

Abstract
Blood-brain barrier (BBB) dysfunction is a potential mechanism involved in progressive striatal damage induced by the mitochondrial excitotoxin, 3-nitropropionic acid (3-NP). After activation by proteases and free radicals, matrix metalloproteinases (MMPs), particularly MMP-9 and -2, can digest the endothelial basal lamina leading to BBB opening. Using CD-1 mice, we show that MMP-9 expression by zymography is increased in the injured striatum compared with the contralateral striatum 2 hr after 3-NP injection [133.50 ± 57.17 vs 50.25 ± 13.56; mean ± SD of optical densities in arbitrary units (A.U.); p < 0.005] and remains elevated until 24 hr (179.33 ± 78.24 A.U.). After 4 hr, MMP-9 expression and activation are accompanied by an increase in BBB permeability. MMP inhibition attenuates BBB disruption, swelling, and lesion volume compared with vehicle-treated controls. There is a clear spatial relationship between MMP-9 expression and oxidized hydroethidine, indicating reactive oxygen species (ROS) production. Furthermore, transgenic mice that overexpress copper/zinc-superoxide dismutase (SOD1) show decreased lesion size and edema along with decreased immunoreactivity for MMP-9, compared with wild-type littermates (lesion: 38.8 ± 15.1 and 53.3 ± 10.3, respectively, p ≤ 0.05; edema: 21.8 ± 11.2 and 35.28 ± 11, respectively, p ≤ 0.05; MMP-9-positive cells: 352 ± 57 and 510 ± 45, respectively, p ≤ 0.005), whereas knock-out mice deficient in SOD1 display significantly greater swelling (48.65 ± 17; p ≤ 0.05). We conclude that early expression and activation of MMP-9 by ROS may be involved in early BBB disruption and progressive striatal damage after 3-NP treatment.
Keywords: matrix metalloproteinase, 3-nitropropionic acid, blood-brain barrier, Evans blue extravasation, oxidative stress, striatum
Introduction
The neurotoxin, 3-nitropropionic acid (3-NP), an inhibitor of mitochondrial complex II, has been used as a model for Huntington's disease (HD) because of its ability to cause selective striatal degeneration, thus mimicking clinical and pathological aspects of this neurodegenerative disorder (Coles et al., 1979; Ludolph et al., 1991; He et al., 1995). The precise mechanism of this degeneration is unknown; however, oxidative stress and mitochondrial dysfunction-linked excitotoxicity have been proposed as key events in the pathophysiology of HD (Albin and Greenamyre, 1992; Coyle and Puttfarcken, 1993; Beal, 1995, 2000). Blood-brain barrier (BBB) disruption has also been implicated in the pathogenesis of progressive neurodegenerative disorders such as Alzheimer's disease (AD), as well as in acute neurodegeneration syndromes such as ischemic and hemorrhagic stroke (Hardy et al., 1986; Kalaria et al., 1991; Dietrich et al., 1992; Nag, 1992; Mayhan and Didion, 1996; Miller et al., 1996; Miyakawa et al., 2000).
To our knowledge, there are no reports of a direct correlation between HD, oxidative damage, and BBB disruption; however, mutant mice with a partial deficiency in manganese-superoxide dismutase show significantly larger striatal lesions (Andreassen et al., 2001; Kim and Chan, 2002), whereas transgenic (Tg) mice that overexpress copper/zinc-superoxide dismutase (SOD1-Tg) show significantly decreased striatal lesions (Beal et al., 1995) after 3-NP treatment, suggesting that reactive oxygen species (ROS) play an important role in the neurotoxicity of HD. Other experimental studies support the hypothesis that excitotoxicity and oxidative stress, together with BBB disruption and astrocyte dysfunction, represent the main mechanisms of 3-NP-induced striatal damage (Hamilton and Gould, 1987; Beal, 1995; Schulz et al., 1996; Nishino et al., 1997, 2000; Fukuda et al., 1998; Greene et al., 1998; Kim et al., 2000a).
Some key mediators of BBB disruption that implicate oxidative stress and excitotoxicity are matrix metalloproteinases (MMPs) (Rosenberg et al., 1998; Gasche et al., 2001). These zinc-dependent proteolytic enzymes are secreted as zymogens and cleave to their active form. MMPs are involved in degradation and remodeling of the extracellular matrix under various physiological and pathological conditions. Among MMPs, gelatinase A (MMP-2) and gelatinase B (MMP-9) are able to digest the endothelial basal lamina leading to the opening of the BBB (Rosenberg et al., 1998). MMPs play an active role in secondary brain injury after focal ischemia (Romanic et al., 1998; Rosenberg et al., 1998; Gasche et al., 1999, 2001; Gu et al., 2002), but they also have been implicated in neurodegenerative diseases such as amyotrophic lateral sclerosis (Lim et al., 1996) and AD (Deb and Gottschall, 1996).
To address the question of ROS and MMP involvement in 3-NP-induced striatal injury, we investigated expression of MMP-9 and the level of BBB disruption in the injured striatum after local injection of 3-NP in CD-1 mice. We also evaluated the efficacy of MMP inhibition preventing BBB leakage in the 3-NP-injured striatum. Finally, we determined the effects of 3-NP administration on infarct size, edema, and MMP-9 expression in SOD1-Tg animals or in knock-out (KO) mice deficient in the cytosolic ROS scavenger SOD1 (SOD1-KO).
Materials and Methods
Three-month-old male CD-1 mice (35-40 gm; Charles River, Wilmington, MA), SOD1-Tg mice (Sugawara et al., 2002), SOD1-KO mice (Reaume et al., 1996), and the appropriate wild-type (WT) littermates were used in these studies. All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by Stanford University's Administrative Panel on Laboratory Animal Care. Each mouse was placed in a stereotaxic frame (Stoelting Co., Wood Dale, IL) under general anesthesia. They were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a mask (a gift from Stoelting Co.). During the procedure, blood gases were monitored, and rectal temperature was controlled at 37°C ± 5°C with a homeothermic blanket. The scalp was incised on the midline, and the skull was exposed.
