

Abstract
Tau-positive inclusions in oligodendrocytes are consistent neuropathological features of corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementias with Parkinsonism linked to chromosome 17. Here we show by immunohistochemistry that tau-positive oligodendroglial inclusion bodies also contain the small heat-shock protein (HSP) αB-crystallin but not HSP70. To study the molecular mechanisms underlying inclusion body formation, we engineered an oligodendroglia cell line (OLN-t40) to overexpress the longest human tau isoform. Treatment of OLN-t40 cells with okadaic acid (OA), an inhibitor of protein phosphatase 2A, caused tau hyperphosphorylation and a decrease in the binding of tau to microtubules. Simultaneously, tau-positive aggregates that also stained with the amyloid-binding dye thioflavin-S as well as with antibodies to tau and αB-crystallin were detected. However, they were only transiently expressed and were degraded within 24 hr. When the proteasomal apparatus was inhibited by carbobenzoxy-l-leucyl-l-leucyl-l-leucinal (MG-132) after OA treatment, the aggregates were stabilized and were still detectable after 18 hr in the absence of OA. Incubation with MG-132 alone inhibited tau proteolysis and led to the induction of HSPs, including αB-crystallin and to its translocation to the perinuclear region, but did not induce the formation of thioflavin-S-positive aggregates. Hence, although tau hyperphosphorylation induced by protein phosphatase inhibition contributes to pathological aggregate formation, only hyperphosporylation of tau followed by proteasome inhibition leads to stable fibrillary deposits of tau similar to those observed in neurodegenerative diseases.
Keywords: αB-crystallin, cytoskeleton, tau phosphorylation, coiled bodies, glial inclusions, proteasome, thioflavin-S
Introduction
Intracellular accumulations of the microtubule (MT)-associated protein tau are the defining neuropathological characteristics of Alzheimer's disease (AD), and they also occur in a variety of dementias and movement disorders collectively named tauopathies (Lee et al., 2001). In AD, fibrillary tau accumulations preferentially form in nerve cells, but abundant neuronal and glial filamentous tau inclusions are prominent in familial multiple system tauopathy, which belongs to a group of frontotemporal dementias with Parkinsonism linked to chromosome 17 (FTDP-17) (Goedert et al., 1998). Tau-positive glial inclusions are consistent features in the brains of patients with FTDP-17, progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) (Chin and Goldman, 1996; Goedert et al., 1998; Komori, 1999; Berry et al., 2001). Oligodendroglial lesions and white-matter pathology are characterized by the occurrence of glial fibrillary tangles or “coiled bodies” containing tau, and tau as well as α-synuclein-positive glial cytoplasmic inclusions (GCIs) in oligodendroglia are the histological hallmarks of multiple system atrophy (MSA). GCIs are composed of a meshwork of loosely packed filaments and stained with antibodies against tau, ubiquitin, α-synuclein, and the small heat-shock protein (HSP) αB-crystallin, although α-synuclein is the major building block of these inclusions (Chin and Goldman, 1996; Tu et al., 1998; Komori, 1999).
Oligodendrocytes are rich in MTs and express tau (Gorath et al., 2001). Tau is abundant in the CNS and modulates MT stability and organization. The molecular mechanisms leading to the formation of insoluble aggregates of tau, which under normal conditions is a highly soluble protein, are not yet resolved. Although tau hyperphosphorylation might be a prerequisite for aggregate formation, evidence has accumulated suggesting that hyperphosphorylation is not directly responsible for pathological aggregation of tau and cell death (Schneider et al., 1999), thus indicating that other factors might contribute to the formation of neurofibrillary tau pathology.
Protein aggregates do not accumulate in healthy, unstressed cells, and HSPs act as molecular chaperones and prevent the aggregation or accumulation of abnormal proteins (Sherman and Goldberg, 2001). If this protective mechanism fails, misfolded proteins are ubiquitinated and degraded by the proteasomal machinery (Schwartz and Ciechanover, 1999). Inclusion bodies or aggregates are formed when the protein degradation machinery is exceeded or impaired. Small aggregates can be sequestered around the MT-organizing center, because of MT-dependent retrograde transport, and assemble to large aggresomes, which are also enriched in molecular chaperones (Johnston et al., 1998; Kopito, 2000).
In the present study, we show by immunohistochemistry that inclusion bodies in oligodendrocytes of the brains of patients with CBD, PSP, or FTDP-17, which are formed by tau filaments rather than α-synuclein, are also positively labeled by antibodies against the small HSP αB-crystallin, whereas HSP70 is not detectable. To investigate whether tau hyperphosphorylation is a necessary requirement for the formation of cytoplasmic aggregates in glia cells, we examined the effects of the protein phosphatase 2A (PP2A) inhibitor okadaic acid (OA) on transfected oligodendroglia cell line OLN-93 cells that were engineered to stably express the longest human tau isoform. OA leads to the transient formation of thioflavin S-positive cytoplasmic inclusions, which can be stabilized by the proteasome inhibitor carbobenzoxy-l-leucyl-l-leucyl-l-leucinal (MG-132). Our studies show that these experimentally induced oligodendroglial inclusion bodies are associated with tau aggregate formation, and that they also accumulate the small HSP αB-crystallin as observed in authentic coiled bodies in the brains of patients with tauopathies.
Materials and Methods
Materials and antibodies. Cell culture media were purchased from Invitrogen (Grand Island, NY). Okadaic acid, MG-132, and proteolytic substrates Z-Leu-Leu-Glu-AMC (S2) and Suc-Leu-Leu-Val-Tyr-AMC (S3) were purchased from Calbiochem (Bad Soden, Germany). Lithium chloride, Taxol, GTP, and ATP were purchased from Sigma (St. Louis, MO).
For Western blot analysis, the following panel of tau antibodies and the working dilutions were used, as described previously (Vogelsberg-Ragaglia et al., 2000): tau 17026 (1:2000), a phosphorylation-independent rabbit polyclonal antibody made against the largest human recombinant tau; monoclonal antibody (MAb) tau-1 (1:1000), specific for nonphosphorylated epitope located in amino acid residues 189-209; MAb PHF-1 (1:500; generously provided by Peter Davies, Albert Einstein College of Medicine, Bronx, NY), specific for phosphorylated serine 396/404; and MAb 12E8 (1:250), specific for phosphorylated serine 262. The anti-α-tubulin MAb (1:1000) was obtained from Sigma. HRP-conjugated anti-mouse IgG was obtained from Amersham Biosciences (Freiburg, Germany), and anti-rabbit IgG was obtained from Bio-Rad (Munich, Germany). Polyclonal anti-αB-crystallin (1:500), polyclonal anti-HSP32/HO-1 (1:1000), polyclonal anti-HSP40 (1:1000), MAb anti-HSP60 (1:1000), MAb anti-HSP70 (1:1000), MAb anti-HSP/HSC70 (1:1000), and MAb anti-HSP90 (1:1000) were obtained from StressGen (Victoria, Canada). Antibodies to the proteasome subunits anti-20Sα (1:1000) and anti-20Sβ (1:1000) were purchased from Calbiochem.
