

Abstract
The tumor suppressor and transcription factor p53 is a key modulator of cellular stress responses, and activation of p53 precedes apoptosis in many cell types. Controversial reports exist on the role of the transcription factor nuclear factor-κB (NF-κB) in p53-mediated apoptosis, depending on the cell type and experimental conditions. Therefore, we sought to elucidate the role of NF-κB in p53-mediated neuron death. In cultured neurons DNA damaging compounds induced activation of p53, whereas NF-κB activity declined significantly. The p53 inhibitor pifithrin-α (PFT) preserved NF-κB activity and protected neurons against apoptosis. Immunoprecipitation experiments revealed enhanced p53 binding to the transcriptional cofactor p300 after induction of DNA damage, whereas binding of p300 to NF-κB was reduced. In contrast, PFT blocked the interaction of p53 with the cofactor, whereas NF-κB binding to p300 was enhanced. Most interestingly, similar results were observed after oxygen glucose deprivation in cultured neurons and in ischemic brain tissue. Ischemiainduced repression of NF-κB activity was prevented and brain damage was reduced by the p53 inhibitor PFT in a dose-dependent manner. It is concluded that a balanced competitive interaction of p53 and NF-κB with the transcriptional cofactor p300 exists in neurons. Exposure of neurons to lethal stress activates p53 and disrupts NF-κB binding to p300, thereby blocking NF-κB-mediated survival signaling. Inhibitors of p53 provide pronounced neuroprotective effects because they block p53-mediated induction of cell death and concomitantly enhance NF-κB-induced survival signaling.
Keywords: pifithrin, apoptosis, p300, DNA damage, cerebral ischemia, hippocampal cultures
Introduction
Several prominent neurological disorders including stroke and Alzheimer's disease manifest symptoms that result from degeneration and death of neurons (Jenner and Olanow, 1998; Dirnagl et al., 1999; Mattson, 2000). Although the genetic and environmental factors that initiate neurodegeneration differ among diseases, a shared biochemical cascade of events appears to perform the cell death process. This cascade involves increased oxidative stress, disruption of calcium homeostasis, and activation of a death program called apoptosis (Green and Reed, 1998; Mattson, 2000). Neuronal apoptosis involves mitochondrial ion permeability changes, cytochrome c release, and activation of caspases (Chan and Mattson, 1999; Lipton, 1999). An important regulatory step in apoptosis occurs at mitochondrial membranes where members of the Bcl-2 family of proteins either promote (Bax, Bid) or prevent [Bcl-2, Bcl-x long (bcl-xl)] membrane permeability transition (Hengartner, 2000; Mattson and Kroemer, 2003). Signaling events that act upstream of mitochondrial changes in neuronal apoptosis are not fully established but may include DNA damage initiating the activation of the tumor suppressor protein p53 (Hou et al., 1997; Hughes et al., 1997; Uberti et al., 1998; Cregan et al., 1999; Culmsee et al., 2001a,c).
Recent findings implicated a role for p53 in neuron death that occurs in stroke (Crumrine et al., 1994; Li et al., 1994), traumatic brain injury (Napieralski et al., 1999), and Alzheimer's disease (De la Monte et al., 1997). Cell culture studies have established strong correlations between p53 expression and neuronal death induced by DNA damaging agents and glutamate (Hughes et al., 1997; Sakhi et al., 1997; Xiang et al., 1998; Cregan et al., 1999), and our previous work using new p53 inhibitors confirmed the essential role of p53 in neuronal apoptosis in models relevant to various neurodegenerative disorders (Culmsee et al., 2001c; Duan et al., 2002; Zhu et al., 2002).
The proapoptotic function of p53 involves its function as transcription factor inducing the synthesis and mitochondrial translocation of Bax and other p53-inducible genes (Wood and Youle, 1995; Enokido et al., 1996). Moreover, mechanisms independent of p53 transcriptional activity may be involved in p53-mediated apoptosis (Caelles et al., 1994; Gao and Tsuchida, 1999; Mihara et al., 2003). For example, a role for p53 in blocking the activity of other transcription factors such as nuclear factor-κB (NF-κB) has been proposed as a crucial mechanism involved in p53-mediated cell death (Wadgaonkar et al., 1999; Webster and Perkins, 1999; Ikeda et al., 2000). In neurons, NF-κB supported survival signaling by inducing the expression of anti-apoptotic factors, for example anti-apoptotic bcl-2 family members, manganese superoxide dismutase (MnSOD), and inhibitors of apoptosis (IAP) (Mattson et al., 2000; Bui et al., 2002; Culmsee et al., 2002). The role of NF-κB in neuron death is controversial, however, and some reports indicated that NF-κB supported apoptosis and was substantially involved in p53-mediated (neuronal) cell death (Grilli and Memo, 1999; Ryan et al., 2000; Uberti et al., 2000). Because the function of NF-κB in p53-mediated neuron cell death remained unclear, we sought to clarify this issue in cultured neurons exposed to apoptotic insults and in ischemic brain tissue.
Materials and Methods
Animals. All efforts were made to minimize the suffering and the number of animals according to the guidelines of the German animal protection law and derived guidelines on the ethical use of animals. Male Wistar rats, female Sprague Dawley rats (gestation day 18), and male Naval Medical Research Institute (NMRI) mice were obtained from Charles-River (Sulzfeld, Germany). NF-κB-luciferase reporter mice (Voll et al., 2000) were bred and used for the measurement of changes in NF-κB transcriptional activity as described previously (Culmsee et al., 2002). The NF-κB-luciferase transgene contained the firefly luciferase gene, driven by two NF-κB sites responding to p65/p50, p50/cRel, and other dimer combinations of NF-κB as established in previous studies (Millet et al., 2000; Voll et al., 2000). In neurons, the luciferase assay reflected well the enhanced synthesis of NF-κB-dependent protective bcl-2 family genes such as bl-2 and bcl-xL (Culmsee et al., 2002). All animals were kept under controlled light and environmental conditions (12 hr dark/light cycle; 23 ± 1°C; 55 ± 5% relative humidity) and had ad libitum access to food (Altromin, Lage, Germany) and water.
Embryonic neuronal cultures. Hippocampi and cortices were removed from the brains of embryonic day (E) 18 rats or E14 mice, and cells were dissociated by mild trypsinization and trituration as described previously (Mattson et al., 1993). Hippocampal cells were then seeded onto polyethyleneimine-coated 35 mm culture dishes (for survival analysis) or 60 mm culture dishes (for protein analysis) and grown in Neurobasal medium (Invitrogen, Karlsruhe, Germany) supplemented with 5 mm HEPES, 1.2 mm glutamine, B27 supplement (Invitrogen, Karlsruhe, Germany) (20 ml/l), and gentamicin (0.1 mg/ml). All experimental treatments were performed on 7- to 8-d-old cultures, at which time they contained <5% astrocytes as determined by GFAP immunocytochemistry. PFT or cyclicPFT (cPFT) (Tocris, Cologne, Germany) was added to the cultures at a final concentration of 1–100 nm. Camptothecin (5–10μm; Sigma, Deisenhofen, Germany) or homocysteine (200–250 μm; Sigma) was added 1 hr after pretreatment with the p53 inhibitors. Dimethylsulfoxide (DMSO, Sigma) at a final concentration of 0.1% was used as the vehicle for PFT and camptothecin. To block NF-kB activation, 5 μm double-strand oligonucleotide with a specific NF-kB-binding consensus sequence (decoy, 5′-AGTTGAGGGACTTTCCCAGGC-3′; MWG-Biotech AG, Munich, Germany) was added to the medium of the cultures 2 hr before and remained in the medium after the onset of respective treatments. As a control, the single–base mutated double-strand oligonucleotide (mutant, 5′-AGTTGAGCGACTTTCCCAGGC-3′) was added to the medium at the same concentration.
