examined were not attributable to alterations in passive properties or Rs within the acceptable ranges defined above. In a sample of recorded cells, GnRH neuron phenotype was confirmed post hoc by immunocytochemical recovery of biocytin (included in internal recording solution at 4 mm) with streptavidin-Cy3 and immunodetection of GnRH peptide within the same cell as described previously (Suter et al., 2000).
PSC analysis. Stored 180 sec traces of current activity were analyzed offline using custom event detection software to identify PSCs (events). Threshold for event detection was set manually for each 180 sec record. Events were confirmed by eye, and detection errors were corrected manually. Mean event frequency (in hertz) from three to five 180 sec records was calculated for each cell to obtain mean PSC frequency in each treatment group. Group means were compared using one-way ANOVA, followed by post hoc analysis with Fisher's protected least significant difference test and Student–Newman–Keuls test for pairwise comparisons when appropriate (p < 0.05). Averaged PSC waveforms were generated for each cell after aligning events on the rising phase. Event rate of rise, peak amplitude, 90–10% decay time, and interevent interval calculated by the program were exported and further analyzed in a spreadsheet (Microsoft Excel; Microsoft, Redmond, WA). To compare PSCs among in vivo treatment groups, cumulative probability plots for each parameter in each treatment group were generated as described previously (DeFazio and Hablitz, 1998) using 100 randomly selected events per cell or all events if fewer than 100 occurred (see Fig. 3) (Van Sickle and Tietz, 2002). To compare PSCs within a recording for in vitro treatments, all events from an individual neuron were included in the distribution, and a distribution was constructed for each individual cell for analysis (see Fig. 4). Distributions among treatments were compared with the Komolgorov–Smirnov goodness of fit (KS) test (SPLUS Professional 2 data analysis software; MathSoft, Cambridge, MA) with significance at p < 0.05. Percentage changes among treatment groups were calculated for each parameter and are reported as mean ± SEM.
Figure 3.

In vivo leptin alters the postsynaptic response of GnRH neurons to activation of GABAA receptors. A, B, sPSCs from a representative GnRH neuron in each treatment group were averaged (A) to show differences in amplitude and normalized by amplitude (B) to show differences in decay time. C, Percentage change in sPSC rate of rise, amplitude, and decay time compared with fed controls in animals that were fasted (black bars) or fasted and treated in vivo with leptin (white bars). Values are calculated from the mean of those events randomly selected for analysis from each group. D–F, Cumulative probability distributions of rate of rise (D), amplitude (E), and decay time (F) in each of the three treatment groups. Distributions were constructed from randomly selected events from each cell in each in vivo treatment group. Recordings of sPSCs were done in the presence of APV and CNQX to block glutamatergic currents. *p < 0.01 versus fed; KS test.
Figure 4.

Changes in mPSC properties are attributable to leptin signaling through JAK2/3.A, B, mPSC traces from an individual, representative GnRH neuron before and during treatment with leptin in the bath solution and after subsequent addition of AG490. All mPSCs in each treatment group were averaged (A) to show differences in amplitude and normalized by amplitude (B) to show differences in decay time. C–E, Cumulative probability distributions of rate of rise (C), amplitude (D), and decay time (E) of mPSCs from an individual GnRH neuron treated in vitro with leptin and AG490. Recordings of mPSCs were done in the presence of APV and CNQX to block glutamatergic currents, as well as tetrodotoxin to block synaptic action potentials and thus minimize presynaptic effects. *p < 0.01 versus bath; KS test.
Results
Serum glucose levels reflect nutritional status
To confirm that a 48 hr fast altered the nutritional status of mice, we measured serum glucose levels before each experiment. Serum glucose levels in fed animals (8.3 ± 0.8 mmol/l; n = 7) were significantly greater (p < 0.01) than in fasted (4.4 ± 0.2 mmol/l; n = 10) and leptin-treated fasted (3.5 ± 0.7 mmol/l; n = 4) animals. Mean glucose levels in fasted and leptin-treated animals were not statistically different (p = 0.13).