Histochemistry and immunohistochemistry. The human tissue was fixed in 10% formalin, paraffin-embedded, and cut into 6 μm thick sections. Immunohistochemistry was performed as described previously (Schmidt et al., 1987) using the ABC method (Vectastain ABC Kit; Vector Laboratories, Burlingame, CA) and 3, 3′-diaminobenzidine (DAB) as chromogen. The following primary antibodies were used: MAb αB-crystallin (1:2500), HSP70 (1:200), and PHF-1 (1:2000). Double-labeling immunofluorescence studies were performed by coincubating sections with antibodies specific for N-tau and αB-crystallin. N-tau is a rabbit polyclonal antibody generated by immunizing rabbits with the synthetic peptide AEPRQEFEVMEC, which corresponds to the amino terminal 12 amino acids (Babco, Richmond, CA). After extensive washes, sections were labeled using AlexaFluor 488 and 594-conjugated secondary antibodies (Molecular Probes, Eugene, OR) and washed and mounted using Vectashield-4′,6′-diamidino-2-phenylindole (DAPI)-mounting medium (Vector Laboratories). The sections were viewed with an Olympus PX51 microscope (Olympus Optical, Tokyo, Japan) equipped with bright-field and fluorescence light sources. Both bright-field and fluorescent images were obtained from the same field using a ProGres C14 camera (Laser Optik System; Jenoptik, Jena, Germany).
Stable expression of the longest human tau isoform (T40) in transfected OLN-93 cells. OLN-93 cells were kept in DMEM supplemented with 10% heat-inactivated fetal calf serum, 2 mm glutamine, 50 U/ml of penicillin, and 50 μg/ml of streptomycin (Richter-Landsberg and Heinrich, 1996). OLN-93 cells were cotransfected with tau40 cDNA and pcDNA3 containing the neomycin gene by using the calcium phosphate precipitation method (Chen and Okayama, 1987). After selection in DMEM containing 1 mg/ml of G418, the cells were screened for tau expression by Western blot and indirect immunofluorescence. A stable cell line was established, designated OLN-t40, and used in the studies reported below.
Immunoblot analysis. Cellular monolayers of control and treated cells were washed with PBS once, scraped off in sample buffer containing 1% SDS, and boiled for 10 min. Protein contents in the samples were determined according to Neuhoff et al. (1979). For immunoblotting, total cellular extracts (5-20 μg of protein per lane) were separated by one-dimensional SDS-PAGE using 7.5 or 12.5% polyacrylamide gels and transferred to nitrocellulose membranes (0.45 μm; Schleicher & Schuell, Dassel, Germany) according to Towbin et al. (1979). The blots were saturated with TBS-T (20 mm Tris, pH 7.5, 136.8 mm NaCl, 0.1% v/v Tween 20) containing 5% dry milk and incubated with the individual antibodies overnight at 4°C. After washing, incubation with HRP-conjugated anti-mouse (1:2000) or anti-rabbit IgG (1:5000) was performed for 1 hr, and blots were visualized by the enhanced chemiluminescence procedure as described by the manufacturer (Amersham Biosciences). All experiments were performed at least three times with similar results.
Measurement of proteasome activity. Proteasome activity was determined using a fluorescence assay (Keller et al., 2000). Chymotrypsin- and postglutamyl-peptidase-hydrolase peptide hydrolyzing activities were assayed by fluorimetric measurement of the release of 7-amido-4-methylcoumarin (AMC) from two synthetic substrates: Z-Leu-Leu-Glu-AMC (proteasome substrate II, S2) and Suc-Leu-Leu-Val-Tyr-AMC (proteasome substrate III, S3) at 37°C for 60 min. S2 was used for determination of the postglutamyl-peptidase-hydrolase function, and S3 was used for the chymotrypsin-like peptidase function of the proteasome. OLN-t40 cells were treated as described and harvested in ice-cold proteolysis assay buffer containing 10 mm Tris-HCl, pH 7.2, 0.035% SDS, 5 mm MgCl2,and5mm ATP. Protein concentrations of the resulting lysates were determined by the bicinchoninic acid method (Pierce, Rockford, IL) using bovine serum albumin as a standard. Aliquots of 350 μl each, with a protein concentration of 1 μg/μl, were incubated with 3.5 μl of S2 or S3 (5 mm) for 30 min at 37°C. Fluorescence was determined at 15 min intervals for 1 hr at 360nm excitation and 410nm emission in a fluorescent microplate reader (FluoroCount; Packard, Meridian, CT). Proteasomal activity was determined as an increase in fluorescence of the reaction products.
MT-binding assay of tau from OLN-t40 cells. To determine whether OA (5-20 nm) or lithium chloride (20 mm) alters the interaction of tau with tubulin in the MTs of intact cells, an MT-binding assay was performed as described previously (Bramblett et al., 1993; Merrick et al., 1997). In brief, OLN-t40 cells were harvested in high-salt reassembly buffer (0.1 m Tris, 0.5 mm MgSO4,1mm EGTA, 2 mm dithiothreitol, and 0.75 m NaCl, pH 6.8) supplemented with 0.1% Triton X-100, 20 μm Taxol, 2 mm GTP, and a mixture of protease inhibitors (2 mm phenylmethylsulfonyl fluoride, l-1-chloro-3-(4-tosylamido)-4-phenyl-2-butanon, l-1-chloro-3-(4-tosylamido)-7-amino-2-heptanone-hydrochloride, leupeptin, pepstatin, and soy bean trypsin inhibitor; each at 1 μg/ml) at 37°C. Cell lysates were homogenized with 15 strokes in a warm Dounce homogenizer and then immediately centrifuged for 20 min at 50,000 × g at 25°C. The supernatant (S) containing unbound tau was removed, and the protein concentration was determined. The remaining pellet (P) was resuspended in a 2× volume of sample buffer corresponding to the total volume of supernatant after normalizing to total protein. The samples were subjected to immunoblot analysis as above. The ratio of tau bound to MTs (P) versus soluble or unbound tau (S) was assessed by comparing the tau immunoreactivities in these two fractions.
Immunofluorescence. Cells were cultured on poly-l-lysine-coated glass coverslips (2 × 104 cells per 35 mm dish) for 24 hr in DMEM/10% FCS followed for 3 d in DMEM/0.5% FCS and then subjected to OA or MG-132 as indicated. After washing with PBS, cells were fixed with 3% paraformaldehyde and permeabilized with 0.1% Triton X-100 for 15 min. The coverslips were washed three times and incubated overnight at 4°C with rabbit polyclonal anti-tau (17026) antibodies (1:1000) or mouse MAb αΒ-crystallin (1:100) or PHF-1 (1:100) antibodies. After washing with PBS, cells were incubated for 1 hr with tetramethylrhodamine isothiocyanate-conjugated (1:100) and FITC-conjugated (1:100) secondary antibodies (Jackson ImmunoResearch, West Grove, PA), washed with PBS, and mounted. Nuclei were stained by including DAPI (1 μg/ml) to the mounting medium. Fluorescent labeling was studied using a Zeiss (Oberkochen, Germany) epifluorescence microscope equipped with a digital camera using a plan-neofluar objective (100×).