Oxygen glucose deprivation. For oxygen glucose deprivation (OGD), glucose-free Locke's medium (Locke's without glucose) containing (in mm): 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 5 HEPES, pH 7.2, supplemented with gentamicin (5 mg/l) was degassed by a mixture of N2/CO2 (95%/5%) for 1 hr. The cultured cells were then washed three times with oxygen-deprived Locke's without glucose and incubated for 2 or 4 hr in an oxygen-free N2/CO2 (95%/5%) atmosphere. Control cultures were incubated in Locke's with glucose (10 mm) or in oxygen-deprived Locke's with glucose (OD). After 2 or 4 hr the medium was replaced with Neurobasal medium. At indicated times after the onset of OGD, cell extracts were obtained for luciferase activity measurements.
Neuron survival was quantified by methods described previously (Mattson et al., 1995; Culmsee et al., 2002). Briefly, viable neurons in premarked fields (10× objective) were counted before experimental treatment and 24 hr after treatment. Neurons that died in the intervals between examination points were usually absent, and the viability of the remaining neurons was assessed by morphological criteria. Neurons with intact neurites of uniform diameter and soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and vacuolated cell bodies were considered nonviable. Approximately 300 cells per culture in at least four separate cultures per treatment condition were counted, and the percentage of neuronal survival was determined. In separate experiments, cell death was determined by the trypan blue exclusion method and after staining the nuclei with the DNA-binding fluorochrome Hoechst 33258 (Molecular Probes, Göttingen, Germany) according to standard protocols as described previously (Culmsee et al., 2001d, 2002). Experiments were repeated at least twice, and analyses were performed without knowledge of the treatment history of the cultures.
Luciferase assay. Luciferase activity was measured in protein extracts from cultured neurons or brain tissue obtained from E14 NF-κB-luciferase reporter mice (Millet et al., 2000) using a luciferase detection kit according to the manufacturer's protocol (Promega, Madison, WI) as described previously (Culmsee et al., 2002). Briefly, after treatment intervals of 1–24 hr, the cells were scraped in ice-cold PBS and centrifuged at 600 × g for 10 min at 4°C, and the pellet was homogenized in 100–150 μl of luciferase reporter lysis buffer. In addition, cortical and hippocampal brain tissue from NF-κB reporter mice was homogenized in 500 μl reporter lysis buffer 1–24 hr after PFT treatment or onset of focal cerebral ischemia. Brain and cell homogenates were centrifuged at 14,000 × g for 10 min at 4°C, and total protein content in the supernatant was determined using the Pierce BCA kit (Perbio Science, Bonn, Germany). Aliquots of 30 μl of the protein extracts were added to 100 μl of luciferase assay buffer containing the luciferase assay substrate and immediately measured in a luminometer. Luminescence was expressed in an arbitrary scale as relative light units (RLU). Because some analyses were performed in parallel with different sets of cultures and different batches of luciferin reagent, values were expressed as RLU as percentage of control values. Double measurements were performed with each sample, and every experiment was repeated at least twice to confirm the results. All analyses were performed without knowledge of the treatment history.
Immunoprecipitation and immunoblots. Protein extracts were obtained from embryonic cortical neurons treated with vehicle (0.1% DMSO) and PFT or camptothecin, or both. The cells were washed in ice-cold PBS and then collected in 200 μl of PBS containing protease inhibitors [protease inhibitor mixture (Sigma) with aminoethyl-benzenesulfonyl fluoride, aprotinin, leupeptin, betastatin, pepstatin A, and E-64] and phosphatase inhibitors [phosphatase inhibitor mixtures I and II (Sigma) with inhibitors of serine/threonine phosphatases and alkaline phosphatases, i.e., cantharidin, bromotetramisole, and microcystin and inhibitors of tyrosine protein phosphatases, acid, and alkaline phosphatases, i.e., sodium salts of orthovanadate, orthomolybdate, tartrate, and imidazole, respectively]. After determination of the protein content, 200 μg of protein was diluted to 250 μl with collecting buffer, and 5 μl monoclonal anti-p300 antibody (Upstate Biotechnology, Lake Placid, NY) was added. After 2 hr incubation at 4°C, 30 μl of fast flow protein G Sepharose (Upstate Biotechnology) was added. Before use, the Sepharose was washed several times with collecting buffer. The immunoprecipitation mixture was then incubated with rocking overnight at 4°C. Afterward, the Sepharose beads were spun down and washed three times in ice-cold collecting buffer. The pellet was resuspended in 30 μl collecting buffer supplemented with (130 mm Tris, pH 6.8, 10% SDS, 10% 2-mercaptoethanol, 20% glycerin, 0.06% bromophenol blue), incubated at 95°C for 5 min, and then loaded onto a 7.5% polyacrylamide gel. After electrophoresis, the proteins were blotted onto a nitrocellulose membrane according to standard protocols (Culmsee et al., 2001d). After blocking in 5% nonfat milk, the membrane was incubated with polyclonal anti-p53 antibodies (Cell Signaling, Beverly, MA) or anti-NF-κB/p65 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. The membrane was then processed using horseradish-peroxidase-conjugated secondary anti-rabbit antibody followed by a chemiluminescence detection method (ECL, Amersham Biosciences, Arlington Heights, IL).
Immunocytochemistry. Immunocytochemistry was performed as described previously (Culmsee et al., 2001b, 2002). Cells were fixed in 4% paraformaldehyde, membranes were permeabilized by exposure to 0.2% Triton X-100 in PBS for 5 min, and cells were placed in blocking serum (5% FCS in PBS) for 30 min. Cells were then exposed to a polyclonal anti-phospho-p53 antibody (1:500; Cell Signaling) overnight at 4°C, followed by an incubation for 1 hr with biotinylated anti-rabbit IgG antibody (1:200; Vector Labs, Burlingame, CA) and 30 min in the presence of Oregon green-streptavidin (Molecular Probes). Images were acquired using a confocal laser scanning microscope with a 40× oil immersion objective (488 nm excitation and 510 nm emission). All images of the different treatment times were acquired randomly using the same laser intensity and photodetector gain in a single analysis session of 3–5 hr to allow comparisons of relative levels of immunoreactivity between the sister cultures within one experiment. Levels of immunoreactivity (integrated optical density per neuron) were quantified as described previously (Culmsee et al., 2001b; Junker et al., 2002).
Gel shift assay. Nuclear extracts were prepared as described previously (Culmsee et al., 2001c), and protein content was determined using the Bradford method (Bio-Rad Laboratories, Hercules, CA). For gel-shift analysis of p53 DNA-binding activity, equal amounts of protein were incubated in a 20 μl reaction mixture containing 20 mg BSA, 1 mg poly(dI-dC), 2 ml buffer D (20 mm HEPES, pH 7.9, 20% glycerol, 100 mm KCl, 0.5 mm EDTA, 0.25% NP-40, 2 mm dithiothreitol, 0.1% PMSF), 4 ml buffer F (20% Ficoll-400, 100 mm HEPES, pH 7.9, 300 mm KCl, 10 mm dithiothreitol, 0.1% PMSF), and 20,000–50,000 cpm of 32P-labeled oligonucleotide (Geneca Biotechnology, Montreal, Canada) corresponding to a p53 binding site (5′-AGCTGGACATGCCCGGGCATGTC-3′). After 20 min at room temperature, reaction products were separated on a 7% nondenaturing polyacrylamide gel. Radioactivity of the dried gel was detected by exposure to film. To determine the specificity of the complex, competition experiments were performed by incubating extracts with labeled oligonucleotide probe in the presence of 100-fold excess unlabeled SP1 or p53 oligonucleotide.
