

Abstract
Transgenic mice, expressing mutant β-amyloid precursor proteins (βAPPs), have lead to a better understanding of the pathophysiological processes in Alzheimer's disease (AD). In many of these models, however, the temporal development of cognitive decline and the relationship to Aβ deposition and inflammation are unclear. We now report a novel transgenic mouse line, PS2APP (PS2N141I × APPswe), which develops a severe cerebral amyloidosis in discrete brain regions, and present a cross-sectional analysis of these mice at 4, 8, 12, and 16 months of age. Each age cohort was investigated for changes in behavior, electrophysiology of synapse efficacy, ELISA-determined Aβ load, histopathology, and in immunoelectron microscopy. Cognitive deficits were first observed at 8 months when Aβ deposits and inflammation were restricted to discrete brain regions, namely the subiculum and frontolateral (motor and orbital) cortex. As early as 5 months, electron microscopy revealed the presence, in these regions, of pre-plaque, immunogold-labeled extracellular fibrillar Aβ. At the same age, increased levels of insoluble Aβ were detected by ELISA, with Aβ1-40 levels exceeding those of Aβ1-42. Further cognitive decline occurred in an age-related manner, and this was accompanied by the spread of amyloidosis to ultimately affect not only neo- and limbic cortices, but also thalamic and pontine nuclei. Dentate gyrus post-tetanic potentiation was significantly attenuated at 17 months, and there were also significant differences in paired-pulse parameters. This systematic cross-sectional study of the behavioral and pathological changes in the PS2APP mouse indicates that it develops age-related cognitive decline associated with severe amyloidosis and inflammation in discrete brain regions and therefore is suitable for testing a range of potential symptomatic and disease-modifying therapies for AD.
Keywords: Alzheimer, PS2APP, transgenic, amyloid β-protein, amyloid precursor protein, presenilin 2, plaque, inflammation, LTP, cognition
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disorder and the major cause of late-onset dementia. There is currently no curative treatment for the disease, only symptomatic treatment. Two histopathological hallmarks are crucial for its postmortem diagnosis, namely the extracellular deposition of neuritic plaques and the intracellular aggregation of neurofibrillary tangles (NFTs) (Braak and Braak, 1997). The former are composed of fibrillar amyloid (A)-β peptide, a 39-43 amino acid peptide generated from a larger amyloid precursor protein (βAPP), and the latter consist of paired helical filaments composed of the hyperphosphorylated cytoskeletal protein tau. Recent research on AD (for review, see Selkoe, 2001; Sisodia and St. George-Hyslop, 2002) has identified genetic factors that either are causal or predispose to the disease. These include mutations in βAPP, presenilins 1 and 2 (PS1, PS2), as well as apolipoprotein E4 polymorphism.
The development of animal models of AD (for review, see Janus and Westaway, 2001; Higgins and Jacobsen, 2003) is of great value not only for the understanding of the pathophysiological process but also to test potential symptomatic or disease-modifying therapies for AD. Transgenic mice overexpressing mutated forms of human βAPP (termed PDAPP, Tg2576, APP23, and CRND8 mice) were among the first to be described (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997; Chishti et al., 2001, respectively). These mice develop diffuse and neuritic plaques and dystrophic neurites, as well as an inflammatory response (microglioses and astroglioses), but no NFT pathology. Accelerated Aβ deposition was achieved in mice (e.g., PSAPP) coexpressing mutated human PS1 and βAPP proteins (Duff et al., 1996; Borchelt et al., 1997; Holcomb et al., 1998). There are relatively fewer tau-based transgenics reported (e.g., JNPL3) (Lewis et al., 2000). The crossing of these mice with Tg2576 mice resulted in the double transgenic mouse (TAPP), which uniquely develops both amyloid plaques and NFT pathology (Lewis et al., 2001). Despite these advances, relatively few studies to date have described, for the same cohorts, the temporal development of cognitive decline relative to plaque formation and accompanying inflammation.
We have created a double transgenic mouse line PS2APP, overexpressing mutant forms of human PS2 (N141I) and human βAPP (K670N, M671L), which develops, within the studied age range, a severe cerebral amyloidosis exclusively in neo- and limbic cortices, including the hippocampal formation and amygdala, as well as thalamic and pontine nuclei, but sparing the olfactory bulb, striatum, hypothalamus, pons, cerebellum, brainstem, spinal cord, pituitary, and brain vasculature. We selected the PS2 transgene to allow a comparison with the two primary PSAPP (i.e., PS1 × APP) lines already published (Borchelt et al., 1997; Holcomb et al., 1998); to the best of our knowledge, no PS2 × APP double transgenic has yet been characterized in detail. To determine the temporal development of this amyloidosis and its possible functional correlates, we performed immunochemical, histopathological, immunoelectron microscopical, electrophysiological, and behavioral analyses of such mice, as well as age-matched nontransgenic controls, at 4, 8, 12, and 16 months of age. Already at 8 months, PS2APP mice develop age-related cognitive deficits and correlative amyloid deposits with inflammation in discrete brain regions.
Materials and Methods
Transgene constructs
APPswe construct. The cDNA encoding human βAPP751 was donated by Prof. D. Goldgaber (State University of New York Health Science Center, Stony Brook, NY). The Swedish familial AD double mutation (SFAD, K670N, M671L substitution) was introduced by site-directed mutagenesis. The Thy-1.2-hrβAPP751SFAD transgene was generated by insertion of the 2.4 kbp hβAPP751 SFAD cDNA fragment (corresponding to nucleotides 32-2400 of AC: X06989) into the XhoI site of an expression vector containing mouse Thy1.2 glycoprotein gene and promoter (AC:M12379). This expression vector has a 6.8 kbp NotI fragment comprising mouse Thy-1.2 gene (Vidal et al., 1990; Andra et al., 1996) in which the 1.5 kbp BanI-XhoI fragment located on exons 2 and 4 was replaced by the XhoI cloning site.
PS2mut construct. The FAD mutation N141I was introduced into a human PS2 cDNA (AC: L43964). For expression in transgenic mice, a vector based on the mouse prion gene was used (pPrPHG) (Fischer et al., 1996; Borchelt et al., 1996). A KpnI-NarI fragment, encompassing the prion gene coding region, was deleted and replaced by a unique SceI site. The presenilin 2 coding sequence was inserted by blunt-end ligation and verified by sequencing of the insertion sites.
Generation of transgenic mice and controls
B6D2F1 (i.e., C57BL/6J × DBA/2 cross), B6CBAF1, and C57BL/6J mice were obtained from RCC (Füllinsdorf, Switzerland). Transgenic animals were generated essentially as described previously (Hogan and Lacy, 1995). Vector-free linear fragments, carrying Thy1-huAPP751SFAD or Prp-huPS2(N141I) constructs, respectively, were microinjected into male pronuclei of B6D2F1 zygotes. After microinjection, viable zygotes were implanted into the oviducts of pseudopregnant B6CBAF1 foster mothers. Integration of the transgene was detected both by genomic Southern blot and PCR analysis, using DNA isolated from tail biopsies of 2- to 3-week-old mice.
Microinjection of the APPswe construct resulted in the generation of nine positive transgenic founder mice. Transgene founders were crossed with wild-type C57BL/6J mice to establish heterozygous offspring. The heterozygous mouse line 71, which expressed hβAPP751 several fold higher than endogenous (murine) βAPP, as assessed by Western blotting (Malherbe et al., 1997), was intercrossed to generate a homozygous transgenic line.
Transgenic founders for Prp-huPS2(N141I) were crossed with C57BL/6J to establish single transgenic lines. The expression levels of the transgene were monitored by Northern blot analysis of total brain RNA using a probe corresponding to amino acids Leu2 to Pro73 of the human PS2 coding region and by a combination of immunoprecipitation and Western blotting using monoclonal antibody PST-20 (Loetscher et al., 1997). The high expressing line HuPS2.30 was intercrossed to homozygosity and used for the current investigation. Double transgenic offspring were generated by crossing APPswe (line 71) males with PS2mut (line 30) females. A small cohort of heterozygous APPswe (line 71) mice aged 16-18 months were taken for morphometric determinations of cerebral plaque load and compared with age-matched PS2APP mice.
For the cross-sectional study, ∼20 PS2APP mice and an equivalent number of nontransgenic controls (C57BL/6J × DBA/2 F1), abbreviated B6D2F1, were used per age group investigated. Only male mice were included in this study to minimize potential gender differences in behavior and pathology. Consequently we used B6D2F1 controls to enable the efficient supply of age- and sex-matched controls to the PS2APP mice. Although the PS2APP transgenics are on a mixed C57BL/6J and DBA/2 background, unlike the B6D2F1 mice they are not isogenic; however, because there were no apparent group differences on cognitive performance at baseline (i.e., 4 months), and on the basis of published evidence that water maze and avoidance learning seem stable in both C57BL/6 and B6D2F1 lines over age range examined in the present study (Fordyce and Wehner, 1993; Dawson et al., 1999; King and Arendash, 2002; Kelly et al., 2003; this study), we predict that any age-related behavioral changes would be attributable to the transgene rather than any subtle difference in the genetic background that may emerge with age. It should also be noted that each transgenic group was derived from multiple litters, preserving random combinations of background C57BL/6J and DBA/2 genes, which should result in a more accurate assessment of the transgene impact on phenotype (Crawley et al., 1997). At the completion of behavioral testing (duration 1 month), the mice were processed either for subsequent electrophysiological and immunochemical analyses or for histological and electron microscopical analyses.
Detection of Aβ1-40 and Aβ1-42 peptides by ELISA
Brains were removed from fluothane-anesthetized mice and halved. Cortex, hippocampus, and cerebellum were isolated, weighed, and frozen in dry-ice until use. Diethanolamine (DEA)-soluble amyloid was prepared as described previously (Savage et al., 1998). The insoluble amyloid was extracted after further incubation of brain homogenates in 9 m urea buffer. Both types of supernatants (collected after either DEA or urea extraction) were tested in ELISA on plates coated with the 6E10 monoclonal antibody (mAb) (Senetek, Maryland Heights, MO) specific for the 17 amino acid residues of the human Aβ. The specific detection of Aβ1-40 and Aβ1-42 bound to microtiter plates was achieved with the horseradish peroxidase (HRP)-coupled mAbs BAP-24 and BAP-15, respectively (Brockhaus et al., 1998). Results are expressed as nanograms of amyloid peptide per milligram wet tissue referring to an internal standard curve. For the comparison of Aβ1-40 and Aβ1-42 levels in single transgenic hAPPswe and PS2APP mice, we measured insoluble Aβ.
