

Abstract
Mechanisms of oligodendrocyte death after spinal cord injury (SCI) were evaluated by T9 cord level hemisection in wild-type mice (C57BL/6J and Bax+/+ mice), Wlds mice in which severed axons remain viable for 2 weeks, and mice deficient in the proapoptotic protein Bax (Bax-/-). In the lateral white-matter tracts, substantial oligodendrocyte death was evident in the ipsilateral white matter 3-7 mm rostral and caudal to the hemisection site 8 d after injury. Ultrastructural analysis and expression of anti-activated caspase-3 characterized the ongoing oligodendrocyte death at 8 d as primarily apoptotic. Oligodendrocytes were selectively preserved in Wlds mice compared with C57BL/6J mice at 8 d after injury, when severed axons remained viable as verified by antereograde labeling of the lateral vestibular spinal tract. However, 30 d after injury when the severed axons in Wlds animals were already degenerated, the oligodendrocytes preserved at 8 d were lost, and numbers were then equivalent to control C57BL/6J mice.
In contrast, oligodendrocyte death was prevented at both time points in Bax-/- mice. When cultured oligodendrocytes were exposed to staurosporine or cyclosporin A, drugs known to stimulate apoptosis in oligodendrocytes, those from Bax-/- mice but not from Bax+/+ or Bax+/- mice were resistant to the apoptotic death. In contrast, the three groups were equally vulnerable to excitotoxic necrosis death induced by kainate. On the basis of these data, we hypothesize that the Wallerian degeneration of white matter axons that follows SCI removes axonal support and induces apoptotic death in oligodendrocytes by triggering Bax expression.
Keywords: apoptosis, spinal cord hemisection, oligodendrocyte death, Wlds mutation, Bax deficiency, Bax-/-, Wallerian degeneration, immunohistochemistry
Introduction
Spinal cord injury (SCI) produces a secondary protracted wave of oligodendrocyte death in degenerating white-matter tracts distant from the injury site for weeks after the initial event (Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997; Abe et al., 1999; Li et al., 1999; Springer et al., 1999; Casha et al., 2001). When judged primarily by anatomical criteria (for review, see Beattie et al., 2000), the predominant type of oligodendrocyte death appears to be apoptosis.
Genetically altered mice provide a powerful tool for examining the mechanisms of oligodendrocyte death after SCI. Here, we use Wlds (Wallerian degeneration, slow) mice (Perry et al., 1989, 1990, 1991; Brown et al., 1991, 1992; Ludwin and Bisby, 1992; Zhang et al., 1996, 1998; Zhang and Guth, 1997; Gillingwater and Ribchester, 2001) and Bax-deficient (Bax-/-) mice (Knudson et al., 1995; Deckwerth et al., 1996; White et al., 1998; Martin and Liu, 2002) in a lateral cord hemisection model of SCI to test the hypothesis that the protracted wave of apoptotic death of oligodendrocytes may be dependent on axonal degeneration and Bax activation.
Wlds is an autosomal dominant single-gene mutation in the C57BL/6J mouse that associates with a remarkable delay in the onset of Wallerian degeneration in damaged axons. In normal mice, Wallerian degeneration begins 1-2 d after axotomy. In Wlds mice, the distal ends of severed axons survive and function for 2-3 weeks, and the damaged axons continue to generate action potentials and exhibit axonal transport (Perry et al., 1989, 1990, 1991). Because axon-derived factors are thought to be important for oligodendrocyte viability (Barres et al., 1993), we hypothesized that the persistence of functional but severed axons in Wlds mice after hemisection would prevent the delayed death of oligodendrocytes distant from the injury site, but this effect would be transient and eventually the oligodendrocytes would die in association with the expected long-term loss of severed axons in Wlds mice.
Bax is a member of the Bcl-2 family of cell death regulators, and it is implicated as a key promoter of programmed cell death (apoptosis) (Boise et al., 1993; Chittenden et al., 1995; Korsmeyer, 1995; Yang et al., 1995; Chen et al., 1997; Schlesinger et al., 1997; Korsmeyer, 1999). The Bax gene plays an important role in developmental cell death (Chittenden et al., 1995; Mosinger Ogilvie et al., 1998) and in CNS injury (Gillardon et al., 1995; Krajewski et al., 1995; Antonawich et al., 1998; Lou et al., 1998; Bar-Peled et al., 1999). Apoptosis and the neuronal cell loss that occurs during normal nervous system development and in response to trophic factor deprivation is attenuated in Bax-/- mice (Deckwerth et al., 1996; White et al., 1998). However, the role of Bax expression in oligodendrocyte survival and death is unknown.
Here, we investigate the death of oligodendrocytes after spinal cord hemisection in both normal and mutant mice that carry the Wlds mutation, or Bax-deficient mice by comparison of quantitative analysis of numbers of oligodendrocytes and neurons rostral and caudal to the injured hemisection side, compared with the noninjured contralateral side as a function of time after injury (8 vs 30 d survival). In addition to these genetic determinants, we also examined expression of anti-activated caspase-3 and ultrastructural features of the oligodendrocyte death.
Materials and Methods
Experimental animals. For in vivo studies, 75 adult mice 9-14 weeks of age weighing 25-30 gm each were used: 27 C57BL/6J mice from Harlan (Indianapolis, IN), 24 Wlds C57BL/6J mice from Harlan Olac (Bicester, UK), and 14 Bax-/- mice and 10 Bax+/+ mice (provided by S.K.) were purchased from Jackson Laboratory (Bar Harbor, ME). Using a Bax-targeting vector, S.K.'s group substituted PGK-Neo for exons 2-5, deleting BH1, BH2, and the capacity for a functional Bax protein. Two of 89 G418-resistant clones underwent homologous recombination in RW-4 embryonic stem cells. The disrupted Bax allele was ultimately transmitted through the germ line. Heterozygous mice appeared normal, and mating resulted in the expected Mendelian frequency of Bax-/- mice that grew to adulthood and were externally indistinguishable from wild-type mice (Knudson et al., 1995). An additional 16 mouse pups (4 Bax-/-, 5 Bax-/+, 7 Bax+/+) were used as sources of cultured oligodendrocyte.
Surgical procedures. Mice were anesthetized with a ketamine-medetomidine (25 mg per 0.5 mg) mixture of 1.25 ml/kg, and the skin overlying the thoracic cord was shaved and disinfected with benzalkonium chloride. After the thorax was incised along the midline, the T9 vertebrae were laminectomized to expose the spinal cord. With a scalpel, the cord was transected on the right side only (Kalderon and Fuks, 1996). The wound was sutured-closed in layers. During surgery, the rectal temperature was maintained at 37 ± 0.5°C by a thermostatically regulated heating pad (Versa-Therm 2156; Cole-Parmer, Chicago, IL). During recovery, the mice were placed in a temperature- and humidity-controlled chamber (Thermocare, Incline Village, NV). All surgical interventions and animal care were provided in accordance with the Laboratory Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals (National Institutes of Health), and Guidelines and Policies for Rodent Survival Surgery (Washington University School of Medicine Animal Studies Committee). In mice with compromised bladder function (a rare complication), the bladder was expressed manually three times daily until the emptying reflex was established.