Focal cerebral ischemia in mice. Permanent middle cerebral artery (MCA) occlusion was performed in male NF-κB reporter mice and NMRI mice according to the method described by Welsh et al. (1987) with modifications described previously (Junker et al., 2002). Briefly, mice were anesthetized with tribromoethanol (600 mg/kg, i.p.), and an incision was made between the left eye and the ear. After trepanation of the skull, the left MCA and the two major branches distal of the bifurcation were permanently occluded by electrocoagulation. Body temperature was maintained at 37°C with a heating lamp during the surgical procedure. Afterward, mice were kept under an environmental temperature of 30°C for 2 hr followed by an additional period of 2–7 d at 23 ± 1°C in their home cages. After 2–7 d the mice were anesthetized again with tribromoethanol and perfused intraperitoneally with a solution of the dye neutral red (1.5%), and the brains were removed and fixed in a phosphate-buffered, pH 7.4, formalin solution (4%). The unstained tissue region was calculated as infarct area (square millimeters) using an image analyzing system (Scion Image, Scion Corp., Frederick, MD) according to Backhauß et al. (1992). For measurements of luciferase activity, brain tissue of κB reporter mice was removed 1–24 hr after PFT treatment or MCA occlusion and homogenized immediately in reporter lysis buffer and analyzed as described previously (Culmsee et al., 2002). Animals received PFT or vehicle via intraperitoneal injection 1 hr before or up to 6 hr after MCA occlusion.
Transient forebrain ischemia in rats. Transient forebrain ischemia (TFI) in male Wistar rats (250–300 gm) was performed as described previously (Zhu et al., 1998). Briefly, ischemia was induced under halothane/N2O/O2 (1.5/30/70%) anesthesia by occlusion of both common carotid arteries combined with depression of the mean arterial blood pressure to 40 mmHg by trimethaphancamsylate (5 mg/kg, i.v.) and central venous exsanguination. After 10 min the artery clips were removed and blood was rapidly reinjected to restore blood pressure. Arterial pH, PCO2, PO2, blood pressure, and plasma glucose were monitored throughout the surgical procedure. After surgery, animals were kept at an environmental temperature of 30°C for 2 hr. Seven days after ischemia the animals were anesthetized deeply and perfused transcardially with 4% phosphate-buffered formalin. The brains were removed and embedded in paraffin, and coronal sections (8 μm) of the hippocampal region were prepared. The sections were stained with 1% celestine blue and 1% acid fuchsin for histological evaluation. CA1 neurons with clear red staining, pyknotic cell shape, and nuclear condensation were considered damaged, whereas surviving neurons did not show red staining but exposed round-shaped, blue-stained nuclei. On the basis of this quantification, neuronal injury in the CA1 subfield of the hippocampus was calculated as the percentage of damaged neurons of the total number of CA1 neurons, as also established in our previous work (Zhu et al., 1998; Culmsee et al., 2001d). The counts of CA1 neurons were performed without knowledge of the treatment history.
Statistics. All values are given as means ± SD. One-way ANOVA combined with Scheffé's test was used for multiple comparisons in cell culture experiments, and Student's t test or ANOVA combined with Duncan's test was performed in the analysis of animal models. Statistical differences are presented at probability levels of p < 0.05, p < 0.01, and p < 0.001.
Results
NF-κB transcriptional activity declines in DNA damage-induced neuron death and is preserved by the p53 inhibitor pifithrin-α
Cultured hippocampal neurons of NF-κB reporter mice were exposed to camptothecin (10 μM), a topoisomerase inhibitor that has been shown to cause DNA damage and p53-dependent apoptosis in neurons (Takimoto et al., 1999; Culmsee et al., 2001c). Camptothecin-induced apoptosis was accompanied by a significant decrease of NF-κB transcriptional activity as compared with the basal NF-κB activity in control sister cultures (Fig. 1 A). The pronounced decline of NF-κB transcriptional activity was detected in the neurons within 3–6 hr after exposure to camptothecin and clearly preceded morphological signs of apoptosis such as nuclear condensation, cell blebbing, or fragmentation of dendritic processes. The NF-κB activity partly recovered but did not reach control levels after 24 hr (Fig. 1 A). Similar results were obtained in cultured cortical neurons of NF-κB reporter mice (data not shown). The p53 inhibitor PFT (100 nm) attenuated the camptothecin-induced decline of NF-κB activity (Fig. 1 B). Interestingly, the p53 inhibitor also enhanced NF-κB transcriptional activity in neuronal cultures not exposed to camptothecin. This result indicated that p53 partly blocked constitutive NF-κB activity under basal culture conditions. This action of p53 was further enhanced after DNA damage resulting in accumulation of p53 and a near total block of NF-κB activity (Fig. 1 A).
Figure 1.

The p53 inhibitor PFT prevents the repression of NF-κB transcriptional activity and apoptosis induced by DNA damage. A, Hippocampal neurons of NF-κB-luciferase reporter mice were incubated with 10 μm camptothecin. After the indicated times, protein extracts were prepared and luciferase activity was determined. The values of the relative light units (RLU) representing NF-κB activity are given as mean percentage of control ± SD from three independent experiments. ***p < 0.001 as compared with controls and #p < 0.05 as compared with 6 hr of camptothecin treatment. B, NF-κB transcriptional activity in hippocampal neurons of NF-κB reporter mice was quantified 6 hr after exposure with 100 nm pifithrin-α (PFT), 10 μm camptothecin (CPT), or a combination of PFT and CPT(CPT/PFT). Values of RLU from three cultures per group are given as percentage of the vehicle-treated control. Note the induction of NF-κB transcriptional activity by PFT under control conditions. Moreover, PFT attenuated the decline in NF-κBactivityin CPT-treated cells. *p<0.05 as compared with controls;###p<0.001 CPT-treated cells compared with controls; ++p < 0.01 as compared with CPT-treated neurons (ANOVA; Scheffé's test). C, Embryonic hippocampal neurons were pretreated with PFT (100 nm) 1 hr before adding camptothecin (CPT, 10 μm). Viable neurons in premarked fields (10× objective) were counted before and 24 hr after experimental treatment. Neurons that died in the intervals between examination points were usually absent, and the viability of the remaining neurons was assessed by morphological criteria. The percentage of surviving neurons was calculated as mean ± SD of five culture dishes per group. ###p < 0.001 CPT-treated cells compared with controls; **p < 0.01 as compared with CPT-treated neurons (ANOVA; Scheffé's test).
In line with the observed attenuation of NF-κB transcriptional activity by PFT, the p53 inhibitor partially protected embryonic hippocampal neurons against camptothecin-induced cell death (Fig. 1C). Similar effects on NF-κB transcriptional activity and neuroprotection against camptothecin-induced apoptosis were found in cultured hippocampal and cortical neurons treated with the PFT analog cyclic-PFT (10 nm-1 μm; data not shown).
Involvement of p300 in NF-κB/p53 interaction after DNA damage
Because our findings suggested an inhibition of NF-κB activity by p53 under basal conditions and more so after induction of apoptosis, we further investigated the mechanism underlying this interaction between the transcription factors. A possible bottleneck for the transcriptional activity of both, p53 and NF-κB may be the competitive binding to cotranscription factors such as p300. Therefore, we investigated whether the accumulation of p53 in the nucleus after induction of DNA damage affected binding of p53 or NF-κB to p300 and whether this was affected by the p53 inhibitor PFT. We performed p53-directed gel shift assays and immunoprecipitation experiments with anti-p300 antibodies after treatment of hippocampal neurons with camptothecin, PFT, or a combination of both. The gel shift assay confirmed an increase in p53 DNA binding activity in nuclear protein extracts within 6 hr of exposure to camptothecin (Fig. 2A). Treatment of the cultures with PFT completely suppressed the camptothecin-induced increase in p53 DNA binding (Fig. 2A). Similar results were obtained from the coimmunoprecipitation studies: in cells treated with camptothecin, p53 binding to p300 was found to be enhanced (Fig. 2B, top panel). In line with the reduced DNA binding activity of p53 in cells treated with PFT, p53 binding to p300 was reduced to control levels in neurons treated with the p53 inhibitor (Fig. 2B, top panel).
Figure 2.