Western blot analysis
Brain tissues from transgenic or nontransgenic littermate control mice were homogenized on ice in 0.32 m sucrose solution containing protease inhibitors (Complete, Roche Applied Science, Mannheim, Germany) using FastPrep homogenization Tubes (Qbiogene, Carlsbad, CA). APP and Aβ detection was done as described previously (Wirths et al., 2001).
Histochemistry and immunohistochemistry
Fluothane-anesthetized mice (n = 3-5 per age group) were fixed by transcardiac perfusion of 4% formaldehyde in PBS at 22°C after which the brains were removed, halved in a sagittal plane, and immersed in the same fixative for 24 hr. One brain half was then cryoprotected overnight in 30% sucrose before freezing in dry-ice, and the other half was dehydrated and embedded in paraffin wax. Cryostat or paraffin sections were cut at 12 μm.
Fibrillar Aβ deposits were detected with Accustain amyloid stain, Congo red (Sigma Diagnostics HT60, Sigma, St. Louis, MO). A modified cupric-silver technique of De Olmos (Carlsen and De Olmos, 1981) was used to reveal dystrophic neurites.
The following antibodies were used to detect antigenic sites with a Vectastain ABC Elite kit and peroxidase (ImmunoPure metal-enhanced DAB substrate; Pierce 34065): BAP-2 (monoclonal mouse IgG1 raised against Aβ1-40; recognizes amino acids 4-7) (Dr. M. Brockhaus); CD45 (protein-tyrosine phosphatase in activated microglia; clone IBL-3/16; Serotec MCA 1388); glial fibrillary acidic protein [(GFAP) in activated astroglia; clone G-A-5; BioMakor 6077]; apoE antibody to the C-terminus region of apolipoprotein E (apoE; Chemicon AB 947); AT-8 (specific for the phosphorylated Ser-202 residue of tau protein, clone AT-8; Innogenetics BR-03); phospho-p38 [p38 MAP kinase (Thr180/Tyr182), Cell Signaling 9211L]; interleukin (IL)-6 (clone MP5-20F3, Pharmingen International 554400, San Diego, CA); tumor necrosis factor (TNF)-α (clone MP6-XT22, Pharmingen International 20231C).
For controls, we used a peptide block or relevant isotype for polyclonal and monoclonal antibodies, respectively. Digital imaging and processing of stained sections were performed with a ProgRes 3012 high-resolution scanner camera (Kontron, Zeiss) and Adobe Photoshop software, respectively.
Plaque load measurements in BAP-2-immunostained brain sections from hAPPswe and PS2APP mice
BAP-2-immunostained brain sections were prepared as described above. An image of the top half of each section (to include the whole of the neocortex plus hippocampal formation) was taken using a ProgRes 3012 camera and Wild M420 macroscope using the highest camera resolution (4608 × 3480). The images were stored in uncompressed TIFF format, 24 bit color. An image of an area of a standard (unstained) slide was taken as well for the area calibration of the MCID M2 image analysis software (Imaging Research, St. Catherines, Ontario, Canada). After the area calibration, a brain image was imported. Image processing filters were used to distinguish the plaques from the other tissue structures. Three filters were applied sequentially: target accent (increasing contrast), binarization (black and white conversion), and median (removing noise). The few remaining erratically positively identified targets were removed manually before quantification (using minimum-maximum size and form factor criteria).
Peptide and receptor binding assays in vitro
Parasagittal cryostat sections of fresh-frozen brains (n = 3-5 per age group) were mounted on Superfrost Plus slides and stored at -20°C. The following radioligands were used: 125I-Aβ1-40 Amersham (for seeds of fibrillar Aβ) (Maggio et al., 1992) and 1-(2-chlorophenyl)-N-methyl-N-(1-methyl-propyl)-3-isoquinoline carboxamide (3H-PK11195) New England Nuclear (for peripheral benzodiazepine binding sites in activated microglia) (Benavides et al., 1990).
The binding assays were performed as described previously. Dried sections were exposed, together with calibration standards (either 14C or 3H microscales), to tritium-sensitive imaging plates (BAS-TR2025) for up to 8 d and subsequently to Hyperfilm Tritium (Amersham) for up to 4 weeks at 4°C. The plates were scanned in a Fujifilm BAS-5000 high-resolution phosphor imager and measured with an MCID M2 image analysis system. Sections radiolabeled with 125I-Aβ1-40 were postfixed in 2% glutaraldehyde in PBS, dipped in liquid emulsion (K5, Ilford), and finally counterstained with Cresyl violet for microscopic radioautography.
Immunoelectron microscopy
Tissue preparation. Perfusion-fixed brain tissue, while immersed in fixative, was cut into ∼100-μm-thick sections using a vibratome and cryoprotected by immersion in increasing concentrations of glycerol (10-20-30% v/v) in 10 mm phosphate buffer, pH 7.4, for 0.5 hr per concentration. Vibratome sections were carefully picked up by forceps and plunged into liquid ethane cooled by liquid nitrogen. Frozen tissue slices were stored in liquid nitrogen before being transferred to the precooled chamber (-90°C) of an automated freeze-substitution apparatus (Reichert AFS; Leica, Wetzlar, Germany). The tissue was immersed overnight at -90°C in anhydrous methanol containing 0.5% (w/v) uranyl acetate. The temperature was allowed to rise to -45°C, at a rate of 6.7°C per hour. Samples were washed three times with anhydrous methanol for 30 min each to remove residual water and excess uranyl acetate before infiltration with Lowicryl HM20 resin (Chemische Werke Lowi; Waldkraiburg, Germany). Infiltration with resin was done in mixtures of HM20 and methanol at volumes of 1:1 and 2:1, each for 2 hr, and pure HM20 for 2, 16, and 2 hr. Samples were processed in the flow-through system of the Reichert AFS apparatus and polymerized by indirect UV irradiation (360 nm) for 24 hr at -45°C, followed by UV irradiation for 1 d at room temperature to achieve complete resin polymerization.
Immunolabeling of thin resin sections. Ultrathin sections were prepared on a Reichert Ultracut S using a diamond knife (Diatome) and collected onto formvar/carbon-coated 200-mesh nickel grids. The sections were floated on 0.05 mol/l glycine in PBS for 15 min to inactivate free aldehyde groups and then on 2.5% (w/v) hen egg white albumin (Fluka) with 2.5% (w/v) bovine serum albumin (BSA) (fraction V; Roche Applied Science, Mannheim, Germany) in PBS for 15 min to block nonspecific binding sites. Sections were incubated with 10 μg/ml mAb BAP-2 in 2% BSA/PBS for 1 hr. After six washes in BSA/PBS, the sections were incubated with a secondary goat anti-mouse IgG (Amersham, Arlington Heights, IL), conjugated to 10 nm gold at 1:20 dilution in 2% BSA with 0.1% Tween 20 in PBS for 1 hr, and washed in BSA/PBS. For controls, we used sections treated with normal mouse serum, which resulted in a negligible background of not more than 10 gold particles in an area of 10 μm2. The sections were postfixed in 2% glutaaldehyde in PBS for 5 min, washed, and stained with 4% aqueous uranyl acetate for 10 min, followed by lead citrate for 90 sec. Electron micrographs were taken with a JEOL 1210 at 100 kV.
Electrophysiology
Mouse hippocampal slices (400 μm) were cut with a Sorvall tissue chopper. Slices were perfused at 35°C with a simple salt solution containing (in mm): 124 NaCl, 2.5 KCl, 2 MgSO4, 2.5 CaCl2, 1.25 KH2PO4, 26 NaHCO3, 10 glucose, 4 sucrose, gassed with 95% O2/5% CO2, pH 7.4. Depending on the experimental protocol, either the CA1 stratum radiatum or the perforant path input to the mid-molecular layer of the dentate gyrus was stimulated (0.033 Hz, 100 μsec) at a strength adjusted to evoke field EPSPs (fEPSPs) equal to 30 or 40% of the relative maximum amplitudes without superimposed population spike, respectively. Field EPSPs were recorded with a glass micropipette (1-3 MΩ) containing 2 m NaCl from either the CA1 stratum radiatum or the mid-molecular layer of the dentate gyrus and were averaged over a 2 min period (each data point is a mean of four stimuli). After stable baseline recordings were attained, input-output relationships and paired-pulse parameters were assessed. Input-output parameters were assayed by measuring evoked fEPSP slopes in response to stepwise increases in stimulus intensity. For paired stimulation (50, 100, or 200 msec interstimulus interval), data are expressed as a ratio of amplitudes fEPSP2/fEPSP1 for each stimulus pair. For long-term potentiation (LTP) assays, fEPSP amplitudes are expressed as the percentage of mean stable baseline values recorded for 10 min before the start of the experiment. LTP was induced in the CA1 using a theta burst stimulation paradigm consisting of two stimulus patterns spaced by 8 sec. Each pattern consisted of 10, 50 msec stimulus trains at 100 Hz, each separated by 150 msec. The duration of the stimulation pulses was doubled during the tetanus. In the dentate gyrus, LTP was elicited by eight 50 msec stimulus trains at 200 Hz, each separated by 2 sec. Picrotoxin (100 μm) was included in the perfusion solution for all dentate gyrus LTP experiments. Results are expressed as means ± SEM of the field EPSP slope as percentage of the baseline values recorded 10 min before tetanus.
Behavioral analyses
Four cohorts of male, age-matched mice were studied (n = 15-20 mice per group). Age groups selected for study were 4, 8, 12, and 16 months ± 2 weeks at the start of testing. An identical test sequence was used for each age group, i.e., neurological test, Y-maze test, locomotor activity test, water maze test, active avoidance test, light-dark test. Tests of prepulse inhibition and 24 hr locomotor activity (over a 12 hr light/dark period) were also conducted in the 16 month age group. Testing took 4 weeks per group. All mice were individually housed and familiarized to the holding room conditions for at least 2 months before commencement of testing. Food and water were available ad libitum, and all testing was conducted during the animals' light cycle phase (12 hr light/dark cycle; lights on 6 A.M. to 6 P.M.). All behavioral experiments complied with Swiss Federal and Cantonal laws on animal research. For each test, the experimenter was blind to genotype but not age.
Tests of neurological function and locomotor activity. The mice were examined initially in a battery of tests that checked body temperature, overt behavioral signs (coat appearance, body posture, secretory signs, flexion, and righting reflex), wire maneuver, grip strength, 1 m swim test, and rotarod. Secretory signs, i.e., salivation and lacrymation, were assessed by visual observation. Further details of each test are described in Higgins et al. (2001) and in the legend to Table 1. At the completion of these neurological tests, the mice were placed in a novel test chamber (Plexiglas box; dimensions: 20 × 20 × 27 cm; Omnitech Electronics, Columbus, OH) containing sawdust bedding, and activity was recorded over a 1 hr period.