Experiment 1: rapid GABA excites GnRH neurons
Recent evidence demonstrates that activation of the GABAA receptor is excitatory in GnRH neurons (DeFazio et al., 2002). In the suprachiasmatic nucleus, circadian shifts in firing rate have been reported to be attributable to changes in the direction of response to GABAA receptor activation (Wagner et al., 1997, 2001; De Jeu and Pennartz, 2002). We thus tested whether reproductive inhibition induced by negative energy balance alters chloride homeostasis in GnRH neurons to an extent that GABAA receptor activation inhibits rather than excites these cells. The response of GFP-identified GnRH neurons to local, rapid pressure application of GABA (1 mm in HEPES-buffered solution) was evaluated in both fed diestrous mice and mice fasted for 48 hr beginning on diestrous day 1, a regimen that suppresses reproductive cyclicity (Ahima et al., 1996; Sullivan et al., 2002). Reproductive inhibition was confirmed by fasting-induced prolongation of diestrus. The on-cell recording configuration was used to avoid disruption of the existing intracellular chloride milieu. In all GnRH neurons tested, and with either the high- or low-chloride pipette solution, rapid GABA application elicited robust action currents (n = 7 of 7 fed; n = 12 of 12 fasted) (Fig. 1, left column). These currents are the extracellular currents generated by action potentials. In contrast, GnRH neurons did not respond to rapid pressure application of glycine (n = 2 fed, 2 fasted, 3 fasted with in vivo leptin) (Fig. 1, right column), indicating that the principal ligand-activated chloride channel in GnRH neurons is the GABAA receptor. Furthermore, the lack of response to either glycine or HEPES buffer alone (n = 3 fed, 3 fasted; data not shown) demonstrate that mechanical disruption of the cell did not account for the action currents fired in response to GABA. Although we did not determine intracellular chloride ion concentrations in GnRH neurons from fasted animals, these results suggest that fasting does not alter GnRH neuron chloride homeostasis sufficiently to prevent excitation of these cells by GABAA receptor activation.
Figure 1.

Activation of GABAA, but not glycine, receptors excites GnRH neurons from both fed (top) and fasted (bottom) female mice. Response of GnRH neurons to rapid pressure application (arrow) of GABA (1 mm; left) or glycine (1 mm, right), which also gates ionotropic chloride channels, was recorded using the on-cell recording configuration. Deflections in response to GABA are action currents, i.e., extracellular recordings of action potentials. There was no response to rapid pressure application of HEPES medium alone (data not shown).
Experiment 2: frequency of GABAergic sPSCs in GnRH neurons is modulated by metabolic state
Because GABAA receptor activation was excitatory in both fed and fasted animals, we next tested whether reproductive inhibition by fasting alters GABAergic drive to GnRH neurons. Fasting inhibits reproduction at least in part by reducing the frequency and amplitude of GnRH secretion (I'Anson et al., 2000). One mechanism by which this may occur is via reduced excitatory GABAergic drive to GnRH neurons; we thus hypothesized that fasting decreases the frequency of spontaneous GABAergic postsynaptic currents (sPSCs) in GnRH neurons. The whole-cell voltage-clamp recording configuration was used with high intracellular chloride to record sPSCs from GnRH neurons from both fed and fasted diestrous female mice. Mean sPSC frequency recorded from GnRH neurons (n = 10) from fasted animals was significantly lower than that from fed animals (n = 8 cells; 0.26 ± 0.09 Hz fasted vs 1.38 ± 0.34 Hz fed; p < 0.05) (Fig. 2A, B). This suggests that negative energy balance induced by fasting decreases presynaptic GABAergic drive to GnRH neurons.
Figure 2.