Pifithrin-α (PFT) inhibits binding of p53 to DNA or p300, whereas it enhances p300 binding to the NF-κB/p65 subunit. A, Hippocampal neurons were exposed to vehicle (control), camptothecin (CPT), pifithrin-α (PFT), or a combination of PFT and CPT (CPT/PFT) for 6 hr. Nuclear extracts were prepared and subjected to gel-shift analysis of p53 DNA-binding activity. Previous experiments showed that only the lower, most intense shifted band (arrow) was eliminated after incubation of the extracts with a p53 antibody, demonstrating that it contains p53 protein. B, Protein extracts of hippocampal neurons exposed to vehicle (control) and PFT or camptothecin, or both, were subjected to immunoprecipitation with anti-p300 antibodies. Western blot analysis of the immunoprecipitated p300 revealed enhanced binding of p53 to p300 in hippocampal neurons exposed to camptothecin for 6 hr as compared with controls (B, top panel). PFT prevented p53 binding to p300 and significantly enhanced NF-κB/p65 binding to p300 at the same time. Note that in line with the activity assays, PFT also enhanced NF-κB/p65 binding to p300 within 6 hr under basal conditions (B, bottom panel).
In contrast, NF-κB/p65 binding to p300 significantly declined in hippocampal neurons within 6 hr after exposure to camptothecin (Fig. 2B, bottom panel). In line with the results from NF-κB reporter neurons (Fig. 1A), the coimmunoprecipitation study further revealed that the camptothecin-mediated decline of NF-κB binding to p300 was prevented in cells pretreated with PFT. Moreover, PFT induced a pronounced binding of NF-κBto p300 within 6 hr as compared with controls. Overall, these findings indicated that inhibition of p53 allowed enhanced binding of NF-κB to the cotranscription factor p300 (Fig. 2B) resulting in enhanced NF-κB transcriptional activity, as also demonstrated in the NF-κB reporter cells (Fig. 1A).
We next explored whether the preserved NF-κB transcriptional activity contributed to the neuroprotective effect of p53 inhibitors against camptothecin-induced neuron death. To address this issue, rat embryonic hippocampal neurons were pretreated with NF-κB decoy DNA or mutant decoy DNA before PFT treatment. The NF-κB decoy DNA blocked the neuroprotective effect of PFT against camptothecin-induced damage (Fig. 3A). The mutant decoy DNA, which was used as a control in sister cultures, affected neither camptothecin-induced cell death nor PFT-mediated neuroprotection (Fig. 3A). Similar results were obtained with pyrrolidine dithiocarbamate (PDTC), another well established inhibitor of NF-κB, which prevented neuroprotection by PFT in a similar manner as the κB-decoy DNA (Fig. 2B). Neither NF-κB decoy DNA nor PDTC alone affected camptothecin-induced neuronal apoptosis. Overall these results indicated that the preservation of NF-κB activity substantially contributed to the neuroprotective effects of p53 inhibitors.
Figure 3.
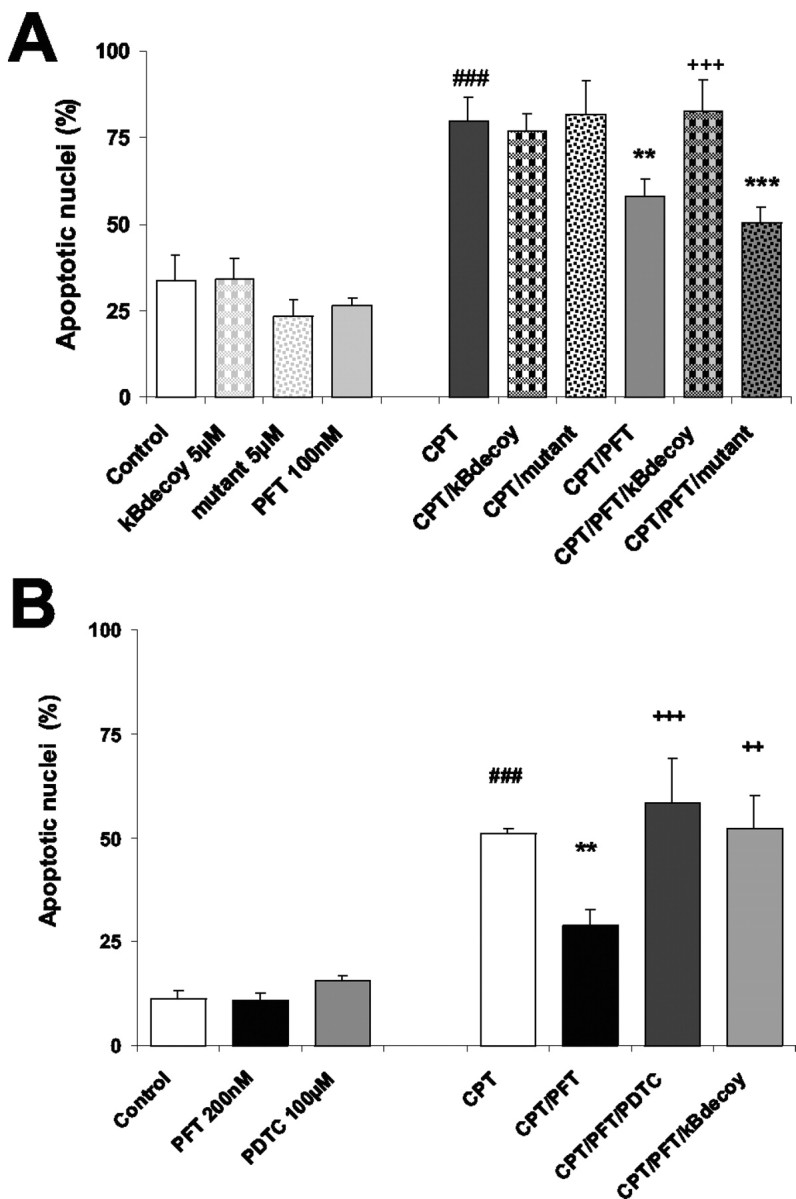
Essential contribution of NF-κB activity in neuroprotection by PFT. A, To evaluate the role of NF-κB activation in the neuroprotective effect of PFT against DNA damage,κBdecoy and mutant DNA oligonucleotides (5μm) were added to the cultures in addition to PFT (100 nm) and camptothecin (CPT; 10μm). Twenty-four hours later, the cells were fixed with paraformaldehyde, and percentages of apoptotic nuclei were evaluated after Hoechst 33258 staining as means ± SD from five cultures per group. Pyknotic or fragmented nuclei exposing an intense fluorescent staining were considered apoptotic in comparison with smooth, round-shaped nuclei exposing low fluorescence of undamaged neurons. ###p < 0.001 as compared with controls; **p < 0.01 and ***p < 0.001 as compared with CPT treatment; and +++p < 0.001 as compared with PFT/CPT cotreatment. B, Embryonic hippocampal cells were treated with the NF-κB inhibitor PDTC (100μm) before exposure to PFT (100 nm) and CPT (10μm). The cultures were fixed in paraformaldehyde stained with Hoechst 33258 24 hr after induction of CPT-mediated apoptosis. The percentage of apoptotic nuclei exposing intense fluorescence and pyknotic and fragmented morphology is presented as mean ± SD of five cultures per group. ###p < 0.001 as compared with controls; **p < 0.01 as compared with CPT treatment; and ++p < 0.01 and +++p < 0.001 as compared with PFT/CPT cotreatment. Note that PDTC blocked the protective effect of PFT in a similar manner as the κBdecoy oligonucleotide, whereas the mutant oligonucleotide did not affect PFT-mediated neuroprotection.
Reciprocal regulation of p53 and NF-κB inhomocysteine-induced neuron death
To test whether the decline of NF-κB activity was a general phenomenon in DNA damage-induced neuronal apoptosis, we used homocysteine, which induced neuron death involving rapid DNA damage and activation of p53 as demonstrated in our previous work (Kruman et al., 2000). Like camptothecin (Culmsee et al., 2001c), homocysteine enhanced the levels of phosphorylated p53 in cultured hippocampal neurons within 5–6 hr of exposure (Fig. 4A). In the same time frame, homocysteine significantly reduced NF-κB transcriptional activity, confirming a reciprocal regulation of both transcription factors in neurons undergoing DNA damage-induced apoptosis. The downregulation of NF-κB transcriptional activity in hippocampal neurons after exposure to homocysteine was prevented by the p53 inhibitors PFT (Fig. 4B) and cPFT (data not shown). Moreover, both p53 inhibitors, PFT and cPFT, prevented neuronal death induced by homocysteine (Fig. 4C). Notably, the preservation of NF-κB transcriptional activity at control levels by PFT in neurons exposed to homocysteine correlated well with the pronounced neuroprotective effect of the p53 inhibitors, which completely prevented homocysteine-induced apoptosis. Overall, these results demonstrated that homocysteine-induced cell death was associated with the activation of p53 and a concomitant decline of NF-κB activity. The p53 inhibitor preserved NF-κB activity and protected neurons in this model of DNA damage-induced apoptosis.