Thioflavin-S staining. Cells were fixed and subjected to indirect immunofluorescence staining as described above. Before mounting, coverslips were immersed in thioflavin-S solution (0.005%) for 5 min. Thereafter, cells were washed three times in ethanol (70%), one time in water, and then mounted.
Immunoelectron microscopy of OLN-t40 cells. Immunoelectron microscopy (immuno-EM) was performed on representative samples of OLN-t40 cells expressing wild-type tau after fixation with 4% paraformaldehyde and 0.25% glutaraldehyde in PBS as described previously (Vogelsberg-Ragaglia et al., 2000). The anti-tau antiserum 17026 (dilution, 1:500) in 0.1% BSA and PBS was used as the primary antibody, and biotinylated goat anti-rabbit IgG (1:100; Vector Laboratories) was used as a secondary antibody. DAB plus silver-gold enhancement (Nanoprobes, Yaphank, NY) immuno-EM method was conducted as described previously (Vogelsberg-Ragaglia et al., 2000). After visualizing the DAB-positive cells labeled by routine immuno-EM methods, silver-gold intensification was performed, and the cells were examined with a JEM1010 electron microscope (Jeol, Peabody, MA) at 80 kV.
Results
Tau-positive inclusions in oligodendrocytes of brains from patients with tauopathies
Immunohistochemical analysis of sections of the globus pallidus from the brains of patients with CBD, PSP, and FTDP-17 was performed using antibodies against PHF-1, HSP70, and αB-crystallin, respectively, and was compared with the globus pallidus of age-matched control patients with the clinical diagnosis of schizophrenia. Sections of CBD, PSP, and FTDP-17 brains revealed abundant tau-positive pathology (Fig. 1A--C). Adjacent sections showed intense αB-crystallin staining, which was specifically observed in oligodendroglia (Fig. 1E--G). The cells are characterized by their scant cytoplasm and inconspicuous cellular processes as well as small round nuclei, and the inclusions referred to as coiled bodies are identified as intensely tau-positive cytoplasmic aggregates that often exhibit a fibrillary appearance by light microscopy (Komori, 1999). No coiled bodies were observable in sections derived from the brain of a schizophrenia patient (Fig. 1D,H), and in all samples, only some baseline staining was seen for HSP70, which represents the inducible form of the HSP70 family of HSPs (Fig. 1I--L). Double-immunofluorescence labeling also confirms the colocalization of tau and αB-crystallin in coiled bodies in the globus pallidus of a CBD patient (Fig. 1M,N, arrows). Thus, oligodendrocytes in tauopathy brains harbor tau-positive inclusion bodies, which are associated with αB-crystallin.
Figure 1.
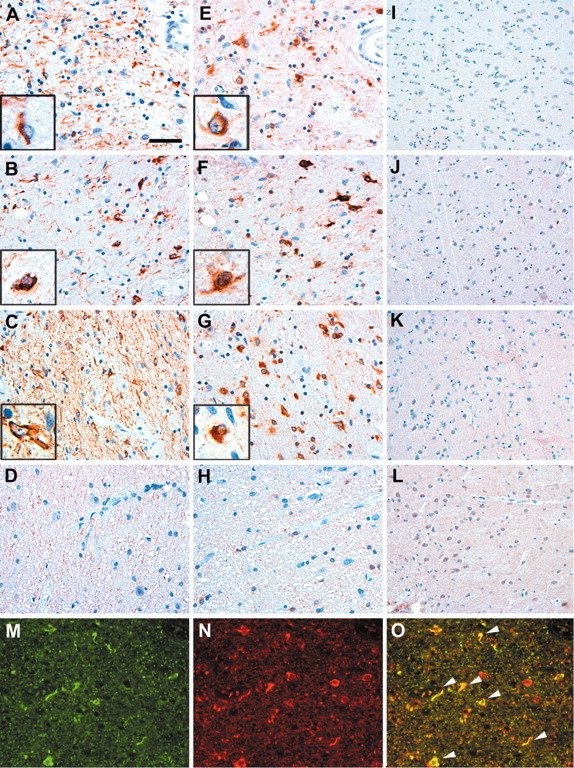
Colocalization of tau and αB-crystallin in oligodendrocytes from patients with tauopathies. A-L, Immunohistochemistry on sections from the globus pallidus of CBD (A, E, I), PSP (B, F, J), FTDP-17 (C, G, K), and schizophrenia (D, H, L) brains were stained with antibodies to PHF-1 (A-D), αB-crystallin (E-H), and HSP70 (I-L). Insets show high-power magnification of tau pathology (A-C) and αB-crystallin staining (E-G) in oligodendrocytes obtained from adjacent sections. Note the abundant tau pathology in the brains of patients with CBD, PSP, FTDP-17 (A-C), and αB-crystallin immunoreactivity (E-G), respectively, which was not observable in sections of schizophrenia brains (D, H). Only baseline levels of the inducible HSP70 could be detected (I-L). Double-immunofluorescence staining of the globus pallidus from a CBD brain shows the individual labeling of tau (M) and αB-crystallin (N) as well as the colocalization of both (O, arrowheads). Staining was performed with polyclonal rabbit anti-tau antibody N-tau (green) and MAb anti-αB-crystallin antibody (red). Scale bars: A-H, M-O, 50 μm; I-L, 100 μm.
Okadaic acid induces tau hyperphosphorylation, a reduction in tau-MT-binding capacity, and the occurrence of thioflavin-S-positive inclusions
To explore the mechanisms underlying oligodendroglia-inclusion body formation, OLN-t40 cells were established, a stably transfected cell line of oligodendroglia origin (Richter-Landsberg and Heinrich, 1996) overexpressing the longest human tau isoform. Wild-type OLN-93 cells do not express tau, and tau transfection per se did not cause the induction of HSPs. The cells were treated with OA at a concentration that specifically leads to the inhibition of PP2A (Favre et al., 1997). Treatment with OA (20 nm) resulted in the hyperphosphorylation of tau, as detected by the phosphorylation-specific antibody tau-1, 12E8, and PHF-1 (Fig. 2). The MAb tau-1 recognizes unphosphorylated tau; hence, a decrease in tau-1 immunoreactivity reflects an increase in phosphorylation. The phosphorylation-dependent MAbs 12E8 and PHF-1 detect tau phosphorylated at Ser262 and Ser396/404, respectively. After treatment with OA, neither HSP70 nor αB-crystallin was induced, and the expression of the constitutive HSC70 isoform was unchanged in all samples (Fig. 2). However, expression of HSP32, a small HSP with enzymatic activity also known as heme oxygenase-1, was induced (Fig. 2). HSP32 is a sensor and regulator of many forms of oxidative stress (Schipper, 2000) and, in oligodendrocytes, is highly inducible by hydrogen peroxide (Goldbaum and Richter-Landsberg, 2001).