Lateral vestibulospinal tract labeling. Deeply anesthetized animals (8 Wlds and 8 C57BL/6J) were placed in a mouse stereotaxic frame (Stoelting, Wood Dale, IL), and the right lateral vestibular nucleus was injected with 1 μl of biotin-dextran-amine (BDA; 10,000 molecular weight) using the following coordinates from bregma: anteroposterior, 6.0 mm; lateral, 1.2 mm; dorsoventral, 2.7 mm. Five days after BDA injection, the right cord was hemisected; the mice were killed 8 d after hemisection. The vestibulospinal projections were visualized immunohistochemically using methods described previously (Herzog and Brosamle, 1997). The animals were perfused with rinse buffer, 0.1 m PBS (0.9% NaCl), containing 100,000 IU/l of heparin and 0.25% NaNO2. They were then perfused with 4% paraformaldehyde in 0.1 m PBS for 5 min. Finally, they were perfused with the same fixative containing 5% sucrose for an additional 5 min. Brains and spinal cords were removed rapidly, postfixed for 2 hr, and kept overnight in 0.1 m PBS containing 30% sucrose. Twenty-five micrometer longitudinal horizontal sections of T6-T12 spinal cord were cut using a cryostat. For diaminobenzidine (DAB) staining, the sections were first washed in 50 mm Tris-buffered saline (plus 1.5 T, pH 8.0) containing 0.5% Triton X-100 (TBST) three times for 30 min each. The sections were incubated overnight at 4°C with avidin-biotin-peroxidase complex (ABC Elite Kit; Vector Laboratories, Burlingame, CA) in TBST buffer, washed three times for 30 min in TBST, and then reacted with 0.5% DAB (Sigma, St. Louis, MO) and 0.01% H2O2 in 0.1 m PB. The reaction was monitored with a microscope and stopped by extensive washing in PBS. Sections were counterstained with cresyl violet.
Immunohistochemical staining. Eight or 30 d after hemisection, 32 animals (10 Wlds and 10 C57BL/6J for the 8 d time point; 6 Wlds and 6 C57BL/6J for the 30 d time point) were deeply anesthetized with sodium pentobarbital (50 mg/kg) and perfused transcardially with heparinized saline, followed by 4% paraformaldehyde in 0.1 m PB, pH 7.4. Spinal cords were removed and stored in the same fixative for 3 hr at 4°C and kept overnight in 30% sucrose in PBS buffer. Spinal cords were embedded in OCT compound (Tissue-Tek, Torrance, CA) and frozen in dry ice; 25 μm sections were taken on a cryostat in the horizontal plane and mounted on gelatin-coated slides. Sections were washed three times with 0.01 m PBS, permeabilized for 10 min using 0.3% Triton X-100 in PBS, and preincubated in 5% normal horse serum. Primary antibodies included caspase-3 (1:200, rabbit polyclonal, PharMingen, San Diego, CA; 1:500, rabbit polyclonal, Serotec, Raleigh, NC), adenomatous polyposis coli CC-1 (APC CC-1) (1:400, mouse monoclonal, Calbiochem, Oncogene Sciences, La Jolla, CA), and neuronal-specific nuclear protein (NeuN) (1:500, mouse monoclonal, Chemicon, Temecula, CA). The antibodies were applied for 24 hr at 4°C, followed by 2 hr incubation in biotinylated anti-mouse or rabbit IgG. Immunoreactivity was visualized by using either an avidin-biotin complex (ABC Elite Kit; Vector Laboratories) and DAB or cyamine dye (Cy3)-conjugated donkey anti-rabbit secondary antiserum (1:400; Jackson ImmunoResearch, West Grove, PA) for immunofluorescence.
Transmission electron microscopy. Eight days after hemisection, three C57BL/6J mice were perfused with 2.5% paraformaldehyde and 2.5% glutaraldehyde in PBS, pH 7.4. The 1 cm span of spinal cord surrounding the hemisection was sectioned with a vibratome (500 μm thickness). Tissue sections were postfixed in 1% osmium tetroxide in phosphate buffer, pH 7.4. After being rinsed in buffer only, the sections were dehydrated in a graded series of ethanol, cleared in propylene oxide, and embedded in Polybed 812 (Polysciences, Warrington, PA). Semithin (1 μm) and ultrathin (75-90 Å) sections were taken on a Leica Ultracut E ultramicrotome (Leica, Deerfield, IL). Ultrathin sections were mounted on 200-mesh grids and stained with 3% uranyl acetate for 20 min and then lead citrate for 5 min. The stained sections were examined and photographed on a Jeol (Peabody, MA) 100C× electron microscope.
Counting cells and fibers. APC CC-1 immunoreactive oligodendrocytes in lateral white matter and NeuN-immunoreactive neurons in gray matter were counted in 25 μm thick horizontal longitudinal sections. Nine sections from dorsal, intermediate, and ventral levels (three sections from each level; 150-200 μm between each level) were chosen from each animal. In each section, counts were obtained from regions that were 2-7 mm rostral and caudal to the center of the transection site (total, 10 mm) (see Fig. 1 A). The 2 mm segments adjacent to the hemisection were excluded because of nonuniformity in cord continuity and the presence of pan-necrosis. Each 1 mm segment was a measuring unit, and two fields per segment were counted under 20× magnification (0.5 mm2 fields were counted in each 1 mm segment). The numbers of APP CC-1 or NeuN in the nine sections were averaged.
Figure 1.
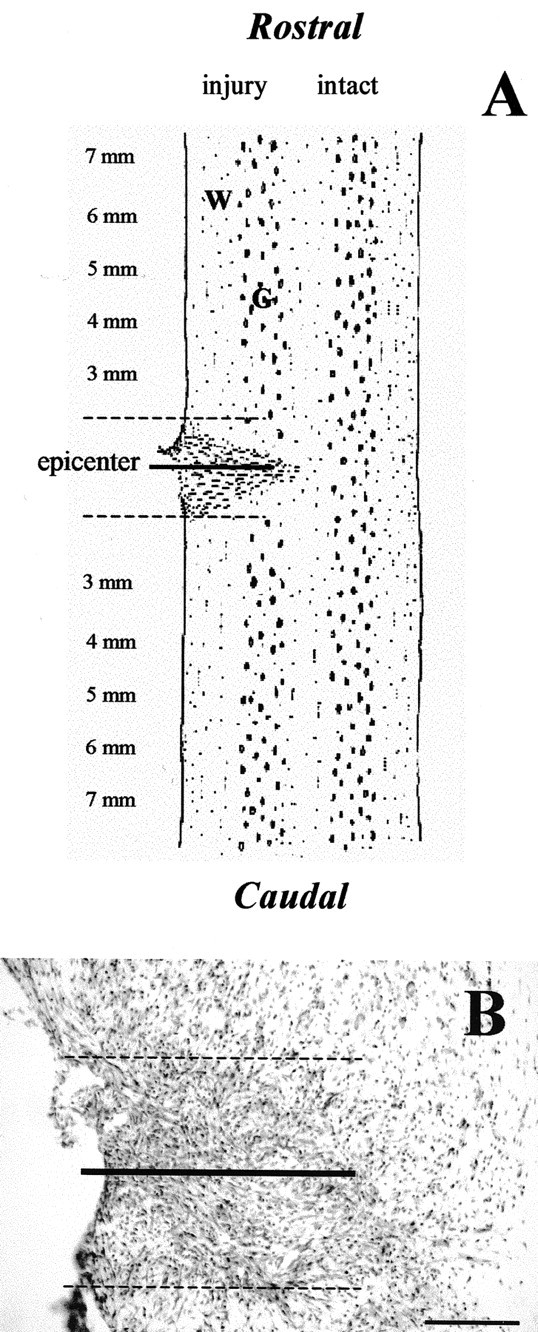
A, Schematic horizontal longitudinal section illustrating the injury resulting from a lateral spinal cord hemisection at the T9 level. All cell counts were measured in the rostral-caudal areas of the lateral white or gray matter outside the immediate pan-necrosis epicenter area surrounding the hemisection. B, Representative Nissl-stained horizontal longitudinal section at the injury epicenter from adult C57BL/6J mice, whose spinal cord was hemisected on the right side 8 d previously. The long line represents hemisection location. Within the dashed lines is the injury epicenter, which represents a distance of ∼1-2 mm on either side of the hemisection site. Analysis was performed on animals with confirmed complete hemisection. Scale bar: (in B) 100 μm.
Lateral vestibulospinal tract (LVST) fibers and related glia were counted in horizontal longitudinal sections labeled with BDA and were stained with Nissl. First, we counted the BDA-labeled cells in the right lateral vestibular nucleus in both the experimental and control animals. For subsequent analyses of the lateral vestibulospinal tract and glia, we selected only those animals that had similar numbers of labeled cells in the lateral vestibular nucleus. From 16 animals, only 8 (4 Wlds and 4 C57BL/6J) qualified for additional investigation. We analyzed three sections from the right lateral vestibulospinal tract from each animal. All BDA-filled fibers and related glia in rostral and caudal regions that were within 5 mm from the injury epicenter were counted under 20× magnification. Two individuals blinded to the group conditions independently replicated all counts, and the data were averaged.
