

Abstract
Impairment of axonal transport leads to neurodegeneration and synapse loss. Glutamate and amyloid β-protein (Aβ) have critical roles in the pathogenesis of Alzheimer's disease (AD). Here we show that both agents rapidly inhibit fast axonal transport in cultured rat hippocampal neurons. The effect of glutamate (100 μm), but not of Aβ25-35 (20 μm), was reversible, was mimicked by NMDA or AMPA, and was blocked by NMDA and AMPA antagonists and by removal of extracellular Ca2+. The effect of Aβ25-35 was progressive and irreversible, was prevented by the actin-depolymerizing agent latrunculin B, and was mimicked by the actin-polymerizing agent jasplakinolide. Aβ25-35 induced intracellular actin aggregation, which was prevented by latrunculin B. Aβ31-35 but not Aβ15-20 exerted effects similar to those of Aβ25-35. Full-length Aβ1-42 incubated for 7 d, which specifically contained 30-100 kDa molecular weight assemblies, also caused an inhibition of axonal transport associated with intracellular actin aggregation, whereas freshly dissolved Aβ1-40, incubated Aβ1-40, and fresh Aβ1-42 had no effect. These results suggest that glutamate inhibits axonal transport via activation of NMDA and AMPA receptors and Ca2+ influx, whereas Aβ exerts its inhibitory effect via actin polymerization and aggregation. The ability of Aβ to inhibit axonal transport seems to require active amino acid residues, which is probably present in the 31-35 sequence. Full-length Aβ may be effective when it represents a structure in which these active residues can access the cell membrane. Our results may provide insight into the early pathogenetic mechanisms of AD.
Keywords: axonal transport, glutamate, amyloid β-protein, actin, Ca2+, cultured hippocampal neurons
Introduction
Impairment of axonal transport leads to neuronal degeneration (Moretto and Sabri, 1988; Yonekawa et al., 1998), and thus it may be an early event in the pathogenesis of some neurodegenerative diseases (Brimijoin, 1982; Warita et al., 1999; Williamson and Cleveland, 1999). In Alzheimer's disease (AD) as well as other dementing disorders, a widespread loss of synapses occurs in the brain, and synaptic injury is an early event during the progression of AD (Masliah et al., 1989; Masliah, 1995). One of critical causes of synapse loss is deficiency in axonal transport (Cull, 1975). Actually, axonal transport is impaired in the brain of AD patients (Suzuki and Terry, 1967; Younkin et al., 1986; Richard et al., 1989; Praprotnik et al., 1996).
In neurodegenerative diseases including AD, excessive glutamate in extracellular space is recognized as an excitotoxin, playing a role in the development of the diseases (Arias et al., 1998; Lancelot and Beal, 1998). Amyloid β-protein (Aβ), which accumulates in senile plaques in brains of AD patients, is also the toxic agent that causes neuronal degeneration and cell death and is profoundly involved in the pathogenesis of AD (Yankner et al., 1990; Mattson et al., 1992; Kowall, 1994); however, the influence of these agents on axonal transport has rarely been studied. Here, we investigated the effects of glutamate and Aβ25-35, an active fragment of Aβ, on fast axonal transport of membrane-bound particles in cultured rat hippocampal neurons using video-enhanced microscopy. Our data suggest that glutamate and Aβ25-35 rapidly inhibit fast axonal transport. The effect of glutamate is mediated by activation of NMDA and AMPA receptors and subsequent Ca2+ influx, whereas the effect of Aβ25-35 occurs via polymerization and aggregation of intracellular actin. Furthermore, we indicate that specific residues and structures are required for the naturally occurring Aβ to inhibit axonal transport.
Materials and Methods
Cell culture. The experimental protocol was approved by the Animal Experimentation and Ethics Committee of Kitasato University School of Medicine. Hippocampi were removed from fetal Wistar rats on day 16 of gestation, and were immediately immersed in 4°C Leibovitz's L-15 medium (Invitrogen, Carlsbad, CA) containing 0.5 mm l-glutamine (Wako, Osaka, Japan). These hippocampal tissues were dissected and then incubated for 15 min at 37°C in Ca2+, Mg2+-free PBS (Invitrogen) containing 0.2 mg/ml dl-cysteine hydrochloride (Sigma, St. Louis, MO), 0.2 mg/ml albumin bovine serum (Sigma), 5 mg/ml d-(+)-glucose (Wako), 0.55 mg/ml papain (Worthington Biochemicals, Freehold, NJ), and 0.01% DNase (Sigma). Then fetal calf serum was added (30% v/v of PBS) to inhibit enzyme activity. Cells were isolated from the tissues using fire-polished pipettes (0.2-0.5 mm inner diameter). Subsequently, the isolated cells were plated onto polylysine-coated glass coverslips (30 × 40 mm, 50 μm thickness) and cultured for 7 d in serum-free Neurobasal medium (Invitrogen) containing 0.5 mm l-glutamine (Invitrogen) and 0.04% B-27 supplement (Wako) at 37°C in a humidified 5% CO2 atmosphere.
Experimental preparations. The coverslip on which cells were cultured was attached with waterproof tape to the underside of a 0.5-mm-thick stainless-steel chamber (50 × 80 mm) with a lozenge-shaped hole (25 × 35 mm). The volume of the chamber was ∼0.45 ml. The topside of the chamber was covered with another coverslip, leaving small openings on both sides to perfuse cells with a solution. The culture medium was then replaced with HEPES-buffered salt solution (see below; 37°C). The chamber was mounted onto the stage of an inverted Zeiss Axiomat microscope equipped with an oil-immersed Plan-Apochromat 63× objective (Carl Zeiss, Oberkochen, Germany). The stage was maintained at 37°C by a thermocontroller. The drug-containing solution (3 ml) was injected into one side opening using a Pasteur pipette, and the solution spilling from the other side opening was removed by an infusion pump.
Solutions and drugs. The composition of HEPES-buffered salt solution, pH 7.4, was (in mm): 120 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 25 d-(+)-glucose (all from Wako). Ca2+-free solution consisted of (in mm): 120 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, 25 d-(+)-glucose, and 2 EGTA (Wako). Amyloid β-protein fragments 25-35 (Aβ25-35; Sigma) and 31-35 (Aβ31-35; Sigma) were dissolved in distilled water to a concentration of 2 or 10 mm. Fragments 1-40 (Aβ1-40; Sigma) and 1-42 (Aβ1-42; Sigma), and acetyl fragment 15-20 amide (Aβ15-20 amide; Sigma) were initially dissolved in 10 mm NaOH, because they are less soluble in water, and then diluted with distilled water to a concentration of 0.5 mm for Aβ1-40 and Aβ1-42 and 1 mm for Aβ15-20 amide. Simultaneously, the pH of these solutions was adjusted to 7.4 using either NaOH or HCl. Then these solutions were further diluted at concentrations indicated in Results with HEPES-buffered salt solution and used for experiments on axonal transport and rhodamine-phalloidin stainings. The solutions (0.5 mm) of Aβ1-40 and Aβ1-42 incubated for 7 d at room temperature were also used after dilution with HEPES-buffered salt solution. The solutions of 1 mm Aβ15-20 amide, 2 mm Aβ31-35, 2 mm Aβ25-35, 0.5 mm Aβ1-40, and 0.5 mm Aβ1-42 were subjected to SDS-PAGE. l-glutamic acid hydrochloride (glutamate; Wako), NMDA (Sigma), AMPA (Sigma), and (±)-MK-801 (Sigma) were dissolved directly in HEPES-buffered salt solution. CNQX (Sigma) and latrunculin B (Biomol, Plymouth Meeting, PA) were each dissolved in dimethyl sulfoxide (DMSO; Wako), and jasplakinolide (Molecular Probes, Eugene, OR) was dissolved in methanol. These organic solutions were then diluted with HEPES-buffered solution. The concentration of DMSO and methanol was 0.01%, and at this concentration these organic solvents had no effect on axonal transport.