Figure 2.

Effect of okadaic acid on tau phosphorylation and heat-shock protein expression. OLN-t40 cells were treated with 20 nm OA for the indicated times. Lysates of control (Co) and OA-treated cells were prepared and subjected to immunoblot analysis using phosphorylation-independent antibody 17026 reacting with total tau, and phosphorylation-dependent antibodies 12E8, Tau-1, and PHF-1, recognizing nonphosphorylated and phosphorylated tau proteins, respectively, and MAb anti-α-tubulin antibodies. Heat-shock proteins were identified by antibodies reacting with HSP/HSC70, HSP32, and αB-crystallin, respectively.
Hyperphosphorylation of tau in OLN-t40 cells led to a decrease in MT-binding activity (Fig. 3). MTs were isolated from cells after stabilization with taxol, and the amount of tau was determined in the pellet (MT fraction) and supernatant (soluble fraction) by Western blot analysis and compared with the immunoreactivity of tubulin in the same fractions. As shown in Figure 3, after the 24 hr treatment, the amount of tau bound to MT, recovered from the pellet, decreased with higher concentrations of OA and was maximal at 20 nm. Treatment with LiCl (20 mm), which is an inhibitor of glycogen synthase kinase 3β (GSK3β) leading to tau dephosphorylation (Hong et al., 1997), in contrast led to an increase in the binding of tau to MTs as reflected by the high proportion of tau in the pellet (Fig. 3). The LiCl-induced alteration in tau phosphorylation causes the appearance of a prominent double band (Fig. 3).
Figure 3.

Okadaic acid decreases the binding of tau to microtubules in OLN-t40 cells. Cells were treated with OA or LiCl as indicated for 24 hr. Cell lysates were separated into cytoskeletal (P) and soluble (S) fractions after MT assembly and subjected to immunoblot analysis using polyclonal tau 17026 and MAb anti-α-tubulin antibodies. OA leads to the detachment of tau from the MTs, as indicated by an increase of tau in the soluble fraction and a decrease in the cytoskeletal fraction. LiCl increases the binding affinity of tau to MTs, as indicated by an increase of tau in the cytoskeletal fraction.
To assess whether aggregates are formed in OLN-t40 cells, indirect immunofluorescence was performed using the polyclonal phosphorylation-independent antibody to tau (17026) followed by thioflavin-S staining. The treatment with OA caused severe morphological changes (e.g., the cytoplasmic processes were retracted); however, cells did not die by programmed cell death, as indicated by the absence of an apoptotic DNA ladder (data not shown) and the integrity of the cellular nuclei (Fig. 4). In addition, in OLN-t40 cells, tau is abundant in both the cell soma and processes (Fig. 4). In control cells and after 3 hr treatment with OA (20 nm), thioflavin-S-positive deposits were rare (<1% of the cells). After treatment for 6 hr, thioflavin-S-positive aggregates containing tau formed in ∼10% of the cells (Fig. 4). However, these aggregates were only transiently expressed and, after 24 hr, cultures resembled the untreated controls. Thus, OA led to the hyperphosphorylation of tau, a decrease in MT-binding capability, and the transient induction of intracytoplasmic inclusions, suggesting that the proteasomal machinery of the cells was capable of degrading the insoluble proteinaceous material. In this study, we tested whether an impairment of the proteasomes contributes to or stabilizes inclusion body formation.
Figure 4.

Okadaic acid leads to thioflavin-S-positive aggregates in OLN-t40 cells. Control (a-c) cells and cells treated with 20 nm OA for 3 hr (d-f) or 6 hr (g-i) were subjected to indirect immunofluorescence staining using polyclonal anti-tau 17026 antibodies, followed by staining with thioflavin-S. For nuclear staining (blue), DAPI was included in the mounting medium. Scale bar, 20 μm.
Inhibition of proteasomes by MG-132 induces the accumulation of HSPs and inhibits tau degradation
MG-132 belongs to a class of recently developed peptide proteasomal inhibitors (Lee and Goldberg, 1998), and treatment of OLN-t40 cells with MG-132 inhibited proteasomal activity effectively. After 4 hr incubation with 0.5 μm MG-132, the proteasomal activity was reduced to ∼40% of untreated controls and almost completely inhibited with 10 μm MG-132 (Fig. 5a). This treatment, as analyzed by immunoblotting, did not impair the proteasomal subunits 20Sα and 20Sβ, even after 18 hr of incubation with MG-132 at a concentration of 10 μm (Fig. 5b). Hence, loss of proteasome activity was not attributable to decreased levels of proteasome subunits. MG-132 caused the induction of HSPs, specifically HSP70, HSP32, and αB-crystallin, in a time- and concentration-dependent manner (Fig. 6). The accumulation of αB-crystallin and HSP32 was observed after treatment with 0.5 μm MG-132 (Fig. 6a) and was maximal after 18 hr at a concentration of 10 μm (Fig. 6b). Western blot analysis of cell extracts also showed that MG-132 could prevent the degradation of tau proteins and led to an increase in PHF-1 immunoreactivity without impairing the level of tubulin (Fig. 7). Hence, MG-132 in OLN-t40 cells leads to an induction of αB-crystallin and an impairment of tau proteolysis.
Figure 5.