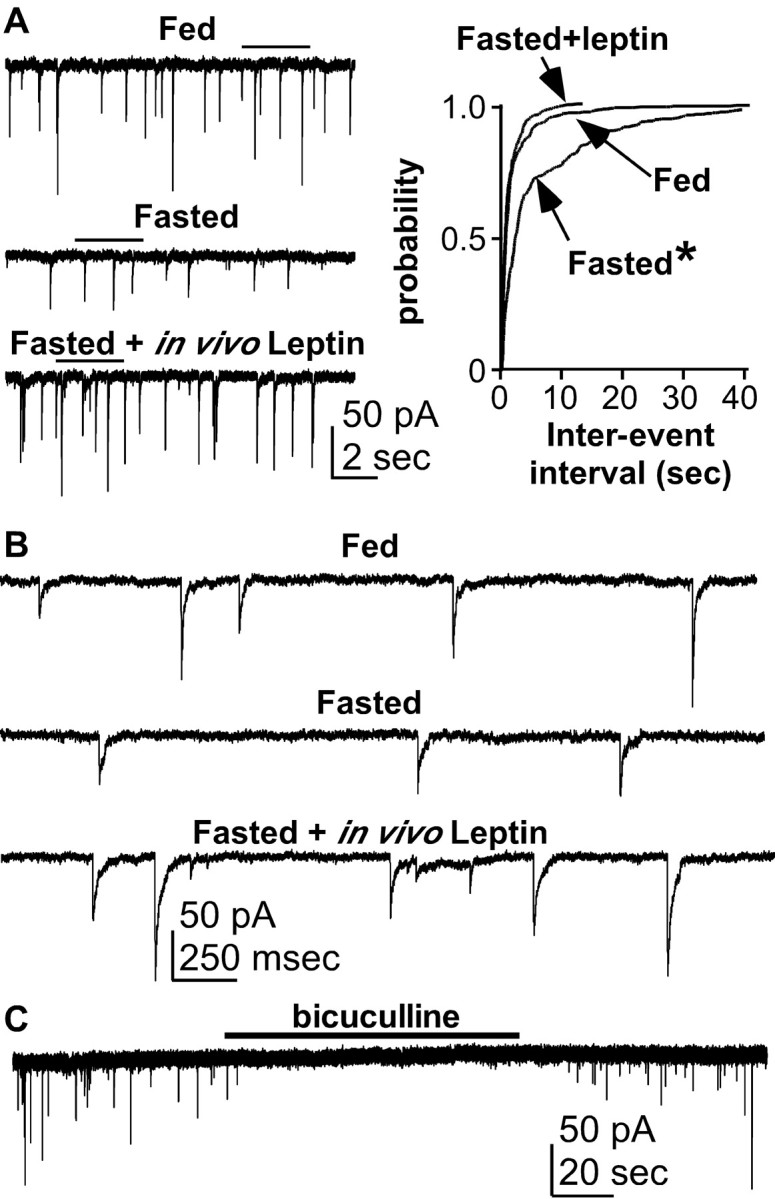
Fasting reduces GABAergic drive to GnRH neurons. A, On the left are representative traces of sPSCs recorded from GnRH neurons from fed (top), fasted (middle), and fasted with in vivo leptin-treated females (bottom). On the right, the cumulative probability distribution of interval between successive sPSCs (interevent interval) in each of three treatment groups. B, A portion of each recording trace in A (indicated by solid line above the trace in A) is expanded to better illustrate individual sPSC events. C, Representative trace of sPSCs recorded from a GnRH neuron from a fasted with in vivo leptin-treated female showing reversible elimination of all sPSC events by the GABAA receptor antagonist bicuculline. *p < 0.01 versus fed; KS test.
We next tested the hypothesis that leptin treatment sufficient to rescue fertility in fasting mice, as measured by estrous cyclicity (Ahima et al., 1996; Sullivan et al., 2002), would also restore sPSC frequency. A third group of diestrous females was fasted as above and treated in vivo with leptin (1 mg/kg, i.p., two times per day) during the fast. Although leptin was not present during electrophysiological recordings, treatment with leptin in vivo prevented the fasting-induced decrease in sPSC frequency (n = 8 cells): mean frequency from GnRH neurons in this group was not different from that in fed animals (0.95 ± 0.18 Hz; p = 0.48) (Fig. 2A, B) but was greater (p < 0.02) than in untreated fasted animals. All sPSC activity was reversibly eliminated by either the GABAA receptor antagonist bicuculline (20 μm; n = 2 fed, 2 fasted, 3 fasted with in vivo leptin) (Fig. 2C) or stepping to the reversal potential for chloride (ECl of 0 mV; data not shown). Together, these data suggest that leptin signals are communicated to GnRH neurons at least in part by regulating the activity or connectivity of presynaptic GABAergic afferents. It cannot be determined from this experiment, however, whether this leptin effect occurred via leptin action on GABAergic neurons directly presynaptic to GnRH neurons and/or via other leptin-receptive neurons that ultimately increased the output of GABAergic afferents.
Effects of fasting and in vivo leptin on sPSC properties
To determine the possibility that fasting and/or in vivo leptin also acted directly on GnRH neurons to alter the postsynaptic response to GABAA receptor activation, we determined differences in sPSC properties among the three treatment groups. Because this set of experiments was not done in the presence of tetrodotoxin to block action potential-dependent neurosecretion, the possibility that any changes we saw were attributable to presynaptic actions could not be eliminated. Nonetheless, this comparison was useful in determining whether additional experiments to examine possible postsynaptic effects were warranted. To evaluate effects of fasting and in vivo leptin treatment on sPSC properties, we analyzed rate of rise (a measure of receptor on-rate), peak amplitude (a measure of total channel conductance), and decay time (a measure of receptor affinity). Fasting alone did not alter any of these parameters compared with fed animals (rate of rise, p > 0.13; amplitude, p > 0.82; decay time, p > 0.13) (Fig. 3, Table 1). In contrast, in vivo leptin administration to fasted mice independently increased (p < 0.01) all three measures of postsynaptic response to GABA compared with fed controls (Fig. 3). Without TTX present to block synaptic input, a presynaptic mechanism for these in vivo leptin effects, such as increased transmitter release, cannot be ruled out. Importantly, however, the data do not eliminate the possibility that leptin may, at least in part, act directly on GnRH neurons to functionally alter the postsynaptic response to GABAA receptor activation.