Administration of 3-NP and the MMP inhibitor. 3-NP (Aldrich Chemical Co., Milwaukee, WI) was dissolved in saline to a concentration of 71.4 mg/ml, adjusted to pH 7.4, and passed though a 0.2 μm filter. A 5 μl Hamilton syringe fitted with a 30 gauge blunt-tipped needle was placed into the right striatum at coordinates of 0.5 mm anterior, 2 mm lateral, and 3.3 mm deep to the bregma after a small hole was made with a dental drill under surgical microscopy. A total of 300 nmol of 3-NP in a volume of 0.5 μl was injected over 7 min, and the needle was left in place for 7 min before being slowly withdrawn. An equal volume of normal saline was administered to the control animals. To evaluate the involvement of MMPs in BBB disruption, the CD-1 mice were randomly assigned in a blinded manner to intraventricularly receive an MMP inhibitor or a placebo. We treated the animals with a broad-range MMP inhibitor (50 μg/μl in PBS of 4-Abz-Gly-Pro-d-Leu-d-Ala-NHOH; Calbiochem, La Jolla, CA) or a second inhibitor that works similarly (p-aminobenzoyl-Gly-Pro-d-Leu-d-Ala-hydoxamate; ICN Biomedicals, Aurora, OH), or the vehicle (PBS) 30 min before 3-NP injection and administered intraventricularly (2 μl, 1.0. mm lateral, 0.2 mm posterior, 3.1 mm deep to the bregma).
Analysis of MMP expression using zymography after 3-NP treatment. To analyze the gelatinase activity in the injured and contralateral striatum, gelatinases were first extracted from the tissue of CD-1 mice as described previously (Gasche et al., 1999). Both the injured and contralateral striatum were sampled and snap-frozen 1, 2, 4, 8, and 24 hr after injection of 3-NP or PBS. The brain tissue was homogenized with a 2 ml Teflon-glass homogenizer in a lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl2, 0.05% BRIJ-35, 0.02% NaN3, 1% Triton X-100). Aliquots (10 μl) of the homogenates were saved for total protein measurement (BCA kit; Pierce, Rockford, IL). The homogenates were centrifuged at 12,000 × g for 5 min. The supernatants were recovered and incubated for 60 min with gelatin-Sepharose 4B (Pharmacia Biotech, Uppsala, Sweden) with constant shaking. After incubation, the samples were centrifuged at 500 × g for 2 min. The pellets were washed with a buffer containing 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl2, 0.05% BRIJ-35, 0.02% NaN3. After a second centrifugation, the pellets were resuspended in 80 μl of elution buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl2, 0.05% BRIJ-35, 0.02% NaN3, dimethylsulfoxide 10%) for 30 min. The samples were then subjected to zymography, performed according to a previously reported method, with some modifications (Kleiner and Stetler-Stevenson, 1994). Ten microliters of a nonreducing sample buffer (0.4 m Tris, pH 6.8, 5% SDS, 20% glycerol, 0.05% bromophenol blue) were added to the sample volume, which was corrected for protein concentration. Samples were loaded on 10% SDS-polyacrylamide electrophoresis gels in which 0.1% porcine skin gelatin (Novex, San Diego, CA) was co-polymerized. The gels were run at 25 mA for the appropriate time. After migration, the gels were incubated twice with 2.5% Triton-X 100 for 1 hr at room temperature, washed for 10 min in Tris-HCl/NaCl/CaCl2 buffer (50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 5 mm CaCl2), and further incubated for 16 hr in Tris-HCl/NaCl/CaCl2 in a water bath at 37°C. The gels were stained for 90 min in Coomassie blue (1% Coomassie brilliant blue, 30% methanol, 10% acetic acid) and destained in 30% methanol/10% acetic acid, four times for 5, 15, 30, and 60 min, respectively. White bands on a blue background indicated zones of digestion corresponding to the presence of different MMPs, identified on the basis of their molecular weight. Ten microliters of human pro-MMP-9 (10 ng/ml; Oncogene Research, San Diego, CA) and human pro-MMP-2 and activated MMP-2 standards (15 ng/ml; Oncogene Research) were loaded onto each gel to allow comparison between the different gels. The bands were scanned using a densitometer (GS-700; Bio-Rad Laboratories, Hercules, CA), and quantification was performed using Multi-Analyst 1.0.2 software (Bio-Rad).
Immunohistochemistry for MMP-9. Anesthetized CD-1 mice were transcardially perfused with normal saline containing 10 U/ml of heparin 4 and 24 hr after 3-NP injection. The brains were removed, postfixed for 12 hr in 3.7% formaldehyde, sectioned at 30 μm with a vibratome, and processed for immunohistochemistry. The sections were incubated with a blocking solution containing 20% normal goat serum (Vector Laboratories, Burlingame, CA) and with an anti-MMP-9 polyclonal antibody (Anawa, Wangen, Switzerland) at a dilution of 1:50. Immunohistochemistry was performed using the avidin-biotin technique as described previously (Gasche et al., 1999), and then the nuclei were counterstained with a methyl green solution for 10 min. As a negative control, the sections were incubated without the primary antibody. To study the relationship between the production of ROS and MMP-9 expression, immunofluorescence staining of MMP-9 was performed in the samples used for hydroethidine (HEt) in situ detection. These samples were incubated with a biotinylated secondary antibody, followed by Fluorescein Avidin DCS (35 μg/ml; Vector) for 30 min. Mounted slides were examined with a fluorescence microscope (Zeiss, Thornwood, NY). The same technique was used to investigate the spatial relationship between MMP-9 expression and Evans blue extravasation. Digitalized images taken after double exposure were analyzed using image analysis software (AxioVision, 2.2; Zeiss).
Western blot for MMP. To further evaluate the expression of MMP-9 in the samples from the injured and contralateral striatum of the CD-1 mice, Western blot analysis was performed as described previously (Gasche et al., 1999; Morita-Fujimura et al., 2000). Extracted samples, as well as the MMP-9 standard, were loaded onto SDS-polyacrylamide gels under nonreducing conditions. After migration, the proteins were electrotransferred onto a polyvinylidine difluoride membrane (Novex), which was blocked overnight in 5% dry milk (Bio-Rad) PBS/0.1% Tween 20 at 4°C. After washing in PBS/0.1% Tween 20, the membranes were incubated for 90 min at 37°C with a rabbit anti-human MMP-9 polyclonal antibody (1:500; Anawa). Then they were washed thee times and incubated for 60 min at 37°C with peroxidase-labeled anti-rabbit IgG (1:12,500 for MMP-9) using the Roche Diagnostics (Indianapolis, IN) chemiluminescence system. The bands were visualized with an enhanced chemiluminescence reagent (ECL plus Western blot analysis system; Amersham Biosciences, Piscataway, NJ).