Table 1.
Summary of studies examining PS2APP mice and age-matched controls in tests of neurological function
|
Age in months |
|
Body weight |
Body temperature |
Shock threshold |
Locomotor activity |
Grip strength |
|
One meter swim |
Rotarod |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
Flinch
|
Jump
|
LMA
|
Rears
|
Wire test
|
16 rpm
|
32 rpm
|
||||||||
| 4 | WT | 30.8 ± 0.5 | 38.5 ± 0.1 | ND | ND | 4950 ± 257 | 3020 ± 277 | 117 ± 4 | 26 (9-31) | 2.8 ± 0.1 | 120 (100-120) | 25 (20-66) | |||
| PS2APP | 27.9 ± 0.6* | 38.5 ± 0.1 | ND | ND | 7311 ± 584* | 2598 ± 211 | 73 ± 4* | 11 (6-17)* | 3.5 ± 0.2* | 120 (93-120) | 38 (26-57) | ||||
| 8 | WT | 40.3 ± 0.2 | 38.4 ± 0.1 | 0.13 ± 0.01 | 0.44 ± 0.03 | 3752 ± 367 | 1093 ± 157 | 121 ± 3 | 24 (7-41) | 2.7 ± 0.1 | 65 (45-104) | 21 (14-25) | |||
| PS2APP | 37.6 ± 0.8 | 38.4 ± 0.1 | 0.18 ± 0.01* | 0.61 ± 0.02* | 5495 ± 488* | 806 ± 77 | 73 ± 6* | 22 (12-37) | 3.7 ± 0.1* | 38 (28-71) | 15 (11-21) | ||||
| 12 | WT | 44.5 ± 1.3 | 38.6 ± 0.2 | 0.13 ± 0.01 | 0.60 ± 0.06 | 3005 ± 316 | 1971 ± 213 | 122 ± 4 | 10 (4-15) | 3.1 ± 0.1 | 35 (21-92) | 19 (14-27) | |||
| PS2APP | 42.9 ± 2.0 | 38.3 ± 0.2 | 0.19 ± 0.01* | 0.70 ± 0.06* | 4169 ± 343* | 1815 ± 145 | 107 ± 7 | 10 (4-18) | 3.9 ± 0.1* | 78 (16-104) | 11 (8-17) | ||||
| 16 | WT | 49.0 ± 1.9 | 38.0 ± 0.2 | 0.13 ± 0.01 | 0.38 ± 0.04 | 2542 ± 188 | 2246 ± 525 | 118 ± 6 | 9 (7-14) | 3.7 ± 0.2 | 20 (12-43) | 10 (7-13) | |||
|
|
PS2APP
|
36.9 ± 1.9*
|
37.7 ± 0.1
|
0.18 ± 0.04*
|
0.52 ± 0.03*
|
4395 ± 476*
|
2544 ± 422
|
89 ± 3*
|
3 (2-5)*
|
4.2 ± 0.2*
|
73 (68-81)*
|
31 (17-54)*
|
|||
n = 13-18 per group. Locomotor activity (LMA)/rearing: Mice were placed in a novel activity chamber for 60 min. Wire maneuver: Mice were placed by forepaws on an elevated wire rod and latency to fall is noted. Cut-off time was 60 sec; best score from three attempts is recorded. Grip strength: Mice were forced to pull on a strain gauge; release point is recorded. Best score from five attempts is shown. One meter swim test: Mice were placed in a straight swim tank (1 m long × 6 cm wide). Latency to swim distance and climb onto platform is noted. Mice were given three trials × 3 d; fastest time is recorded. Rotarod: Mice were placed on a constant speed rotorod; latency to fall noted. Cut-off time was 120 sec. Best score from three trials is taken. Two speeds were used: 16 rev/min and 32 rev/min. In addition, rectal body temperature, coat appearance, secretory signs, and body posture were also noted. Data are expressed as means ± SEM or median and interquartile range (wire and rotorod test).
p < 0.05 versus wild-type (WT) mice at equivalent age.
Y-maze. The dimensions of the Y-maze arms were 0.66 × 0.3 × 0.12 m (length × width × height). Mice were placed singly into one of the arms, and the arm entry sequence was recorded during a 5 min test period. The dependant measures were total arm entries (entry being classified as entire body within that arm) and the percentage of alternations (Anderson and Higgins, 1997).
Morris water maze. The water maze consisted of a gray circular tank (1 m diameter) filled with water made opaque by the addition of a white dye (E-308; Induchem, Voletswil, Switzerland). Pool temperature was 21 ± 1°C. For the hidden platform task, the escape platform (8 cm diameter) was positioned 1 cm below water level in the center of one of the pool quadrants (target quadrant). For the cued task, platform position was signaled by the addition of a small black flag positioned in the center of the submerged platform. The walls surrounding the water maze were hung with posters and flags that served as visual cues and were visible during all stages of training and testing. Movement of the mice within the pool was tracked and analyzed with a computerized tracking system (HVS Image, Hampton, UK).
For initial cued training, 12 PS2APP mice and 12 nontransgenic controls were placed in the pool facing the edge at one of four start positions (NE, SE, SW, NW) and required to locate a flagged platform, the position of which varied across trials. Each mouse received a total of 12 trials (3 trials per block, 2 blocks per day, 2 d duration). The intertrial interval averaged 10 min, and maximum trial length was 60 sec. If a mouse failed to find the platform within 60 sec, it was guided to its position by the experimenter. The cued task was followed by the place task, in which the mice were required to locate a submerged hidden platform, the position of which remained fixed throughout training. Platform location was balanced within groups. Each mouse received eight blocks of training trials over 4 consecutive days (three trials per block, timing as per cued test) in which they were placed in the pool at one of four start positions and allowed to locate the hidden platform. Assessment of spatial learning was conducted in probe trials performed both 30 min after block 4, and 1 hr and 5 d after the final trial. In each probe trial the platform was removed from the pool, and the path that each mouse swam was recorded over a 60 sec period.
Active avoidance. Mice were placed into a two-compartment chamber within which they could freely pass between compartments (San Diego Instruments). Each trial began with the side currently occupied by the mouse being illuminated by a 10 sec light [conditioned stimulus (CS)] that was used to signal a foot shock (0.2 mA) with a maximum duration of 20 sec that was initiated by offset of the CS. It is important to note that mice never received this shock duration because they either escaped within 1 sec to the other (unshocked) compartment or learned to avoid the shock altogether. A variable timeout period (mean 20 sec, range 15-25 sec) (no light) in which the mouse could freely explore the chamber was introduced between each trial. After each timeout, the next trial began. Shock could be avoided either by a shuttle to the next compartment during the CS period, (i.e., avoidance) or escape at any time during the shock presentation. Ten consecutive daily test sessions were run with each session consisting of 20 trials. The dependant measure was percentage avoidance, although avoidance and escape latencies were also noted.
During the course of testing the 8, 12, and 16 month age groups, it emerged that the PS2APP mice showed signs of impairment in avoidance learning. Accordingly, shock threshold experiments were conducted on mice from the same cohort but not used in the avoidance test, i.e., they were naive to shock. Briefly, the threshold shock required to elicit a flinch, jump, and vocalization response was noted. Foot shock was applied for 1 sec in 0.05 mA steps from a starting level of 0.05 mA. A 30 sec interval was applied between successive stimuli.
Light/dark and prepulse inhibition tests. The methods and apparatus used to study these behaviors in the wild-type and transgenic groups were as described in Kew et al. (2000).
Results
ELISA determination of Aβ levels
Levels of DEA-soluble and insoluble Aβ40 and Aβ42 were measured in the brains of PS2APP mice and from age-matched nontransgenic controls (B6D2F1). No significant level of amyloid peptides was detected in control mice tested from 5 to 17 months of age (data not shown). In PS2APP mice, Aβ peptides were found in the cortex and in the hippocampus from the age of 5 months (earliest time point tested in this study) but were never detected in the cerebellum even at the age of 17 months (data not shown). A comparison of 16- to 18-month-old PS2APP mice and age-matched APPswe single transgenics revealed considerably greater insoluble Aβ40 and Aβ42 in the PS2APP cohort.
In PS2APP mice, increased levels of DEA-soluble Aβ40 were detected only after the age of 9 months. Mean values of 0.6 ng/mg in the cortex and 21 ng/mg in the hippocampus, and of 1.5 ng/mg in the cortex and 43 ng/mg in the hippocampus were measured at 13 and 17 months, respectively. Levels of DEA-soluble Aβ42 increased steadily up to 0.6 ng/mg in the cortex and to 5 ng/mg in the hippocampus as measured at 17 months.
Amyloid deposition in the brain was quantified by the levels of DEA-insoluble but urea-soluble amyloid. The same kinetics of deposition was observed in the cortex and hippocampus. DEA-insoluble amyloid was present in PS2APP mice already at the age of 5 months (Fig. 1a), with mean values of ∼1 ng/mg tissue for both Aβ40 and Aβ42. Aβ40 deposits increased 6- to 12-fold at the age of 9 months and up to 60- to 100-fold at the age of 13 months when they reached a plateau. In comparison, the level of Aβ42 deposition increased 20- to 40-fold between the age of 5 and 17 months. When the load of amyloid deposition was analyzed in individual mice at a given time point, a large variability was observed. At the age of 9 months, only five of the nine mice had increased Aβ40 deposition when compared with 5-month-old mice. At the age of 13 months, levels of amyloid deposition were increased in the cortex of all the mice tested, but these levels varied >100-fold for Aβ40 and >10-fold for Aβ42. This variation was less pronounced at the age of 17 months, with approximately a maximum of 10-fold difference in the Aβ load (Aβ40 and Aβ42). These results indicate that the rate of progression of amyloid deposition varies considerably between individual animals.
Figure 1.

a, Measurement of human Aβ1-40 (□) and Aβ1-42 (▪) in cortex and hippocampus of PS2APP mice at ages 5, 9, 13, and 17 months. Soluble (top bar graphs) and insoluble (bottom bar graphs) peptides obtained after DEA and urea extractions, respectively, were quantified by ELISA. The mean and SEM values of 6-10 mice are represented. b, Western blot analysis of APP, 12 kDa β-cleaved fragment of APP (C-99), and Aβ (Abeta) in brain homogenate of individual PS2APP mice at ages 4, 6, 9, 11, 14, and 16 months and a 16 month nontransgenic control. The data show an age-related accumulation of Aβ, with some inter-animal variability; e.g., compare both lanes at age 14 months.