Table 1.
Leptin alters sPSC properties in GnRH neurons
|
Group |
Rise rate (pA/msec) |
Amplitude (pA) |
Decay (msec) |
|---|---|---|---|
| Fed | 62.3 ± 2.5 | 47.6 ± 1.4 | 12.10 ± 0.50 |
| Fasted | 67.8 ± 2.6 | 48.1 ± 1.4 | 13.43 ± 0.07 |
| Fasted with leptin
|
85.5 ± 3.0*
|
63.8 ± 1.8*
|
14.40 ± 0.04**
|
Mean ± SEM rate of rise (a measure of receptor on-rate), amplitude (a measure of conductance), and decay time (a measure of receptor off-rate/affinity) of GABAergic sPSCs in GnRH neurons from fed, fasted (48 hr), and in vivo leptin-treated fasted mice. Recordings were done in the presence of APV and CNQX to block glutamatergic currents but without synaptic action potential blockade by tetrodotoxin. *p < 0.01 versus fed and fasted; **p < 0.001 versus fed.
Experiment 3: acute effects of in vitro leptin on miniature PSC properties in GnRH neurons from fed animals
On the basis of the changes in sPSC properties observed in experiment 2, we next tested whether leptin altered PSC properties acutely and independently from fasting when postsynaptic effects were isolated. We assessed the acute effects of leptin, applied in vitro to the extracellular solution, on miniature PSCs (mPSCs). Miniature PSCs are recorded in the presence of tetrodotoxin to minimize activity-dependent presynaptic influences on PSC properties. Because they occur in the absence of action potentials, mPSCs provide a direct measure of the postsynaptic effects of a drug without confounds of presynaptic actions. The leptin dose used (50 nm) was chosen to achieve close to a maximal leptin response based on the EC50 for leptin in brain slice preparations (5.9 nm) (Cowley et al., 2001). Recordings were obtained from GnRH neurons from fed, diestrous mice to eliminate effects of fasting. Leptin did not change mPSC frequency (-2.8 ± 7.2% from baseline; p = 0.453), consistent with a lack of effect on the random release processes in the presynaptic neurons that drive mPSCs. Leptin did, however, increase mPSC rate of rise (24.6 ± 4.1%; p < 0.001), amplitude (22.3 ± 4.0%; p < 0.001), and decay time (61.8 ± 14.3%; p < 0.001) within 5–8 min of application (n = 15) (Figs. 4, 5, 6); these effects were reversed by washout of leptin from the bath solution (data not shown).
Figure 5.

Pretreatment with the JAK2/3 inhibitor AG490 blocks leptin action on mPSCs. Changes in mPSC rate of rise (A), amplitude (B), and decay time (C) are shown for each cell to illustrate consistency of response to treatment, as well as individual cell variability in baseline mPSC properties. Circles show cells that were not pretreated with AG490 before recording; squares show cells that were pretreated with AG490 before recording. Open symbols designate baseline values for each parameter (○, baseline without AG490 pretreatment; □, baseline during AG490 pretreatment); filled symbols designate values for each parameter after in vitro leptin treatment (•, unpretreated baseline with leptin; ▪, leptin after AG490 pretreatment). Means for each parameter in each condition are also indicated.
Figure 6.

Summary of leptin action on mPSC properties in GnRH neurons. Mean ± SEM percentage change from baseline in mPSC rate of rise, amplitude, and decay time after various in vitro treatments. leptin+AG490 refers to reversal of leptin effect by AG490; AG490+leptin refers to blockade of leptin action by pretreatment with AG490. *p < 0.05 versus untreated baseline.