Detection of oxidative stress by in situ HEt detection and 8-hydroxyguanosine immunohistochemistry. After 3-NP injection, the temporal and spatial production of striatal ROS were investigated using a previously reported method of in situ detection of oxidized HEt with slight modifications (Chan et al., 1998; Kim et al., 2000a,b, 2002; Kim and Chan, 2001). HEt is oxidized to ethidium by superoxide anion radicals (Bindokas et al., 1996; Benov et al., 1998). HEt (stock solution 100 mg/ml in dimethyl sulfoxide; Molecular Probes, Eugene, OR) was diluted to 1 mg/ml in PBS just before use, transferred to a 1 ml foiled syringe, and sonicated in a water bath. The animals were anesthetized, and 200 μl of HEt was administered intravenously through the jugular vein 1 and 4 hr after 3-NP or PBS injection. Two hours after HEt injection the animals were killed by transcardial perfusion with 200 ml of heparinized saline and 200 ml of 3.7% formaldehyde in PBS. After overnight postfixation, the brains were sectioned 50 μm thick with a vibratome at the level of the mid-striatum and placed on glass slides. The brain sections were incubated with 2.5 × 10-3 mg/ml Hoechst 33258 (Molecular Probes) in PBS for 15 min in a dark chamber and then rinsed with distilled H2O and mounted with Aquamount (ThermoShandon, Pittsburgh, PA). These sections were observed with a fluorescent microscope. Fluorescence was assessed microscopically at excitation = 510-550 nm and emission >580 nm for ethidium detection. The intensity and expression pattern of the oxidized HEt were observed and compared in the injured and contralateral striatum using image analysis software (AxioVision 2.2; Zeiss). To semiquantify ROS production, cells in the cytosol with oxidized HEt were counted and averaged in the entire field after random determination of four different sites under high magnification (400×) using the image analysis program. The percentage of these cells to the total cells stained with Hoechst nuclear staining was calculated. To obtain further evidence of oxidative stress in the injured striatum after 3-NP treatment, we evaluated DNA oxidation using a monoclonal antibody against 8-hydroxyguanosine (8OHG) according to a previously reported method (Kim and Chan, 2002). Sections 20 μm thick, mounted and frozen, were dried overnight at 45°C. They were immersed for 10 min in 70% ethanol at -20°C and then washed in PBS. Some samples were treated for 1 hr at 37°C with RNase or DNase or with a mixture of DNase plus RNase diluted in 50 mm Tris-HCl/10 mm MgCl2 buffer. Samples were denatured in 4N HCl for 7 min and neutralized with 50 mm Tris-base. After washing, the samples were incubated with a mouse monoclonal antibody for 8OHG (1:300; QED Bioscience, San Diego, CA) after biotinylation and a blocking procedure that followed the manufacturer's protocol to avoid cross-reaction between the secondary antibody and mouse IgG in the tissue (DAKO ARK; Dako, Carpinteria, CA). After development with diaminobenzidine, the sections were mounted on slides and counterstained with a methyl green solution.
Measurement of BBB disruption, lesion volume, and striatal swelling after 3-NP injection. Four, 8, or 24 hr after 3-NP or saline injection, BBB disruption was evaluated in the vehicle- and MMP inhibitor-treated mice (n = 62). Four hours before the animals were killed, 2.5 ml/kg of 4% Evans blue (Sigma, St. Louis, MO) in 0.9% saline was injected into every animal. The animals were anesthetized and perfused with 200 ml of heparinized saline. For quantitative measurement of Evans blue leakage, the brains were removed, and the striatum was separated and homogenized in 400 μl of N, N-dimethylformamide (Sigma) and then incubated for 72 hr in a water bath at 55°C and centrifuged at 20,000 × g for 20 min (Kim et al., 2001). The Evans blue level was quantitatively determined in the supernatants using a spectrophotometer (Molecular Devices, Sunnyvale, CA). The results are presented as milligrams of Evans blue per striatum. For qualitative examination of Evans blue extravasation, the brains were fixed in formaldehyde as described above and sectioned 20 μm thick. Nuclear counterstaining was achieved by incubating the sections with 2 μg/ml Hoechst 33258 (Molecular Probes) for 10 min. The sections were mounted and observed with a fluorescence microscope. To further evaluate the involvement of MMP in striatal tissue disruption after 3-NP treatment, the brains of the CD-1 mice treated with the MMP inhibitor were removed 1 d after 3-NP injection and sectioned 20 μm thick on a cryostat (n = 16). At 300 μm intervals, the sections were stained with cresyl violet to measure the lesion volume, using a published method (Kim et al., 2000a). The entire injured striatum, the unstained area of the injured striatum, and the contralateral striatum were each measured with a densitometer (Model GS-700; Bio-Rad) and quantified with an image analysis system (Bio-Rad). The lesion volume of the injured striatum, corrected for edema by the volume of the contralateral striatum, was calculated by multiplying each measured area by the distance between the sections. For the study using SOD1-Tg and SOD1-KO mice, the brains were paraffin embedded, cut in 6 μm coronal sections at 150 μm intervals, and stained with hematoxylin and eosin for histological assessment and determination of lesion size. Adjacent coronal sections were used for MMP immunohistochemistry (see below). The area of injury was expressed as percentage of the total area of the ipsilateral hemisphere and corrected for edema (Grzeschik et al., 2003).