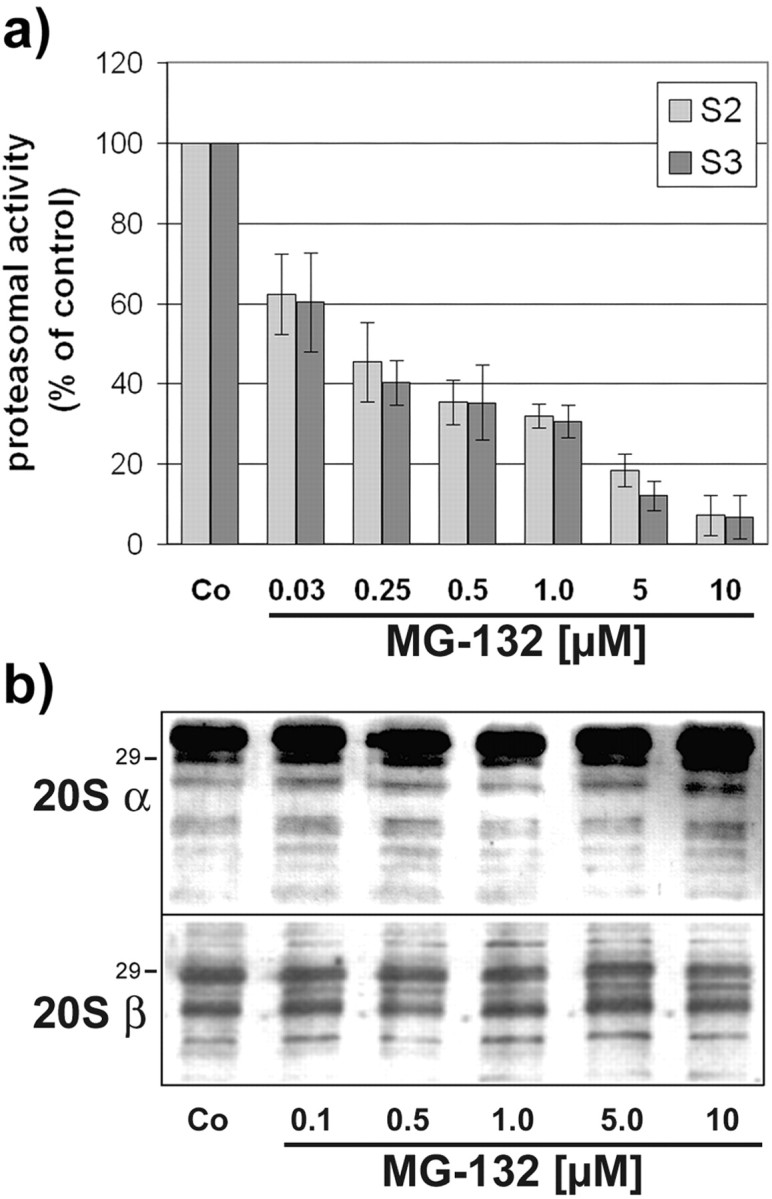
Inhibition of proteasome activity by MG-132 occurs without decreasing the levels of proteasomal subunits. a, OLN-t40 cells were incubated with increasing concentrations of MG-132 as indicated for 4 hr, and postglutamyl-peptidase-hydrolase and chymotrypsin-like proteasome activity were determined using fluorogenic substrate S2 and S3, respectively (see Materials and Methods for details). The cleavage of both substrates is inhibited by MG-132 in a concentration-dependent manner. Data are expressed as a percentage of the untreated control and represent the means of three independent experiments. b, Immunoblot analysis of proteasome subunits. OLN-t40 cells were incubated with increasing concentrations of MG-132 as indicated for 18 hr. Cell lysates were prepared and immunoblot analysis was performed using antibodies against the proteasome subunit 20Sα and 20Sβ, respectively. No decrease in the individual subunits was observed. Numbers on the right indicate the molecular weight in kilodaltons.
Figure 6.

a, b, Induction of heat-shock proteins by MG-132. OLN-t40 cells were treated with MG-132 at various concentrations for 18 hr (a) or with 10 μm MG-132 for 1-24 hr (b). Cell lysates were prepared and subjected to immunoblot analysis using a panel of antibodies against the following after-heat-shock proteins: HSP90, HSP70, HSP60, HSP40, HSP32, and αB-crystallin. In comparison, the immunoreactivity of α-tubulin was determined using MAb anti-α-tubulin. Co, Untreated control.
Figure 7.

MG-132 inhibits tau proteolysis. OLN-t40 cells were incubated with 10 μm MG-132 for 1-24 hr, and cell lysates were prepared. Immunoblot analysis was performed using phosphorylation-independent tau 17026 antibody reacting with total tau and phosphorylation-dependent antibodies tau-1 and PHF-1, recognizing nonphosphorylated and phosphorylated tau proteins, respectively, and MAb anti-α-tubulin antibodies. MG-132 inhibits the formation of proteolytic tau fragments. Co, Untreated control.
MG-132 stabilizes hyperphosphorylated tau
Exposure of OLN-t40 cells to OA (20 nm) caused a reversible hyperphosphorylation of tau (Fig. 2). Sixteen hours after removal of OA, tau phosphorylation resembled the untreated control (Fig. 8). Comparison of hyperphosphorylated tau after incubation with OA or MG-132 alone, or OA followed by MG-132 (Figs. 2, 8), demonstrates that MG-132 leads to the specific accumulation of hyperphosphorylated tau, detected by 12E8, PHF-1, and tau-1 antibodies.
Figure 8.

Hyperphosphorylated tau is stabilized in OLN-t40 cells by MG-132. OLN-t40 cells were treated with MG-132 alone (MG; 0.5 μm; 18 hr), with OA (20 nm; 6 hr) followed by MG-132 (0.5 μm; 18 hr) in the absence of OA (OA-MG), or with OA (20 nm; 6 hr) followed by a recovery period of 18 hr in the absence of OA (OA). Co, Untreated control. Cell lysates were analyzed by immunoblot procedure using a panel of phosphorylation-dependent and phosphorylation-independent tau antibodies, antibodies against HSPs, and antibodies against tubulin, as described in Figure 2.
OA-induced cytoplasmic inclusions are stabilized by MG-132 and contain hyperphosphorylated filamentous tau
Exposure of OLN-t40 cells to MG-132 (0.5 μm; 24 hr) led to the accumulation of αB-crystallin in the perinuclear region; however, these aggregates were not stained by thioflavin-S. Treatment with OA alone (24 hr) also did not lead to detectable aggregates (Fig. 9). In contrast, when cells were treated with OA (20 nm; 6 hr) first and then subjected to MG-132 (0.5 μm) for an additional 18 hr, thioflavin-S-positive aggregates were observed in a cellular location distinct from the MG-132-induced αB-crystallin accumulations (Fig. 9). Quantitative evaluation showed that incubation with OA (20 nm; 6 hr) followed by MG-132 resulted in 6% of OLN-t40 cells containing thioflavin-S-positive aggregates. Hence, MG-132 led to the stabilization of OA-induced aggregates. Additional characterization of the inclusion bodies by indirect immunofluorescence staining, using antibodies against tau or αB-crystallin, followed by thioflavin-S staining, demonstrates that tau and αB-crystallin are present in these aggregates (Fig. 10). Furthermore, the inclusions contain phosphorylated tau, as demonstrated by PHF-1 immunoreactivity (Fig. 10d--f). Double labeling, using antibodies against αB-crystallin and tau, confirmed the colocalization of both proteins in aggregate-like inclusions (Fig. 10j--l).
Figure 9.

Accumulation of cytoplasmic inclusions by proteasome inhibition after OA treatment. OLN-t40 cells were subjected to 0.5 μm MG-132 for 24 hr (d-f), to 20 nm OA for 24 hr (g-i), or to 20 nm OA for 6 hr followed by 0.5 μm MG-132 for 18 hr in the absence of OA (j-l). Indirect immunofluorescence was performed using MAb anti-αB-crystallin (red) followed by thioflavin-S staining (green). For nuclear staining (blue), DAPI was included in the mounting medium. a-c, Untreated control. a, d, g, j,αB-crystallin (red). b, e, h, k, Thioflavin-S (green). c, f, i, l, Overlay with DAPI. Scale bar, 20 μm.
Figure 10.