Western blot analysis of transgene expression and processing
Brain homogenates of PS2APP mice at different ages were analyzed by Western blot for APP and Aβ expression using the monoclonal antibody WO-2 (Fig. 1b). This antibody readily detects human APP and Aβ. Expression levels of full-length human APP levels showed a certain variability in individual transgenic mice; this variability was also reflected in the quantity of the 12 kDa APP C-terminal fragment (C-99) that is generated. There was a strong accumulation of Aβ in PS2APP mice starting as early as the age of 5 months, parallel to the appearance of insoluble Aβ deposits (see above). The strong increase of Aβ detected by Western blot reflects Aβ accumulated in amyloid plaques.
Histopathological analyses
Detection of Aβ
Various methods (histochemical, immunohistochemical, and in vitro binding) were used to detect deposits of Aβ in mouse brain sections. The nonsaturable binding of monomeric 125I-Aβ to templates of Aβ fibrils has been reported to occur from a concentration as low as 10-11m (Maggio et al., 1992), thereby attesting to the sensitivity of the assay. Although rare deposits were detected in brains of 5-month-old PS2APP mice, there was a temporal development thereafter, beginning at 9 months with their appearance in discrete brain regions, namely the subiculum and frontolateral (motor and orbital) cortices (Fig. 2a-f). The number and size of these deposits increased with age to include, by 13 and 17 months, not only most of the neocortex, hippocampal formation (particularly the subiculum and dentate gyrus), and amygdala, but also thalamic and pontine nuclei. Other brain regions (striatum, hypothalamus, pons, medulla, cerebellum), spinal cord, pituitary, and brain vasculature were consistently devoid of plaques. No binding was observed in tissue sections of nontransgenic controls at all ages investigated (Fig. 2a).
Figure 2.

Fibrillar Aβ deposits, i.e., diffuse and mature plaques, revealed by in vitro binding of monomeric 125I-Aβ1-40 to parasagittal brain sections followed by film radioautography. a, Nontransgenic control; 5 (b)-, 9 (c)-, 13 (d)-, and 17 (e)-month-old transgenic mice, respectively. Note the absence of deposits, i.e., binding, in the control, rare deposits in the subiculum (arrowhead) and frontolateral cortex (arrow) of a 5-month-old mouse, and their gradual, age-related increase in number and distribution at subsequent time points. f, Emulsion radioautography of a radiolabeled brain section of a 17-month-old mouse revealing the cellular distribution of Aβ deposits (black silver grains) in the hippocampal formation. Note, inter alia, the markedly reduced number of granule cells (arrowhead) in the vicinity of one cluster of silver grains, i.e., plaque. CA1, Ammon's horn layer 1; cb, cerebellum; CPu, caudate putamen; gran, dentate gyrus granule cell layer; flc, frontolateral cortex; s, subiculum; t, thalamus. Scale bars: a-e, 1 mm; f, 200 μm.
Congo red histochemistry and BAP-2 immunohistochemistry confirmed these findings. Congo red-stained Aβ deposits (Fig. 3a,b) were birefringent when observed by polarization microscopy (Fig. 3c). The distribution and abundance of Congo red-stained deposits corresponded with that of BAP-2-immunoreactive plaques (Fig. 3d). Moreover, the modified cupric-silver technique of De Olmos (Carlsen and De Olmos, 1981) revealed the presence of plaque-associated dystrophic neurites (degenerating axons and dendrites) in the affected brain regions (Fig. 3e). Among the plaque-associated antigenic sites detected were apoE and phospho-tau (Fig. 3f,g); however, appropriate controls indicated false-positive immune reactions for phospho-p38, IL6, and TNFα.
Figure 3.
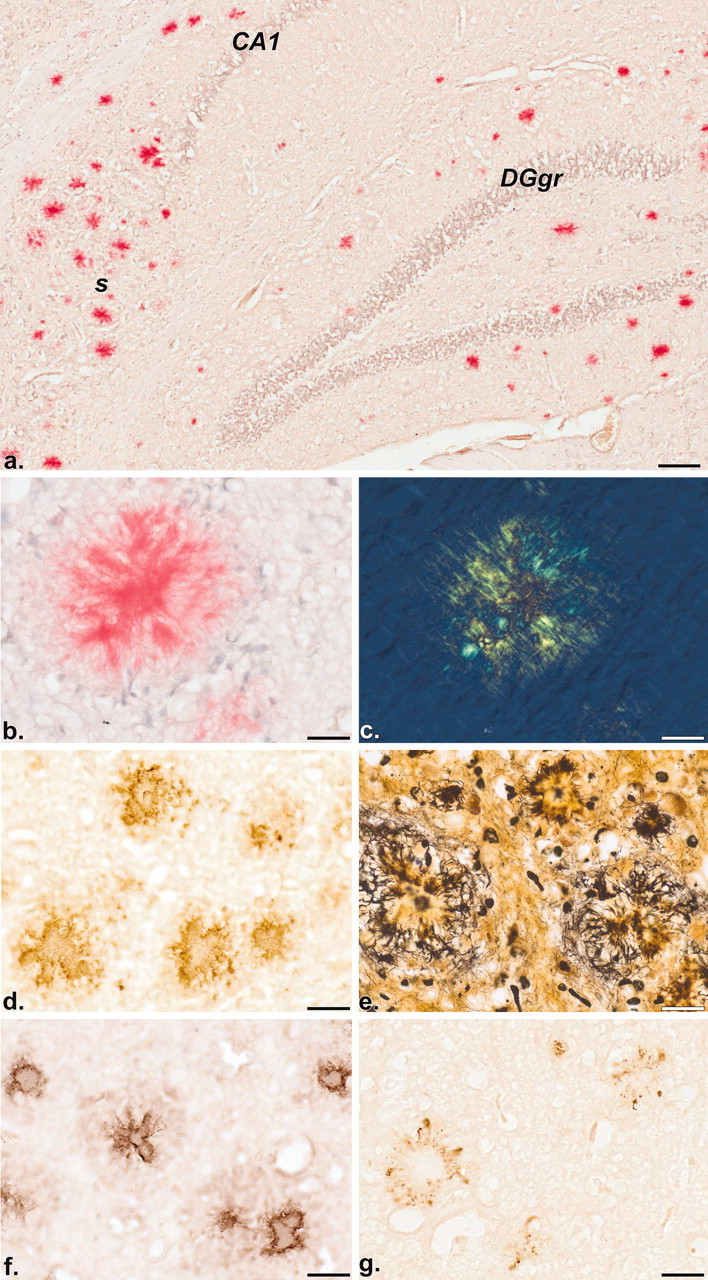
Distribution of Aβ deposits revealed by histochemistry (a-c; Congo red with hemalum counterstain) and immunohistochemistry (d; BAP-2 antibody). Note the discrete congophilic staining of Aβ deposits in the hippocampal formation of a 17-month-old mouse (bright-field image a). The birefringent nature of a congophilic deposit (b) is demonstrated by polarization microscopy (c). Immunoreactive (BAP-2 positive) deposits in the neocortex of a 9-month-old mouse are illustrated in d. Plaque-associated dystrophic neurites are visualized by a modified cupric-silver staining procedure of de Olmos (Carlsen and De Olmos, 1981) (e). Immunoreactivities in f and g reveal the co-distribution of apoE and phospo-tau, respectively, with plaques. CA1, Ammon's horn layer 1; DGgr, dentate gyrus granule cell layer;. s, subiculum. Scale bars: a, 100 μm; b-g, 25 μm.
Finally, we compared the amyloid burden (defined as percentage area occupied by BAP-2 immunoreactivity in neocortex plus hippocampus) in a separate cohort of 17-18 month APPswe and PS2APP mice. Mean plaque burden was 5.5 ± 1.5% in the PS2APP line compared with 0.5 ± 0.2% in the APPswe single transgenic line, although considerable interanimal variability was evident.
Detection of activated microglia and astroglia
Inflammatory response markers for microglioses and astroglioses, namely the protein-tyrosine phosphatase CD45 and GFAP, respectively, revealed a co-distribution with deposits of Aβ (Fig. 4). This was demonstrated most clearly in transgenic mice of 9 and 13 months of age in which both plaques and inflammatory responses were invariably restricted to the subiculum (Fig. 4a-c) and frontolateral cortex (Fig. 4d-f) before spreading to other areas.
Figure 4.

Immunohistochemical localization of the inflammatory response markers CD45 (a-c) and GFAP (d-f) in the hippocampal formation and frontolateral cortex, respectively, of a nontransgenic control (a, d) and a 13-month-old transgenic mouse (b, c, e, f). Note the absence of CD45 and GFAP immunoreactivities, i.e., microglioses and astroglioses, respectively, in the control but their discrete localization in the transgenic brain. CA1, Ammon's horn layer 1; cc, corpus callosum; Ctx, neocortex; DGgr, dentate gyrus granular layer; s, subiculum. Scale bars: a, b, d, e, 200 μm; c, f, 50 μm.
In vitro binding of 3H-PK11195
PK11195, the high-affinity ligand for peripheral benzodiazepine binding sites in mitochondria of reactive microglia, showed a distinctly patchy appearance in transgenic mice when compared with nontransgenic controls (Fig. 5a,b). The distribution of patches corresponded with that of amyloid deposits and CD45 immunoreactivity. Quantitative radioautography (Fig. 5c) revealed a significantly increased number and binding intensity of patches in the subiculum of PS2APP mice relative to age-matched controls.
Figure 5.

Distribution of in vitro binding sites for 3H-PK11195 in the brain of a 17-month-old nontransgenic control (a) and a transgenic mouse (b) revealed by film radioautography. Note the additional patchy distribution of binding sites in b, particularly in the neocortex and hippocampal formation (subiculum). In the histogram (c), quantitative receptor radioautography reveals increased binding in the subiculum of 13- and 17-month-old mice. Similar trends were also observed in other brain regions (frontolateral cortex, lacunosum moleculare, and dentate gyrus). cb, Cerebellum; CPu, caudate putamen; gran, dentate gyrus granule cell layer; flc, frontolateral cortex; s, subiculum; t, thalamus. Scale bar: a, b, 1 mm.
Ultrastructural localization of Aβ
Immunoelectron microscopy (EM) was performed on low temperature-embedded mouse brain tissue. The method combines the advantage of superior preservation of ultrastructure and antigenicity for efficient immunocytochemical staining.