Quantitative analysis of MMP expression by immunocytochemistry in SOD1-Tg and SOD1-KO mice and WT littermates. Tissue sections were deparaffinized, treated for endogenous peroxidases, and blocked for avidin-biotin. Sections were incubated with a primary antibody (anti-MMP-9 and anti-MMP-2 polyclonal antibody, 1:100 dilution; Oncogene Science, Cambridge, MA) at 4°C for 72 hr followed by a secondary antibody at 25°C for 1 hr. Between steps, sections were washed three times in PBS or PBST, plus 1% blocking serum. Antibodies were detected using the Vector ABC kit and colorized with diaminobenzidine or with the Vector VIP kit (Vector). Both colorizing agents were used in the identification of different cell types or for colocalizing protein expression, which requires double labeling. Negative controls were run in parallel using adjacent sections incubated without the primary antibody. The number of MMP-9- and MMP-2-positive cells was counted in the entire ipsilateral striatum (level 3, ∼0.4 mm anterior to the bregma) of representative animals from each group (n = 4 per group). The coronal section of the striatum was divided into image areas that were digitalized in a Zeiss Axioplan 2 microscope, with a 10-fold objective using a Zeiss Axiocam and then imported into AxioVision. The images were subsequently viewed at 175% of the original 10× images with Adobe Photoshop 5.5 software (Adobe Systems Inc., San Jose, CA) for counting. In some sections, double immunohistochemistry of MMP-9 and glial fibrillary acidic protein (GFAP) for glial cells (Santa Cruz Biotechnology, Santa Cruz, CA; goat polyclonal antibody; 1:200) or CD31 for endothelial cells (Santa Cruz; goat polyclonal antibody; 1:100) was performed. After reaction of MMP-9 antibody, samples were incubated with a biotinylated secondary antibody, followed by Fluorescein Avidin DCS (35 μg/ml; Vector) for 30 min. After MMP-9 immunohistochemistry, GFAP or CD31 immunohistochemistry was performed as described above and visualized with Texas Red avidin (30 μg/ml; Vector) for 20 min. Mounted slides were examined with a fluorescence microscope (Zeiss). Fluorescence of Texas Red was observed at excitation of 510 nm and emission of >580 nm. Fluorescence of fluorescein was observed at excitation of 495 nm and emission of >515 nm.
Statistical analysis. Data are expressed as mean ± SD. The statistical comparisons among multiple groups were made using an ANOVA followed by Fisher's post hoc protected least-significant difference test, whereas comparisons between two groups were performed using the unpaired t test (StatView, version 5. 01; SAS Institute Inc, Cary, NC).
Results
Neurological outcome
After 3-NP treatment the mice showed initial hyperactivity by rotating toward the contralateral side of the injured striatum (left) for 2-3 hr and then to the injured side (right). After 6-8 hr the mice became hypoactive. There was no mortality after the injection. The control mice injected with saline, or only with the MMP inhibitor or the intraventricular vehicle without 3-NP, did not display rotation or abnormal clinical signs. Twenty-four hours after 3-NP injection, the mice pretreated with the MMP inhibitor displayed variable behavior, such as decreased activity (18 of 38) or dystonic posture with hypoactivity (20 of 38), whereas all of the mice pretreated with the vehicle showed dystonic posture with hypoactivity (40 of 40).
Oxidative stress occurs early in the injured striatum after 3-NP injection
To determine whether oxidative stress occurs in the striatum after 3-NP injection, we evaluated the in situ level of HEt oxidation. As illustrated in Figure 1A, oxidized HEt signals were detected as small red cytosolic particles in the striatal lesion 1 hr after 3-NP injection (Fig. 1A, arrow). In the injured striatum 4 hr after 3-NP injection, oxidized HEt-positive cells with red signals extending into the cytosol were distributed more widely compared with the 1 hr samples (Fig. 1B). In the contralateral striatum, there were a few oxidized HEt signals, visible as small red particles, mainly in the perinuclear area (Fig. 1C, arrow). In some injured areas, morphologically identified endothelial cells in the microvessels showed oxidized HEt signals in the cytosol (Fig. 1D, arrowheads). Next, we quantified ROS production after 3-NP injection. Four hours after 3-NP injection, the percentage of cells with oxidized HEt was significantly increased in the injured striatum compared with the contralateral intact striatum or the injured striatum 1 hr after 3-NP injection (contralateral side, 3.77 ± 1.29; 1 hr, 10.26 ± 1.34; 4 hr, 62.74 ± 2.25; mean ± SD, percentage of cells with cytosolic oxidized HEt per total cells, p < 0.001).
Figure 1.
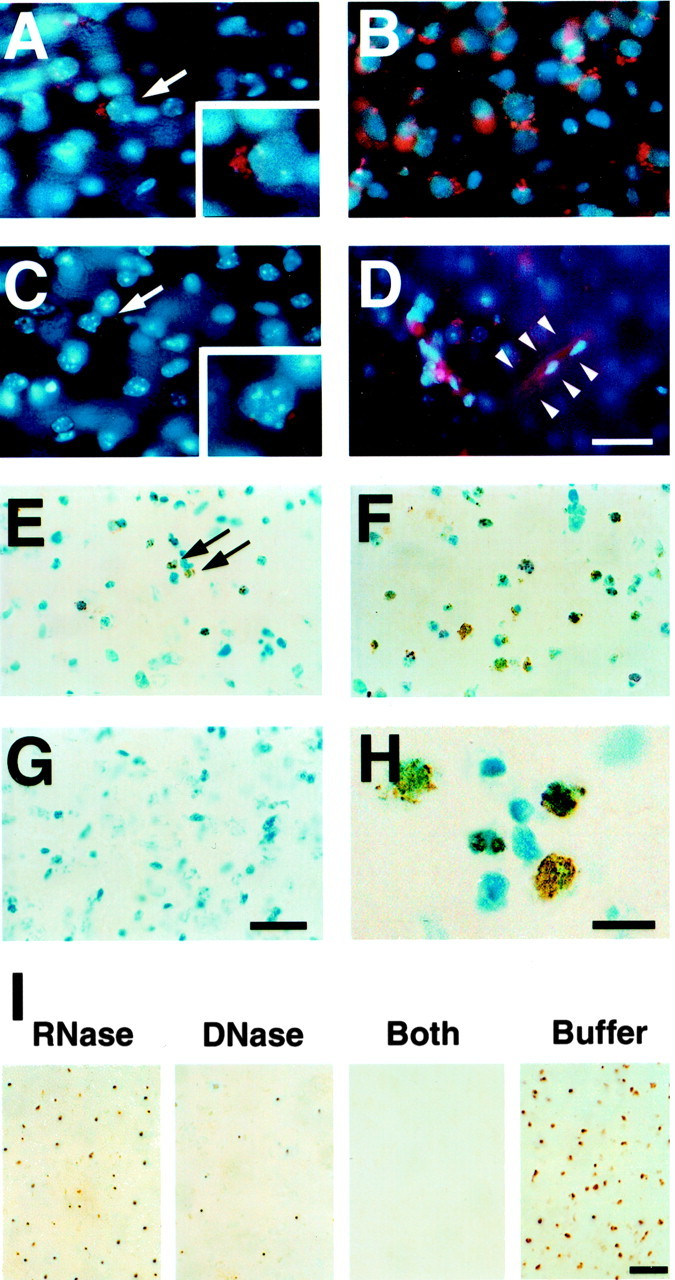
Representative findings for oxidative stress after 3-NP treatment. A-D, HEt in situ detection. A, Oxidized HEt appeared in the cytosol as red particles and was detected in the striatal lesion 1 hr after insult (arrow). In a detailed view, oxidized HEt extended into the cytosol (inset). B, Cytosolic oxidized HEt appeared as intense and widely distributed red signals 4 hr after 3-NP treatment. C, In the contralateral striatum, oxidized HEt (perinuclear location) was barely detected (arrow). In a detailed view, perinuclear oxidized HEt was seen (inset). D, Morphologically identified endothelial cells in microvessels showed oxidized HEt signals (arrowheads). E-I, 8OHG immunohistochemistry. E, 8OHG-positive cells were detected in the striatal lesion 4 hr after 3-NP treatment. F, Twenty-four hours after 3-NP administration, 8OHG-positive cells were widely detected. G, The corresponding contralateral striatum did not show any 8OHG immunoreactivity. H, Under high magnification, 8OHG immunoreactivity was present in a granular pattern in the cytosol and nucleus. I, Representative findings of 8OHG immunoreactivity on samples treated with RNase or DNase 24 hr after 3-NP administration. In the RNase-treated samples, 8OHG immunoreactivity was more intense than in the DNase-treated samples. The samples treated with RNase plus DNase did not show any immunoreactivity, whereas the buffer-treated samples had intense 8OHG immunoreactivity. RNase, RNase treatment; DNase, DNase treatment; Both, RNase plus DNase treatment; Buffer, buffer treatment. Scale bars: A-D, 20 μm; insets, 12 μm; E-G, 50 μm; H, 15 μm; I, 100 μm. For detailed values and comparisons, see Results.