Video-enhanced microscopy system. Nomarski images acquired by an inverted microscope were transformed into video images by a video camera (Harpicon, Hamamatsu Photonics, Hamamatsu, Japan) and a camera controller (C2741, Hamamatsu Photonics). The video images were digitalized and enhanced by a video image enhancement system (DVS-20, Hamamatsu Photonics), displayed in real-time on a video monitor (C1864, Hamamatsu Photonics), and stored on a video recorder (PVW-2800, Sony, Tokyo, Japan). This processing provided a 10,000-fold final magnification on the video monitor.
Analysis of axonal transport. Axonal transport was analyzed from the video replay. The numbers of particles (diameter ≥50 nm) moving toward the axon terminal (anterograde) and back to the cell body (retrograde) were counted before, during, and after application of the drug. In the control extracellular medium (HEPES-buffered salt solution, pH 7.4, 37°C), the mean numbers of particles (per minute) transported in anterograde and retrograde directions were 59.7 ± 19.7 (mean ± SD; n = 105) and 57.5 ± 19.7 (n = 105), respectively. Averaged data in each experiment are expressed as mean (± SD) percentage of the control value that was obtained before the drug application. ANOVA was used to evaluate the statistical significance of fluctuations over time. Differences between control and test conditions were examined for statistical significance using the Student's paired t test.
Rhodamine-phalloidin staining. Hippocampal cells grown on coverslips were treated for 30 min with drugs (glutamate, Aβ fragments, latrunculin B, and a combination of Aβ25-35 and latrunculin B) dissolved in a HEPES-buffered solution. Then, cells were fixed with 4% paraformaldehyde for 3 min at room temperature. After fixation, they were washed three times with 0.025 m PBS containing 0.3% Triton X-100 (PBST). The cells were incubated for 30 min at room temperature in 5 U/ml rhodamine-phalloidin (Molecular Probes) dissolved in PBST and washed three times with PBS. The stained cells were examined with a Zeiss Axiovert 135 TV microscope equipped with a 546 nm excitation filter and a 590 nm emission filter.
SDS-PAGE. Ten micrograms of each Aβ in a solution was mixed with Tris-HCl SDS sample buffer, loaded onto 5-20% gradient acrylamide gel (ATTO, Tokyo, Japan), and electrophoresed. Proteins were visualized with SimplyBlue SafeStain (Invitrogen).
Results
Effects of glutamate on axonal transport
Video-enhanced microscopy displayed movement of particles in neurites of cultured rat hippocampal neurons. Continuous application (26 min) of 100 μm glutamate significantly reduced the number of particles moving in anterograde and retrograde directions (Fig. 1A). The number began to decrease within 2 min and reached a plateau at 40-50% of the number before the application (control) 20 min after the start of application. The decreased number was reversed to the control level after a 30 min washout of glutamate. A higher concentration (1000 μm) of glutamate produced similar effects. The number of particles transported in both directions was decreased to 40-50% of control by 1000 μm glutamate (Fig. 1B) and was restored to ∼85% of control 30 min after removal of the drug (data not shown; n = 5). The effects of glutamate were concentration dependent from 1 to 1000 μm (Fig. 1B). Application of NMDA at a concentration of 100 μm reduced the number of particles (∼50% of control) transported in anterograde and retrograde directions in a similar manner to 100 μm glutamate (Fig. 1C,D). AMPA at a concentration of 100 μm also rapidly decreased the number of particles to ∼60% of control in both directions (Fig. 1E,F). The inhibitory effects induced by each 100 μm NMDA and 100 μm AMPA were reversed to control levels 30 min after washout of the drugs (Fig. 1C,E). The effects of NMDA and AMPA were concentration dependent from 1 to 1000 μm (Fig. 1D,F). Because both NMDA and AMPA mimicked the effects of glutamate, these types of receptors could possibly mediate the effect of glutamate on axonal transport. Video-enhanced microscopy also revealed the progressive formation of membrane blebs in neurites during 26 min application of glutamate at concentrations ≥100 μm (see Fig. 3A). The size of each bleb was decreased considerably after washout for 30 min.
Figure 1.

Effects of glutamate and its agonists on the number of particles transported in neurites of cultured rat hippocampal neurons. A, C, E, Percentage changes in the number of transported particles of control (the value before application) induced by 26 min application of 100 μm glutamate (A), 100 μm NMDA (C), and 100 μm AMPA (E). Note that the number of transported particles was rapidly reduced by these drugs and restored by removal of the drugs. B, D, F, Concentration dependence of reduction in the number of transported particles induced by glutamate (B), NMDA (D), and AMPA (F). The value obtained at 20 min after each application of the drugs is expressed as a percentage of control (the value before application). Data in all panels are expressed as mean ± SD of five neurons. Error bars represent SD. *p < 0.05; **p < 0.005; ***p < 0.0005 compared with control (100%).
Figure 3.

Morphological changes induced by glutamate and Aβ25-35 during video-enhanced microscopic recordings. Video images of neurites were acquired before (Control) and 20 min after treatment with 100 μm glutamate (A), and before (Control) and 20 min after treatment with 20 μm Aβ25-35 (B). Note that the membrane in the neurite was blebbing in glutamate-treated neurons, whereas the neurite was shrunken and microtubules were clearly visible in the Aβ25-35-treated neuron. Scale bar, 2 μm.
Effects of Aβ25-35 on axonal transport
Application of Aβ25-35 at a concentration of 20 μm resulted in a rapid and progressive decrease in the number of particles transported in anterograde and retrograde directions during 26 min application (Fig. 2A). The effect of Aβ25-35 was concentration dependent from 0.2 to 200 μm (Fig. 2B). Even after washout, the decreasing effect of 20 μm Aβ25-35 was progressive and irreversible (Fig. 2A), although the effect induced by a lower concentration, 2 μm, of Aβ25-35 applied for 26 min was restored to the control level within 30 min after washout (data not shown; n = 5). We also found that neurites were shrunken by exposure to 20 μm Aβ25-35, and longitudinal microtubules in neurites became clearly visible (Fig. 3B). This phenomenon was irreversible even after washout. These morphological features were extremely different from those observed in glutamate-treated neurons (Fig. 3A).
Figure 2.

Effects of Aβ25-35 and jasplakinolide, an actin-polymerizing agent, on the number of particles transported in neurites of cultured rat hippocampal neurons. A, C, Percentage changes in the number of transported particles of control (the value before application) induced by 26 min application of 20 μm Aβ25-35 (A) and 0.5 μm jasplakinolide (C). Note that the number of transported particles was rapidly and progressively reduced by both drugs and not restored after removal of the drugs. B, Concentration dependence of reduction in the number of transported particles induced by Aβ25-35. The value obtained at 20 min after each application of various concentrations of Aβ25-35 is expressed as a percentage of control (the value before application). Data in all panels are expressed as mean ± SD of five neurons. Error bars represent SD. *p < 0.05; **p < 0.005; ***p < 0.0005 compared with control (100%).