Oligodendrocyte cultures and toxicity experiments. Oligodendrocytes from the meninges-free forebrain of postnatal day (P) 2 mice were plated onto an astrocyte monolayer in a single step, as described previously (McDonald et al., 1996, 1998). Tissue from individual genotyped mice was minced into 1 mm cubes and incubated in 0.25% trypsin in HBSS (Invitrogen, San Diego, CA) for 30 min at 37°C. After the mixture was centrifuged for 5 min at 500 gm, the supernatant was discarded and the pellet was resuspended. Cells were plated into 24-well primaria plates (two hemispheres per plate) with medium containing MEM, Earle's salts, 10% heat-inactivated fetal bovine serum, 10% horse serum, and 15 ng/ml of basic fibroblast growth factor. The latter (15 ng/ml) was included in the oligodendrocyte-plating medium for 7 d in vitro (DIV) to promote cell division and inhibit differentiation of oligodendrocyte progenitor cells (Bogler et al., 1990; McKinnon et al., 1990).
After maintaining the mixed oligodendrocyte-astrocyte cultures for 12-14 DIV, we exposed sister cultures to 10 nm staurosporine, 20 μm cyclosporin A, or 300 nm kainate in medium stock (MEM with added glucose and bicarbonate buffer) for 24 hr at 37°C as described previously (McDonald et al., 1996). Each experiment was repeated three times. Cyclosporin A was a gift from Sandoz (Basel, Switzerland) and was prepared as 30 mm stock in DMSO.
Oligodendrocyte survival or death was assessed by comparing the number of viable galactocerebroside (Gal-C)-immunoreactive cells present after the injury paradigm with the number present in sham-washed sister cultures (controls). Anti-Gal-C immunoreactivity (mouse IgG1, 1:100; Boehringer Mannheim, Indianapolis, IN) was measured as described previously (McDonald et al., 1996). Viable Gal-C+ cells were defined as Gal-C+ cell bodies with two or more processes, the lengths of which equaled or exceeded the diameter of the cell body (McDonald et al., 1998). Ten adjacent fields were counted in each well at 400× magnification. Cell death was confirmed indirectly using dye exclusion (propidium iodide, 5 μg/ml for 10 min).
Bax genotyping (+/+, +/-, -/-) was completed with standard techniques using tail clippings from P1 littermates as described previously (Deckwerth et al., 1996). Briefly, tail DNA was prepared and typed by a single PCR using a set of three primers: Bax exon 5 forward primer (5′-TGATCAGAACCATCATG-3′), Bax intron 5 reverse primer (5′-GTTGACCAGAGTGGCGTAGG-3′), and Neo reverse primer (5′-CCGCTTCCATTGCTCAGCGG-3′). Cycling parameters were 5 min at 94°C for one cycle, and 1 min at 94°C, 1 min at 55°C, and 1 min at 74°C for a total of 30-35 cycles. PCR products were sequenced to ensure correct genotyping of the Bax-deficient animals.
Statistical analysis. All in vivo data were analyzed using ANOVA. The investigated areas (hemisection side: rostral 3-7 mm, caudal 3-7 mm segments; intact side: rostral 3-7 mm, caudal 3-7 mm segments) were used as the between-group factors. All post hoc analyses used the Fishers protected least significant difference (PLSD) test. A result was considered significant when p < 0.05 (two-tailed).
Results
Delayed apoptotic death of oligodendrocytes occurs after hemisection SCI in mice
In Nissl-stained sections, a connective tissue scar was seen around the hemisection site in all groups of mice. In C57BL/6J mice, the lesion was hypervascularized and composed predominately of large macrophages and fibroblasts, as noted in previous studies of rodent SCI (Zhang et al., 1996; Schwab and Bartholdi, 1997) (Fig. 1B).
Previous studies of contusion SCI in rats have detected a protracted wave of oligodendrocyte death occurring primarily by apoptosis (Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997; Emery et al., 1998; Abe et al., 1999; Casha et al., 2001). In the present study, cell death was examined 8 d after hemisection at the T9 level, because that time point coincides with the peak of delayed oligodendrocyte death in the rodent contusion injury models (Liu et al., 1997). The current data on anti-activated caspase-3 immunohistochemistry and ultrastructural assessment suggest that delayed death of oligodendrocytes also occurs in the mouse spinal cord 8 d after hemisection (Fig. 2). Eight days after T9 segment hemisection in C57BL/6J mice, we found that anti-activated caspase-3 staining was expressed in oligodendrocytes that were 3-7 mm rostral and caudal to the hemisection epicenter center but not in those on the intact side of the cord or in control sections without primary antibody (Fig. 2A). From double-labeling APC CC-1 and activated caspase-3, we found that ∼10-15% of APC+ oligodendrocytes express activated caspase-3 at 8 d after injury (Fig. 2B,C). However, expression of activated caspase-3 was not present in APC+ oligodendrocytes from Bax-/- mice at 8 d (Fig. 2D) after injury. Anti-activated caspase-3 expression is an accurate predictor of cell death in this model, because histological examination 30 d after injury detected substantial additional oligodendrocyte cell loss in areas expressing anti-activated caspase-3 (Fig. 3A). In addition, ultrastructural examination demonstrated prominent apoptotic features in white-matter oligodendrocytes that were 5-7 mm from the lesion epicenter (Fig. 2E,F). Dying oligodendrocytes contained highly condensed chromatin masses, intact cell membranes, shrunken cytoplasm, apoptotic bodies, and associated myelin products. However, infiltration of macrophages and neutrophils was not evident in white matter at these distal sites.
Figure 2.
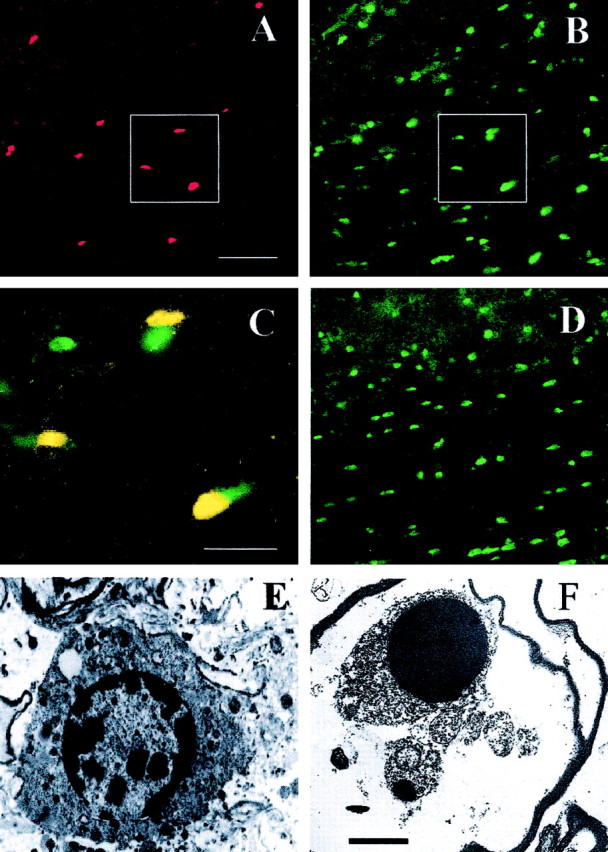
Oligodendrocytes in white matter ipsilateral to hemisection express anti-activated caspase-3 immunoreactivity and ultrastructural features consistent with apoptosis when examined 8 d after hemisection injury. Anti-activated caspase-3 (A, red) and APC (B, green) immunofluorence labeling and double labeling (C, yellow) in a representative longitudinal section from a C57BL/6J mouse are shown. C is the immunofluorescence overlay of insets in A and B. No specific anti-caspase-3 (A, red) immunofluorescence was evident in Bax-/- mice; APC is shown in D (green). All pictures were taken in white matter examined 5-7 mm from the injury epicenter. E-F, In C57BL/6J mice with hemisections, early (E) and late (F) stage ultrastructural features of dying oligodendrocytes are consistent with apoptosis. Nuclei are highly condensed and exhibit adjacent apoptotic bodies (arrow). Scale bars: A-D, 100 μm; E-F, 2 μm.