To further confirm the oxidative stress induced by 3-NP injection, we used immunohistochemistry in the presence of 8OHG on nucleic acids (Zhang et al., 1999). 8OHG-positive cells were first detected in the injured striatum 4 hr after 3-NP treatment (9.40 ± 1.11; mean ± SD, percentage of 8OHG-positive cells to total cells) (Fig. 1E, arrows). 8OHG-positive cells were detected more extensively in the injured striatum 24 hr after 3-NP treatment (60.47 ± 2.91; mean ± SD, percentage of 8OHG-positive cells to total cells) (Fig. 1F). In contrast, in the contralateral striatum, no 8OHG-positive cells were detected (Fig. 1G). At high magnification, 8OHG immunoreactivity was observed as a granular pattern in the nucleus and cytoplasm (Fig. 1H). To identify the targets of 8OHG oxidation, we treated sections obtained from brains subjected to 3-NP treatment for 24 hr with RNase, DNase, and RNase plus DNase and performed 8OHG immunohistochemistry. As illustrated in Figure 1I, the RNase-treated samples showed an increased density of 8OHG immunoreactivity at low magnification compared with the DNase-treated samples. The RNase plus DNase-treated samples had no 8OHG immunoreactivity, whereas the untreated sections showed an intense density of 8OHG immunoreactivity. These data confirm that nucleic acids, and DNA in particular, were targets of ROS after 3-NP injection.
MMPs are involved in 3-NP-induced BBB disruption
Four hours after 3-NP injection, Evans blue leakage was detected as a red fluorescence in the injured striatum (Fig. 2A). The intensity and extension of Evans blue extravasation was more pronounced 8 hr after 3-NP treatment (Fig. 2B) and remained so until 24 hr (data not shown). In contrast, we observed only a mild leakage of Evans blue at 8 hr in the injured striatum of mice treated with the MMP inhibitor (Fig. 2C), and results at other time points were no different (data not shown). We did not detect any Evans blue leakage in the contralateral striatum (Fig. 2D). As illustrated in Figure 2E, quantitative measurement of Evans blue revealed low levels of extravasation in the control saline-injected mice (4 hr, 0.78 ± 0.09; 8 hr, 0.98 ± 0.77; 24 hr, 0.95 ± 0.16; mean ± SD of μg/striatum; no significant difference at each time point, by ANOVA). Four hours after 3-NP injection, the vehicle-treated mice showed a significantly increased amount of Evans blue leakage, whereas the inhibitor-treated mice had no significant change in Evans blue extravasation compared with the controls (vehicle, 3.65 ± 0.84; MMP inhibitor, 0.85 ± 0.24; mean ± SD of μg/striatum, p < 0.001) (Fig. 2E). In the vehicle-treated mice, Evans blue extravasation was more intense at 8 hr (4.40 ± 0.72; mean ± SD of μg/striatum, p < 0.001) and persisted 24 hr (3.69 ± 0.69; mean ± SD of μg/striatum, p < 0.001) after 3-NP injection. The inhibitor-treated mice had no significant change in Evans blue extravasation over time in the injured striatum (8 hr, 0.659 ± 0.12; 24 hr, 0.718 ± 0.17; mean ± SD of μg/striatum, p > 0.05) (Fig. 2E). At each time point, the MMP inhibitor significantly attenuated Evans blue extravasation in the injured striatum compared with vehicle treatment (ANOVA) (Fig. 2E).
Figure 2.

Representative findings of Evans blue extravasation in the striatum after 3-NP treatment. A, Evans blue extravasation in fixed samples. Evans blue extravasation was widely detected as a red fluorescence in the injured striatum 4 hr after 3-NP treatment. B, Eight hours after 3-NP treatment, Evans blue leakage intensified and extended to the entire striatum. All cells were stained with Evans blue. C, Inhibitor-treated mice showed only a weak Evans blue extravasation, located mainly around the vessels. D, Corresponding contralateral striatum did not show any Evans blue leakage. E, Quantitative assay of Evans blue leakage in the striatum 4, 8, and 24 hr after 3-NP injection. Values are mean ± SD of the amount of Evans blue in the striatum. Control (Ctrl) amount of Evans blue in the striatum 4, 8, and 24 hr after saline injection. Data in respective time points indicate the amount of Evans blue in the injured striatum of the vehicle- or MMP inhibitor-treated mice (n = 6-10). Asterisks indicate a significant decrease in Evans blue leakage in the MMP inhibitor-treated mice compared with the vehicle-treated animals (p < 0.001; Fisher's protected least-significant difference test). Scale bar, 100 μm.