Immunogold labeling, using a monoclonal antibody against the N-terminal portion of the Aβ-peptide (BAP-2), revealed almost exclusively fibrillar Aβ, either as dense-core/mature (Fig. 6a) or diffuse plaques, confirming that most of the Aβ is present in its polymerized fibrillar state (Fig. 6b). Dystrophic neurites were found at the periphery of both types of plaques. Also, microglia were frequently observed in close contact with fibrillar plaques, forming an interdigitating complex of fibrils and the microglial cytoplasm.
Figure 6.

On-section immunogold labeling after cryofixation using an anti-Aβ monoclonal antibody (BAP-2) visualized by a goat anti-mouse secondary antibody conjugated to 10 nm colloidal gold. Overviews and details (a-d) of electron micrographs from sections through 5 (c)- or 9 (a, b, d)-month-old transgenic mouse brains in which Aβ molecules have been labeled with anti-Aβ antibodies (BAP-2) bound to tiny colloidal gold spheres (each seen as a black dot). Immunoreactivity reveals the localization of Aβ. P, Plaque core; AF, fibrils of Aβ; M, microglia; DN, dystrophic neurites; NF, nonfibrillar Aβ. Scale bars: a, 10 μm; b, 200 nm; c, 250 nm; d, 500 nm.
Aβ fibrils were also detected intracellularly in cells, showing a dense cytoplasm, that resemble degenerating neurites (Fig. 6c). An intriguing observation was that occasionally fibrils appeared to penetrate through the cell membrane, which suggests a direct interaction of Aβ fibrils with the cell surface.
In some of the investigated cells, immunoreactivity on nonfibrillar deposits in electron-dense organelles of neurons was visible (Fig. 6d). The detection of intracellular nonfibrillar, i.e., amorphous Aβ, might indicate an early event in the intracellular deposition of Aβ. Another explanation is that the N-terminal-specific anti-Aβ (BAP-2) antibody used also recognizes APP that is extensively expressed and catabolized in the neurons of transgenic mice.
Electrophysiology
Hippocampal synaptic transmission and plasticity were assayed in control and PS2APP mice in four parallel cohorts at 5, 9, 13, and 17 months of age (Figs. 7, 8) using both the Schaffer collateral-CA1 pathway and medial-perforant path input to the dentate gyrus. Input-output parameters were assayed by plotting evoked field EPSP slope in response to stepwise increases in stimulus intensity. Short- and long-term plasticity were assayed by a paired-pulse paradigm with paired-pulse intervals of 50, 100, and 200 msec and by tetanus-induced long-term potentiation, respectively.
Figure 7.

Cross-sectional study of Schaffer collateral-CA1 pathway synaptic transmission and plasticity in control (○,□) and PS2APP (•, ▪) mice. Experiments were performed using parallel cohorts of mice at 5, 9, 13, and 17 months of age. For each cohort, input-output parameters (left inset), paired-pulse plasticity (right inset), and LTP (*p < 0.05; ANOVA; 4-20 min after tetanus) were assayed (see Materials and Methods).
Figure 8.

Cross-sectional study of dentate gyrus medial-perforant pathway synaptic transmission and plasticity in control (○, □) and PS2APP (•, ▪) mice. Experiments were performed using parallel cohorts of mice at 5, 9, 13, and 17 months of age. For each cohort, input-output parameters (left inset), paired-pulse plasticity (right inset) (*p < 0.01; t test), and LTP (*p < 0.05; ANOVA; 4-20 min after tetanus) were assayed (see Materials and Methods).
At the Schaffer collateral-CA1 pathway (Fig. 7), no significant differences in the input-output relationship or relative paired-pulse facilitation were observed across all of the age groups assayed. A significant attenuation of the immediate post-tetanic potentiation (PTP) was observed in transgenic mice in the 9 month age group (ANOVA; p < 0.05; 4-20 min after tetanus), although there was no significant difference in LTP at 70-90 min after tetanus (ANOVA; p = 0.20; NS); however, there was no significant difference in either PTP or LTP elicited in the 13 or 17 month age groups. A general decline in the efficacy of LTP with age was observed in both control and transgenic animals, such that the relative potentiation at 90 min after tetanus fell from 172 ± 6 and 156 ± 12% at 5 months to 127 ± 6 and 120 ± 13% at 17 months, respectively.
At the dentate gyrus medial-perforant pathway (Fig. 8), there were no significant differences in the input-output relationships between control and transgenic animals across all the age groups assayed, although from the 5-9 month time point onward transgenic animals exhibited a trend toward an enhanced output relative to controls. There were no significant differences observed in relative paired-pulse depression or LTP in the 5, 9, and 13 month age groups. At 17 months, however, transgenic animals exhibited significantly enhanced paired-pulse depression at the 50 and 100 msec interpulse intervals (p < 0.01; t test) and reduced PTP (ANOVA; p < 0.05, 4-20 min after tetanus; p = 0.07, 70-90 min after tetanus). As in the CA1, an age-related reduction in LTP was observed in both control and transgenic animals such that the relative potentiation at 90 min after tetanus fell from 163 ± 8 and 173 ± 8% at 3 months to 148 ± 8 and 121 ± 14% at 17 months, respectively.
Behavior
Four cohorts of mice were evaluated at 4, 8, 12, and 16 months of age, and independent groups were used at each time point. All testing was conducted by the same experimenter (A.O.) in an attempt to retain consistency of measurement between cohorts.
Age 4 months
The general appearance and all reflexes of the PS2APP mice were similar to controls, except that body weight was ∼10% less in the transgenic group. Some subtle differences were noted in the neurological tests, with the transgenic mice showing higher locomotor activity in a 60 min test, reduced grip strength, and slower swim speed in a 1 m swim test (Table 1). Water maze performance in both the cued and place learning tasks was similar between the PS2APP mice and controls (Fig. 9, Table 2). For example, no main effect of genotype was recorded in either task (cued: F(1,22) = 1.7, NS; place: F(1,22) = 1.9, NS). Active avoidance tests also identified no main effect of genotype on percentage avoidance (F(1,11) = 2.9; NS) or genotype × days interaction (F(9,117) = 1.6; NS). Escape latencies were similar between groups (F(1,11) = 0.4; NS) (Table 3). Y-maze and light/dark test data are summarized in Table 3. The only changes noted in these tests were reduced total Y-maze arm entries and percentage alternation in the PS2APP mice compared with controls.
Figure 9.
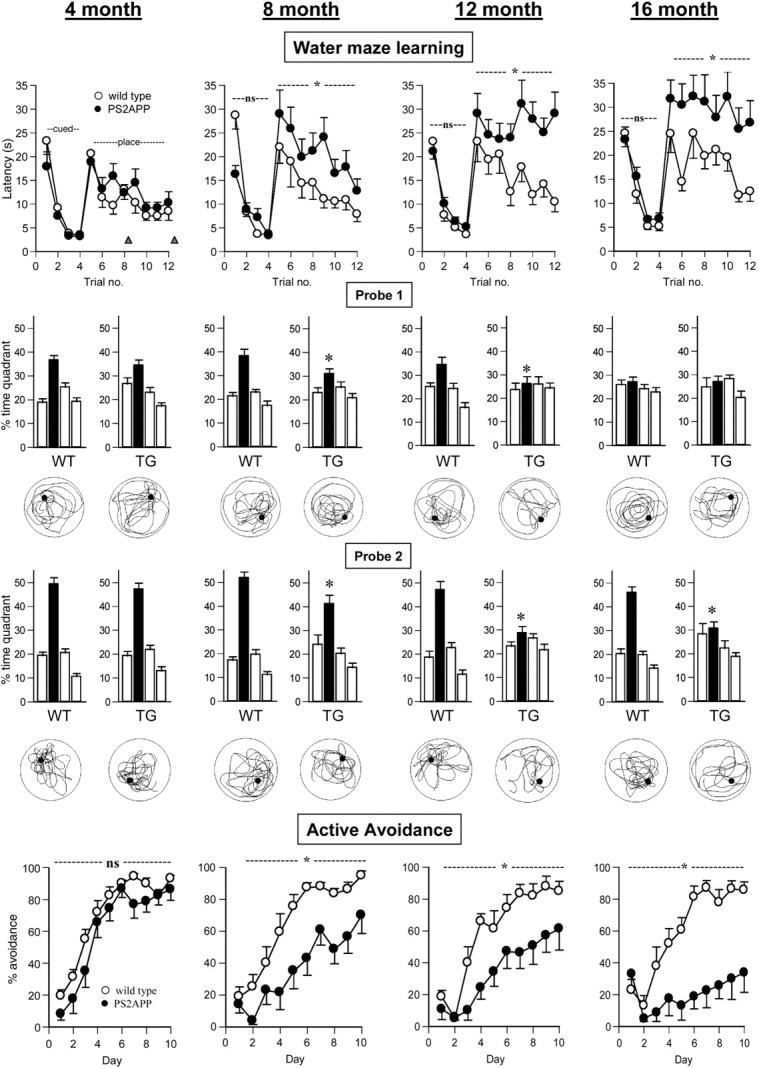
Cross-sectional study of the performance of control (WT; ○) and PS2APP (TG; •) mice at age 4, 8, 12, and 16 months. Independent groups were used at each time point. n = 12 per genotype (water maze study); n = 7-8 per genotype (active avoidance study). At each time point the data are presented in an identical manner to aid comparison across ages. Water maze learning: Latency to locate the escape platform (in the target quadrant) during the cued test phase (trial blocks 1-4, platform visible, variable location) and place test phase (trial blocks 5-12, platform hidden, fixed location). The timing of probe tests 1 and 2 is indicated by the triangles. ns, No significant difference; *p < 0.05 between groups over days (ANOVA). Probe 1: Probe test 1 conducted after fourth place learning trial. Quadrant preference expressed as percentage time per quadrant over a 60 sec test in which the platform was removed from the pool. For each group the four histobars represent adjacent left, target quadrant (filled bar), adjacent right, and opposite quadrant. The swim plot for the median performing animal (factor percentage time in target quadrant) in each group is shown below the relevant histobar. *p < 0.05 versus control target quadrant preference. Probe 2: Probe test 2 conducted after the completion of place learning. Note for the sake of space that probe 3 data are presented in Table 3. Active avoidance: Active avoidance learning as represented by percentage avoidance over 10 consecutive training days.
Table 2.