These results suggest that leptin acts, at least in part, postsynaptically to directly alter the response of GnRH neurons to GABAA receptor activation. In that regard, the time course of these effects is consistent with the possibility that leptin modulates GABAA receptor-mediated currents in GnRH neurons through activation of leptin receptors expressed in these cells. The long isoform of the leptin receptor, which mediates most leptin actions and is the most abundant isoform in the mouse hypothalamus, activates the JAK (Janus kinase)–STAT (signal transducers and activators of transcription) intracellular pathway (Strosberg and Isaad, 1999; Chehab, 2000). To test the hypothesis that leptin modulation of GABAA receptors occurred via activation of this pathway, possibly through functional leptin receptors in GnRH neurons, mPSCs were recorded from GnRH neurons from fed, diestrous females in the presence of in vitro leptin and the JAK2/3 inhibitor AG490 (10 μm). AG490 reversed leptin-induced increases in rate of rise, amplitude, and decay time back to baseline values (-0.3 ± 2.3%, p > 0.9; 0.4 ± 2.1%, p > 0.7; and 12.6 ± 5.7%, p > 0.1, from baseline, respectively; n = 8) (Figs. 4, 6). Additionally, pretreatment with AG490 (5–10 min) eliminated the response to in vitro leptin; mPSC rate of rise, amplitude, and duration remained at baseline levels (-3.1 ± 3.6%, p > 0.7; -4.7 ± 2.8%, p > 0.2; and -0.3 ± 8.9%, p > 0.9, change from baseline, respectively; n = 5) (Figs. 5, 6). Neither AG490 alone (n = 11; rate of rise, p > 0.9; peak, p > 0.8; decay time, p > 0.1) nor its DMSO vehicle (n = 6; rate of rise, p > 0.6; peak, p > 0.3; decay time, p > 0.2) altered these mPSC parameters (Fig. 6). Together, these results provide functional evidence for the possibility of leptin receptor expression in GnRH neurons and suggest a mechanism by which postsynaptic leptin receptor activation may alter GABAergic input to these cells.
Discussion
It is well established that fasting inhibits reproductive function in mammals by ultimately altering GnRH neuron function, but the precise central mechanism(s) for this have not yet been elucidated. The present studies are the first to examine neuronal responses to fasting directly at the GnRH neuron. GABAA receptor activation in mature GnRH neurons is excitatory (DeFazio et al., 2002). Although negative energy balance induced by fasting did not alter this excitatory response of GnRH neurons, it altered GABAergic communication with GnRH neurons in multiple ways. Specifically, fasting decreased the frequency of GABAA receptor-mediated sPSCs in GnRH neurons, supporting the hypothesis that negative energy balance has presynaptic actions that are conveyed by a reduction in excitatory GABAergic drive onto GnRH neurons. Leptin treatment sufficient to maintain reproductive cyclicity during fasting (Ahima et al., 1996; Sullivan et al., 2002) prevented this reduction in sPSC frequency, suggesting that leptin acted at least in part presynaptically to restore afferent GABAergic drive to GnRH neurons in fasted animals and supporting the hypothesis that GABAergic inputs help convey leptin signals to the GnRH neurosecretory axis. It could not be determined from these experiments whether these presynaptic effects of fasting and in vivo leptin occurred via actions on the GABAergic neurons directly afferent to GnRH neurons and/or on other neuronal phenotypes that subsequently altered hypothalamic GABAergic transmission.
Neuroendocrine responses to fasting include stimulation of food intake and inhibition of reproductive function mediated at least in part by decreased leptin-mediated signaling. It has been suggested that leptin has a dual role in the hypothalamus, inhibiting some neural subpopulations (Spanswick et al., 1997) and stimulating others (Cowley et al., 2001). Subpopulations of hypothalamic GABAergic neurons express leptin receptors (Ovesjo et al., 2001); one possibility is that these GABAergic subpopulations are excited by leptin and that they include cells with projections to GnRH neurons. In that case, elevated leptin levels during a fed state would stimulate GABA release, contributing to increased excitatory GABAergic drive to the GnRH axis. During fasting the reverse would occur, i.e., diminished leptin levels result in decreased GABA release and thus decreased excitatory drive onto GnRH neurons. Such leptin effects would not rule out, of course, the possibility of leptin actions on other neuronal subpopulations that, in turn, also alter hypothalamic GABAergic neurotransmission and ultimately GnRH neuron activity. Determining the response of GABAergic and other cell types to leptin will thus be of interest to deciphering presynaptic actions of this hormone on the reproductive axis.
We show that in vivo leptin, but not fasting, also altered the postsynaptic response of GnRH cells to GABAA-receptor activation, suggesting the possibility that leptin may act directly on GnRH neurons to alter postsynaptic responsiveness to GABA. Interestingly, analysis of the postsynaptic response showed no effect of fasting on sPSC properties compared with fed animals. Because fasting has been shown to decrease leptin levels (Ahima et al., 1996), one might expect sPSC properties in GnRH neurons to be diminished in fasted compared with fed mice. There are several possible explanations for why this did not occur. First, it is possible that leptin-injected fasted animals attained supraphysiological hormone levels that were responsible for potentiation of GABAergic input in this group. In that regard, leptin levels could not be determined because of the small amount of total serum available from a mouse, but this leptin regimen has been shown previously to restore circulating levels back to control fed values (Ahima et al., 1996). Second, it is possible that, during a fast, peripheral leptin is decreased, but central levels are maintained. In that case, no differences in sPSC properties would be expected in fasted versus fed animals. Studies in humans argue against maintenance of central leptin levels during fasting (Hagan et al., 1999). It also follows that no leptin-dependent reduction in sPSC frequency such as we observed should have occurred if central leptin levels were maintained. Finally, it is important to point out that spontaneous PSCs are subject to a myriad of presynaptic influences. It is thus likely that, besides leptin, other neurohormones and neurotransmitters altered by negative energy balance affected sPSC properties, thereby masking any direct leptin actions. For this reason, it was more appropriate to test postsynaptic leptin effects using fed animals to eliminate influences of fasting and to compare miniature PSCs, which are recorded after blockade of action potential-dependent transmitter release to isolate postsynaptic effects. This was done in the last series of experiments in this report, discussed below.