Figure 3.

Selective but transient preservation of APC+ oligodendrocytes is present on the hemisection side in Wlds mice compared with C57BL/6J mice. In Wlds mice, oligodendrocytes preserved at 8 d after injury are lost by 30 d. A, In C57BL/6J mice, the number of APC+ cells was significantly less on the hemisected side of the cord compared with the contralateral intact cord 8 d after injury (Fig. 5A), but there was a trend for additional loss of APC+ cells from 8-30 d. B, In Wlds mice, the number of APC+ cells on the injury side was primarily normal 8 d after hemisection (Fig. 5B). However, this preservation was transient because APC+ cells were uniformly depleted on the hemisection side by 30 d after injury, a time interval in which the severed axons that survived in Wlds mice during the first 2 weeks would have degenerated by 30 d after injury. C, Comparison of APC+ cells on the side of hemisection 8 d after injury in wild-type C57BL mice and Wlds mice demonstrated selective loss of cells in the wild-type group. D, By 30 d, the number of APC+ cells was similar in C57BL mice and Wlds mice on the injured side, indicating a transient preservation of APC+ cells in Wlds mice. Data represent mean ± SEM (n = 6). *p < 0.05; **p < 0.01; 30 versus 8 d (top); C57BL versus Wlds (bottom); ANOVA with Fishers PLSD post hoc test.
Reduced oligodendrocyte death in Wlds mice
Two discrete regions of cell death were evident in the hemisected cords of mice. The epicenter area (1-2 mm) surrounding the hemisection exhibited signs of pan-necrosis, with loss of both oligodendrocytes (APC+ cells) and neurons (NeuN+ cells) (Bhat et al., 1996; Wolf et al., 1996; McDonald et al., 1999). Similar patterns of pan-necrosis cell death were seen in C57BL/6J mice and Wlds mice at the hemisection level, and we therefore did not quantify and compare within this zone. In contrast, selective loss of oligodendrocytes was observed in a second region that lay 3-7 mm from the hemisection epicenter in C57BL control mice at 8 d. In C57BL/6J mice, APC+ cell counts 8 d after injury were significantly less on the injured side in lateral white-matter columns than on the intact contralateral side for up to 5 mm from the injury epicenter (p < 0.01; two-way ANOVA) (Figs. 4B,5A). The number of NeuN+ cells (neurons) did not decrease in the second area (4-7 mm from the lesion site) in the gray matter of C57BL/6J mice (Fig. 6A).
Figure 4.

Anti-APC immunostaining demonstrates intrafasicular oligodendrocytes in representative horizontal longitudinal sections through spinal cords that were hemisected on the right side 8 d previously (the images are from lateral white matter 4 mm caudal to the hemisection). A, B, White matter contralateral (A) and ipsilateral (B) to the hemisection in a representative C57BL/6J mouse. C, White matter ipsilateral to the hemisection in a Wlds mouse. Arrows indicate small train-like clusters of intrafasicular, APC CC-1+ oligodendrocytes in white matter. Note that the number of APC+ cells in the hemisected side is markedly less in the wild-type C57BL mice compared with Wlds mice. Scale bar, 50 μm.
Figure 5.

A, In C57B/6J mice, the number of APC+ cells in lateral white matter rostral and caudal to the hemisection was significantly less than in the corresponding intact contralateral white matter 8 d after hemisection injury. B, In Wlds mice, there were similar numbers of APC+ cells contralateral and ipsilateral to the hemisection, except for a trend for lower numbers at the injury epicenter (1-2 mm). C, APC+ oligodendrocytes on the side of hemisection were selectively preserved in Wlds mice compared with C57BL/6J mice. D, The two groups had similar numbers of cells on the intact side. Data represent mean ± SEM (n = 6). *p < 0.05; **p < 0.01; injured side versus noninjured side (top); C57BL versus Wlds (bottom); two-way ANOVA with Fishers PLSD post hoc test.
Figure 6.

Within- and between-group comparisons showed that the numbers of NeuN+ neurons in gray matter of C57BL/6J and Wlds mice were decreased only in the 1-3 mm area surrounding the hemisection epicenter. Data represent mean ± SEM (n = 6). *p < 0.05; **p < 0.01; injured side versus noninjured side (top); C57BL versus Wlds (bottom); ANOVA with Fishers PLSD post hoc test.
In contrast, little loss of APC+ oligodendrocytes was evident in the white-matter zone distal to the hemisection in Wlds mice, and numbers of APC+ oligodendrocytes were similar on the hemisected and intact contralateral sides of the cord (p > 0.05) (Figs. 4C,5B). Intergroup comparisons demonstrated a substantial preservation of APC+ cells on the injured side when Wlds mice were compared with C57BL/6J mice (p < 0.05) (Fig. 5C). The two groups did not differ significantly in the number of APC+ cells in the intact side of the cord (Fig. 5D). We confirmed that loss of APC staining represents oligodendrocyte cell death, rather than mere downregulation of APC expression which has been noted previously after optic nerve transection (Friedman et al., 1989; Daston and Ratner, 1994), by analyzing two other oligodendrocyte markers, anti-receptor-interacting protein (RIP) and anti-myelin basic protein (MBP). Qualitative results similar to those found with APC immunolabeling were obtained with both additional markers of oligodendrocytes.
Preservation of severed axons accompanies reduced oligodendrocyte death in Wlds mice
To confirm that the damaged axons of Wlds mice do indeed survive for 8 d after hemisection, we submitted a subset of C57BL/6J mice and Wlds mice to antereograde BDA labeling of the LVST 5 d before the SCI. Preliminary work in control mice showed that this 5 d interval is sufficient for labeling the entire length of the LVST, and that the label persists for at least 8 d after hemisection. In C57BL/6J mice and Wlds mice, we observed persistent and complete labeling of fibers in the lateral columns of white matter in the intact side of the cord 13 d after labeling (8 d after hemisection). On the hemisected side, the number of labeled caudal fibers was markedly reduced in C57BL/6J mice but remained normal in Wlds mice (p < 0.05; two-way ANOVA; each group, n = 4, C57BL/6J mice vs Wlds mice) (Fig. 7B,C). The persistence of distal segments of severed LVST fibers in Wlds mice is consistent with previous work demonstrating that distal cut axons persist for >14 d in mutant Wlds mice (Perry et al., 1990, 1991; Ludwin et al., 1992). The preservation of LVST fibers caudal to the hemisection site in Wlds mice compared with C57BL/6J mice paralleled the preservation of APC+ and Nissl-stained glia cells in white matter surrounding the LVST fibers (Fig. 7C).
Figure 7.