Summary of performance measures from the probe tests conducted at various phases during the water maze experiment
|
|
|
Probe 1 (0.5 hr post-trial block 4) |
Probe 2 (1 hr post-trial block 8) |
Probe 3 (5 d post-training) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age in months
|
|
Swim speed (cm/sec)
|
%TQ (time)
|
Platform site crossing (n)
|
Swim speed (cm/sec)
|
%TQ (time)
|
Platform site crossing (n)
|
Swim speed (cm/sec)
|
%TQ (time)
|
Platform site crossing (n)
|
||||||
| 4 | WT | 24.1 ± 0.5 | 37 ± 2 | 9 ± 1 | 22.7 ± 0.5 | 49 ± 2 | 11 ± 1 | 25.2 ± 0.9 | 40 ± 3 | 10 ± 1 | ||||||
| PS2APP | 22.3 ± 0.4* | 34 ± 2 | 8 ± 1 | 21.3 ± 0.6 | 47 ± 2 | 10 ± 1 | 23.5 ± 0.5 | 38 ± 3 | 9 ± 1 | |||||||
| 8 | WT | 22.2 ± 0.7 | 38 ± 3 | 9 ± 1 | 21.1 ± 0.6 | 52 ± 3 | 12 ± 1 | 23.6 ± 0.5 | 38 ± 3 | 9 ± 1 | ||||||
| PS2APP | 21.1 ± 0.5 | 31 ± 2 | 6 ± 1** | 19.8 ± 0.7 | 41 ± 4* | 7 ± 1** | 21.3 ± 0.8* | 30 ± 2 | 5 ± 1** | |||||||
| 12 | WT | 21.2 ± 0.4 | 34 ± 3 | 8 ± 1 | 20.1 ± 0.3 | 47 ± 4 | 9 ± 1 | 22.1 ± 0.2 | 39 ± 2 | 8 ± 1 | ||||||
| PS2APP | 17.6 ± 0.4** | 26 ± 3* | 3 ± 1** | 17.1 ± 0.7** | 29 ± 3** | 5 ± 1** | 19.5 ± 0.6** | 26 ± 2** | 5 ± 1** | |||||||
| 16 | WT | 20.3 ± 1.0 | 27 ± 2 | 6 ± 1 | 19.5 ± 0.6 | 46 ± 3 | 10 ± 1 | 21.1 ± 0.7 | 38 ± 3 | 8 ± 1 | ||||||
|
|
PS2APP
|
14.5 ± 1.5**
|
27 ± 3
|
4 ± 1*
|
14.7 ± 1.5**
|
31 ± 3**
|
5 ± 1**
|
16.7 ± 1.9*
|
26 ± 3**
|
4 ± 1**
|
||||||
Data are expressed as means ± SEM. Note that swim speed is not adjusted for floating rate, however, because the genotype-induced difference in 1 m swim speed did not vary with age (Table 1). The reduced swim speed evident in the PS2APP mice at the 12 and 16 month time points is essentially attributable to increased time floating (i.e., swim speed <5 cm/sec). For instance, in the 16 month age group, percentage of swim speed time was <5 cm/sec during the 60 sec probe test: Probe 1: WT, 2 ± 1%; TG, 15 ± 7%; Probe 2: WT,2 ± 1%; TG, 13 ± 6%; Probe 3:WT, 1 ± 1%; TG, 13 ± 8%; *p < 0.05; **p < 0.01 at each probe test. WT, Wild type, % TQ, percentage of time spent in target quadrant (quadrant containing an escape platform).
Table 3.
Summary of behavioral test data
|
Age in months |
|
Y-maze test |
Active avoidance test escape latency (sec) |
Light/dark test |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
Arm entry (n)
|
Alternations (%)
|
Day 1
|
Day 5
|
Day 10
|
Transitions (n)
|
Time in light (sec)
|
|||||
| 4 | WT | 18 ± 1 | 70 ± 4 | 0.5 ± 0.1 | 0.7 ± 0.2 | 0.4 ± 0.1 | 6 ± 1 | 46 ± 10 | ||||
| PS2APP | 13 ± 1* | 52 ± 7* | 0.8 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 | 4 ± 1 | 56 ± 21 | |||||
| 8 | WT | 18 ± 1 | 70 ± 5 | 0.4 ± 0.1 | 0.3 ± 0.1 | 0.4 ± 0.1 | 6 ± 1 | 49 ± 9 | ||||
| PS2APP | 16 ± 2 | 61 ± 3 | 1.0 ± 0.1 | 0.7 ± 0.1 | 0.9 ± 0.2 | 9 ± 1 | 60 ± 7 | |||||
| 12 | WT | 17 ± 2 | 64 ± 4 | 0.6 ± 0.1 | 0.6 ± 0.2 | 0.4 ± 0.1 | 8 ± 1 | 75 ± 8 | ||||
| PS2APP | 20 ± 2 | 63 ± 3 | 1.4 ± 0.4 | 0.7 ± 0.2 | 1.4 ± 0.6 | 8 ± 1 | 83 ± 11 | |||||
| 16 | WT | 13 ± 1 | 71 ± 4 | 0.8 ± 0.1 | 0.4 ± 0.1 | 1.9 ± 1.0 | 9 ± 1 | 81 ± 16 | ||||
|
|
PS2APP
|
19 ± 1*
|
66 ± 3
|
1.3 ± 0.4
|
1.3 ± 0.4
|
1.4 ± 0.6
|
9 ± 1
|
90 ± 11
|
||||
Summary data taken from the Y-maze, active avoidance, and light/dark tests. All data are expressed as means ± SEM; *p < 0.05 versus age-matched controls. Escape latency data from days 1, 5, and 10 are included as a performance measure from the active avoidance test. In all groups and all time points the level of escape failures was <5%. WT, Wild type.
Age 8 months
In the cued phase of the Morris water maze test, the factor genotype was of borderline significance (F(1,22) = 4.3; p = 0.05), reflecting slower swim latencies of wild-type compared with transgenic mice on trial block 1. In the place test phase, a main effect of genotype (F(1,22) = 6.5; p < 0.05) but not genotype × trial block interaction (F(7,154) = 0.3; NS) reflected the fact that although both groups showed a trial dependent decrease in latency, the transgenic group was slower overall. Analysis of probe test data revealed target quadrant preferences in both groups, but the preference score in the transgenic group was significantly less than that of the control group (Fig. 9, Table 2). In the active avoidance test, a main effect of genotype was found on percentage avoidance (F(1,13) = 16.1; p < 0.01), although no genotype × days interaction was recorded (F(9,117) = 1.6; NS) (Fig. 9). These data reflected slower acquisition of the avoidance behavior in the transgenic group. There was no significant main effect on escape latency (F(1,13) = 4.9; NS), although there was a trend for the transgenic group to be slower on this measure (Table 3). A shock threshold experiment on mice not previously exposed to shock revealed the PS2APP mice to have a lower reactivity to shock, indicated by higher threshold to elicit flinch and vocalization response (Table 1). Behavior in the Y-maze and light/dark tests were similar between groups (Table 3). In the neurological tests, findings essentially similar to those of the 4 month group were obtained (Table 1).
Age 12 months
No group differences were found on cued learning in Morris water maze (F(1,22) = 0.4; NS), yet on place learning a significant main effect of genotype was revealed (F(1,22) = 17.6; p < 0.01), although no genotype × trial block interaction (F(7,154) = 1.4; NS). On probe testing, a genotype × target quadrant interaction emerged (probe 1: F(3,66) = 2.4, p = 0.07; probe 2: F(3,66) = 9.2, p < 0.01; probe 3: F(3,66) = 27.9, p < 0.01) that reflected the fact that the transgenic group failed to develop any quadrant preference compared with controls, which had a significant target quadrant preference at each time point (Fig. 9). These differences were also reflected by other performance changes, such as reduced platform site crossings and increased float time (i.e., swim speed <5 cm/sec) in the transgenic group that accounted for the reduced overall swim speed (Table 2). In the active avoidance test, a main effect of genotype was recorded on percentage avoidance (F(1,11) = 5.9; p < 0.05) but not a genotype × days interaction (F(9,99) = 1.6; NS), suggesting that with extended training the transgenic group would reach the final avoidance level of controls. Escape latency did not significantly differ (F(1,11) = 1.7; NS), although again a trend toward slower scores in the transgenic group was apparent (Fig. 9, Table 3). In a shock threshold test, significant group differences were seen on flinch but not threshold to vocalization (Table 1). Y-maze and light/dark test behavior were similar between genotypes (Table 3). Neurological examination revealed qualitatively similar differences between the transgenic and control group compared with earlier time points (Table 1).
Age 16 months
Similar to other age groups, neurological testing showed that the PS2APP mice exhibited higher locomotor activity, lower body weight, reduced grip strength, and slower swim speed compared with controls. Of note, body weight at this time point was reduced by ∼24% in the transgenic group, being lower than that recorded in the 12 month age group; however, reflexes and general appearance were similar between groups (Table 1). In the water maze test, cued learning was similar (F(1,22) = 1.0; NS), yet again a significant main effect of genotype on place learning was recorded (F(1,22) = 8.4; p < 0.01) (Fig. 9). Only on probe 2 was a significant genotype × target quadrant interaction evident (F(3,63) = 4.2; p < 0.01). On probe test 1 and 3, neither group demonstrated a preference for the island quadrant, thus perhaps reflecting an emergence of an age-related cognitive decline in the control group, because at the earlier ages tested each control group had significant quadrant preferences at each probe test (Fig. 10). Other measures from the water maze test further emphasized a cognitive deficit in the transgenic group; thus platform site crossings were reduced and percentage float time increased in these animals compared with age-matched controls (Table 2). Active avoidance testing again showed a main effect of genotype on percentage avoidance (F(1,13) = 21.4; p < 0.01) and a genotype × days interaction (F(9,117) = 8.4; p < 0.01), reflecting a severe learning impairment in the PS2APP mice (Fig. 9). Escape latencies were similar between groups (F(1,13) = 1.8; NS) (Table 3). A shock threshold test again revealed that higher thresholds were required to elicit flinch and vocalization in the transgenic group (Table 1). Performances in the light/dark and Y-maze tests were essentially similar between the groups, although the PS2APP mice made more total arm entries (Table 3). Tests of paired-pulse inhibition (PPI) and 24 hr circadian activity failed to reveal any group differences at this time point (data not shown).
Figure 10.

Mean probe score (MPS) from probe tests 1-3 for each group as a function of age (Westerman et al., 2002). *p < 0.05 versus wild-type group of equivalent age; +p < 0.05 versus equivalent genotype at age 4 months. Controls (□; PS2APP (▪).