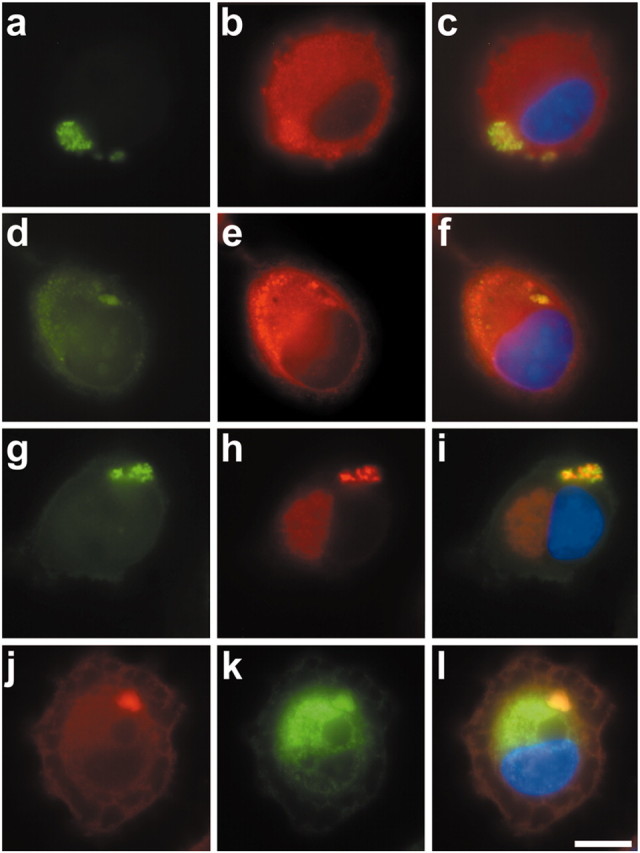
Cytoplasmic inclusions contained tau and αB-crystallin. OLN-t40 cells were treated with 20 nm OA for 6 hr, followed by 0.5 μm MG-132 for 18 hr in the absence of OA. Cells were subjected to indirect immunofluorescence using polyclonal tau antibody 17026 (a-c), MAb PHF-1 (d-f), or MAb αB-crystallin (g-i), followed by thioflavin-S staining (green). To demonstrate tau and αB-crystallin colocalization, double labeling was performed using polyclonal tau antibody 17026 (red) and αB-crystallin (green) (j-l). For nuclear staining (blue), DAPI was included in the mounting medium. Scale bar, 10 μm.
To assess whether the thioflavin-S-positive aggregates contain tau filaments, immuno-EM was conducted on OLN-t40 cells treated with OA (20 nm; 6 hr) followed by MG-132 (Fig. 11). Using DAB plus silver enhancement for immunolabeling with anti-tau antibody, ∼30% of the transfected OLN-t40 cells contained tau-positive cytoplasmic aggregates (Fig. 11a), and in ∼10% of the aggregates, tau-positive staining was localized to filamentous structures (Fig. 11b,c). Thus, the tau aggregates found in OLN-t40 cells after OA and MG-132 treatment resemble authentic tau aggregates in the brains of patients with tauopathies.
Figure 11.
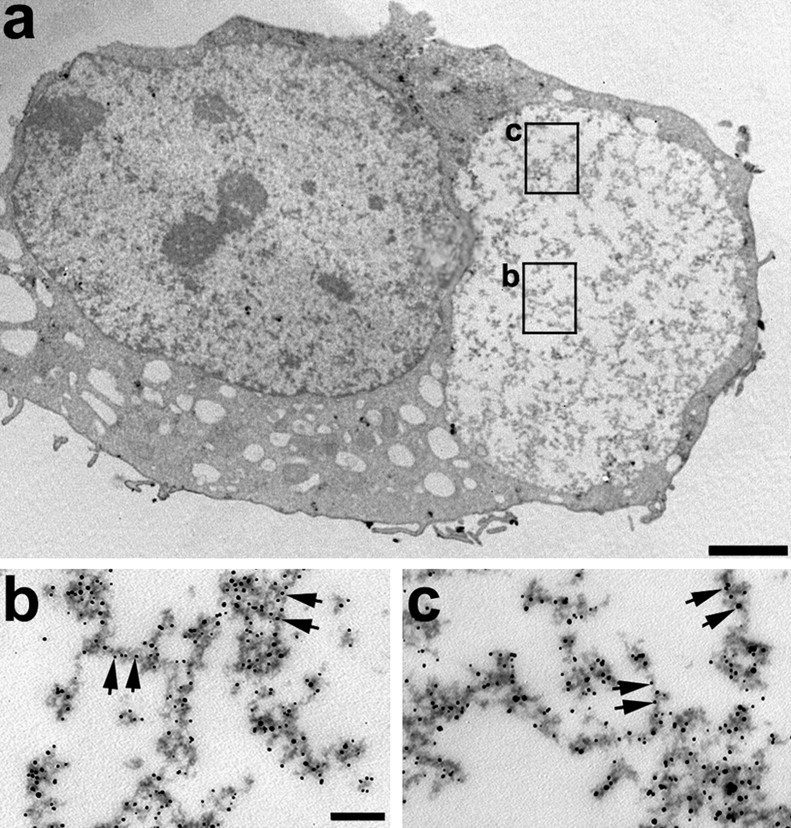
Cytoplasmic inclusions in OLN-t40 cells are composed of filamentous aggregated tau. Immuno-EM was performed on OLN-t40 cells treated with OA followed by MG-132. Cells were stained with rabbit anti-recombinant tau (17026) as the primary antibody visualized with nanogold-conjugated secondary antibody. Squares in a mark the areas in the inclusion body magnified in b and c. Arrows indicate tau-positive filaments. Scale bars: a, 2 μm; b, c, 100 nm.
Discussion
The importance of glial pathology has become recognized only recently. Insoluble tau is often more abundant in white matter than in gray matter (Forman et al., 2002; Zhukareva et al., 2002), and tau-positive glial inclusions in oligodendrocytes are specifically prominent in PSP, CBD, and Pick's disease (Komori, 1999). Tau pathology in AD is characterized by the occurrence of hyperphosphorylated tau predominantly in the intracytoplasmic aggregates of neurons, which are identified by a number of phosphorylation-dependent antibodies, including PHF-1 (for review, see Bueé et al., 2000; Lee et al., 2001). The inclusion bodies found in oligodendrocytes of CBD, PSP, and FTDP-17 brains in our study also contained PHF-1 immunoreactivity, indicating the presence of hyperphosphorylated tau. This contrasts with the GCIs found in white-matter oligodendrocytes of MSA brains, which are composed primarily of α-synuclein and could be immunolabeled only with phosphorylation-independent tau antibodies or MAb tau-1, recognizing unphosphorylated tau (Cairns et al., 1997; Tu et al., 1998).