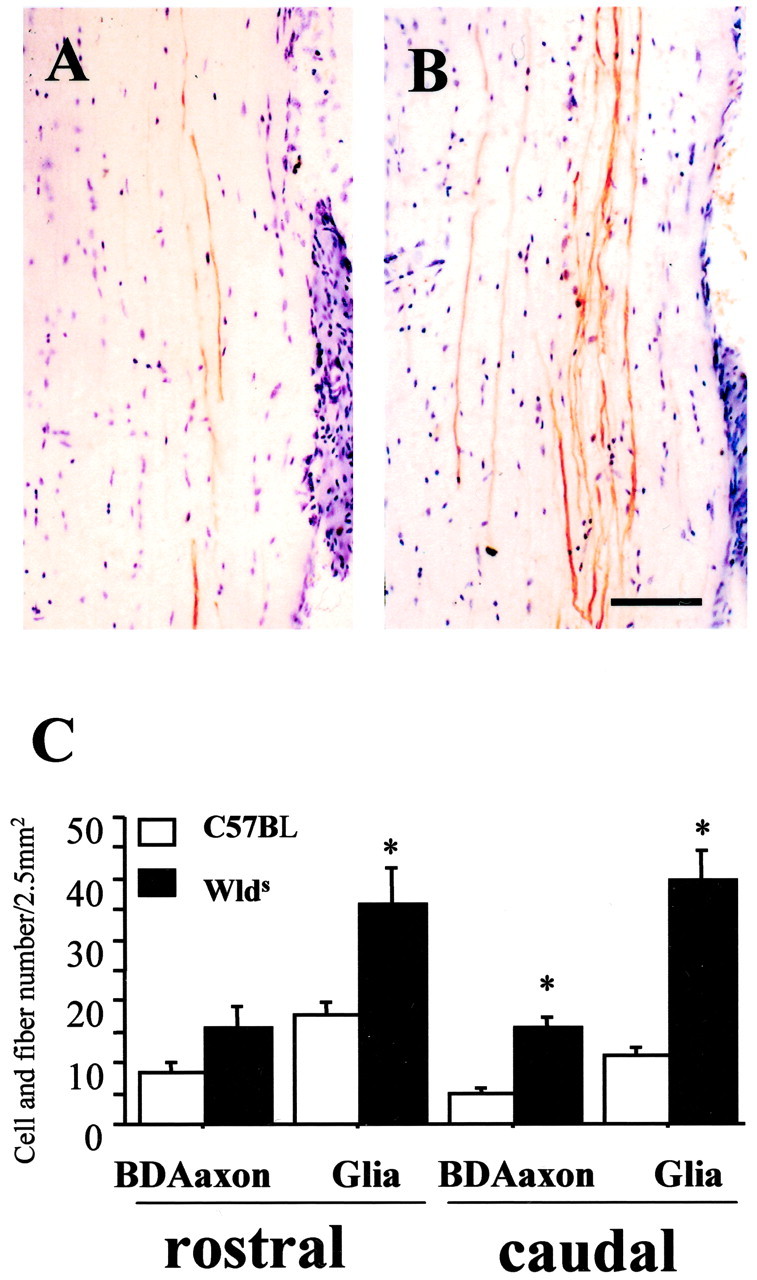
Representative horizontal longitudinal sections illustrating antereograde BDA-labeled lateral vestibular spinal tract axons in lateral white matter 2-5 mm caudal to an ipsilateral hemisection (8 d earlier) in C57BL/6J (A) and Wlds (B) mice. The mean number of BDA-labeled fibers in the intact side of the cord was 19.5 ± 3 per section. There were consistently fewer BDA-labeled fibers on the side distal to the injury in C57BL/6J mice (mean, 5 ± 1.5; A, C) than in similarly treated Wlds mice (mean, 16 ± 2; B, C), which had a more normal complement of labeled fibers. Significant differences between groups existed 5 mm rostral and caudal to the hemisection injury site for both BDA-labeled fibers and glial numbers (C; data represent mean ± SEM; n = 4; p < 0.05; Wlds vs corresponding C57BL/6J group; ANOVA with Fishers PLSD post hoc test). Scale bar, 50 μm.
The reduction in oligodendrocyte death in Wlds mice is transient
Parallel groups of hemisected C57BL/6J mice and Wlds mice were examined as before, but the time to kill was extended from 8 to 30 d after injury. In C57BL/6J mice, delayed oligodendrocyte death appears to continue between 8 and 30 d after hemisection, particularly in distal zones rostral and caudal to the hemisection (Fig 3A). The progressive loss of oligodendrocytes after 8 d of injury is consistent with the continued loss of oligodendrocytes observed in the second week after contusion SCI (Liu et al., 1997). It is possible that delayed oligodendrocyte death continues beyond the 7 mm zone distal to the hemisection measured in the current study.
We predicted that the eventual death of severed axons preserved in the Wlds mice group for at least 8 d after injury would cause the demise of oligodendrocytes that survived while the axons were still alive. A comparison of the Wlds group killed 30 d after hemisection with those killed at 8 d confirmed this prediction (Fig. 3B). The selective preservation of oligodendrocytes on the hemisected side of the cord at 8 d was not seen at 30 d, when the number of oligodendrocytes remaining on the hemisected side of Wlds mice was similar to that in C57BL control mice (Fig. 3C,D). Histological examination revealed fewer LVST axons at 30 d than at 8 d (data not shown).
Delayed oligodendrocyte death is reduced in Bax-deficient mice
As in the above experiments with control mice, the number of APC+ oligodendrocytes but not NeuN+ neurons (Fig. 8A,B) was reduced in regions that were 3-7 mm from the injury site on the injured side in wild-type (Bax+/+) mice (Fig. 9A,C). In contrast, APC+ oligodendrocyte numbers were preserved in Bax-/- mice at 8 d after hemisection injury (Fig. 9B,D). Loss of APC+ oligodendrocytes in region 1 (0-2 mm), immediately surrounding the hemisection epicenter, was not attenuated in Bax-/- mice, as might be expected if the cells died by necrosis after acute mechanical injury. Substantial preservation of APC+ cells (only a 9% loss) was evident in the Bax-/- mice, whereas a 45% loss was observed in the Bax+/+ mice. In additional studies, the preservation of oligodendrocytes on the side of hemisection in Bax-/- mice at 8 d after injury was still present at 30 d after injury (Fig. 10). Therefore, the effect of Bax deletion was not simply to delay oligodendrocyte death, but death appears to have been prevented long-term.
Figure 8.
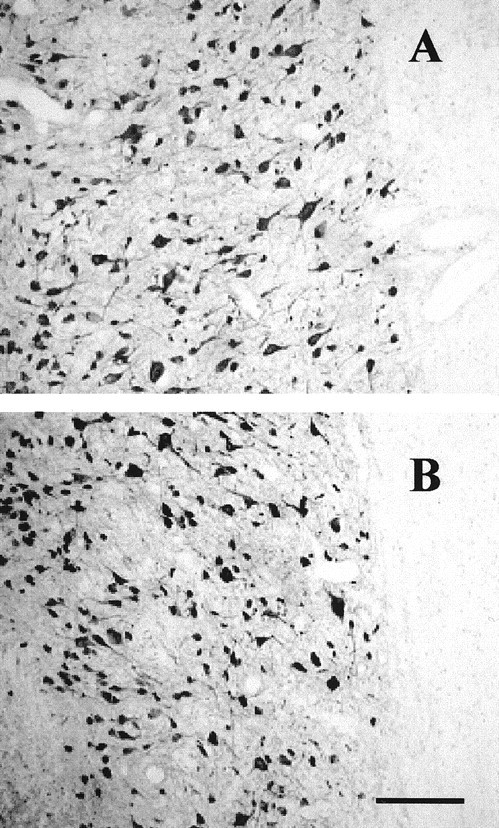
Anti-NeuN immunocytochemistry detected NeuN+ neurons in gray matter on the intact contralateral (A) and ipsilateral hemisected sides of the cord (B) 5 mm caudal to the hemisection in a representative section from a Bax-/- mouse. There was no substantial difference in the number of NeuN+ cells in these representative segments. Images were taken at 5 mm caudal to the site of hemisection. Scale bar, 100 μm.
Figure 9.

In Bax+/+ mice (A) but not Bax-/- mice (B), the number of APC+ oligodendrocytes on the injured side was significantly less than the contralateral intact side 8 d after hemisection. The numbers of NeuN+ cells on injured and intact sides were similar in both Bax+/+ mice (C) and Bax-/- mice (D) after hemisection, except in the area immediately surrounding the injury epicenter. Data represent mean ± SEM (n = 4-7 for each group). *p < 0.05; **p < 0.01; injury versus intact side; ANOVA with Fishers PLSD post hoc test.
Figure 10.