Effects of glutamate and Aβ25-35 under various conditions
It has been reported that Aβ acts on glutamate receptors (Calligaro et al., 1993; Cowburn et al., 1994, 1997; Le et al., 1995) and that both glutamate and Aβ exert their toxic effects by Ca2+ influx (Joseph and Han, 1992; Mattson et al., 1992; Hartley et al., 1993; Weiss et al., 1994; Le et al., 1995; Limbrick et al., 2001). Aβ disrupts the intracellular actin network (Davis et al., 1999), and actin depolymerization prevents neurotoxicity of glutamate and Aβ25-35 (Furukawa and Mattson, 1995; Furukawa et al., 1995). Actin can interact with microtubules, which are the main tracks for fast axonal transport (Nagele et al., 1988; Bearer and Reese, 1999). The above experiments indicated that glutamate and Aβ25-35 induced morphological changes in neurites, implicating the possible involvement of the cytoskeleton. Therefore, to identify the mechanisms mediating the effects of glutamate and Aβ25-35, we investigated the effects of these two agents in the presence of glutamate receptor antagonists or the actin filament depolymerizer, or in the absence of extracellular Ca2+.
As shown in Figure 4A, the inhibitory effect of 100 μm glutamate was completely blocked by pretreatment with both the NMDA receptor antagonist MK-801 (20 μm) and the AMPA receptor antagonist CNQX (100 μm), confirming the involvement of NMDA and AMPA receptors in the effect of glutamate on axonal transport. In contrast, the effect of 20 μm Aβ25-35 was not blocked by pretreatment with a combination of 20 μm MK-801 and 100 μm CNQX (Fig. 4B). In Ca2+-free extracellular medium (with 2 mm EGTA), the inhibitory effect of 100 μm glutamate was abolished completely (Fig. 4C). Similarly, the effects of NMDA (100 μm) and AMPA (100 μm) were each blocked in the absence of extracellular Ca2+ (data not shown; n = 5 each). On the contrary, the effect of 20 μm Aβ25-35 remained in the absence of extracellular Ca2+ (Fig. 4D). Nevertheless, the effect of 20 μm Aβ25-35 was abolished by pretreatment with 5 μm latrunculin B, an actin filament depolymerizer (Fig. 4F). The presence of latrunculin B (5 μm) did not block the decreasing effect of glutamate (Fig. 4E), although the decreased axonal transport tended to recover at the end of the glutamate application period (Fig. 4E). The actin-polymerizing agent jasplakinolide (0.5 μm) mimicked the effect of 20 μm Aβ25-35 (Fig. 2C). From these results, the inhibitory effect of glutamate on axonal transport is mediated mainly by Ca2+ influx after activation of NMDA and AMPA receptors. In contrast, the inhibitory effect of Aβ25-35 is likely caused by actin polymerization but not by glutamate receptor activation or Ca2+ influx. In these experiments, application of 20 μm MK-801 plus 100 μm CNQX (Fig. 4A,B) and of 5 μm latrunculin B (Fig. 4E,F) did not significantly affect axonal transport, although in 2 of 10 neurons treated with latrunculin B, axonal transport in both anterograde and retrograde directions increased to >10% above the control value. In such latrunculin B-sensitive neurons, polymerized or aggregated actin originally present might be depolymerized and dissolved by latrunculin B, allowing axonal transport to increase slightly. We also confirmed that axonal transport was not changed by Ca2+-free solution over a 2 hr experimental period.
Figure 4.

Effects of glutamate and Aβ25-35 under various conditions. Data represent percentage changes in the number of transported particles of control (the value before application). Glutamate (100 μm) (A, C, E) and Aβ25-35 (20 μm) (B, D, F) were applied after treatment with a combination of 20 μm MK-801, an NMDA receptor antagonist, and 100 μm CNQX, an AMPA receptor antagonist (A, B), in Ca2+-free solution (C, D), and after treatment with 5 μm latrunculin B, an actin depolymerizer (E, F). Each data point indicates the mean ± SD of the values obtained from five neurons. Error bars represent SD. *p < 0.05; **p < 0.005; ***p < 0.0005 compared with control (100%).
Rhodamine-phalloidin staining of the actin cytoskeleton
Cultured hippocampal neurons were stained with rhodamine-phalloidin, which specifically binds to filamentous actin, after treatment for 30 min with glutamate (100 μm), Aβ25-35 (20 μm), latrunculin B (5 μm), and a combination of Aβ25-35 (20 μm) and latrunculin B (5 μm), all dissolved in HEPES-buffered salt solution. The effects of washout (for 30 min) after exposure to Aβ25-35 (20 μm) were also examined. Control neurons (treated with HEPES-buffered solution alone) revealed a reticular distribution of actin filaments in cell bodies and neurites (Fig. 5A). In glutamate-treated neurons, the distribution of actin filaments was diffuse and the staining was more intense, in particular in the periphery of the cell body, than in control neurons (Fig. 5B), indicating an increase in fragmented actin filaments. Neurons treated with Aβ25-35 showed characteristic features of actin cytoskeleton (Fig. 5C). These neurons were stained intensely, with brightly stained aggregates of actin filaments in both cell bodies and neurites, indicating hyperpolymerization of actin filaments. Interestingly, formation of filopodia, which results from polymerization of actin filaments (Yamada et al., 1970, 1971; Oldenbourg et al., 2000), was observed frequently in Aβ25-35-treated neurons. The same features were observed in neurons that were washed for 30 min after treatment with Aβ25-35 (Fig. 5D). In contrast, staining in latrunculin B-treated neurons was weak and diffuse in both cell bodies and neurites (Fig. 5E), indicating dissolution of actin filaments. Simultaneous treatment with latrunculin B and Aβ25-35 abolished the characteristic features observed in neurons treated with Aβ25-35 alone (Fig. 5F).
Figure 5.

Rhodamine-phalloidin staining of actin filaments in cultured hippocampal neurons. Neurons were stained with rhodamine-phalloidin after treatment of drugs dissolved in HEPES-buffered solution. A, Control (treatment with HEPES-buffered solution alone for 30 min). B, Treatment with 100 μm glutamate for 30 min. C, Treatment with 20 μm Aβ25-35 for 30 min. D, Treatment with 20 μm Aβ25-35 for 30 min followed by washout for 30 min. E, Treatment with 5 μm latrunculin B for 30 min. F, Simultaneous treatment with 20 μm Aβ25-35 and 5 μm latrunculin B for 30 min. Scale bar, 20 μm.