Figure 4.

Homocysteine-induced neuronal apoptosis involves enhanced activation of p53 and inhibition of NF-κB.A, Exposure of hippocampal neurons to homocysteine (200μm) resulted in a significant increase in levels of p53 within 5–6 hr as detected by the use of an anti-phospho-p53 (Ser15) antibody. The values represent the average level of phospho-p53 immunoreactivity [integrated optical density (IOD)] per cell at the indicated times. Measurements were performed in six cultures (15–20 neurons per culture). **p < 0.01 as compared with controls (ANOVA; Scheffé's test). At similar times after exposure to homocysteine, the activity of NF-κB was determined in protein extracts of hippocampal neurons. Measurements were performed in extracts of three independent cultures at each chosen time point and calculated as relative light units (RLU) compared with control levels. ##p < 0.01 as compared with controls (ANOVA; Scheffé's test). Note that the decline in NF-κB activity also occurred within 3–6 hr after exposure of homocysteine, i.e., in the same time period as the increase in phospho-p53 levels. B, Quantification of NF-κB activity was performed at 3 and 6 hr after treatment of hippocampal neurons with PFT (100 nm; black bars), homocysteine (200 μm; hatched bars), or both (checkered bars). Relative light units were calculated as percentage of control levels as mean ± SD of three to four samples per group. **p < 0.01 and ***p < 0.001 PFT-treated cultures compared with controls; ##p < 0.01 homocysteine-treated cells compared with controls; and ++p < 0.01 as compared with homocysteine-treated cultures at 6 hr (ANOVA; Scheffé'stest). Note that PFT induced a significant increase in NF-κBactivitywithin 3 hr and attenuated NF-κBinhibition in homocysteine-treated cells at 6 hr. C, Hippocampal neurons were treated with PFT or cPFT (100 nm each) 1 hr before 24 hr exposure to homocysteine (200 nm). After 24 hr the cells were stained with Hoechst 33258, and the percentage of apoptotic nuclei was determined as mean ± SD from five cultures per group. ###p < 0.001 as compared with controls and ***p < 0.001 as compared with homocysteine-treated neurons (ANOVA; Scheffé's test).
Protective effects of PFT in models of oxygen glucose deprivation and cerebral ischemia
Next, we explored whether the reciprocal regulation of p53 and NF-κB was also relevant in models of OGD and cerebral ischemia. Therefore, we challenged cultured embryonic neurons in a model of OGD in which oxygen and glucose were withdrawn for 4 hr. Measurement of luciferase activity in NF-κB reporter cells revealed a significant decline in NF-κB transcriptional activity within 6 hr after the onset of OGD (Fig. 5A). Moreover, the p53 inhibitors not only restored but enhanced NF-κB transcriptional activity over control levels (Fig. 5A), comparable with the effects of PFT demonstrated with or without additional induction of DNA damage with camptothecin or homocysteine (Figs. 1B,4B). This result confirmed that potential activation of NF-κB was only transduced into enhanced transcriptional activity while p53 was inhibited, whereas p53 blocked NF-κB activity in neurons undergoing apoptosis. In line with the enhanced NF-κB transcriptional activity by PFT, the p53 inhibitor protected cultured hippocampal neurons against cell death induced by OGD (Fig. 5B), and similar results were also obtained in cortical neurons treated with either PFT or cPFT (data not shown). Moreover, inhibition of NF-κB by decoy DNA blocked neuroprotection by PFT against OGD-mediated neuron death (Fig. 5C). These findings indicated that the preservation of NF-κB activity substantially contributed to the neuroprotective effect of PFT in OGD-induced neuron death.
Figure 5.

PFT protects hippocampal neurons against oxygen-glucose deprivation (OGD). A, Hippocampal cultures from NF-κB-luciferase reporter mice were subjected to OGD for 4 hr in Locke's without glucose. Controls received similar medium changes with Neurobasal medium. PFT (100 nm) was added to sister cultures 1 hr before, during, and after OGD. Luciferase activity was determined from cell extracts after 1–8 hr. Relative light units (RLU) representing NF-κB activity are given as percentage of control (n = 3). Note that NF-κB activity declined after OGD, whereas the PFT-treated cells exhibited a pronounced increase in NF-κB activity as compared with both controls and OGD-treated cells. ***p < 0.001 as compared with controls and OGD (ANOVA; Scheffé's). B, OGD was performed by incubating hippocampal neurons in glucose-free Locke's solution in an oxygen-free incubation chamber in a N2 /CO2 (95/5%) atmosphere. Sister cultures incubated with Locke's with glucose deprived of oxygen (OD) were used as controls. The cultures were treated with 100 nm PFT 1 hr before, during, and up to 24 hr after OGD. Neuron cell death was evaluated 24 hr after onset of OGD by the trypan blue exclusion method. Values show the percentage of trypan blue-positive cells of five cultures in each group. ###p<0.001 as compared with the control and OD; **p<0.01 and ***p<0.001 as compared with OGD (ANOVA; Scheffé's test). C, Inhibition of NF-κB activity was achieved by adding κBdecoy DNA (5 μm) 1 hr before PFT treatment and OGD (4 hr). Control cultures were incubated simultaneously in oxygen-free Locke's with glucose (OD). ###p < 0.001 as compared with OD; ***p < 0.001 as compared with OGD; and +++as compared with OGD/PFT cotreatment (ANOVA; Scheffé test).
We next addressed the question of whether the observed phenomenon of NF-κB activation by the p53 inhibitor was also relevant in vivo and therefore administered PFT to NF-κB reporter mice. We detected enhanced luciferase activity in tissue extracts from cortex and hippocampus within 6–12 hr after intraperitoneal injection of PFT (2 mg/kg), indicating that the p53 inhibitor crossed the blood–brain barrier and induced NF-κB transcriptional activity in brain tissue (Fig. 6). A significant induction of NF-κB transcriptional activity as compared with control levels was detected within 12 hr after PFT treatment (2 mg/kg) in cortical (Fig. 6A) and hippocampal brain tissue (Fig. 6B) of the κB reporter mice. When PFT was administered at a dose of 4 mg/kg, a significant increase of luciferase activity was already measured at 3–6 hr (data not shown). This dose-dependent effect of PFT on NF-κB transcriptional activity was also reflected in the cerebroprotective effect of the compound against ischemic brain damage (Fig. 7A). In the model of permanent MCAO in mice, PFT dose-dependently reduced the cortical infarct area up to 50% as compared with controls at a dose of 4 mg/kg. Most interestingly, PFT (2 mg/kg) also protected brain tissue against ischemic brain damage when applied up to 3 hr after onset of ischemia (Fig. 7B). This effect was also dose dependent because 4 mg/kg PFT still provided significant cerebroprotection when administered up to 6 hr after onset of ischemia (data not shown). In addition, we tested PFT (2 mg/kg) in rats exposed to 10 min of TFI to verify the neuroprotective effects of PFT in this rat model of ischemia-induced delayed hippocampal cell death in vivo. In line with the results obtained in cultured hippocampal neurons, PFT significantly reduced ischemia-induced damage of CA1 neurons by ∼40% (Fig. 7C). PFT did not affect blood pressure or body temperature or any other physiological parameter measured throughout the surgical procedure. In both models of cerebral ischemia, body and brain temperature were regulated during surgery and controlled up to 6 hr after ischemia. Overall, these findings confirmed a pivotal role of p53 in ischemic brain damage, which could be prevented in the present experiments by p53 inhibitors.
Figure 6.