In Bax+/+ mice, the number of APC+ oligodendrocytes on the injured hemisection side was similarly reduced at 8 and 30 d after hemisection (A). In contrast, in Bax-/- mice, the preservation of APC+ numbers at 8 d persisted to 30 d (B). The loss of NeuN+ cells in the central 1-2 mm area surrounding the hemisection site observed at 8 d was similar to numbers at 30 d in both Bax+/+ and Bax-/- mice (C, D). Data represent mean ± SEM (n = 4-7 for each group; 30 vs 8 d; ANOVA with Fishers PLSD post hoc test).
Postnatal Bax-/- mice have more spinal motoneurons than Bax+/+ mice, because they lose fewer neurons during development (White et al., 1998); the Bax mutation might also help regulate glial numbers during development. However, in the present study, quantitative analysis demonstrated that wild-type Bax+/+ mice and Bax-/- mice had similar numbers of NeuN+ neurons and APC+ oligodendrocytes on the intact side of the cord in adult mice (Fig. 9). Previous studies examining developmental regulation of cell numbers compared postnatal animals, and comparisons of adults have not been described previously and appear to differ from postnatal events.
Oligodendrocytes cultured from Bax-deficient mice are resistant to apoptosis but not to excitotoxic necrosis
To further understand the genetic selectivity of Bax deficiency, we examined the vulnerability of sister cultures derived from Bax-/-, Bax-/+, and Bax+/+ mice to agents known to induce apoptosis (staurosporine, cyclosporin A) or necrosis (kainate) in oligodendrocytes (McDonald et al., 1996, 1998). Mixed glial cultures were derived from P2-P3 Bax-/-, Bax+/-, and Bax+/+ mice. Sister cultures (21 DIV) were exposed to 10 μm cyclosporin A, 10 nm staurosporine, 300 μm kainate, or a sham wash for 24 hr, and the number of viable Gal-C+ cells was determined (n = 3-5 cultures; values are expressed as a percentage of viable Gal-C+ cells in corresponding wash-control cultures). Consistent with our previous work, Gal-C+ oligodendrocytes derived from Bax+/+ or Bax-/+ mice but not Bax-/- mice were highly vulnerable to apoptotic death induced by cyclosporin A or staurosporine (Fig. 11) (McDonald et al., 1996). Staurosporine killed the majority of oligodendrocytes in cultures derived from Bax+/+ mice but not Bax-/- mice (p < 0.05; independent Student's t test; Bax+/+ vs Bax-/- cultures; n = 4; Gal-C+ cell survival: 15 ± 5% Bax+/+ cultures survival vs 93 ± 6% Bax-/- cultures survival). In contrast, all three Bax groups were similarly vulnerable to excitotoxic death induced by a 24 hr exposure to 300 μm kainate (Gal-C+ cell survival, 29 ± 7% Bax+/+ vs 32 ± 5% Bax+/- vs 26 ± 7% Bax-/- cultures; n = 4).
Figure 11.

Cultured oligodendrocytes derived from Bax-/- mice were resistant to apoptotic death induced by cyclosporin A (20 μm) or staurosporine (10 nm) but were vulnerable to excitotoxic necrotic death induced by exposure to kainate (300 μm). Oligodendrocyte survival in 21 DIV cultures derived from single postnatal dissections of Bax-/-, Bax+/-, and Bax+/+ littermates after a 24 hr exposure to 20 μm cyclosporin A is shown. The numbers of viable Gal-C+ cells per 200× field were counted (≥10 fields per well were counted). Values are expressed as a percentage of corresponding wash control. *p < 0.05 versus respective wash (control) condition; #p < 0.05 CsA exposure in Bax-/- group versus CsA exposure in wild-type group (n = 3-5; mean ± SEM). Exposure to staurosporine produced a similar pattern of results. In contrast, exposure to kainite caused equivalent oligodendrocyte death across all three groups.
Discussion
This study advanced our understanding of the mechanisms of oligodendrocyte death after SCI. The major findings are that mouse spinal cord oligodendrocytes die by apoptosis over a protracted period after hemisection, that this death is dependent on Bax expression, and it does not occur while severed axons survive (in Wlds mice). However, the preservation of oligodendrocytes in Wlds mice observed at 8 d after hemisection is transient, and oligodendrocytes are eventually lost in association with the predicted loss of severed axons by 30 d after injury. The data demonstrate additional new findings including the following: (1) oligodendrocyte apoptotic death continues to occur from 8-30 d after hemisection injury in control mice, (2) Bax deletion does not appear to regulate oligodendrocyte numbers (or spinal cord neuronal numbers) in the thoracic cord when examined at the adult stage, and (3) oligodendrocytes derived from Bax-/- mice but not Bax+/+ mice are resistant to cyclosporin A or staurosporine (agents that normally induce apoptosis in oligodendrocytes) but remain vulnerable to kainite-induced necrosis death.
We detected a prominent phase of oligodendrocyte death 8 d after injury at sites up to 7 mm distal to the hemisection injury, and the delayed oligodendrocyte death continued in the period of 8-30 d. We used multiple methods including APC, MBP, Rip immunocytochemistry, Nissl staining, and electron microscopy to identify loss of oligodendrocytes to overcome the potential problem of downregulation of immunohistochemical markers resulting from injury, as demonstrated previously for proteolipid protein in mature oligodendrocytes in the optic nerve after nerve transection (McPhilemy et al., 1990, 1991). The mode by which oligodendrocytes were dying was identified using biochemical, genetic, and ultrastructural characteristics and was found to be most consistent with apoptosis: anti-activated caspase-3 expression, prevention by absence of Bax, and typical ultrastructural features of apoptosis. Death of these distal oligodendrocytes was delayed in mice carrying the Wlds mutation, which exhibit markedly delayed Wallerian degeneration of axons. In Wlds mice, distant oligodendrocytes were still alive 8 d after hemisection injury, a time when severed axons remain anatomically, and presumably physiologically, intact. However, this preservation of oligodendrocytes was temporary, because the oligodendrocytes died when the spinal cord was examined 30 d after hemisection, when severed axons degenerated.
In addition, oligodendrocyte death was prevented in Bax-/- mice, both at 8 and 30 d after hemisection injury. Cultured oligodendrocytes derived from Bax-/- mice were resistant to apoptosis induced by cyclosporin A and staurosporine, agents that induce apoptosis in cultured oligodendrocytes (McDonald et al., 1996) but remained vulnerable to excitotoxic necrosis death induced by exposures to kainate. This latter finding also supports the apoptosis nature of oligodendrocyte death observed in vivo, because death of oligodendrocytes was prevented in Bax-/- mice only in the segments distal to hemisection (3-7 mm) but not in the immediate area of necrosis surrounding the hemisection (1-2 mm).
Previous studies (Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997; Casha et al., 2001) have demonstrated that delayed oligodendrocyte death occurs in rats, monkeys, and perhaps humans (Emery et al., 1998) after SCI, and that such death exhibits ultrastructural features of apoptosis. Here, we demonstrate that apoptotic death also occurs in a mouse model of SCI hemisection. However, the immunological and anatomical response of the injured spinal cord is different between mice and rats. In mice, the injury epicenter is limited to a small area surrounding the initial injury and is characterized by hypervascularization, the presence of large macrophages and fibroblast, and the rare formation of a central cavity or cyst (Zhang et al., 1996). In contrast, in rats, the injury epicenter is more broadly distributed, and central cavity formation is common (Schwab and Bartholdi, 1997). Thus, we chose a zone of 3-7 mm outside the epicenter in our mouse model as the secondary injury area. Active cell loss was still evident 8 d after injury, as shown by ultrastructural findings and expression of anti-activated caspase-3. We fulfilled genetic criteria for apoptosis by using Bax-/- mice to demonstrate that oligodendrocyte death required Bax expression. Together, the ultrastructural, biochemical, and genetic data provide the most advanced and convincing evidence for the apoptotic nature of delayed oligodendrocyte death in rodent models of SCI.