Effects of various Aβ fragments
Aβ molecules found mainly in senile plaques are Aβ1-40 and Aβ1-42 (Iwatsubo et al., 1994). Recent studies have shown that Aβ1-40 and Aβ1-42 assemblies, soluble oligomers and fibril intermediates (protofibrils), have potent neurotoxic activities (Lambert et al., 1998; Hartley et al., 1999; Walsh et al., 1999). Because Aβ is secreted from brain cells as a monomeric form (Haass et al., 1992), the process toward the assembly is necessary for neurotoxicity of Aβ1-40 and Aβ1-42. Hydrophobic residues of Aβ are correlated with fibril formation (Li et al., 1999; Petkova et al., 2002). Thus, we further examined the effects of the hydrophobic Aβ fragments, Aβ15-20 amide and Aβ31-35, and full-length Aβ1-40 and Aβ1-42, to identify residues and structures contributing to the inhibition of axonal transport. Aβ fragments were used immediately after dissolution (fresh condition). Because Aβ1-40 and Aβ1-42 assemble to form large structures during incubation (Pike et al., 1991; Kayed et al., 2003), the effects of these agents incubated for 7 d were also investigated. Light microscopic screening and SDS-PAGE were performed simultaneously to examine the solubility and assembly state of Aβ fragments in solution (Fig. 6A-E). The results are summarized in Table 1. Aβ31-35 (20 μm) but not Aβ15-20 amide (20 μm) induced intracellular actin aggregation and inhibition of axonal transport similar to that of Aβ25-35. Thus, the active site in Aβ is probably present in residues 31-35. Among fresh and incubated full-length Aβs, only incubated Aβ1-42 (20 μm) was effective. In contrast, fresh Aβ1-40 (20 μm), incubated Aβ1-40 (20 μm), or fresh Aβ1-42 (20 μm) had no effect. SDS-PAGE analysis revealed that Aβ15-20 amide and Aβ31-35 were present in low molecular weight forms (<1 kDa) (Fig. 6C). Aβ25-35 was composed of both low molecular weight molecules (∼1 kDa) and aggregates that did not enter the SDS-PAGE gel (Fig. 6C), and this fragment formed precipitates detected by light microscopy (Fig. 6A); that is, in these shorter fragments, there was no correlation between their structures and activities. Fresh Aβ1-40 was primarily monomeric (Fig. 6D), whereas incubated Aβ1-40 additionally produced aggregates (Fig. 6E) and precipitates (Fig. 6B). Fresh Aβ1-42 contained monomers, pentamers, and hexamers (Fig. 6D), whereas incubated Aβ1-42 contained 30-100 kDa high molecular weight forms in addition to the lower molecular forms observed in fresh Aβ1-42 (Fig. 6E). Thus, among various states of full-length Aβ observed in this experiment, only high molecular weight assemblies (30-100 kDa) of Aβ1-42 appear to participate in the inhibition of axonal transport, whereas lower molecular weight molecules (monomers, pentamers, and hexamers) of Aβ1-40/Aβ1-42 or aggregates and precipitates of Aβ1-40 do not.
Figure 6.
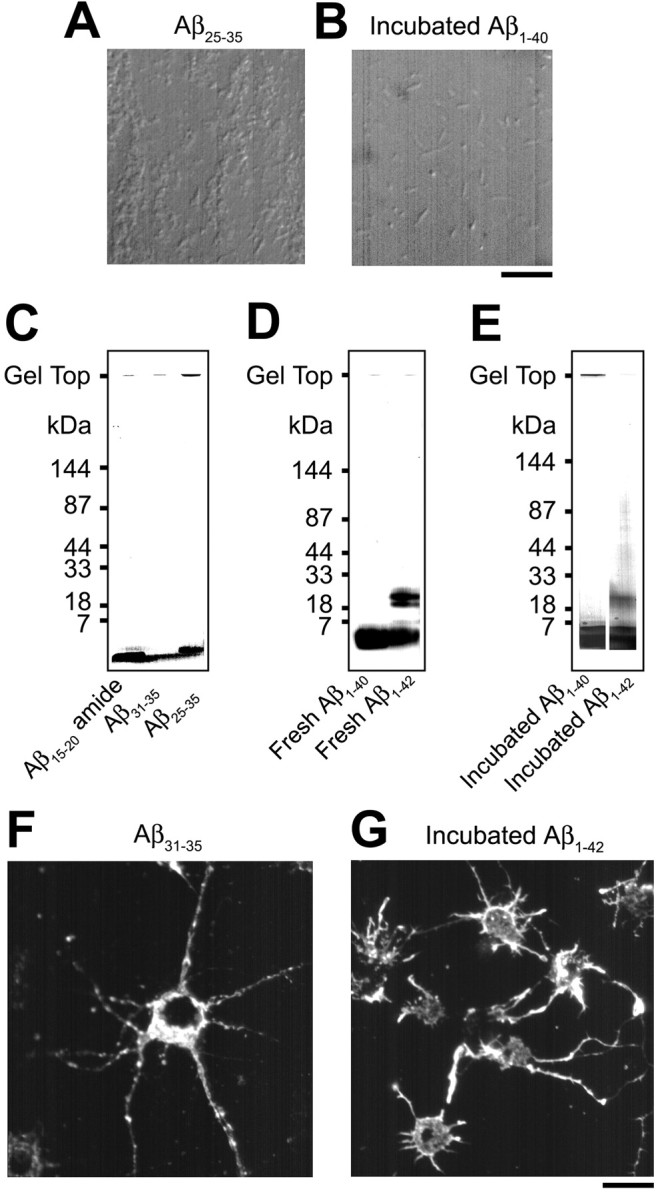
Photographs representing properties of various Aβ fragments. A, B, Precipitates detected under light microscopy in Aβ25-35 solution (A) and in incubated Aβ1-40 solution (B). Aβ25-35 produced cotton-like precipitates (A), and incubated Aβ1-40 formed fiber-like or amorphous precipitates (B). Scale bar, 10 μm. C-E, SDS-PAGE profiles of various Aβ fragments. F, G, Rhodamine-phalloidin staining of intracellular actin filaments in hippocampal neurons treated with Aβ31-35 (F) and with incubated Aβ1-42 (G). Aβ31-35 (20 μm) and incubated Aβ1-42 (20 μm) formed intracellular actin aggregates. Scale bar, 20 μm.
Table 1.
Properties and effects of various Aβ fragments
|
Aβ fragment |
Precipitates detected under light microscopy |
Structural properties analyzed by SDS-PAGE |
Intracellular actin aggregation |
Effects on axonal transport |
|---|---|---|---|---|
| Aβ15-20 amide | − | Low molecular weight forms (<1 kDa) | − | No effect |
| Aβ31-35 | − | Low molecular weight forms (<1 kDa) | + | Inhibitory |
| Aβ25-35 | + | Low molecular weight forms (~1 kDa) | + | Inhibitory |
| Aggregates | ||||
| Fresh Aβ1-40 | − | Monomers | − | No effect |
| Incubated Aβ1-40 | + | Monomers | − | No effect |
| Aggregates | ||||
| Fresh Aβ1-42 | − | Monomers | − | No effect |
| Pentamers | ||||
| Hexamers | ||||
| Incubated Aβ1-42 | − | Monomers | + | Inhibitory |
| Pentamers | ||||
| Hexamers | ||||
|
|
|
High molecular weight forms (30-100 kDa)
|
|
|
Aβ fragments dissolved in HEPES-buffered salt solution, pH 7.4, at a concentration of 20 μM were used for light microscopic analysis, intracellular actin observation with rhodamine-phalloidin, and axonal transport study under video-enhanced microscopy. Ten micrograms of each Aβ fragment dissolved in an aqueous solution were subjected to SDS-PAGE. +, Detected; −, not detected.
Discussion
Inhibitory effects of glutamate and Aβ on axonal transport
We show evidence that glutamate and Aβ25-35 inhibit anterograde and retrograde fast axonal transport in cultured rat hippocampal neurons. Only a few previous reports have described the effects of these agents on axonal transport. One of them has shown that glutamate inhibits axonal transport of neurofilaments (Ackerley et al., 2000). Neurofilaments, carried within axons by slow axonal transport, have recently been found to be transported by similar mechanisms to fast axonal transport, microtubule-based and kinesin and dynein motors-dependent mechanisms, with a long rest period (Roy et al., 2000; Wang et al., 2000). One more report has demonstrated, using immunohistochemistry, that human Aβ1-42 applied in vivo inhibits fast axonal transport of acetylcholinesterase, amyloid precursor protein, vesicular acetylcholine transporter, and synaptophysin in the sciatic nerve of the rat (Kasa et al., 2000). Therefore, the present and previous studies strongly suggest that both glutamate and Aβ are agents that inhibit fast axonal transport. We also found that the response of axonal transport to glutamate and Aβ25-35 was rapid, being detected within a few minutes after the start of stimulation. Thus, axonal transport may be one of the early targets for toxicity of these two agents.