Age-related assessment of cognitive function
To investigate the cognitive changes in each group across age, we determined a mean probe score (MPS) for each animal, according to the methods of Westerman et al. (2002). Thus the mean percentage time for each animal in the target quadrant of the water maze over the three probe trials was determined. Because we conducted these probe tests during the acquisition phase (probe 1, fourth trial block), immediately after training (probe 2, eighth trial block), and during the retention phase (probe 3, 5 d after eighth trial block), the MPS is reflective of no single cognitive domain but rather gives an overall estimate of cognitive performance in the water maze test. Two-way ANOVA with age and genotype as between-subject factors revealed significant main effects of genotype (F(1,21) = 53.5; p < 0.01), age (F(3,63) = 6.7; p < 0.01), and genotype × age interaction (F(3,63) = 2.9; p < 0.05). Post hoc tests confirmed that at the 4 month time point there was no effect of genotype; however, at all later time points the PS2APP mice had significantly poorer performance. Furthermore, an age-dependent cognitive decline was apparent in both groups, although this failed to reach significance in the wild-type group (Fig. 10).
Discussion
Behavioral phenotype of PS2APP mice
An overall assessment of the behavioral phenotype of PS2APP mice supports the conclusion that they demonstrate an age-related cognitive impairment from 8 months. This is seen most clearly in the water maze test. The spatial learning phase was preceded by a series of cued learning trials to identify any potential sensorimotor or motivational phenotypic differences (Crawley, 1999). At each time point, the cued learning performance of the PS2APP mice was similar to controls. In contrast, from the 8 month time point, the PS2APP mice showed a significant impairment in the acquisition of spatial learning, demonstrated by slower acquisition latencies, reduced quadrant preference during probe tests, reduced platform site crossings, and increased float time. Each behavior has been reported in mice after hippocampal or entorhinal cortex-perforant path lesions (Hardman et al., 1997; Logue et al., 1997; Cho et al., 1999; Mohajeri et al., 2003). Further evidence for cognitive impairment in the PS2APP mice was apparent in the active avoidance task. Despite evidence for a heightened threshold to foot shock, the PS2APP mice had similar response latencies to controls, suggesting a similar motivation to avoid the foot shock, yet from the 8 month time point showed poorer acquisition of avoidance learning.
In addition to the active avoidance and water maze tests, spontaneous Y-maze alternation was also included as part of the cognitive test battery; however, the outcome from these studies was essentially the reverse of that seen in the other cognitive tests: namely, the PS2APP mice showed the characteristic cognitive impairment (reduced percentage alternation) only at the 4 month time point. Thus, not all tests yielded similar findings, which may reflect the fact that each taxes different cognitive domains (or other aspects of behavior) or simply reflect a general (in)sensitivity of that test.
Because mid- to late-stage AD is also accompanied by other behavioral disturbances such as agitation, psychosis, and disturbed circadian rhythm (Berg and Morris, 1994), we attempted to examine these behaviors using the light/dark (anxiety test), PPI (psychosis liability), and 24 hr activity tests, respectively, in the oldest cohort. At the 16 month time point, there was no evidence for any such disturbance, the only robust difference being increased overall 24 hr activity in the PS2APP. This result is consistent with the mild hyperactivity recorded in the PS2APP mice at each time point: a feature that seems common to various APP transgenic lines [Huitrón Reséndiz et al., 2002 (PDAPP line); Chapman et al., 1999; King and Arendash, 2002 (Tg2576); Holcomb et al., 1999; Arendash et al., 2001 (PSAPP). Testing at later time points may have revealed more generalized behavioral disturbances.
Hippocampal synaptic transmission and plasticity
A number of previous studies of transgenic mice expressing mutant APP have reported inconsistent changes in hippocampal synaptic transmission and plasticity (Chapman et al., 1999; Hsia et al., 1999; Larson et al., 1999; Moechars et al., 1999; Fitzjohn et al., 2001). In this study, up to the 17 month age group there were no significant differences in input-output parameters and short-term (paired-pulse) plasticity between the groups in both pathways. At 9 months, CA1 PTP was attenuated relative to control; however, this difference was not reproducible with older animals. At 17 months there was a significant attenuation of PTP in the dentate gyrus of transgenic mice relative to controls, with a trend toward attenuation of LTP and also a significantly enhanced paired-pulse depression. In the absence of data from a later time point it was not possible to confirm a sustained or progressive impairment in the transgenic animals; however, it is notable that the onset of this deficit accompanies the elevation of Aβ and the appearance of prominent deposition in the hippocampal formation. In this regard, it would be of interest to assay synaptic parameters in other regions exhibiting pronounced early Aβ deposition, inflammation, and dystrophic neurites such as the subiculum, where any changes might better parallel the onset of cognitive deficits. In both the CA1 and dentate gyrus there were age-related declines in the efficacy of LTP in both control and transgenic animals that interestingly parallel the age-dependent cognitive decline evident in both groups.
Amyloidosis in the PS2APP mouse
The PS2APP mouse develops an age-dependent brain amyloidosis in a region-selective manner as has been observed in other transgenic models that express either human APP alone or in combination with human PS1. The kinetics of amyloid peptide increase as well as the final total amounts in aged PS2APP animals are similar to those described for Tg2576 mice (Kawarabayashi et al., 2001). Insoluble Aβ was the major species, and its deposition was detected in PS2APP transgenics from age 5 months, increasing up to age 17 months with a tendency to plateau. Elevated levels of soluble Aβ were detected only later, starting at 13 months of age and increasing toward the late time point. Increased soluble Aβ was detected predominantly in the hippocampus; the cortical levels remained low throughout the study. In both the soluble and insoluble fractions, Aβ40 levels exceeded Aβ42 by three- to fourfold.
Aβ deposits are an invariant and defining feature of AD pathology; however, there is some controversy as to which form of Aβ is relevant for the pathophysiology of AD. Historically, plaque-associated, insoluble Aβ was the criteria always used for correlation with cognitive impairments and neurodegeneration. More recent studies support the finding that increased soluble Aβ is a major correlate of the degree of severity (Lue et al., 1999; McLean et al., 1999). Similarly, Westerman et al. (2002) explain the inconsistencies in the correlation between plaque load and cognitive deficits in the Tg2576 mouse model of AD, with the idea that small Aβ oligomers might be responsible for neural toxicity or dysfunction leading to memory impairment [see also Van Dam et al. (2003) with respect to the APP23 line]. In our study, deficiencies in cognitive tests became apparent in the 8 months group, which coincides with increases in insoluble Aβ, but occurred before increases in soluble Aβ could be demonstrated; however, direct comparison with the data of Westerman et al. (2002) is difficult because their extraction method for soluble Aβ used 2% SDS, whereas we used a DEA extraction protocol.
Histopathologically, the most significant finding in the current study is that both Aβ deposits and inflammation are initiated, already at 9 months (and even earlier according to our EM study), in well defined and discrete brain regions, namely the subiculum and frontolateral (motor and orbital) cortex. From the evidence presented, using the very sensitive 125I-Aβ binding-aggregation assay, Congo red histochemistry, and BAP-2 immunohistochemistry, it appears that the Aβ plaque deposits in PS2APP mice are of both the mature and diffuse kind.
Dystrophic neurites, mainly dendritic but also axonal profiles, were invariably associated with fibrillar plaques. Plaque-associated phospho-tau immunoreactivity was also detected using the antibody AT-8; however, as in other APP and PS transgenic mice, we failed to detect NFT pathology.
Inflammatory response in PS2APP mice
At the age of 9 months, PS2APP mice show the first signs of an inflammatory response, namely reactive microgliosis and astrogliosis. The distribution and abundance of activated microglia, as revealed by CD45 immunoreactivity and 3H-PK11195 binding, correlate with that of Aβ deposition. CD45, a protein-tyrosine phosphatase, is a cell-surface antigen reported to be induced in AD (Masliah et al., 1991) and in an animal model of neurodegeneration (Karp et al., 1994). It negatively controls microglial activation by various proinflammatory stimuli (Tan et al., 2000). PK11195 has been used to detect and quantify various neuropathological lesions in the CNS (Benavides et al., 1990) and was recently demonstrated in a positron emission tomography study (Cagnin et al., 2001) to be a potential marker for preclinical to early AD (MCI). In that study, 11C-PK11195 binding was reported to increase in brain regions with the highest degree of atrophy (entorhinal, temporal, parietal, and cingulate cortices). Thus, consistent with AD, microglia activation appears to be an early event in the neuropathology evident in the PS2APP mouse. The distribution and abundance of reactive astrocytes, stained with a GFAP antibody, also correlate with the deposition of Aβ, at least at the earlier time points. By 17 months, however, there was a widespread distribution of astrocytes throughout the neocortex and not only spatially associated with plaques.
Summary
In the present study, the functional correlate of the severe cerebral amyloidosis, which develops temporally in the double transgenic mouse line PS2APP, is a marked cognitive deficit. This behavioral change, evaluated by the Morris water maze and active avoidance tests, is initiated at a time when Aβ deposits and accompanying inflammation first appear in essentially two brain regions, namely the subiculum and frontolateral (motor and orbital) cortex. The perforant path-subiculum are the major interfaces between the hippocampal formation and cortex and represent crucial structures for relaying information between these systems. Histopathologically they are also one of the earliest affected regions in AD (Van Hoesen et al., 1991; Gómez Isla et al., 1996; Braak and Braak, 1997). The subsequent age-related increase in severity of cognitive decline in the transgenic mice is paralleled by the spread of Aβ deposition to neo- and limbic cortices, including the hippocampal formation and amygdala, as well as thalamic and pontine nuclei. In common with the published PS1 × APP transgenic lines (Borchelt et al., 1997; Holcomb et al., 1998), the PS2APP line demonstrates an accelerating effect of mutant PS2 on the brain plaque load in the single transgenic APPswe line. Behaviorally, as with the PSAPP mouse, the clearest cognitive impairments in the PS2APP mouse seem to coincide with amyloid plaque deposition (Arendash et al., 2001; Gordon et al., 2001), although a contribution of soluble Aβ to these changes as evidenced in other APP transgenic lines (Westerman et al., 2002; Van Dam et al., 2003) cannot be excluded.
Footnotes
We thank the following colleagues for their excellent technical contributions: Anita Albientz, Francoise Gerber, Fabienne Goepfert, Yolande Lang, Bernard Morand, Marie-Claire Pflimlin, Claudia Richardson, Dieter Reinhard and, in particular, Jürg Messer.
Correspondence should be addressed to Dr. J. Grayson Richards, Pharma Research Biology Discovery, Building 69, Room 252, F. Hoffmann-La Roche Ltd., CH-4070 Basel, Switzerland. E-mail: richards@balcab.ch.