We tested for direct, postsynaptic effects of leptin at the GnRH neuron by recording PSCs in the presence of tetrodotoxin to block presynaptic input (mPSCs) and treating slices acutely with in vitro leptin. Under these conditions, leptin increased mPSC rate of rise, amplitude, and decay time, a response that was reversed or eliminated by the tyrosine kinase inhibitor AG490. The leptin-induced increase in mPSC amplitude indicates adult GnRH neurons contain GABAA receptors that are not saturated by quantal release of GABA (Frerking et al., 1995; Nusser et al., 1997; DeFazio and Hablitz, 1998). Furthermore, the time course of this in vitro leptin effect and its elimination by AG490 suggest that leptin modulated GnRH neuron GABAA receptor function via activation of JAK kinases in these cells, possibly downstream from leptin receptors. Together, these data provide evidence for a direct link between metabolism and fertility through leptin receptor-mediated modulation of the responsiveness of GnRH neurons to activation of GABAA receptors. To our knowledge, these data also provide the first evidence for modulation of current through the GABAA receptor by cytokines in the mammalian CNS. Thus, in addition to the possible physiological significance of the reported leptin actions in the metabolic regulation of fertility, this work suggests a novel mechanism for modulating the cellular response to a dominant neurotransmitter across the CNS, including regions involved in the pathology of neurological diseases.
Allosteric modulators of GABAA receptors can alter GABAergic mPSC properties similarly to the leptin effect seen here (Majewska et al., 1986; Twyman and Macdonald, 1992; Sullivan and Moenter, 2003). Despite this similarity in postsynaptic response, it is unlikely leptin, a 16 kDa molecule, acted as an allosteric modulator. The present results suggest that postsynaptic effects of in vitro leptin were via signaling cascades initiated by leptin receptors in GnRH neurons. Functional leptin receptors belong to the cytokine receptor superfamily that activates JAK tyrosine kinases. Of interest, it has been shown that leptin-induced alterations in neural function in other brain regions depend on intracellular phosphorylation events (Shanley et al., 2001), and phosphorylation is a well established mechanism for functional modulation of the GABAA receptor (Moss et al., 1995; Moss and Smart, 1996; Leidenheimer and Chapell, 1997; Smart, 1997; Fancsik et al., 2000). In the present study, elimination of JAK2/3 activity reversed or eliminated leptin effects on mPSCs, indicating that functional modification of GABAA receptors by leptin was attributable to activation of this kinase, possibly downstream from leptin receptors in GnRH neurons. Although molecular analyses have failed to identify leptin receptors on GnRH neurons, the functional assays used here provide support for leptin receptor action in these cells. In that regard, increased sensitivity of functional over molecular assays has been described previously in this system (DeFazio et al., 2002). In the present study, it is possible that functional tests were necessary to overcome the confounds of identifying message or protein for the leptin receptor specifically within GnRH neurons, because these receptors are abundant throughout the hypothalamus. Although these data provide the first indication of functional leptin receptors in GnRH neurons, the development of a specific leptin receptor antagonist will be necessary to confirm this possibility using electrophysiological approaches.
This report suggests one neural pathway communicating metabolic cues to GnRH neurons via the GABAA receptor system. Metabolic signals adjust GnRH neuron activity and presumably hormone release at least in part by altering GABAergic afferents as well as GnRH neuron response to GABAA receptor activation. Besides fasting-induced infertility, alterations in GABAergic signaling could have implications in other causes of hypothalamic subfertility or infertility. In this regard, two common disorders associated with hypothalamic infertility have been linked to altered functioning of GABAergic systems, specifically some forms of epilepsy (Morrell, 1999), and polycystic ovarian syndrome (Loucks et al., 2002). Precisely defining the neural pathways involved and their susceptibility to cytokine and other manipulations may thus help explain both metabolic control of reproduction and etiologies of these and other clinical disorders, in which the normal physiology of GnRH and other neurons, including hippocampal and cortical cells, is impaired by altered GABAergic signaling.