The effect of glutamate, even when applied at high concentrations (≥100 μm) for 26 min, was reversed after removal of glutamate. These exposure concentrations and duration are sufficient to cause cell death in cultured neurons 24 hr later (Hartley et al., 1993). Despite this, the effect of glutamate on axonal transport was reversible, probably by receptor-mediated mechanisms as described below. In contrast, the effect of Aβ25-35 at 20 μm, a concentration able to kill neuronal cells several days later (Yankner et al., 1990; Weiss et al., 1994), was irreversible and progressive even after washout. Similar prolongation and irreversibility of the effects of Aβ have been reported previously. Aβ, at various concentrations, induces a prolonged activation of pro-apoptotic markers, caspase 3 and annexin V binding (A. R. White et al., 2001), an irreversible inward cation current (Furukawa et al., 1994; Sanderson et al., 1997), and a prolonged glutamate-induced [Ca2+]i increase even after removal of glutamate (Scorziello et al., 1996) in neuronal or neuron-like cells. In astrocytes, Aβ irreversibly produces a reactive morphological change (Kato et al., 1997). Thus, Aβ can cause prolonged and irreversible changes in some cellular structures and functions including axonal transport. The present study using rhodamine-phalloidin revealed that 20 μm Aβ25-35-induced changes in the actin cytoskeleton were irreversible after removal of Aβ25-35. Therefore, persistent intracellular structural changes induced by Aβ25-35 may result in an irreversible impairment of axonal transport.
Different mechanisms of inhibition of axonal transport by glutamate and Aβ
Glutamate can trigger Ca2+ influx after activation of NMDA and AMPA receptors to cause excitotoxicity (Frandsen and Schousboe, 1993; Hartley et al., 1993). In agreement with this, we find that the inhibitory effect of glutamate on axonal transport is mediated by activation of both NMDA and AMPA receptors and subsequent Ca2+ influx. In contrast, the effect of Aβ25-35 is unlikely to be mediated by NMDA or AMPA receptors, or by Ca2+ influx. This seems to be inconsistent with the previously reported mechanisms of Aβ-induced neurodegeneration and cell death. It has been proposed that Aβ has an affinity for the agonist recognition sites of the NMDA receptor (Cowburn et al., 1994; 1997) and increases intracellular Ca2+ through an influx of extracellular Ca2+ in cultured neurons (Joseph and Han, 1992; Weiss et al., 1994). Toxicity of Aβ can be inhibited by the NMDA receptor antagonist (Le et al., 1995) and by Ca2+ channel blockers (Weiss et al., 1994). On the contrary, our proposed mechanisms are compatible with some other previous investigations. Cell death induced by Aβ is not mediated by intracellular early Ca2+ accumulation, because Aβ inhibits spontaneous Ca2+ oscillation and does not affect high K+- and glutamate-evoked Ca2+ increase (Gao et al., 1998). The neurotoxic effect of Aβ is not blocked by glutamate receptor antagonists (Busciglio et al., 1993; Weiss et al., 1994; Gray and Patel, 1995) or by Ca2+ channel blockers (Whitson and Appel, 1995). Collectively considered, Aβ can trigger multiple signaling cascades leading to neuronal cell damage, and one or some of which may be specific for the impairment of axonal transport. Here we found that the inhibitory effect of Aβ25-35 on axonal transport was abolished by the actin-depolymerizing agent latrunculin B. In addition, the actin-polymerizing agent jasplakinolide mimicked the effect of Aβ25-35, and Aβ25-35 induced aggregation of intracellular actin. Because aggregation of actin is supposed to result from actin polymerization (Lee et al., 1998; Fabian et al., 1999; Bai et al., 2002), our results suggest that the inhibitory effect of Aβ25-35 on axonal transport occurs via polymerization and subsequent aggregation of actin. Neurite shrinkage and filopodia formation induced by Aβ25-35, revealed by the present morphological investigation, may also be explained as being caused by polymerization of actin filaments. Actin polymerization dynamics involves changes in cell shape and size. Filopodia are formed by polymerization of actin (Yamada et al., 1970, 1971; Oldenbourg et al., 2000). Such filopodia formations may be involved in the previously reported neurotrophic action of Aβ (Whitson et al., 1990; Yankner et al., 1990; Koo et al., 1993), which appears to contribute to aberrant sprouting of neurites around senile plaques in AD (Whitson et al., 1990; Koo et al., 1993). Furthermore, in support of our conclusion, Aβ has been suggested to cause disruption of the intracellular actin network (Davis et al., 1999), which results from actin polymerization (Senderowicz et al., 1995; Fabian et al., 1999; S. R. White et al., 2001) as well as depolymerization. Hirano bodies, one of pathologic features found frequently in neurites adjacent to a senile plaque in the AD brain, include abundant actin polymers (Goldman, 1983; Galloway et al., 1987). Depolymerization of actin attenuates Aβ neurotoxicity (Furukawa and Mattson, 1995). Thus, actin polymerization appears to relate to some neurotoxic effects of Aβ. Certainly, our results are strongly supported by recent evidence that Aβ induces actin polymerization to form stress fibers in SN1 cells, a septal neuronal cell line, and in cultured mouse hippocampal neurons (Song et al., 2002). Presently, it is not known how extracellular Aβ polymerizes intracellular actin; however, because Aβ forms pores on cell membrane (Furukawa et al., 1994; Sanderson et al., 1997; Kawahara and Kuroda, 2000) and activates intracellular cascades that cause actin polymerization, such as the p38 mitogen-activated protein kinase (MAPK) signaling pathway (Song et al., 2002), Aβ could interact with intracellular actin.
Axonal transport can be modified by the dynamics of the cytoskeleton, including actin filaments. A number of observations indicate the participation of actin in fast axonal transport of organelles. Organelles can be transported axonally along actin filaments (Kuznetsov et al., 1992; Morris and Hollenbeck, 1995). Microtubule-based tracks for fast axonal organelle transport include actin filaments (Bearer and Reese, 1999), and actin is a component of granular, microtubule-associated cross-bridges that connect to other structures, including other microtubules, neurofilaments, organelles, and plasma membrane (Nagele et al., 1988). Changes in intracellular actin states have been shown to modify axonal transport (Goldberg et al., 1980; Goldberg, 1982; Brady et al., 1984; Kuznetsov et al., 1992; Morris and Hollenbeck, 1995; Ligon and Steward, 2000). Several early studies suggest that some actin-depolymerizing agents such as cytochalasins, DNase I, and gelsolin seem to inhibit fast axonal transport (Goldberg et al., 1980; Isenberg et al., 1980; Goldberg, 1982; Brady et al., 1984; Nemhauser and Goldberg, 1985; Kuznetsov et al., 1992), which appears to be inconsistent with the present results indicating that polymerization and aggregation of actin inhibit axonal transport; however, some of these agents aggregate actin, on the other hand. Indeed, fast axonal transport of mitochondria is impeded by cytochalasin D-induced actin aggregates but not affected by latrunculin B (Ligon and Steward, 2000). Nemhauser and Goldberg (1985) have described the possibility of the secondary effects of DNase I on axonal transport, i.e., a substantial part of the inhibition of fast axonal transport caused by DNase I is attributable to depolymerization of microtubules, or DNase I induces a decrease in microtubules by reducing actin filaments. Indeed, polymerization of actin with jasplakinolide inhibits intracellular motility in non-neuronal cells, HL60 cells, and monocytes (Fabian et al., 1999). These previous studies support our proposal that actin polymerization and aggregation induced by Aβ inhibit fast axonal transport.