MMPs contribute to striatal lesion volume and swelling after 3-NP treatment
To investigate the involvement of MMPs in striatal damage caused by 3-NP, we compared striatal lesion volume between the mice treated with the MMP inhibitor and the vehicle 24 hr after 3-NP treatment. Striatal enlargement was also measured to evaluate swelling related to BBB disruption. As illustrated in Figure 3A, the vehicle-treated mice had large lesions widely distributed in the entire striatum, and the MMP inhibitor-treated mice had small lesions localized in the lateral striatum. In contrast, the saline-injected control mice had only needle-track damage at the site of injection (Fig. 3A, arrowheads). Next, we assessed striatal enlargement by calculating the percentage of injured striatal volume to contralateral striatal volume 24 hr after 3-NP treatment. As shown in Figure 3B, the inhibitor-treated mice had significantly less striatal enlargement compared with the vehicle-treated mice (119.43 ± 6.57 and 10.92 ± 0.32; vehicle- and inhibitor-treated mice, respectively; mean ± SD, percentage of striatal enlargement, p < 0.0001). To determine the actual lesion volume after 3-NP injection, we used the percentage of lesion volume to the injured striatal volume to correct for edema. The MMP inhibitor significantly attenuated the actual lesion volume in the injured striatum, although this effect was smaller than the reduction in striatal enlargement (Fig. 3C) (68.19 ± 2.25 or 21.92 ± 0.37; vehicle- or inhibitor-treated mice, respectively; mean ± SD, percentage of lesion volume, p < 0.001).
Figure 3.

Striatal lesion volume and swelling in MMP inhibitor- or vehicle-treated mice 24 hr after 3-NP injection. A, Vehicle-treated mice had a large unstained lesion extending to the entire striatum with cresyl violet staining (left panel). Inhibitor-treated mice had a localized and small unstained lesion with cresyl violet staining (middle panel). Control saline-injected mice showed only restricted damage around the needle track (right panel). B, C, Quantitative assay of striatal swelling and lesion volume in the vehicle- or MMP inhibitor-treated mice after 3-NP treatment. Values are mean ± SD of the percentage of striatal enlargement (B) or lesion volume (C). Data indicate percentage of injured striatal or lesion volume to corresponding contralateral striatal volume (n = 8 each). Asterisks indicate a significant decrease in swelling or lesion volume in MMP inhibitor-treated mice compared with vehicle-treated mice (p < 0.0001 in striatal enlargement; p < 0.001 in lesion volume; unpaired t test). Scale bar, 500 μm.
MMP-9 and -2 zymography in the striatum after 3-NP treatment
Latent (pro-MMP) and activated (MMP) forms were measured using the zymography technique after a gelatin affinity-based purification, in which pro-MMP-9 and MMP-9 appeared as light bands on a blue background with molecular weights of ∼96 kDa and 88 kDa, respectively, whereas pro-MMP-2 was identified at 72 kDa. These are in agreement with our previous report (Gasche et al., 1999). Low levels of pro-MMP-9 as well as pro-MMP-2 were observed in the contralateral or control saline-injected striatum (Fig. 4A). Pro-MMP-9 started to increase 2 hr after 3-NP injection and reached its maximal elevation at 24 hr (Fig. 4A,B). The activated form of MMP-9 appeared only 4 hr after 3-NP and persisted until 24 hr (Fig. 4 A,B). In contrast, the pro-MMP-2 increase was significant only after 8 and 24 hr (Fig. 4A,C), and no activated form of MMP-2 was ever observed (Fig. 4A,C). Mean values of MMP activity are summarized in Table 1.
Figure 4.

Zymographical analysis of the injured striatum after 3-NP injection. A, Representative findings of zymography after 3-NP treatment. Pro-MMP-9 or -2 or MMP-9 or -2 were observed as light bands on a dark background. Low levels of pro-MMP-9 as well as pro-MMP-2 were observed as light bands in the contralateral or control saline-injected striatum. Pro-MMP-9 markedly increased 2 hr after treatment and remained increased until 24 hr, whereas pro-MMP-2 increased 8 and 24 hr after treatment compared with contralateral samples. No activated form of either enzyme was present 1-2 hr after 3-NP treatment. After 4 hr, activated MMP-9 appeared in the injured striatum, but not MMP-2. hST, Human pro- and activated MMP-9 and human pro- and activated MMP-2 standards. B, C, Zymographic measurement of gelatinase activity in the injured striatum after 3-NP injection. Measurement of MMP-9 (B) or MMP-2 (C) activity in the injured striatum after 3-NP injection. Graphs show mean values (± SD) of optical densities (O.D.) expressed in arbitrary units (A.U.) and obtained from quantification of gelatinolytic bands. Asterisks indicate a significant increase in pro-MMP-9 or -2 compared with contralateral samples (n = 6-10 each; *p < 0.005; Fisher's protected least-significant difference test).
Table 1.
MMP-9 and MMP-2 activity in the striatum after 3-NP treatment in mice
|
End point |
Pro-MMP-9 |
MMP-9 |
Pro-MMP-2 |
|---|---|---|---|
| Contralateral | 50.25 (± 13.56) | 0 | 148.75 (± 32.48) |
| 1 hr | 41.25 (± 17.90) | 0 | 156.50 (± 31.99) |
| 2 hr | 133.50 (± 57.17)* | 0 | 225.05 (± 94.11) |
| 4 hr | 122.50 (± 58.51)* | 13.08 (± 11.32) | 212.25 (± 83.57) |
| 8 hr | 141.67 (± 58.51)* | 16.55 (± 12.55) | 301.38 (± 115.45)* |
| 24 hr
|
179.33 (± 78.24)*
|
23.00 (± 31.12)
|
334.13 (± 160.38)*
|
Mean values (± SD) of optical density representing gelatinolytic activity of MMP-9 and MMP-2 found in control samples (contralateral or saline-treated control regions) or 3-NP-treated striatal samples. Values are compared using ANOVA (Fisher's protected least-significant difference test). Pro-MMP-9 activity was significantly higher 2,4,8, and 24 hr after 3-NP treatment than in the control samples (*p<0.005). Pro-MMP-2 was significantly increased 8 and 24 hr after 3-NP treatment (*p<0.005) compared with contralateral.