PFT-induced activation of NF-κB in brain tissue. Luciferase activity was determined from cortical (A) and hippocampal (B) brain tissue extracts at the indicated times after intraperitoneal administration of PFT (2 mg/kg). Relative light units (RLU) representing NF-κB activity were calculated as percentages of control levels. Values are the mean ± SD of three animals per time point. **p < 0.01 as compared with controls (ANOVA; Duncan's test).
Figure 7.

Cerebroprotective effects of pifithrin-α (PFT) in models of cerebral ischemia. A, Mice were given an intraperitoneal injection of vehicle (0.2% dimethylsulfoxide/saline) or PFT(0.4–4 mg/kg) 1 hr before permanent middle cerebral artery occlusion (MCAO). Forty-eight hours after onset of ischemia, the mice were perfused with a solution of neutral red to stain the brain tissue. After the isolated brain was fixed, the unstained tissue region on the cortical surface was evaluated by measurement with an image-processing system. Values are the mean and SD of 12–15 mice per group. ***p < 0.001 compared with controls; ###p < 0.001 as compared with PFT doses of 0.4–2 mg/kg (ANOVA; Duncan's test). B, PFT (2 mg/kg) was administered intraperitoneally 1 hr before, immediately after (at MCAO), and 3–6 hr after permanent middle cerebral artery occlusion in mice. Mice were killed 48 hr later, and the infarct area was determined on the cortical surface of neutral red-stained brains. Data are given as means ± SD of 15 animals. **p < 0.01 as compared with controls (ANOVA; Duncan's test). C, Rats received 2 mg/kg PFT 1 hr before 10 min of transient forebrain ischemia. Seven days later the rats were killed, and the percentage of damaged neurons was quantified in the CA1 subfield of the hippocampus. The values are the mean ± SD of 12–14 rats per group. *p < 0.05 as compared with controls (Student's t test).
To explore whether p53-dependent ischemic brain damage also involved regulation of NF-κB transcriptional activity, we used κB reporter mice in the model of permanent MCAO. For the measurement of NF-κB activity in the ischemic tissue, we extracted protein at given times from defined ipsilateral cortical regions within the supply area of the MCAO, which becomes infarcted after occlusion of the vessel. For comparison, similar regions of the nonischemic contralateral hemisphere of mice receiving MCAO and of both hemispheres in control animals were removed, respectively. Within a few hours after ischemia, pronounced changes in NF-κB activity were observed in the ipsilateral and the contralateral cortical tissue. In the ischemic ipsilateral cortex, a significant decline of NF-κB activity to <20% of control levels occurred within 3–6 hr that only partly recovered within 24 hr (Fig. 8A). The p53 inhibitor PFT not only restored but significantly enhanced NF-κB activity in the ipsilateral cortex within 3–6 hr after MCAO as compared with ischemic controls (Fig. 8C, D). In contrast to the findings on postischemic NF-κB activity in the ipsilateral hemisphere, a pronounced induction of NF-κB transcriptional activity was observed in the contralateral cortex within 6–12 hr after MCAO (Fig. 8B). In addition, PFT significantly accelerated the ischemia-induced NF-κB activity in the contralateral hemisphere (Fig. 8C, D). This revealed activation of NF-κB in nonischemic brain tissue, whereas NF-κB activity rapidly declined in the infarct area, where mechanisms of cell death dominated, which may include accumulation of p53. Similar to the findings in vitro, the p53 inhibitor released the suppression of NF-κB in the ipsilateral ischemic tissue, thereby allowing the transduction of NF-κB activity that correlated with the cerebroprotective effect of PFT.
Figure 8.

Pifithrin-α (PFT) prevents the decline in NF-κB transcriptional activity in the ischemic cortex after middle cerebral artery occlusion. A, B, Luciferase activity was determined in the ipsilateral (A) and contralateral (B) cortical tissue at the indicated times after permanent MCAO in NF-κB-luciferase reporter mice. Relative light units (RLU) resembling NF-κB activity are given as mean percentage ± SD of controls from three to four animals per group. Note the pronounced decline in NF-κB activity at 3–6 hr after ischemia, which recovered within 12–24 hr in the ipsilateral cortex (A). In contrast, a pronounced increase was observed within 6–12 hr after MCAO in the contralateral cortex (B). C, D, PFT (2 mg/kg) was administered 1 hr before MCAO and rapidly induced a pronounced enhancement of NF-κB activity within 3 hr (C) and 6 hr (D) in the ipsilateral and contralateral cortex. Luciferase activity resembling NF-κB activity was determined as mean RLU percentage ± SD of controls from three to four animals per group. **p < 0.01 and ***p < 0.001 as compared with vehicle-treated controls (Student's t test).
Discussion
The present study established a reciprocal regulation of the transcription factors NF-κB and p53 in neurons undergoing cell death induced by DNA damage or ischemia. Both p53-mediated block of NF-κB activity and neuron cell death were attenuated by the p53 inhibitor PFT in vitro and in vivo. The reciprocal activities of the transcription factors appeared to be mediated through competitive binding to the transcriptional cofactor p300. According to earlier studies in tumor cell lines, the transcriptional cofactors p300 and cAMP response element-binding protein (CBP) function as a link between the transcriptional activities of p53 and NF-κB (Ravi et al., 1998; Wadgaonkar et al., 1999; Webster and Perkins, 1999). In these cell lines, NF-κB and p53 competed in binding to p300/CBP, which were proven to be essential for the proper function of both transcription factors (Ravi et al., 1998; Wadgaonkar et al., 1999; Ikeda et al., 2000). In our study, lethal stress induced by DNA damage or ischemia obviously changed the balance of p300 binding to NF-κB or p53 toward an activation of p53 and a concomitant loss of NF-κB activity. Thus, in addition to the activation of an important cell death pathway via p53 activation, the neurons faced the inhibition of NF-κB-dependent survival pathways. This would also explain the observed fast decline of NF-κB activity within the first hours after induction of cell death associated with a rapid activation of p53 in our experiments. It will be of interest to further investigate whether other factors such as CBP are involved in the interaction between NF-κB and p53 in neurons and whether additional survival pathways are blocked in p53-mediated neuron death. Very recently, Gudkov and collegues (Komarova et al., 2003) reported that the p53 inhibitor PFT suppressed heat shock and glucocorticoid pathways in fibroblasts and cancer cell lines. Partly in contrast to our findings in neurons, PFT did not further enhance DNA binding activity of NF-κB induced by TNF-α in HeLa cells, probably because NF-κB activation was already at a maximum because of the TNF treatment; however, in their studies, the concentration of PFT (10–30 μM) exceeded the effective concentrations used in our neuronal cultures (100 nm), and PFT concentrations >1 μM were found to be neurotoxic in our previous studies (Culmsee et al., 2001c). These contrary results suggest that differences in p53-mediated regulation of cell signaling exist between different cell types.
Although considerable evidence indicated a pivotal role for p53 in neuronal death that occurs in disorders ranging from stroke and brain trauma to Alzheimer's disease and Parkinson's disease, no therapeutic agents that target p53 have been available until recently (Komarov et al., 1999; Culmsee et al., 2001c; Zhu et al., 2002). As also demonstrated in our previous work, inhibition of p53 transcriptional activity is a major mechanism of neuroprotection by PFT, which blocks crucial upstream steps of neuronal apoptosis, such as mitochondrial dysfunction and activation of caspases, and therefore provides protection against various insults including DNA damage, oxidative stress, and glutamate toxicity (Culmsee et al., 2001c).
In the present study, PFT was used as a tool to investigate the proposed novel mechanism of neuronal apoptosis that involved p53-mediated inhibition of the transcription factor NF-κB. DNA damage induced by camptothecin or homocysteine and OGD as well as cerebral ischemia have been shown to involve upregulation and activation of p53 in neurons (Kruman et al., 2000; Culmsee et al., 2001c; Zhou et al., 2001). Here we showed that the p53 inhibitor prevented neuron death in these experimental models and concomitantly attenuated the decline of NF-κB transcriptional activity. Furthermore, inhibition of NF-κBby κB-decoy oligonucleotides and PDTC abolished neuroprotection by PFT against camptothecin-induced apoptosis. Thus, the effect of PFT on NF-κB activity was not just a result of enhanced neuronal survival but essentially contributed to the neuroprotective effect. These findings are in line with recent studies demonstrating a crucial role of NF-κB in neuronal survival during brain development and after lethal insults in cultured neurons (Mattson et al., 2000; Bhakar et al., 2002).