Footnotes
This work was supported by National Science Foundation Grant IBN 98058023, Whitehall Foundation Grant 2000-12-43A, and National Institute of Child Health and Human Development—National Institutes of Health Cooperative Agreement U54 HD28934. We thank Catherine Christian, Glenn Harris, and Chun Xu for editorial comments and Dr. Xu-Zhi Xu for expert technical assistance.
Correspondence should be addressed to S. M. Moenter, Internal Medicine and Cell Biology, P.O. Box 800578, Jefferson Park Avenue, University of Virginia, Charlottesville, VA 22908. E-mail: smm4n@virginia.edu.
R. A. DeFazio's present address: University of Miami School of Medicine, Department of Physiology and Biophysics, 1600 Northwest 10th Avenue, Room 4044, Miami, FL 33136. E-mail: RDefazio@med.miami.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238578-08$15.00/0
References
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowel B, Maratos-Flier E, Flier JS ( 1996) Role of leptin in the neuroendocrine response to fasting. Nature 382: 250-252. [DOI] [PubMed] [Google Scholar]
- Barry PH ( 1994) JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods 51: 107-116. [DOI] [PubMed] [Google Scholar]
- Chehab FF ( 2000) Leptin as a regulator of adipose mass and reproduction. Trends Pharmacol Sci 21: 309-314. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ ( 2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411: 480-484. [DOI] [PubMed] [Google Scholar]
- Cunningham MJ, Clifton DK, Steiner RA ( 1999) Leptin's actions on the reproductive axis: perspectives and mechanisms. Biol Reprod 60: 216-222. [DOI] [PubMed] [Google Scholar]
- Darwin C ( 1859) On the origin of species. New York: Oxford UP.
- DeFazio RA, Hablitz JJ ( 1998) Zinc and zolpidem modulate mIPSCs in rat neocortical pyramidal neurons. J Neurophysiology 80: 1670-1677. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Heger S, Ojeda SR, Moenter SM ( 2002) Activation of GABAA receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol 16: 2872-2891. [DOI] [PubMed] [Google Scholar]
- De Jeu M, Pennartz C ( 2002) Circadian modulation of GABA function in the rat suprachiasmatic nucleus: excitatory effects during the night phase. J Neurophysiol 87: 834-844. [DOI] [PubMed] [Google Scholar]
- Dyer RG, Mansfield S, Corbet H, Dean AD ( 1985) Fasting impairs LH secretion in female rats by activating an inhibitory opioid pathway. J Endocrinol 105: 91-97. [DOI] [PubMed] [Google Scholar]
- Fancsik A, Linn DM, Tasker JG ( 2000) Neurosteroid modulation of GABA IPSCs is phosphorylation dependent. J Neurosci 20: 3067-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn PD, Cunningham MJ, Pau KY, Spies HG, Clifton DK, Steiner RA ( 1998) The stimulatory effect of leptin on the neuroendocrine reproductive axis of the monkey. Endocrinology 139: 4652-4662. [DOI] [PubMed] [Google Scholar]
- Frerking M, Borges S, Wilson M ( 1995) Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron 15: 885-895. [DOI] [PubMed] [Google Scholar]
- Hagan MM, Havel PJ, Seeley RJ, Woods SC, Ekhator NN, Baker DG, Hill KK, Wortman MD, Miller AH, Gingerich RL, Geracioti TD ( 1999) Cerebrospinal fluid and plasma leptin measurements: covariability with dopamine and cortisol in fasting humans. J Clin Endocrinol Metab 84: 3579-3585. [DOI] [PubMed] [Google Scholar]
- I'Anson H, Manning JM, Herbosa CG, Pelt J, Friedman CR, Wood RI, Bucholtz DC, Foster DL ( 2000) Central inhibition of gonadotropin-releasing hormone secretion in the growth-restricted hypogonadotropic female sheep. Endocrinology 141: 520-527. [DOI] [PubMed] [Google Scholar]
- Kaila K ( 1994) Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol 42: 489-537. [DOI] [PubMed] [Google Scholar]
- Leidenheimer NJ, Chapell R ( 1997) Effects of PKC activation and receptor desensitization on neurosteroid modulation of GABA(A) receptors. Brain Res Mol Brain Res 52: 173-181. [DOI] [PubMed] [Google Scholar]
- Loucks TL, Rohan LC, Kalro BN, Berga SL ( 2002) Increased γ-aminobutyric acid (GABA) levels in lean women with polycystic ovary syndrome (PCOS). Endocrine Soc Abstr 108-109.