The binding activity of tau to MTs is regulated by phosphorylation, and hyperphosphorylation at certain sites impairs the binding of tau to MTs and destabilizes the MT network (Johnson and Hartigan, 1998; Hong et al., 2000). The phosphorylation state of tau is balanced by the activities of protein kinases, including cyclin-dependent kinase cdk5, p42 and p44 MAP kinases [ERK1 (extracellular signal-regulated kinase 1), ERK2], GSK-3β, and protein phosphatases. PP2A and PP2B efficiently dephosphorylate tau in vitro. The selective inhibition of PP2A by OA induced AD-like phosphorylation and accumulation of tau in the mammalian brain (Gong et al., 2000). In AD brains, a decrease in PP2A activity (Gong et al., 1995) and mRNA expression was detected (Vogelsberg-Ragaglia et al., 2001). The molecular mechanisms underlying GCI formation and tau accumulation in glial cells are primarily unknown.
The data presented here, using a cell culture model system, demonstrate that inhibition of PP2A by OA in oligodendroglial cells overexpressing the longest human tau isoform leads to tau hyperphosphorylation and a decrease in the binding of tau to MTs. Simultaneously, tau- and αB-crystallin-positive aggregates can be detected in the cell soma that stain with thioflavin-S and filamentous aggregated tau. However, these aggregates are removed within 24 hr, which points to an efficient unimpaired proteolytic system. When the proteasomal machinery was inhibited by MG132 after OA treatment, the aggregates were stabilized. Hence, although hyperphosphorylation contributes to pathological aggregation, an age-related proteasomal dysfunction might potentiate this effect. The proteasomal degradation of tau, possibly without the requirement of ubiquitination, has been demonstrated recently (David et al., 2002). A loss of proteasomal functions occurs in aging animals (for review, see Keller et al., 2002), and proteasome inhibition might play a role in neurodegeneration (Keller et al., 2000). As shown in the present study, tau proteolysis is inhibited, and HSPs are induced when the degradative process is blocked by proteasome inhibitors. The induction of HSPs by proteasome inhibitors has been described previously in other cell systems (Lee and Goldberg, 1998). For example, proteasome inhibition by MG-132 induced the accumulation and recruitment of HSP27 and αB-crystallin to aggresomes in U373 MG cells (Ito et al., 2002). In OLN-t40 cells, αB-crystallin was induced in a time- and concentration-dependent way and translocated to the perinuclear region of the cells. Yet, these deposits did not stain with thioflavin-S. Thioflavin-S-positive inclusions were observed only after treatment with OA and when OA-treated cells were subsequently incubated with MG-132. This indicates that tau hyperphosphorylation and the detachment of these forms of tau from the MTs is a prerequisite for the formation of fibrillary inclusions. In contrast, our data indicate that tau hyperphosphorylation alone is not sufficient to induce permanent aggregates. Hyperphosphorylation followed by proteasomal inhibition is necessary for the formation of stable aggregates, which contain hyperphosphorylated tau and αB-crystallin.
We also show that tau-positive oligodendrocytes bearing coiled bodies from patients with tauopathies (i.e., PSP, CBD, and FTDP-17) are positive for αB-crystallin. So far, the association of αB-crystallin has been identified repeatedly only in GCIs in oligodendrocytes from patients with MSA (for review, see Chin and Goldman, 1996; Komori, 1999). αB-crystallin belongs to the group of small HSPs, which interact with the cytoskeleton in normal cells and play a role in inclusion body formation (Head and Goldman, 2000). In the CNS, αB-crystallin is primarily expressed in oligodendrocytes (Head and Goldman, 2000; Goldbaum and Richter-Landsberg, 2001). It exerts cytoprotective functions and is involved in the modulation of intermediate filament organization under conditions of physiological stress and neurodegenerative disease (Head and Goldman, 2000). In AD brains, the appearance of αB-crystallin in neurons has been linked to later severe stages of pathogenesis, when abundant tau-positive neurofibrillary tangles are observed (Mao et al., 2001). It is also a major component of Rosenthal fibers, which are the characteristic inclusion bodies found in astrocytes of patients with Alexander's disease (Head and Goldman, 2000). The introduction of αB-crystallin into inclusion-bearing astrocytes overexpressing glial fibrillary acidic protein (GFAP) caused the dispersion of GFAP accumulations but not the appearance of Rosenthal fibers (Koyama and Goldman, 1999). Hence, αB-crystallin might initially be a protective means against intermediate filament disorganization, and inclusion body formation is a later stage of the pathogenic response.
Oligodendrocytes do not contain intermediate filaments but have a very prominent MT-system (Richter-Landsberg, 2001). Also, the cell line used in this study is devoid of an intermediate filament system (Richter-Landsberg and Heinrich, 1996). An affinity of αB-crystallin for MTs has been suggested previously (Arai and Atomi, 1997), and agents that promote the disassembly of MTs induce its expression (Kato et al., 1996). HSPs constitute the major cellular defense against unfolded proteins (for review, see Sherman and Goldberg, 2001), and their expression is connected to protection of neural cells (for review, see Richter-Landsberg and Goldbaum, 2003). In oligodendrocytes, αB-crystallin is effectively upregulated under conditions of oxidative and heat stress, which eventually lead to the onset of programmed cell death (Goldbaum and Richter-Landsberg, 2001). It seems likely that αB-crystallin is involved in the protection of the cytoskeletal organization in oligodendrocytes and, as a chaperone, is recruited to prevent tubulin and tau aggregation. However, when the damage is beyond repair, protection fails and HSPs most likely contribute to degenerative processes and cell death.
In summary, cytoplasmic glial inclusions are a common feature of a variety of neurodegenerative diseases. Prominent oligodendroglia tau pathology occurs in PSP, CBD, Pick's disease, and FTDP-17. At least a subset of the inclusion bodies in oligodendroglia, as shown in the present study, also contains the small HSP αB-crystallin, indicating that molecular chaperones participate in the cellular responses to stressful situations that cause protein aggregation and the assembly into aggresome-like structures. Tau hyperphosphorylation and proteasomal impairment contribute to aggregate formation in oligodendroglial cells, suggesting that alterations of the proteasomal system, which might occur during aging, are involved in the neuropathological events leading to degeneration.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Ri384/11-3), the Alzheimers Association, and the National Institute on Aging. We gratefully acknowledge the expert technical help from Angelika Spanjer. The 12E8 antibody was generously provided by Dr. Peter Seubert (Elan Pharmaceuticals, San Francisco, CA).