MMP-9 is expressed early in the injured striatum after 3-NP treatment
To evaluate MMP-9 expression in the injured striatum after 3-NP treatment, we performed MMP-9 immunohistochemistry. Using a previously described in vivo method of MMP purification (Zhang and Gottschall, 1997), which allowed us to substantially increase the threshold for MMP-9 detection, our results demonstrated earlier detection of activated MMP-9 in the injured striatum, as well as detection of pro-MMP-9 in control samples compared with contradictory reports (Romanic et al., 1998; Rosenberg et al., 1998). MMP-9 expression was detected in the injured striatum 4 hr after 3-NP injection (Fig. 5A, arrowheads), whereas MMP-9 immunoreactivity was barely detected in the contralateral striatum (Fig. 5B). MMP-9 was observed in morphologically identified endothelial cells in the microvessel lesions (Fig. 5A, arrow). Twenty-four hours after 3-NP treatment, MMP-9 cellular expression was much more intense and extended to the entire striatal lesion (Fig. 5C, arrowheads). Moreover, extracellular localization of the enzyme was also observed in the injured striatum. Under high magnification, nuclear, cytosolic (Fig. 5D), and extracellular (Fig. 5E) MMP-9 was clearly observed. MMP-9 immunoreactivity was not detected in the specimens that were processed without the primary antibody (Fig. 5F). We also investigated the spatial relationship between oxidized HEt and MMP-9 by double staining. Four hours after 3-NP treatment, cells with oxidized HEt extended into the cytosol (Fig. 5G1, arrows) and showed MMP-9 cellular expression (Fig. 5G2, arrows). Next, we evaluated the spatial relationship between Evans blue extravasation and MMP-9. Twenty-four hours after 3-NP treatment, Evans blue extravasation was intensely detected as a red fluorescence, and cells in that area were stained with Evans blue (Fig. 5G3). In the same specimens, increased MMP-9 cellular expression was detected mainly in the Evans blue-stained cells (Fig. 5G4, arrowheads). These data confirm that MMP-9 expression has a spatial relationship with Evans blue leakage.
Figure 5.
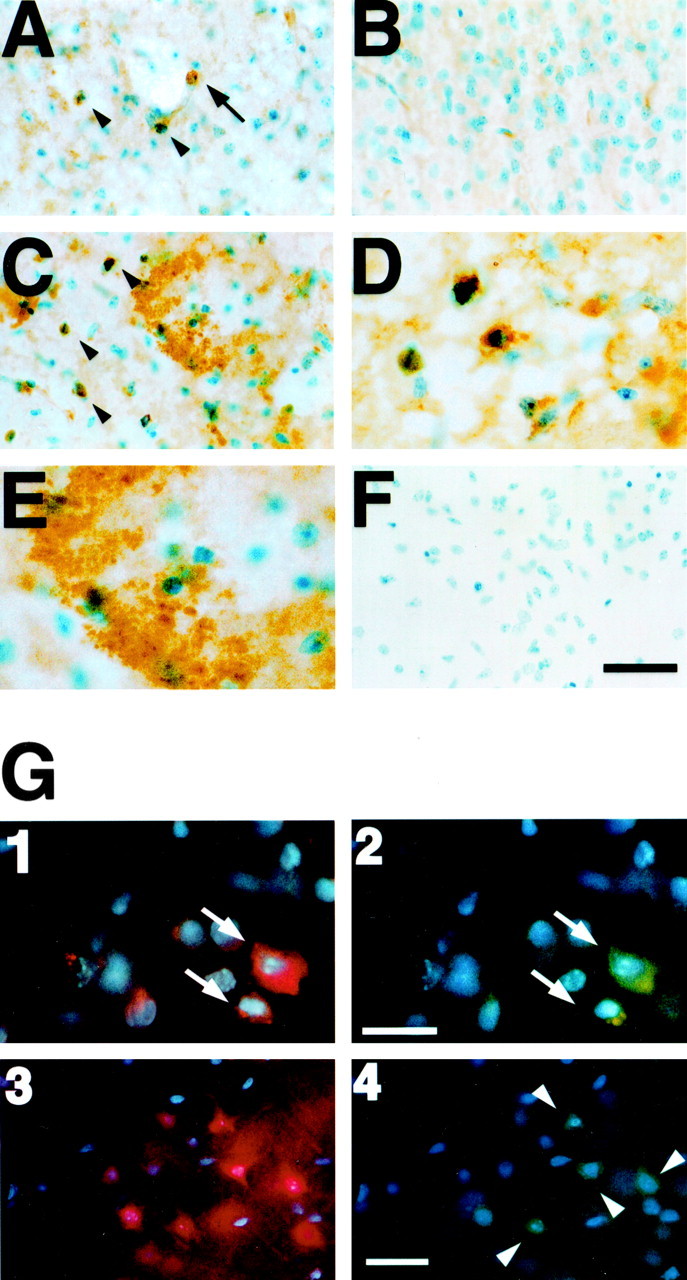
MMP-9 expression after 3-NP treatment and its spatial relationship to oxidized HEt or Evans blue leakage. A, Four hours after 3-NP treatment, increased MMP-9-labeled cells were detected (arrowheads), and some morphologically identified endothelial cells in microvessels were stained with MMP-9 in the injured striatum (arrow). B, In corresponding contralateral striatum, MMP-9 immunoreactivity was barely detected. C, Twenty-four hours after 3-NP treatment, cells with intense MMP-9 immunoreactivity were widely distributed, and extracellular MMP-9 was also observed. D, Under high magnification, cytosolic and cellular expression of MMP-9 was observed. E, Under high magnification, extracellular MMP-9 expression was observed. F, Negative control. G1, G2, Double staining for in situ HEt and MMP-9 in the injured striatum 4 hr after 3-NP treatment. G1, Cells with oxidized HEt extended into the cytosol in the injured striatum (arrows). G2, On the same specimen, increased MMP-9 immunoreactivity was detected in oxidized HEt-positive cells (arrows). G3, G4, Double staining for Evans blue and MMP-9 in the injured striatum 24 hr after 3-NP treatment. G3, Intensive Evans blue leakage was detected in the injured striatum, and cells were stained with Evans blue. G4, On the same specimen, increased MMP-9 immunoreactivity was observed in some cells, which were Evans blue positive. Scale bars: A-C, F, 30 μm; D, E, 15 μm; G1, G2,10 μm; G3, G4, 50 μm.
To further study MMP expression in the injured striatum after 3-NP treatment, we performed Western blot analysis of MMP-9 with the same polyclonal antibody used for immunohistochemistry. As illustrated in Figure 6A, pro-MMP-9 immunoreactivity was evident as bands with a molecular mass of ∼96 kDa in the contralateral striatum and was increased in the samples from the injured striatum 4 and 24 hr after 3-NP treatment. Activated MMP-9, as characterized by an 88 kDa band, was absent from the normal or contralateral striatum but was detected in the 4 hr samples and increased in the 24 hr samples. These data confirm the reactivity of the antibody to MMP-9 used in this study and that the activated form of MMP-9 increased after 3-NP treatment.