It is generally accepted, but unproven, that apoptotic death of oligodendrocytes in white matter relates to Wallerian degeneration of axons damaged by the initial injury (Beattie et al., 2000). The present study offers the first direct evidence that survival of severed axons prevents oligodendrocyte death after hemisection SCI. The Wlds mice served as a unique tool for investigating this question. Whereas axons of wild-type mice begin to degenerate 1-2 d after transection, those of Wlds mice remain functional for 14 d, generating action potentials and exhibiting axonal transport (Perry et al., 1990, 1991). Our antereograde labeling of the LVST in Wlds mice confirmed the predicted persistence of transected axons 8 d after hemisection. We also found near-normal numbers of glia and oligodendrocytes (APC+ cells) in the injured side of the cord at this same interval of 8 d after injury. Therefore, the persistence of functional but severed axons is sufficient to support oligodendrocyte survival, despite the ongoing secondary injury associated with hemisection. All other events initiated by hemisection still occur in Wlds mice: neurons and glia within 1 mm of the lesion undergo acute mechanical death, and an inflammatory cascade ensues. We acknowledge that preservation of severed axons and their oligodendrocytes alters the environment around the injury site, reducing macrophage infiltration and scar production (Zhang et al., 1998). Nevertheless, this model currently provides the best support for the hypothesis that oligodendrocyte death is initiated by the death of severed axons. Because spinal oligodendrocytes myelinate multiple axons (∼10-20), it is interesting to speculate how many axons are required to be lost to initiate oligodendrocyte death.
In this study, we also used Bax-/- adult mice and oligodendrocyte cultures derived from Bax-/- mice to assess the role of Bax in oligodendrocyte death. Considerably fewer oligodendrocytes were lost from the Bax-/- mice than from the Bax+/+ mice. We also evaluated the specificity of the Bax deletion to apoptotic death by exposing cultured oligodendrocytes to cyclosporin A, staurosporine, or kainate. Oligodendrocytes derived from Bax-/- mice and cultured in mixed glial systems were resistant to apoptotic death (cyclosporin A, staurosporine) but not to excitotoxic necrotic death (kainate). These data are consistent with similar protection seen in cortical neurons of Bax-/- mice (Snider et al., 1999). Our in vivo and in vitro experiments with Bax-/- mice provide the first evidence that delayed oligodendrocyte death requires Bax gene expression.
Little is known about the roles of Bax in glial survival and death, either during development or after CNS injury, although previous work has indicated that Bcl-2 overexpression prevents neuronal loss after traumatic brain injury (Raghupathi et al., 1998) or SCI (Lou et al., 1998; Takahashi et al., 1999; Saavedra et al., 2000). Examination of optic and sciatic nerves at P4 from Bax-/- and wild-type littermates uncovered no substantial difference in mean numbers of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive cells (White et al., 1998), suggesting that Bax-/- does not prevent the naturally occurring cell death of non-neuronal cells at P4. These observations are not in keeping with our findings that the numbers of spinal cord oligodendrocytes are not altered in Bax-/- mice compared with Bax+/+ mice. It is therefore possible that Bax has different functions under physiological and pathological conditions in glial cells. Interestingly, we also did not find a change in the number of spinal cord neurons, which is in contrast to the increased numbers of sensory neurons observed previously in postnatal mice (White et al., 1998). This finding may be explained by a preferential developmental effect of Bax on sensory neurons, or that the preservation of the neurons normally dying during development is transient, and they are eventually lost by the adult stage.
Most potential causes of delayed apoptotic death of oligodendrocytes after SCI can be categorized into the following two groups: loss of critical survival factors and production of toxic factors. Maintenance of trophic support is critical to oligodendrocyte survival, and degeneration of damaged axons after SCI would predictably reduce trophic support (Barres et al., 1993). In vivo and in vitro studies have indicated that insulin and insulin-like growth factors, neurotrophin-3, and ciliary-neurotrophic factor are the most important factors for oligodendrocyte survival (Barres et al., 1992, 1993). Potentially toxic molecules released from degenerating axons include TNF-α and glutamate (McDonald et al., 1998; for review, see Beattie et al., 2000). Processes initiated by injury, such as microglial activation and infiltration of inflammatory cells, can also injure white matter (Crowe et al., 1997; Shuman et al., 1997; Popovich et al., 1999).
The current experiments provide direct evidence that delayed oligodendrocyte death relates to axonal loss and depends on Bax expression. Defining the mechanisms underlying this process will be therapeutically important, because such death likely contributes to spinal cord dysfunction via segmental axonal demyelination-dysmyelination (McDonald and Sadowsky, 2002). Because each oligodendrocyte myelinates segments of 10-20 different axons, and because the delayed death of oligodendrocytes occurs in distant and relatively preserved regions of the cord, loss of a single oligodendrocyte will produce segmental demyelination and dysfunction in an exponential number of axons. Additional studies on the long-term survival of oligodendrocytes and the potential impact on functional recovery of preventing delayed oligodendrocyte death will now be important. Although the pathophysiology of oligodendrocyte death is complicated (Ludwin, 1997), our findings offer new ways to prioritize mechanistic investigations. Such studies should permit the rationale design of genetic approaches to reducing delayed cell death to examine the role of demyelination in CNS dysfunction and toward perhaps improving functional outcomes in SCI.
Footnotes
This work was supported by National Institute of Neurologic Disorders and Stroke Grants NS01931 and NS37927 (J.W.M.) and by St. Louis Gateway to a Cure (J.W.M.).
Correspondence should be addressed to Dr. John W. McDonald, Department of Neurology, Restorative Treatment and Research Program, Campus Box 8518, Washington University School of Medicine, 4444 Forest Park Avenue, St. Louis, MO 63108. E-mail: mcdonald@neuro.wustl.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238682-10$15.00/0
References
- Abe Y, Yamamoto T, Sugiyama Y, Watanabe T, Saito N, Kayama H, Kumagai T ( 1999) Apoptotic cells associated with Wallerian degeneration after experimental spinal cord injury: a possible mechanism of oligodendroglial death. J Neurotrauma 16: 945-952. [DOI] [PubMed] [Google Scholar]
- Antonawich FJ, Krajewski S, Reed JC, Davis JN ( 1998) Bcl-x (I) Bax interaction after transient global ischemia. J Cereb Blood Flow Metab 18: 882-886. [DOI] [PubMed] [Google Scholar]
- Bar-Peled O, Knudson M, Korsmeyer SJ, Rothstein JD ( 1999) Motor neuron degeneration is attenuated in Bax-deficient neurons in vitro J Neurosci Res 55: 542-556. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC ( 1992) Cell death and control of cell survival in the oligodendrocyte lineage. Cell 70: 31-46. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC ( 1993) Multiple extracellular signals are required for long-term oligodendrocyte survival. Dev 118: 283-295. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Farooqui AA, Bresnahan JC ( 2000) Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma 17: 915-925. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM ( 1996) Expression of the APC tumor suppressor protein in oligodendroglia. Glia 17: 169-174. [DOI] [PubMed] [Google Scholar]
- Bogler O, Wren D, Barnett SC, Land H, Noble M ( 1990) Cooperation between two growth factors promotes extended self-renewal and inhibits differentiation of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proc Natl Acad Sci USA 87: 6368-6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB ( 1993) Bcl-x, a Bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74: 597-608. [DOI] [PubMed] [Google Scholar]
- Brown MC, Booth CM, Lunn ER, Perry VH ( 1991) Delayed response to denervation in muscles of C57BL/Ola mice. Neuroscience 43: 279-283. [DOI] [PubMed] [Google Scholar]
- Brown MC, Lunn ER, Perry VH ( 1992) Consequences of slow Wallerian degeneration for regenerating motor and sensory axons. J Neurobiol 23: 521-536. [DOI] [PubMed] [Google Scholar]
- Casha S, Yu WR, Fehlings MG ( 2001) Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in rats. Neuroscience 103: 203-218. [DOI] [PubMed] [Google Scholar]
- Chen DF, Schneider GE, Martinou J-C, Tonegawa S ( 1997) Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 385: 434-439. [DOI] [PubMed] [Google Scholar]