In neurons, NF-κB is an important regulator of evolutionarily conserved biochemical and molecular cascades designed to prevent cell death and promote neuronal plasticity (Mattson et al., 2000). Activation of NF-κB can interrupt apoptotic cascades at relatively early steps, before mitochondrial dysfunction and oxyradical production (Mattson and Furukawa, 1996; Mattson and Lindvall, 1997). Gene targets of NF-κB that mediate its anti-apoptotic actions include, for example, the antioxidant enzyme MnSOD (Wong et al., 1989; Mattson et al., 1997), members of the IAPs (Chu et al., 1997), and the calcium binding protein calbindin D28k (Cheng et al., 1994; Mattson et al., 1995).
In contrast, NF-kB can enhance many genes involved in the pathogenesis of cerebral ischemia, for example, inducible nitric oxide synthase or interleukin-1α (Mattson et al., 2000); however, such neurotoxic factors were predominantly expressed in glial cells activated as a reaction to neurodegeneration (Mattson et al., 2000). In addition, our findings imply that NF-κB may be activated and translocated to the nucleus, without exerting transcriptional activity, because of p53 competing for essential transcriptional cofactors. This would also explain why some authors found enhanced NF-κB DNA binding in their gel shift assays of nuclear extracts from apoptotic (neuronal) cells, concluding that enhanced NF-κB activity contributed to (neuron) cell death. For many years it was thought that NF-κB played a role in killing the cells because NF-κB was upregulated and seemed to be activated under conditions in which many neurons die, for example after seizures or ischemic stroke (Salminen et al., 1995; Grilli et al., 1996; Clemens et al., 1997); however, the interpretation that NF-κB plays an active role in killing neurons was in most part on the basis of phenomenology rather than data addressing cause–effect relationships. By using NF-κB reporter mice we now showed clearly that NF-κB activity was significantly blocked during cell death induced by ischemia or DNA damage. Similar results were obtained in osteosarcoma cell lines and lymphocytes in which irradiation-induced apoptosis was accompanied by enhanced NF-κB DNA binding activity in gel shift assays, whereas in the living cells transcriptional activity of NF-κB was blocked (Campbellet al., 2001; D. Zhou, C. Culmsee, and M. P. Mattson, unpublished observations). In lymphocytes as well as in neurons, such potential NF-κB activity was “released” by the p53 inhibitor PFT. A similar effect apparently occurred in cultured neurons, where the p53 inhibitor clearly attenuated the pronounced decline of NF-kB transcriptional activity after exposure to cellular stress induced by OGD, camptothecin, or homocysteine.
Finally, we found that the p53 inhibitors induced NF-κB transcriptional activity in neurons under basal cultured conditions in the absence of exposure to insults, a finding that suggests a possible role for p53 in modulating physiological signaling through NF-κB. Data suggest that NF-κB plays important roles in developmental and synaptic plasticity (Mattson et al., 2000), and when taken together with the recent evidence that p53 is present in synaptic terminals where it can locally modulate mitochondrial function (Gilman et al., 2003), it therefore will be of interest to determine whether p53 can modulate synaptic plasticity.
In conclusion, our data expose the inhibition of NF-κB activity as a pivotal mechanism in p53-mediated neuron death. The fine-tuned equilibrium of these counteracting transcription factors may depend on access to cofactors such as p300 and appears to be highly important in controlling the fate of a cell toward death or survival signaling. Therefore, p53 inhibitors do not simply block important cell death pathways but enhance potent endogenous mechanisms of neuroprotection at the same time.
Footnotes
We thank Sandra Engel, Elke Bauerbach, and Michaela Stumpf for excellent technical assistance. We gratefully acknowledge the kind support of Jay C. Dunn (University of Kentucky, Lexington, KY), who kindly shared the protocol for coimmunoprecipitation of p300 and p53. We also thank Nikolaus Plesnila for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Carsten Culmsee, Biotechnologie, Department Pharmazie, Zentrum für Arzneimittelforschung, Ludwig-Maximilians-Universität, Butenandtstraße 9D, D-81377 München, Germany. E-mail: ccuph@cup.uni-muenchen.de.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238586-10$15.00/0
References
- Backhauß C, Karkoutly C, Welsch M, Krieglstein J ( 1992) A mouse model of focal cerebral ischemia for screening neuroprotective drug effects. J Pharmacol Methods 27: 27-32. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA ( 2002) Constitutive nuclear factor-κ B activity is required for central neuron survival. J Neurosci 22: 8466-8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui TN, König HG, Culmsee C, Bauerbach E, Poppe M, Krieglstein J, Prehn JHM ( 2002) p75 neurotrophin receptor is required for constitutive and NGF-induced survival signaling in PC12 cells and rat hippocampal neurones. J Neurochem 81: 594-605. [DOI] [PubMed] [Google Scholar]
- Caelles C, Helmberg A, Karin M ( 1994) p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 370: 220-223. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Chapman NR, Perkins ND ( 2001) UV stimulation induces nuclear factor kappaB (NF-kappaB) DNA-binding activity but not transcriptional activation. Biochem Soc Trans 29: 688-691. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mattson MP ( 1999) Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res 58: 167-190. [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP ( 1994) Tumor necrosis factors protect neurons against excitotoxic/metabolic insults and promote maintenance of calcium homeostasis. Neuron 12: 139-152. [DOI] [PubMed] [Google Scholar]
- Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW ( 1997) Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-κB control. Proc Natl Acad Sci USA 94: 10057-10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP ( 1997) Global ischemia activates NF-κB in forebrain neurons of rats. Stroke 28: 1073-1080. [DOI] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS ( 1999) Bax-dependent caspase-3 activation is a key determinant in p53-dependent apoptosis in neurons. J Neurosci 19: 7860-7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crumrine RC, Thomas AL, Morgan PF ( 1994) Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab 14: 887-891. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Bondada S, Mattson MP ( 2001a) Hippocampal neurons of mice deficient in DNA-dependent protein kinase exhibit increased vulnerability to DNA damage, oxidative stress and excitotoxicity. Brain Res Mol Brain Res 87: 257-262 [DOI] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp B, Mattson MP ( 2001b) Evidence that AMP activated protein kinase protects against metabolic, excitotoxic and oxidative insults in embryonic hippocampal neurons. J Mol Neurosci 17: 45-58. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP ( 2001c) A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem 77: 220-228. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu Y, Krieglstein J, Mattson MP ( 2001d) Evidence for the involvement of Par-4 in ischemic neuron cell death. J Cereb Blood Flow Metab 21: 334-343. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Junker V, Gerling N, Lehmann M, Nikolova-Karakashian M, Prehn JHM, Mattson MP, Krieglstein J ( 2002) NGF-induced neuroprotection in hippocampal neurons requires the common neurotrophin receptor p75NTR. Neuroscience 115: 1089-1108. [DOI] [PubMed] [Google Scholar]
- De la Monte SM, Sohn YK, Wands JR ( 1997) Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci 152: 73-83. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA ( 1999) Pathobiology of ischemic stroke: an integrated view. Trends Neurosci 22: 391-397. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP ( 2002) p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol 52: 597-606. [DOI] [PubMed] [Google Scholar]
- Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H ( 1996) Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci 8: 1812-1821. [DOI] [PubMed] [Google Scholar]