Aβ residues and structures contributing to the inhibition of axonal transport
Furthermore, we investigated various Aβ fragments including Aβ1-40 and Aβ1-42, the main fragments found in senile plaques in the human AD brain. We obtained the results that Aβ31-35 induced aggregation of actin filaments and inhibition of axonal transport similarly to Aβ25-35, whereas Aβ15-20 amide had no effects. Thus, the active site in full-length Aβ may reside in fragments 31-35, as suggested previously (Yan et al., 1999; Qi and Qiao, 2001; Bond et al., 2003). In these shorter fragments, there was no correlation between structures and activities. In contrast, in full-length Aβ, the activity seems to depend on its structure. Our results indicated that incubated Aβ1-42 produced an inhibition of axonal transport and actin aggregation, whereas fresh Aβ1-40, incubated Aβ1-40, or fresh Aβ1-42 had no effect. Because incubated Aβ1-42 but not fresh Aβ1-42 contained 30-100 kDa molecules corresponding to higher oligomers or protofibrils (or protofilaments), these structures of Aβ1-42 may participate in the impairment of axonal transport. Our data also indicate that Aβ1-40 in monomers, aggregates, or participates has no apparent effect, although the possibility remains that Aβ1-40 in other states is effective. These results are consistent with the current concept that soluble oligomers and protofibrils of full-length Aβ are neurotoxic (Lambert et al., 1998; Hartley et al., 1999; Walsh et al., 1999; Dahlgren et al., 2002; Kim et al., 2003). Furthermore, on the basis of our results, we postulate that residues 31-35 may be the key to understanding the structure dependence of the activity of full-length Aβ. We propose that Aβ is effective when it represents a structure in which the active site can access the cell membrane. This proposal is supported by the recent x-ray diffraction study by Bond et al. (2003). They suggest that residues 31-35 are exposed at the surface of oligomeric Aβ and interact with the cell membrane. Assembly of full-length Aβ can be driven by the formation of β-sheet structures where hydrophobic residues 17-20 are in contact with hydrophobic residues 31-35 (Lee et al., 1995; Li et al., 1999). Monomers of Aβ1-40 and Aβ1-42 exist as random extended chain (Zeng et al., 2001) or α-helix (Barrow and Zagorski, 1991; Coles et al., 1998; Zeng et al., 2001), and dimer and small oligomers can form a β-sheet (Barrow and Zagorski, 1991; Chaney et al., 1998) or an irregular structure that is not α-helix or β-structure (Huang et al., 2000). Higher oligomers form β-sheet (Huang et al., 2000) and appear to combine with each other to build protofibrils (or protofilaments), and then protofibrils (or protofilaments) associate into larger fibrils (Chaney et al., 1998; Walsh et al., 1999; Serpell, 2000; Petkova et al., 2002; Bitan et al., 2003). Because the results of the present study as well as other recent studies (Lambert et al., 1998; Hartley et al., 1999; Walsh et al., 1999; Dahlgren et al., 2002) indicate that full-length Aβ assemblies, such as higher oligomers and protofibrils, are toxic, the β-sheet formation seems to contribute to toxicity in full-length Aβ. In much larger assemblies such as fibrils, however, hydrophobic β-sheets that contain residues 31-35 are associated laterally, and then the β-sheet structures are positioned inside the Aβ assembly or are shielded by hydrophilic residues (Chaney et al., 1998; Petkova et al., 2002). In such states, toxic 31-35 residues can no longer access the cell membrane, and therefore Aβ may become ineffective; however, further structural evidence is needed to confirm this speculation.
In summary, glutamate and Aβ25-35 rapidly inhibit anterograde and retrograde fast axonal transport in cultured hippocampal neurons. The effects of glutamate are mediated by activation of NMDA and AMPA receptors and subsequent Ca2+ influx, which is different from the inhibitory mechanisms of Aβ25-35. The latter is likely mediated by actin polymerization and aggregation. The ability of Aβ to exert these effects seems to require specific amino acid residues and structures. These results may provide insight into the early pathogenetic mechanisms of AD.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research (11670638) from the Ministry of Education, Science, Sports and Culture, Japan, to H.H.
Correspondence should be addressed to Dr. Hiromi Hiruma, Department of Physiology, Kitasato University School of Medicine, 1-15-1 Kitasato, Sagamihara 228-8555, Japan. E-mail: hiruma@med.kitasato-u.ac.jp.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238967-11$15.00/0
References
- Ackerley S, Grierson AJ, Brownlees J, Thornhill P, Anderton BH, Leigh PN, Shaw CE, Miller CC ( 2000) Glutamate slows axonal transport of neurofilaments in transfected neurons. J Cell Biol 150: 165-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C, Becerra-García F, Tapia R ( 1998) Glutamic acid and Alzheimer's disease. Neurobiology 6: 33-43. [PubMed] [Google Scholar]
- Bai R, Covell DG, Liu C, Ghosh AK, Hamel E ( 2002) (-)-Doliculide, a new macrocyclic depsipeptide enhancer of actin assembly. J Biol Chem 277: 32165-32171. [DOI] [PubMed] [Google Scholar]
- Barrow CJ, Zagorski MG ( 1991) Solution structures of β peptide and its constituent fragments: relation to amyloid deposition. Science 253: 179-182. [DOI] [PubMed] [Google Scholar]
- Bearer EL, Reese TS ( 1999) Association of actin filaments with axonal microtubule tracts. J Neurocytol 28: 85-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB ( 2003) Amyloid β-protein (Aβ) assembly: Aβ 40 and Aβ 42 oligomerize through distinct pathways. Proc Natl Acad Sci USA 100: 330-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond JP, Deverin SP, Inouye H, El-Agnaf OM, Teeter MM, Kirschner DA ( 2003) Assemblies of Alzheimer's peptides Aβ25-35 and Aβ31-35: reverse-turn conformation and side-chain interactions revealed by X-ray diffraction. J Struct Biol 141: 156-170. [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD, Yin HL, Stossel TP ( 1984) Gelsolin inhibition of fast axonal transport indicates a requirement for actin microfilaments. Nature 310: 56-58. [DOI] [PubMed] [Google Scholar]
- Brimijoin WS ( 1982) Abnormalities of axonal transport: are they a cause of peripheral nerve disease? Mayo Clin Proc 57: 707-714. [PubMed] [Google Scholar]
- Busciglio J, Yeh J, Yankner BA ( 1993) β-Amyloid neurotoxicity in human cortical culture is not mediated by excitotoxins. J Neurochem 61: 1565-1568. [DOI] [PubMed] [Google Scholar]
- Calligaro DO, O'Malley PJ, Monn JA ( 1993) β-Amyloid (25-35) or sub stance P stimulates [3H]MK-801 binding to rat cortical membranes in the presence of glutamate and glycine. J Neurochem 60: 2297-2303. [DOI] [PubMed] [Google Scholar]
- Chaney MO, Webster SD, Kuo YM, Roher AE ( 1998) Molecular modeling of the Aβ1-42 peptide from Alzheimer's disease. Protein Eng 11: 761-767. [DOI] [PubMed] [Google Scholar]
- Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ ( 1998) Solution structure of amyloid β-peptide(1-40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry 37: 11064-11077. [DOI] [PubMed] [Google Scholar]