- Chittenden T, Harrington EA, O'Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC ( 1995) Induction of apoptosis by the Bcl-2 homologue Bak. Nature 374: 733-736. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS ( 1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3: 73-76. [DOI] [PubMed] [Google Scholar]
- Daston MM, Ratner N ( 1994) Amphoterin (P30, HMG-1) and RIP are early markers of oligodendrocytes in the developing rat spinal cord. J Neurocytology 23: 323-332. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson Jr EM, Snider WD, Korsmeyer SJ ( 1996) Bax is required for neuronal death after trophic factor deprivation and during development. Neuron 17: 401-411. [DOI] [PubMed] [Google Scholar]
- Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, Bethea J, Levi AD ( 1998) Apoptosis after traumatic human spinal cord injury. J Neurosurg 89: 911-920. [DOI] [PubMed] [Google Scholar]
- Friedman B, Hockfield S, Black JA, Woodruff KA, Waxman SG ( 1989) In situ demonstration of mature oligodendrocytes and their processes: an immunocytochemical study with a new monoclonal antibody, rip. Glia 2: 380-390. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Wickert H, Zimmermann M ( 1995) Up-regulation of bax and down-regulation of bcl-2 is associated with kainate-induced apoptosis in mouse brain. Neurosci Lett 192: 85-88. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Ribchester RR ( 2001) Compartmental neurodegeneration and synaptic plasticity in the Wlds mutant mouse. J Physiol (Lond) 534: 627-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog A, Brosamle C ( 1997) “Semifree-floating” treatment: a simple and fast method to process consecutive sections for immunohistochemistry and neuronal tracing. J Neurosci Methods 72: 57-63. [DOI] [PubMed] [Google Scholar]
- Kalderon N, Fuks Z ( 1996) Structural recovery in lesioned adult mammalian spinal cord by x-irradiation of the lesion site. Proc Natl Acad Sci USA 93: 11179-11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CM, Tung KSK, Tourtellotte WG, Brown GA, Korsmeyer SJ ( 1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 95-101. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ ( 1995) Regulators of cell death. Trends Genet 11: 101-105. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ ( 1999) Bcl-2 gene family and the regulation of programmed cell death. Cancer Res 59: 1693-1700. [PubMed] [Google Scholar]
- Krajewski S, Mai JK, Krajewski M, Sikorska M, Mossakowski MJ, Reed JC ( 1995) Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci 15: 6364-6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GL, Farooque M, Holtz A ( 1999) Apoptosis of oligodendrocytes occurs for long distances away from the primary injury after compression trauma to rat spinal cord. Acta Neuropathol 98: 473-480. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW ( 1997) Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci 17: 5395-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J, Lenke LG, Xu F, O'Brien M ( 1998) In vivo Bcl-2 oncogene neuronal expression in the rat spinal cord. Spine 23: 517-523. [DOI] [PubMed] [Google Scholar]
- Ludwin SK ( 1997) The pathobiology of the oligodendrocyte. J Neuropathol Exp Neurol 56: 111-124. [DOI] [PubMed] [Google Scholar]
- Ludwin SK, Bisby MA ( 1992) Delayed Wallerian degeneration in the central nervous system of Ola mice: an ultrastructural study. J Neurol Sci 109: 140-147. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z ( 2002) Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and Bax-dependent. J Neurobiol 50: 181-197. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Sadowsky C ( 2002) Spinal cord injury: doable therapeutics. Lancet 359: 417-425. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Goldberg MP, Gwag BJ, Chi S-I, Choi DW ( 1996) Cyclosporine induces neuronal apoptosis and selective oligodendrocyte death in cortical cultures. Ann Neurol 40: 750-758. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP ( 1998) Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med 4: 291-297. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Liu X-Z, Qu Y, Liu S, Mickey SK, Turetsky D, Gottlieb DI, Choi DW ( 1999) Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med 5: 1410-1412. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA ( 1990) FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron 5: 603-614. [DOI] [PubMed] [Google Scholar]
- McPhilemy K, Mitchell LS, Griffiths IR, Morrison S, Deary AW, Sommer I, Kennedy PGE ( 1990) Effect of optic nerve transection upon myelin protein gene expression by oligodendrocytes: evidence for axonal influences on gene expression. J Neurocytol 19: 494-503. [DOI] [PubMed] [Google Scholar]
- McPhilemy K, Griffiths IR, Mitchell LS, Kennedy PGE ( 1991) Loss of axonal contact causes down-regulation of the PLP gene in oligodendrocytes: evidence from partial lesions of the optic nerve. Neuropathol Appl Neurobiol 17: 275-287. [DOI] [PubMed] [Google Scholar]
- Mosinger Ogilvie J, Deckwerth TL, Knudson CM, Korsmeyer SJ ( 1998) Suppression of developmental retinal cell death but not of photoreceptor degeneration in Bax-deficient mice. Invest Ophthalmol Vis Sci 39: 1713-1720. [PubMed] [Google Scholar]
- Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S ( 1989) Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. Eur J Neurosci 2: 408-413. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER, Tree P, Gordon S ( 1990) Evidence that very slow Wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur J Neurosci 2: 802-808. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER ( 1991) Very slow retrograde and Wallerian degeneration in the CNS of C57BL/Ola mice. Eur J Neurosci 3: 102-105. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT ( 1999) Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 158: 351-365. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Fernandez SC, Murai H, Trusko SP, Scott RW, Nishioka WK, Mcintosh TK ( 1998) BCL-2 overexpression attenuates cortical cell loss after traumatic brain injury in transgenic mice. J Cereb Blood Flow Metab 18: 1259-1269. [DOI] [PubMed] [Google Scholar]
- Saavedra RA, Murray M, de Lacalle S, Tessler A ( 2000) In vivo neuroprotection of injured CNS neurons by a single injection of a DNA plasmid encoding the Bcl-2 gene. Prog Brain Res 128: 365-372. [DOI] [PubMed] [Google Scholar]
- Schlesinger PH, Gross A, Yin X-M, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ ( 1997) Comparison of the ion channel characteristic of proapopptotic BAX and antiapoptotic BCL-2. Proc Natl Acad Sci USA 94: 11357-11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab ME, Bartholdi D ( 1997) Degeneration and regeneration of axons in the lesion spinal cord. Physiol Rev 76: 319-370. [DOI] [PubMed] [Google Scholar]
- Shuman SL, Bresnahan JC, Beattie MS ( 1997) Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res 50: 798-808. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Knapp PE ( 1999) Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med 5: 943-946. [DOI] [PubMed] [Google Scholar]
- Snider BJ, Gottron FJ, Choi DW ( 1999) Apoptosis and necrosis in cerebrovascular disease. Ann NY Acad Sci 893: 243-253. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Schwarz E, Ljubetic C, Murray M, Tessler A, Saavedra RA ( 1999) DNA plasmid that codes for human Bcl-2 gene preserves axotomized Clarke's nucleus neurons and reduces atrophy after spinal cord hemisection in adult rats. J Comp Neurol 404: 159-171. [DOI] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD ( 1998) Widespread elimination of naturally occurring neuronal death in Bax- deficient mice. J Neurosci 18: 1428-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf HK, Buslei R, Schmidt-Kastner R, Schmidt-Kastner PK, Pietsch T, Wiestler OD, Bluhmke I ( 1996) NeuN: a useful neuronal marker for diagnostic histopathology. J Histochem Cytochem 44: 1167-1171. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ ( 1995) Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell 80: 285-291. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Guth L ( 1997) Experimental spinal cord injury: Wallerian degeneration in the dorsal column is followed by revascularization, glial proliferation, and nerve regeneration. Exp Neurol 147: 159-171. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Fujiki M, Guth L, Steward O ( 1996) Genetic influences on cellular reactions to spinal cord injury: a wound-healing response present in normal mice is impaired in mice carrying a mutation (Wlds) that causes delayed Wallerian degeneration. J Comp Neurol 371: 485-495. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Guth L, Steward O ( 1998) Mechanisms of motor recovery after subtotal spinal cord injury: insights form the study of mice carrying a mutation (Wlds) that delays cellular responses to injury. Exp Neurol 149: 221-229. [DOI] [PubMed] [Google Scholar]