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM ( 1986) Steroid hormone metabolites are barbituate-like modulators of the GABA receptor. Science 232: 1004-1007. [DOI] [PubMed] [Google Scholar]
- Morrell MJ ( 1999) Epilepsy in women: the science of why it is special. Neurology 53: S42-S48. [PubMed] [Google Scholar]
- Moss SJ, Smart TG ( 1996) Modulation of amino acid-gated ion channels by protein phosphorylation. Int Rev Neurobiol 39: 1-52. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Gorrie GH, Amato A, Smart TG ( 1995) Modulation of GABAA receptors by tyrosine phosphorylation. Nature 377: 344-348. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Ataike N ( 2002) Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci 22: 4412-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatani S, Guthikonda P, Thompson RC, Tsukamura H, Maeda KI, Foster DL ( 1998) Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology 67: 370-376. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM ( 2002) Estradiol-sensitive afferents modulate long-term episodic firing patterns of GnRH neurons. Endocrinology 143: 2284-2292. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M ( 1997) Differences in synaptic GABAA receptor number underlie variation in GABA mini amplitude. Neuron 19: 697-709. [DOI] [PubMed] [Google Scholar]
- Ohkura S, Tanaka T, Nagatani S, Bucholtz DC, Tsukamura H, Maeda K, Foster DL ( 2000) Central, but not peripheral, glucose-sensing mechanisms mediate glucoprivic suppression of pulsatile luteinizing hormone secretion in the sheep. Endocrinology 141: 4472-4480. [DOI] [PubMed] [Google Scholar]
- Ovesjo ML, Gamstedt M, Collin M, Meister B ( 2001) GABAergic nature of hypothalamic leptin target neurones in the ventromedial arcuate nucleus. J Neuroendocrinol 13: 505-516. [DOI] [PubMed] [Google Scholar]
- Rentsch J, Levens N, Chiesi M ( 1995) Recombinant ob-gene product reduces food intake in fasted mice. Biochem Biophys Res Commun 214: 131-139. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K ( 1999) The K +/Cl - co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251-255. [DOI] [PubMed] [Google Scholar]
- Schneider JE, Zhou D ( 1999) Interactive effects of central leptin and peripheral fuel oxidation on estrous cyclicity. Am J Physiol 277: R1020-R1024. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Harvey J ( 2001) Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci 21: 1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JA, Skynner MJ, Herbison AE ( 2001) Heterogeneity in the basic membrane properties of postnatal gonadotropin-releasing hormone neurons in the mouse. J Neurosci 21: 1067-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart TG ( 1997) Regulation of excitatory and inhibitory neurotransmittergated ion channels by protein phosphorylation. Curr Opin Neurobiol 7: 358-367. [DOI] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML ( 1997) Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390: 521-525. [DOI] [PubMed] [Google Scholar]
- Stoving R, Hangaard J, Hansen-Nord M, Hagen C ( 1999) A review of endocrine changes in anorexia nervosa. J Psychiatr Res 33: 139-152. [DOI] [PubMed] [Google Scholar]
- Strosberg AD, Isaad T ( 1999) The involvement of leptin in humans revealed by mutations in leptin and leptin receptor genes. Trends Pharmacol Sci 20: 227-230. [DOI] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM ( 2003) Direct actions of steroids on GnRH neurons via neurosteroid modulation of GABAA receptor function. Endocrinology, in press.
- Sullivan SD, Howard LC, Clayton AH, Moenter SM ( 2002) Serotonergic activation rescues reproductive function in fasted mice: does serotonin mediate the metabolic effects of leptin on reproduction? Biol Reprod 66: 1702-1706. [DOI] [PubMed] [Google Scholar]
- Suter KJ, Song WJ, Sampson TL, Wuarin JP, Saunders JT, Dudek FE, Moenter SM ( 2000) Genetic targeting of green fluorescent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology 141: 412-419. [DOI] [PubMed] [Google Scholar]
- Twyman RE, Macdonald RL ( 1992) Neurosteroid regulation of GABAA receptor single-channel kinetic properties of mouse spinal cord neurons in culture. J Physiol (Lond) 456: 215-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sickle BJ, Tietz EI ( 2002) Selective enhancement of AMPA receptor-mediated function in hippocampal CA1 neurons from chronic benzodiazepine-treated rats. Neuropharmacology 43: 11-27. [DOI] [PubMed] [Google Scholar]
- Wagner S, Castel M, Gainer H, Yarom Y ( 1997) GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature 387: 598-603. [DOI] [PubMed] [Google Scholar]
- Wagner S, Sagiv N, Yarom Y ( 2001) GABA-induced current and circadian regulation of chloride in neurones of the rat suprachiasmatic nucleus. J Physiol (Lond) 537: 853-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigle D ( 1995) Recombinant ob protein reduces feeding and body weight in the ob/ob mouse. J Clin Invest 96: 2065-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]