Correspondence should be addressed to Dr. Christiane Richter-Landsberg, Department of Biology, Molecular Neurobiology, University of Oldenburg, P.O. Box 2503, D-26111 Oldenburg, Germany. E-mail: Christiane.Richter.Landsberg@Uni-Oldenburg.de.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238872-09$15.00/0
References
- Arai H, Atomi Y ( 1997) Chaperone activity of αB-crystallin suppresses tubulin aggregation through complex formation. Cell Struct Funct 22: 539-544. [DOI] [PubMed] [Google Scholar]
- Berry RW, Quinn B, Johnson N, Binder LI ( 2001) Pathological tau accumulations in neurodegenerative disease: review and case report. Neurochem Int 39: 469-479. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM-Y ( 1993) Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron 10: 1089-1099. [DOI] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR ( 2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33: 95-130. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Atkinson PF, Hanger DP, Anderton BH, Daniel SE, Lantos P ( 1997) Tau protein in glial cytoplasmic inclusions of multiple system atrophy can be distinguished from abnormal tau in Alzheimer's disease. Neurosci Lett 230: 49-52. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H ( 1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7: 2745-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin SS-M, Goldman JE ( 1996) Glial inclusions in CNS degenerative diseases. J Neuropathol Exp Neurol 55: 499-508. [DOI] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG ( 2002) Proteasomal degradation of tau protein. J Neurochem 83: 176-185. [DOI] [PubMed] [Google Scholar]
- Favre B, Turowski P, Hemmings BA ( 1997) Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J Biol Chem 272: 13856-13863. [DOI] [PubMed] [Google Scholar]
- Forman MS, Zhukareva V, Bergeron C, Chin SS-M, Grossman M, Clark C, Lee VM-Y, Trojanowski JQ ( 2002) Signature tau neuropathology in gray and white matter of corticobasal degeneration. Am J Pathol 160: 2045-2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Davies SW ( 1998) Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol 8: 619-632. [DOI] [PubMed] [Google Scholar]
- Goldbaum O, Richter-Landsberg C ( 2001) Stress proteins in oligodendrocytes: differential effects of heat shock and oxidative stress. J Neurochem 78: 1233-1242. [DOI] [PubMed] [Google Scholar]
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K ( 1995) Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem 65: 732-738. [DOI] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K ( 2000) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in the mammalian brain. J Biol Chem 275: 5535-5544. [DOI] [PubMed] [Google Scholar]
- Gorath M, Stahnke T, Mronga T, Goldbaum O, Richter-Landsberg C ( 2001) Developmental changes of tau protein and mRNA in cultured rat brain oligodendrocytes. Glia 36: 89-101. [DOI] [PubMed] [Google Scholar]
- Head MW, Goldman JE ( 2000) Small heat shock proteins, the cytoskeleton and inclusion body formation. Neuropathol Appl Neurobiol 26: 304-312. [DOI] [PubMed] [Google Scholar]
- Hong M, Chen DCR, Klein PS, Lee VM-Y ( 1997) Lithium reduces tau phosphorylation by inhibition of glycogen synthase-3. J Biol Chem 272: 25326-25332. [DOI] [PubMed] [Google Scholar]
- Hong M, Trojanowski JQ, Lee VM-Y ( 2000) Tau-based neurofibrillary lesions. In: Neurodegenerative dementias (Clark CM, Trojanowski JQ, eds), pp 161-175. New York: McGraw Hill.
- Ito H, Kamei K, Iwamamoto I, Inaguma Y, Garcia-Mata R, Sztul E, Kato K ( 2002) Inhibition of proteasomes induces accumulation, phosphorylation and recruitment of HSP27 and αB-crystallin to aggresomes. J Biochem 131: 593-603. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Hartigan JA ( 1998) Tau protein in normal and Alzheimer's disease brain: an update. J Alzheimers Dis 1: 329-351. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR ( 1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Ito H, Inaguma Y, Okamoto K, Saga S ( 1996) Synthesis and accumulation of αB-crystallin in C6 glioma cells is induced by agents that promote the disassembly of microtubules. J Biol Chem 271: 26989-26994. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR ( 2000) Impaired proteasome function in Alzheimer's disease. J Neurochem 75: 436-439. [DOI] [PubMed] [Google Scholar]
- Keller JN, Gee J, Ding Q ( 2002) The proteasome in brain aging. Ageing Res Rev 1: 279-293. [DOI] [PubMed] [Google Scholar]
- Komori T ( 1999) Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick's disease. Brain Pathol 9: 663-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR ( 2000) Aggresomes, inclusion bodies, and protein aggregation. Trends Cell Biol 10: 524-530. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Goldman JE ( 1999) Formation of GFAP cytoplasmic inclusions in astrocytes and their disaggregation by αB-crystallin. Am J Pathol 154: 1563-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL ( 1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 8: 397-403. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ ( 2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Mao JJ, Katayama S, Watanabe C, Harada Y, Noda K, Yamamura Y, Nakamura S ( 2001) The relationship between αB-crystallin and neurofibrillary tangles in Alzheimer's disease. Neuropathol Appl Neurobiol 27: 180-188. [DOI] [PubMed] [Google Scholar]
- Merrick SE, Trojanowski JQ, Lee VM-Y ( 1997) Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons. J Neurosci 17: 5726-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhoff V, Philipp K, Zimmer HG, Mesecke S ( 1979) A simple, versatile, sensitive and volume-independent method for quantitative protein determination which is independent of other external influences. Hoppe Seylers Z Physiol Chem 360: 1657-1670. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C ( 2001) Organization and functional roles of the cytoskeleton in oligodendrocytes. Microsc Res Tech 53: 628-636. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C, Goldbaum O ( 2003) Stress proteins in neural cells: functional roles in health and disease. Cell Mol Life Sci 60: 337-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Landsberg C, Heinrich M ( 1996) OLN-93: a new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J Neurosci Res 45: 161-173. [DOI] [PubMed] [Google Scholar]
- Schipper HM ( 2000) Heme oxygenase-1: role in brain aging and neurodegeneration. Exp Gerontol 35: 821-830. [DOI] [PubMed] [Google Scholar]
- Schmidt ML, Carden MJ, Lee VM, Trojanowski JQ ( 1987) Phosphate dependent and independent neurofilament epitopes in the axonal swellings of patients with motor neuron disease and controls. Lab Invest 56: 282-294. [PubMed] [Google Scholar]
- Schneider A, Biernat J, Von Bergen M, Mandelkow E, Mandelkow EM ( 1999) Phosphorylation that detaches tau protein from microtubules (Ser261/Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38: 3549-3558. [DOI] [PubMed] [Google Scholar]
- Schwartz AL, Ciechanover A ( 1999) The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med 50: 57-74. [DOI] [PubMed] [Google Scholar]
- Sherman MY, Goldberg AL ( 2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29: 15-32. [DOI] [PubMed] [Google Scholar]
- Towbin HTM, Staehelin TM, Gordon J ( 1979) Electrophoretic transfer of proteins from polyacryamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsuho T, Trojanowski JQ, Lee VM-Y ( 1998) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann Neurol 44: 415-422. [DOI] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, Zhang B, Hong M, Trojanowski J, Lee VM-Y ( 2000) Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell 11: 4093-4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM-Y ( 2001) PP2A mRNA expression is quantitatively decreased in Alzheimer's disease hippocampus. Exp Neurol 168: 402-412. [DOI] [PubMed] [Google Scholar]
- Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, Grossman M, Miller BI, Hulette C, Feinstein SC, Trojanowski JQ, Lee VM-Y ( 2002) Sporadic Pick's disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol 51: 730-739. [DOI] [PubMed] [Google Scholar]