- Cowburn RF, Messamore E, Li ML, Winblad B, Sundström E ( 1994) β-Amyloid related peptides exert differential effects on [3H]MK-801 binding to rat cortical membranes. NeuroReport 5: 405-408. [DOI] [PubMed] [Google Scholar]
- Cowburn RF, Wiehager B, Trief E, Li-Li M, Sundström E ( 1997) Effects of β-amyloid-(25-35) peptides on radioligand binding to excitatory amino acid receptors and voltage-dependent calcium channels: evidence for a selective affinity for the glutamate and glycine recognition sites of the NMDA receptor. Neurochem Res 22: 1437-1442. [DOI] [PubMed] [Google Scholar]
- Cull RE ( 1975) Role of axonal transport in maintaining central synaptic connections. Exp Brain Res 24: 97-101. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ ( 2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem 277: 32046-32053. [DOI] [PubMed] [Google Scholar]
- Davis J, Cribbs DH, Cotman CW, Van Nostrand WE ( 1999) Pathogenic amyloid β-protein induces apoptosis in cultured human cerebrovascular smooth muscle cells. Amyloid 6: 157-164. [DOI] [PubMed] [Google Scholar]
- Fabian I, Halperin D, Lefter S, Mittelman L, Altstock RT, Seaon O, Tsarfaty I ( 1999) Alteration of actin organization by jaspamide inhibits ruffling, but not phagocytosis or oxidative burst, in HL-60 cells and human monocytes. Blood 93: 3994-4005. [PubMed] [Google Scholar]
- Frandsen A, Schousboe A ( 1993) Excitatory amino acid-mediated cytotoxicity and calcium homeostasis in cultured neurons. J Neurochem 60: 1202-1211. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Mattson MP ( 1995) Cytochalasins protect hippocampal neurons against amyloid β-peptide toxicity: evidence that actin depolymerization suppresses Ca2+ influx. J Neurochem 65: 1061-1068. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Abe Y, Akaike N ( 1994) Amyloid β protein-induced irreversible current in rat cortical neurones. NeuroReport 5: 2016-2018. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Smith-Swintosky VL, Mattson MP ( 1995) Evidence that actin depolymerization protects hippocampal neurons against excitotoxicity by stabilizing [Ca2+]i Exp Neurol 133: 153-163. [DOI] [PubMed] [Google Scholar]
- Galloway PG, Perry G, Gambetti P ( 1987) Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol 46: 185-199. [DOI] [PubMed] [Google Scholar]
- Gao ZY, Collins HW, Matschinsky FM, Lee VM, Wolf BA ( 1998) Cytotoxic effect of β-amyloid on a human differentiated neuron is not mediated by cytoplasmic Ca2+ accumulation. J Neurochem 70: 1394-1400. [DOI] [PubMed] [Google Scholar]
- Goldberg DJ ( 1982) Microinjection into an identified axon to study the mechanism of fast axonal transport. Proc Natl Acad Sci USA 79: 4818-4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg DJ, Harris DA, Lubit BW, Schwartz JH ( 1980) Analysis of the mechanism of fast axonal transport by intracellular injection of potentially inhibitory macromolecules: evidence for a possible role of actin filaments. Proc Natl Acad Sci USA 77: 7448-7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JE ( 1983) The association of actin with Hirano bodies. J Neuropathol Exp Neurol 42: 146-152. [DOI] [PubMed] [Google Scholar]
- Gray CW, Patel AJ ( 1995) Neurodegeneration mediated by glutamate and β-amyloid peptide: a comparison and possible interaction. Brain Res 691: 169-179. [DOI] [PubMed] [Google Scholar]
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ ( 1992) Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359: 322-325. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW ( 1993) Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci 13: 1993-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ ( 1999) Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19: 8876-8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TH, Yang DS, Plaskos NP, Go S, Yip CM, Fraser PE, Chakrabartty A ( 2000) Structural studies of soluble oligomers of the Alzheimer β-amyloid peptide. J Mol Biol 297: 73-87. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Schubert P, Kreutzberg GW ( 1980) Experimental approach to test the role of actin in axonal transport. Brain Res 194: 588-593. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y ( 1994) Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13: 45-53. [DOI] [PubMed] [Google Scholar]
- Joseph R, Han E ( 1992) Amyloid β-protein fragment 25-35 causes activation of cytoplasmic calcium in neurons. Biochem Biophys Res Commun 184: 1441-1447. [DOI] [PubMed] [Google Scholar]
- Kasa P, Papp H, Kovacs I, Forgon M, Penke B, Yamaguchi H ( 2000) Human amyloid-β1-42 applied in vivo inhibits the fast axonal transport of proteins in the sciatic nerve of rat. Neurosci Lett 278: 117-119. [DOI] [PubMed] [Google Scholar]
- Kato M, Saito H, Abe K ( 1997) Nanomolar amyloid β protein-induced inhibition of cellular redox activity in cultured astrocytes. J Neurochem 68: 1889-1895. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y ( 2000) Molecular mechanism of neurodegeneration induced by Alzheimer's β-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res Bull 53: 389-397. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG ( 2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486-489. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Chae SC, Lee DK, Chromy B, Lee SC, Park YC, Klein WL, Krafft GA, Hong ST ( 2003) Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J 17: 118-120. [DOI] [PubMed] [Google Scholar]
- Koo EH, Park L, Selkoe DJ ( 1993) Amyloid β-protein as a substrate interacts with extracellular matrix to promote neurite outgrowth. Proc Natl Acad Sci USA 90: 4748-4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowall NW ( 1994) Beta amyloid neurotoxicity and neuronal degeneration in Alzheimer's disease. Neurobiol Aging 15: 257-258. [DOI] [PubMed] [Google Scholar]
- Kuznetsov SA, Langford GM, Weiss DG ( 1992) Actin-dependent organelle movement in squid axoplasm. Nature 356: 722-725. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL ( 1998) Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95: 6448-6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancelot E, Beal MF ( 1998) Glutamate toxicity in chronic neurodegenerative disease. Prog Brain Res 116: 331-347. [DOI] [PubMed] [Google Scholar]
- Le WD, Colom LV, Xie WJ, Smith RG, Alexianu M, Appel SH ( 1995) Cell death induced by β-amyloid 1-40 in MES 23.5 hybrid clone: the role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res 686: 49-60. [DOI] [PubMed] [Google Scholar]
- Lee E, Shelden EA, Knecht DA ( 1998) Formation of F-actin aggregates in cells treated with actin stabilizing drugs. Cell Motil Cytoskeleton 39: 122-133. [DOI] [PubMed] [Google Scholar]
- Lee JP, Stimson ER, Ghilardi JR, Mantyh PW, Lu YA, Felix AM, Llanos W, Behbin A, Cummings M, Van Criekinge M, Timms W, Maggio JE ( 1995) 1H NMR of Aβ amyloid peptide congeners in water solution. Conformational changes correlate with plaque competence. Biochemistry 34: 5191-5200. [DOI] [PubMed] [Google Scholar]
- Li L, Darden TA, Bartolotti L, Kominos D, Pedersen LG ( 1999) An atomic model for the pleated β-sheet structure of Aβ amyloid protofilaments. Biophys J 76: 2871-2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon LA, Steward O ( 2000) Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol 427: 351-361. [DOI] [PubMed] [Google Scholar]