- Gao C, Tsuchida N ( 1999) Activation of caspases in p53-induced transactivation-independent apoptosis. Jpn J Cancer Res 90: 180-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman CP, Chan SL, Guo Z, Zhu X, Greig NH, Mattson MP ( 2003) p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromol Med 3: 159-172. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC ( 1998) Mitochondria and apoptosis. Science 28: 1309-1312. [DOI] [PubMed] [Google Scholar]
- Grilli M, Memo M ( 1999) Possible role of NF-kappaB and p53 in the glutamate-induced pro-apoptotic neuronal pathway. Cell Death Differ 6: 22-27. [DOI] [PubMed] [Google Scholar]
- Grilli M, Pizzi M, Memo M, Spano P ( 1996) Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science 274: 1383-1385. [DOI] [PubMed] [Google Scholar]
- Hengartner MO ( 2000) The biochemistry of apoptosis. Nature 407: 770-776. [DOI] [PubMed] [Google Scholar]
- Hou ST, Tu Y, Buchan AM, Huang Z, Preston E, Rasquina I, Robertson GS, MacManus JP ( 1997) Increases in DNA lesions and the DNA damage indicator Gadd45 following transient cerebral ischemia. Biochem Cell Biol 75: 383-392. [PubMed] [Google Scholar]
- Hughes PE, Alexi T, Schreiber SS ( 1997) A role for the tumour suppressor gene p53 in regulating neuronal apoptosis. NeuroReport 8: v-xii. [PubMed] [Google Scholar]
- Ikeda A, Sun X, Li Y, Zhang Y, Eckner R, Doi TS, Takahashi T, Obata Y, Yoshioka K, Yamamoto K ( 2000) p300/CBP-dependent and -independent transcriptional interference between NF-kappaB RelA and p53. Biochem Biophys Res Commun 272: 375-379. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW ( 1998) Understanding cell death in Parkinson's disease. Ann Neurol 44: S72-S84. [DOI] [PubMed] [Google Scholar]
- Junker V, Becker A, Hühne R, Zembatov M, Ravati A, Culmsee C, Krieglstein J ( 2002) Stimulation of β-adrenoceptors activates astrocytes and provides neuroprotection. Eur J Pharmacol 446: 25-36. [DOI] [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV ( 1999) A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285: 1733-1737. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Neznanov N, Komarov PG, Chernov MV, Wang K, Gudkov AV ( 2003) p53 Inhibitor pifithrin alpha can suppress heat shock and glucocorticoid signaling pathways. J Biol Chem 278: 15465-15468. [DOI] [PubMed] [Google Scholar]
- Kruman II, Culmsee C, Chan S, Kruman Y, Penix L, Mattson MP ( 2000) Homocysteine induces apoptosis and accelerates oxidative and excitotoxic damage in hippocampal neurons. J Neurosci 20: 6920-6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chopp M, Zhang ZG, Zaloga C, Niewenhuis L, Gautam S ( 1994) p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke 25: 849-855. [DOI] [PubMed] [Google Scholar]
- Lipton P ( 1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. [DOI] [PubMed] [Google Scholar]
- Mattson MP ( 2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1: 120-129. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Furukawa K ( 1996) Programmed cell life: anti-apoptotic signaling and therapeutic strategies for neurodegenerative disorders. Restor Neurol Neurosci 9: 191-205. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Kroemer G ( 2003) Mitochondria in cell death: novel targets for neuro- and cardio-protection. Trends Mol Med 9: 196-205. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Lindvall O ( 1997) Neurotrophic factor and cytokine signaling in the aging brain. In: Advances in cell aging and gerontology, Vol 2, The aging brain (Mattson MP, Geddes JW, eds), pp 299-345. Greenwich, CT: JAI. [Google Scholar]
- Mattson MP, Zhang Y, Bose S ( 1993) Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol 121: 1-13. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Baldwin S, Smith-Swintosky VL, Keller J, Geddes JW, Scheff SW, Christakos S ( 1995) Brain injury and tumor necrosis factors induce expression of calbindin D-28k in astrocytes: a cytoprotective response. J Neurosci Res 42: 357-370. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K ( 1997) Activation of NF-κB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of MN-SOD and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res 49: 681-697. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Culmsee C, Yu ZF, Camandola S ( 2000) Roles of NF-κB in neuronal survival and plasticity. J Neurochem 74: 443-456. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM ( 2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577-590. [DOI] [PubMed] [Google Scholar]
- Millet I, Phillips RJ, Sherwin RS, Ghosh S, Voll RE, Flavell RA, Vignery A, Rincon M ( 2000) Inhibition of NF-κB activity and enhancement of apoptosis by the neuropeptide calcitonin gene-related peptide. J Biol Chem 275: 15114-15121. [DOI] [PubMed] [Google Scholar]
- Napieralski JA, Raghupathi R, McIntosh TK ( 1999) The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Mol Brain Res 71: 78-86. [DOI] [PubMed] [Google Scholar]
- Ravi R, Mookerjee B, van Hensbergen Y, Bedi GC, Giordano A, El-Deiry WS, Fuchs EJ, Bedi A ( 1998) p53-mediated repression of nuclear factor-kappaB RelA via the transcriptional integrator p300. Cancer Res 58: 4531-4536. [PubMed] [Google Scholar]
- Ryan KM, Ernst MK, Rice NR, Vousden KH ( 2000) Role of NF-κB in p53-mediated programmed cell death. Nature 892-897. [DOI] [PubMed]
- Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS ( 1997) Induction of tumor suppressor p53 and DNA fragmentation in organotypic hippocampal cultures following excitotoxin treatment. Exp Neurol 145: 81-88. [DOI] [PubMed] [Google Scholar]
- Salminen A, Liu PK, Hsu CY ( 1995) Alteration of transcription factor binding activities in the ischemic brain. Biochem Biophys Res Commun 212: 939-944. [DOI] [PubMed] [Google Scholar]
- Takimoto CH, Kieffer LV, Kieffer ME, Arbuck SG, Wright J ( 1999) DNA topoisomerase I poisons. Cancer Chemother Biol Response Modif 18: 81-124. [PubMed] [Google Scholar]
- Uberti D, Belloni M, Grilli M, Spano P, Memo M ( 1998) Induction of tumour-suppressor phosphoprotein p53 in the apoptosis of cultured rat cerebellar neurones triggered by excitatory amino acids. Eur J Neurosci 10: 246-254. [DOI] [PubMed] [Google Scholar]
- Uberti D, Grilli M, Memo M ( 2000) Contribution of NF-kappaB and p53 in the glutamate-induced apoptosis. Int J Dev Neurosci 18: 447-454. [DOI] [PubMed] [Google Scholar]
- Voll RE, Jimi E, Phillips RJ, Barber DF, Rincon M, Hayday AC, Flavell RA, Ghosh S ( 2000) NF-kappa B activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity 13: 677-689. [DOI] [PubMed] [Google Scholar]
- Wadgaonkar R, Phelps KM, Haque Z, Williams AJ, Silverman ES, Collins T ( 1999) CREB-binding protein is a nuclear integrator of nuclear factor-kappaB and p53 signaling. J Biol Chem 274: 1879-1882. [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND ( 1999) Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol 19: 3485-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh FA, Sakamoto T, McKee AE, Sims RE ( 1987) Effect of lactacidosis on pyridine nucleotide stability during ischemia in mouse brain. J Neurochem 49: 846-851. [DOI] [PubMed] [Google Scholar]
- Wong GH, Elwell JH, Oberley LW, Goeddell DV ( 1989) Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell 58: 923-931. [DOI] [PubMed] [Google Scholar]
- Wood KA, Youle RJ ( 1995) The role of free radicals and p53 in neuron apoptosis in vivo J Neurosci 15: 5851-5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS ( 1998) Bax involvement in p53-mediated neuronal cell death. J Neurosci 18: 1363-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fu Y, Tang XC ( 2001) Huperzine A protects rat pheochromocytoma cells against oxygen-glucose deprivation. NeuroReport 12: 2073-2077. [DOI] [PubMed] [Google Scholar]
- Zhu X, Yu QS, Cutler R, Culmsee C, Holloway HW, Mattson MP, Greig NH ( 2002) Design and synthesis of novel p53 inhibitors as neuroprotective agents. J Med Chem 45: 5090-5097. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Culmsee C, Semkova I, Krieglstein J ( 1998) Stimulation of beta2-adrenoceptors inhibits apoptosis in rat brain after transient forebrain ischemia. J Cereb Blood Flow Metab 18: 1032-1039. [DOI] [PubMed] [Google Scholar]