J. G. Richards's present address: Rheintalweg 86A, CH-4125 Riehen BS, Switzerland.
G. A. Higgins's present address: CNS/CV Biol Research, Schering-Plough Research Institute, K-15-2-2600, 2015 Galloping Hill Road, Kenilworth, NJ 07033.
A.-M. Ouagazzal's present address: IGBMC, Mouse Clinical Institute, BP 10142, 1 rue Laurent Fries, 67404 Illkirch, France.
J. N. C. Kew's present address: GlaxoSmithKline, Psychiatry Centre for Excellence, New Frontiers Science Park, Third Avenue, CM19 5AW Harlow, UK.
G. Huber's present address: Fachhochschule Solothurn Nordwestschweiz, Leitung Forschung/Entwicklung und Dienstleistungen, Bittertenstrasse 15, 4702 Oensingen, Switzerland.
J. A. Kemp's present address: Evotec Neurosciences GmbH, Schnackenburgallee 114, D-22525 Hamburg, Germany.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238989-15$15.00/0
J.G.R. and G.A.H. contributed equally to this study.
References
- Anderson R, Higgins GA ( 1997) Absence of central cholinergic deficits in ApoE knockout mice. Psychopharmacology 132: 135-144. [DOI] [PubMed] [Google Scholar]
- Andra K, Abramowski D, Duke M, Probst A, Wiederhold KH, Burki K, Goedert M, Sommer B, Staufenbiel M ( 1996) Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol Aging 17: 183-190. [DOI] [PubMed] [Google Scholar]
- Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM ( 2001) Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res 891: 42-53. [DOI] [PubMed] [Google Scholar]
- Benavides J, Capdeville C, Dauphin F, Dubois A, Duverger D, Fage D, Gotti B, MacKenzie ET, Scatton B ( 1990) The quantification of brain lesions with an omega 3 site ligand: a critical analysis of animal models of cerebral ischaemia and neurodegeneration. Brain Res 522: 275-289. [DOI] [PubMed] [Google Scholar]
- Berg L, Morris JC ( 1994) Diagnosis. In: Alzheimer disease (Terry RD, Katzman R, Bick KL, eds), pp 197-229. New York: Raven.
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL ( 1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal 13: 159-163. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, Van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS ( 1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19: 939-945. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E ( 1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18: 351-357. [DOI] [PubMed] [Google Scholar]
- Brockhaus M, Grünberg J, Röhrig S, Loetscher H, Wittenburg N, Baumeister R, Jacobsen H, Haass C ( 1998) Caspase-mediated cleavage is not required for the activity of presenilins in amyloidogenesis and NOTCH signaling. NeuroReport 9: 1481-1486. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB ( 2001) In-vivo measurement of activated microglia in dementia. Lancet 358: 461-467. [DOI] [PubMed] [Google Scholar]
- Carlsen J, De Olmos JS ( 1981) A modified cupric-silver technique for the impregnation of degenerating neurons and their processes. Brain Res 208: 426-431. [DOI] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao K ( 1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: 271-276. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George Hyslop PS, Westaway D ( 2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276: 21562-21570. [DOI] [PubMed] [Google Scholar]
- Cho YH, Friedman E, Silva AJ ( 1999) Ibotenate lesions of the hippocampus impair spatial learning but not contextual fear conditioning in mice. Behav Brain Res 98: 77-87. [DOI] [PubMed] [Google Scholar]
- Crawley JN ( 1999) Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res 835: 18-26. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Belknap JK, Collins AC, Crabbe JC, Frankel W, Henderson N, Hitzemann RJ, Maxson SC, Miner LL, Silva AJ, Wehner JM, Wynshaw-Boris A, Paylor R ( 1997) Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology 132: 107-124. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van Der Ploeg LHT, Sirinathsinghji DJS ( 1999) Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the β-amyloid precursor protein. Neuroscience 90: 1-13. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S ( 1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383: 710-713. [DOI] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C ( 1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15: 1255-1264. [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR ( 2001) Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci 21: 4691-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce DE, Wehner JM ( 1993) Effects of aging on spatial learning and hippocampal protein kinase C in mice. Neurobiol Aging 14: 309-317. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagoplan S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, et al. ( 1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373: 523-527. [DOI] [PubMed] [Google Scholar]
- Gómez Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT ( 1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16: 4491-4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MN, King DL, Diamond DM, Jantzen PT, Boyett KV, Hope CE, Hatcher JM, DiCarlo G, Gottschall WPE, Morgan D, Arendash GW ( 2001) Correlation between cognitive deficits and Aβ deposits in transgenic APP + PS1 mice. Neurobiol Aging 22: 377-386. [DOI] [PubMed] [Google Scholar]
- Hardman R, Evans DJ, Fellows L, Hayes B, Rupniak HT, Barnes JC, Higgins GA ( 1997) Evidence for recovery of spatial learning following entorhinal cortex lesions in mice. Brain Res 758: 187-200. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Jacobsen H ( 2003) Transgenic mouse models of Alzheimer disease: phenotype and application. Behav Pharmacol 14: 419-438. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Grottick AJ, Ballard TM, Richards JG, Messer J, Takeshima H, Pauly Evers M, Jenck F, Adam G, Wichmann J ( 2001) Influence of the selective ORL1 receptor agonist, Ro64-6198, on rodent neurological function. Neuropharmacology 41: 97-107. [DOI] [PubMed] [Google Scholar]
- Hogan CF, Lacy E ( 1995) Manipulating the mouse embryo: a laboratory manual, Ed 2. Plainview, NY: Cold Spring Harbor Laboratory.
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K ( 1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97-100. [DOI] [PubMed] [Google Scholar]
- Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D ( 1999) Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet 29: 177-185. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L ( 1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96: 3228-3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G ( 1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Huitrón Reséndiz S, Sánchez Alavez M, Gallegos R, Berg G, Crawford E, Giacchino Jeannie L, Games D, Henriksen Steven J, Criado José R ( 2002) Age-independent and age-related deficits in visuospatial learning, sleep-wake states, thermoregulation and motor activity in PDAPP mice. Brain Res 928: 126-137. [DOI] [PubMed] [Google Scholar]
- Janus C, Westaway D ( 2001) Transgenic mouse models of Alzheimer's disease. Physiol Behav 73: 873-886. [DOI] [PubMed] [Google Scholar]
- Karp HL, Tillotson ML, Soria J, Reich C, Wood JG ( 1994) Microglial tyrosine phosphorylation systems in normal and degenerating brain. Glia 11: 284-290. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG ( 2001) Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. Ann Neurol 21: 372-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PH, Bondolfi L, Hunziker D, Schlecht H-P, Carver K, Maguire E, Abramowski D, Weiderhold K-H, Sturchler-Pierrat C, Jucker M, Bergmann R, Staufenbiel M, Sommer B ( 2003) Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Neurobiol Aging 24: 365-378. [DOI] [PubMed] [Google Scholar]
- Kew JNC, Koester A, Moreau JL, Jenck F, Ouagazzal AM, Mutel V, Richards JG, Trube G, Fischer G, Montkowski A, Hundt W, Reinscheid RK, Pauly Evers M, Kemp JA, Bluethmann H ( 2000) Functional consequences of reduction in NMDA receptor glycine affinity in mice carrying targeted point mutations in the glycine binding site. J Neurosci 20: 4037-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DL, Arendash GW ( 2002) Behavioral characterization of the Tg2576 transgenic model of Alzheimer's disease through 19 months. Physiol Behav 75: 627-642. [DOI] [PubMed] [Google Scholar]
- Larson J, Lynch G, Games D, Seubert P ( 1999) Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res 840: 23-35. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van-Slegtenhorst M, Gwinn Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M ( 2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: 402-405. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E ( 2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293: 1487-1491. [DOI] [PubMed] [Google Scholar]
- Loetscher H, Deuschle U, Brockhaus M, Reinhardt D, Nelboeck P, Mous J, Grünberg J, Haass C, Jacobsen H ( 1997) Presenilins are processed by caspase-type proteases. J Biol Chem 272: 20655-20659. [DOI] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM, et al ( 1997) Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci 111: 104-113. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J ( 1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155: 853-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio JE, Stimson ER, Ghilardi JR, Allen CJ, Dahl CE, Whitcomb DC, Vigna SR, Vinters HV, Labenski ME, Mantyh PW ( 1992) Reversible in vitro growth of Alzheimer disease beta-amyloid plaques by deposition of labeled amyloid peptide. Proc Natl Acad Sci USA 89: 5462-5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe P, Richards JG, Bluethmann H, Martin JR, Bleuel Z, Thomas B, Fischer C, Diener C, Huber G ( 1997) Transgenic mice overexpressing three isoforms of human mutant amyloid precursor protein driven by the neuron-specific elements of the thy-1 gene promoter. Soc Neurosci Abstr 23: 1636. [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T ( 1991) Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer's disease. Acta Neuropathol 83: 12-20. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL ( 1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860-866. [DOI] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F ( 1999) Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 274: 6483-6492. [DOI] [PubMed] [Google Scholar]
- Mohajeri MH, Saini K, Li H, Crameri A, Lipp H-P, Wolfer DP, Nitsch RM ( 2003) Intact spatial memory in mice with seizure-induced partial loss of hippocampal pyramidal neurons. Neurobiol Dis 12: 174-181. [DOI] [PubMed] [Google Scholar]
- Savage MJ, Trusko SP, Howland DS, Pinsker LR, Mistretta S, Reaume AG, Greenberg BD, Siman R, Scott RW ( 1998) Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J Neurosci 18: 1743-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ ( 2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741-766. [DOI] [PubMed] [Google Scholar]
- Sisodia S, St. George Hyslop PH ( 2002) γ-Secretase, Notch, Abeta and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci 3: 281-290. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B ( 1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94: 13287-13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Mori T, Wu Y, Saxe M, Crawford F, Mullan M ( 2000) CD45 opposes β-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase. J Neurosci 20: 7587-7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP ( 2003) Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci 17: 388-396. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Hyman BT, Damasio AR ( 1991) Entorhinal cortex pathology in Alzheimer's disease. Hippocampus 1: 1-8. [DOI] [PubMed] [Google Scholar]
- Vidal M, Morris R, Grosveld F, Spanopoulou E ( 1990) Tissue-specific control elements of the Thy-1 gene. EMBO J 9: 833-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman MA, Cooper Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin Linda H, Carlson George A, Younkin Steven G, Ashe KH ( 2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 22: 1858-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Tremp G, Pradier L, Beyreuther K, Bayer TA ( 2001) Reelin in plaques of beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 316: 145-148. [DOI] [PubMed] [Google Scholar]