- Limbrick Jr DD, Pal S, DeLorenzo RJ ( 2001) Hippocampal neurons exhibit both persistent Ca2+ influx and impairment of Ca2+ sequestration/extrusion mechanisms following excitotoxic glutamate exposure. Brain Res 894: 56-67. [DOI] [PubMed] [Google Scholar]
- Masliah E ( 1995) Mechanisms of synaptic dysfunction in Alzheimer's disease. Histol Histopathol 10: 509-519. [PubMed] [Google Scholar]
- Masliah E, Terry RD, DeTeresa RM, Hansen LA ( 1989) Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett 103: 234-239. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE ( 1992) β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12: 376-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretto A, Sabri MI ( 1988) Progressive deficits in retrograde axon transport precede degeneration of motor axons in acrylamide neuropathy. Brain Res 440: 18-24. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ ( 1995) Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol 131: 1315-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele RG, Kosciuk MC, Hunter ET, Bush KT, Lee H ( 1988) Immunoelectron microscopic localization of actin in neurites of cultured embryonic chick dorsal root ganglia: actin is a component of granular, microtubule-associated cross-bridges. Brain Res 474: 279-286. [DOI] [PubMed] [Google Scholar]
- Nemhauser I, Goldberg DJ ( 1985) Structural effects in axoplasm of DNase I, an actin depolymerizer that blocks fast axonal transport. Brain Res 334: 47-58. [DOI] [PubMed] [Google Scholar]
- Oldenbourg R, Katoh K, Danuser G ( 2000) Mechanism of lateral movement of filopodia and radial actin bundles across neuronal growth cones. Biophys J 78: 1176-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R ( 2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA 99: 16742-16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW ( 1991) In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res 563: 311-314. [DOI] [PubMed] [Google Scholar]
- Praprotnik D, Smith MA. Richey PL, Vinters HV, Perry G ( 1996) Filament heterogeneity within the dystrophic neurites of senile plaques suggests blockage of fast axonal transport in Alzheimer's disease. Acta Neuropathol (Berl) 91: 226-235. [DOI] [PubMed] [Google Scholar]
- Qi JS, Qiao JT ( 2001) Amyloid β-protein fragment 31-35 forms ion channels in membrane patches excised from rat hippocampal neurons. Neuroscience 105: 845-852. [DOI] [PubMed] [Google Scholar]
- Richard S, Brion JP, Couck AM, Flament-Durand J ( 1989) Accumulation of smooth endoplasmic reticulum in Alzheimer's disease: new morphological evidence of axoplasmic flow disturbances. J Submicrosc Cytol Pathol 21: 461-467. [PubMed] [Google Scholar]
- Roy S, Coffee P, Smith G, Liem RK, Brady ST, Black MM ( 2000) Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci 20: 6849-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson KL, Butler L, Ingram VM ( 1997) Aggregates of a β-amyloid peptide are required to induce calcium currents in neuron-like human teratocarcinoma cells: relation to Alzheimer's disease. Brain Res 744: 7-14. [DOI] [PubMed] [Google Scholar]
- Scorziello A, Meucci O, Florio T, Fattore M, Forloni G, Salmona M, Schettini G ( 1996) β25-35 alters calcium homeostasis and induces neurotoxicity in cerebellar granule cells. J Neurochem 66: 1995-2003. [DOI] [PubMed] [Google Scholar]
- Senderowicz AM, Kaur G, Sainz E, Laing C, Inman WD, Rodri águez J, Crews P, Malspeis L, Grever MR, Sausville EA ( 1995) Jasplakinolide's inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J Natl Cancer Inst 87: 46-51. [DOI] [PubMed] [Google Scholar]
- Serpell LC ( 2000) Alzheimer's amyloid fibrils: structure and assembly. Biochim Biophys Acta 1502: 16-30. [DOI] [PubMed] [Google Scholar]
- Song C, Perides G, Wang D, Liu YF ( 2002) β-Amyloid peptide induces formation of actin stress fibers through p38 mitogen-activated protein kinase. J Neurochem 83: 828-836. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Terry RD ( 1967) Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. Acta Neuropathol (Berl) 8: 276-284. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB ( 1999) Amyloid β-protein fibrillo-genesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem 274: 25945-25952. [DOI] [PubMed] [Google Scholar]
- Wang L, Ho CL, Sun D, Liem RK, Brown A ( 2000) Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol 2: 137-141. [DOI] [PubMed] [Google Scholar]
- Warita H, Itoyama Y, Abe K ( 1999) Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res 819: 120-131. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Pike CJ, Cotman CW ( 1994) Ca2+ channel blockers attenuate β-amyloid peptide toxicity to cortical neurons in culture. J Neurochem 62: 372-375. [DOI] [PubMed] [Google Scholar]
- White AR, Guirguis R, Brazier MW, Jobling MF, Hill AF, Beyreuther K, Barrow CJ, Masters CL, Collins SJ, Cappai R ( 2001) Sublethal concentrations of prion peptide PrP106-126 or the amyloid beta peptide of Alzheimer's disease activates expression of proapoptotic markers in primary cortical neurons. Neurobiol Dis 8: 299-316. [DOI] [PubMed] [Google Scholar]
- White SR, Williams P, Wojcik KR, Sun S, Hiemstra PS, Rabe KF, Dorscheid DR ( 2001) Initiation of apoptosis by actin cytoskeletal derangement in human airway epithelial cells. Am J Respir Cell Mol Biol 24: 282-294. [DOI] [PubMed] [Google Scholar]
- Whitson JS, Appel SH ( 1995) Neurotoxicity of A β amyloid protein in vitro is not altered by calcium channel blockade. Neurobiol Aging 16: 5-10. [DOI] [PubMed] [Google Scholar]
- Whitson JS, Glabe CG, Shintani E, Abcar A, Cotman CW ( 1990) β-Amyloid protein promotes neuritic branching in hippocampal cultures. Neurosci Lett 110: 319-324. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW ( 1999) Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2: 50-56. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Spooner BS, Wessells NK ( 1970) Axon growth: roles of microfilaments and microtubules. Proc Natl Acad Sci USA 66: 1206-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Spooner BS, Wessells NK ( 1971) Ultrastructure and function of growth cones and axons of cultured nerve cells. J Cell Biol 49: 614-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan XZ, Qiao JT, Dou Y, Qiao ZD ( 1999) β-amyloid peptide fragment 31-35 induces apoptosis in cultured cortical neurons. Neuroscience 92: 177-184. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Duffy LK, Kirschner DA ( 1990) Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science 250: 279-282. [DOI] [PubMed] [Google Scholar]
- Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N ( 1998) Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol 141: 431-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younkin SG, Goodridge B, Katz J, Lockett G, Nafziger D, Usiak MF, Younkin LH ( 1986) Molecular forms of acetylcholinesterases in Alzheimer's disease. Fed Proc 45: 2982-2988. [PubMed] [Google Scholar]
- Zeng H, Zhang Y, Peng L, Shao H, Menon NK, Yang J, Salomon AR, Freidland RP, Zagorski MG ( 2001) Nicotine and amyloid formation. Biol Psychiatry 49: 248-257. [DOI] [PubMed] [Google Scholar]