Figure 6.
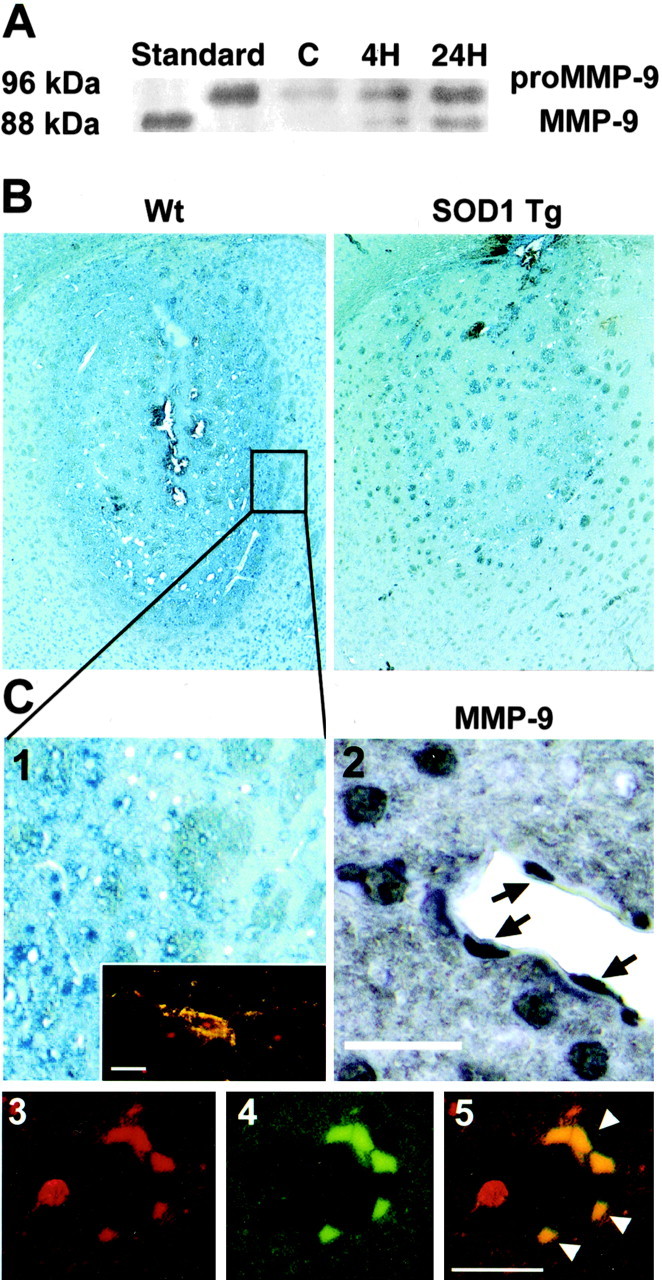
A, Western blot analysis of MMP-9 after 3-NP treatment. Analysis was performed with a rabbit polyclonal antibody against human MMP-9, and the results confirm the cross-reactivity with mouse MMP-9. Pro-MMP-9 was evident as 96 kDa characteristic bands in both the control and injured striatum 4 and 24 hr after 3-NP treatment. Activated MMP-9, as shown by the 88 kDa bands, was absent in the control but was detected in the injured striatum 4 and 24 hr after 3-NP administration. Similar results were obtained from independent studies (n = 4). C, Control (contralateral or saline-injected striatum). B, Photomicrographs of representative 3-NP-induced lesions (24 hr after intrastriatal injection) show that SOD1-Tg mice had significantly smaller lesions than their WT littermates. C1, Inset shows MMP-9 immunoreactivity (dark blue) in the outer rim of the lesion, which was significantly greater in the WT mice compared with the SOD1-Tg animals. C2, MMP-9 immunoreactivity (blue) in a WT animal 24 hr after 3-NP injection. C1, Inset shows double immunoreactivity (yellow) of MMP-9 and GFAP in the stellate glial cells. C3-C5, Double immunohistochemistry of CD31 (red) and MMP-9 (green). C3, CD31; C4, GFAP; C5, yellow (arrowheads) indicates immunoreactivity in endothelial cells. Scale bars: C1, C2, 25 μm; C1 inset, C3-5, 20 μm.
SOD1 protects against striatal swelling and injury with decreased MMP-9 expression after 3-NP treatment
To confirm the involvement of ROS in MMP-9-related striatal swelling and damage after 3-NP treatment, we first compared striatal lesion size and edema levels among the SOD1-Tg mice, SOD1-KO mice, and WT littermates 24 hr after insult. The SOD1-Tg mice had a significantly smaller lesion size compared with the WT littermates (38.8 ± 15.1 and 53.3 ± 10.3, respectively; p ≤ 0.05) (Fig. 6B), whereas the SOD1-KO mice showed a trend, albeit not statistically significant, toward increased damage (60.0 ± 14.0; p = 0.3). Edema levels were also significantly reduced in the SOD1-Tg mice compared with the WT littermates (21.8 ± 11.2 and 35.28 ± 11, respectively; p ≤ 0.05), whereas the SOD1-KO mice displayed significantly greater swelling (48.65 ± 17; p ≤ 0.05). Furthermore, normalized to lesion size, the SOD1-Tg animals showed a significantly smaller number of MMP-9- and MMP-2-positive cells 24 hr after 3-NP injection compared with the WT animals (MMP-9, 352 ± 57 and 510 ± 45, respectively, p ≤ 0.005; MMP-2, 379 ± 44 and 482 ± 39, respectively, p ≤ 0.005). MMP-9 immunoreactivity was clearly observed in endothelial cells and astrocytes (Fig. 6C1-5), as well as in neutrophils (data not shown).
Discussion
Our data demonstrate that ROS play an important role in 3-NP-induced striatal damage and that MMPs can promote BBB disruption and subsequent damage in this model. Furthermore, our results suggest that among MMPs, gelatinase B is the enzyme responsible for this pathophysiological involvement. Our conclusions are on the basis of the following: (1) MMP inhibition attenuated early Evans blue leakage, striatal swelling, and lesion volume in mice subjected to 3-NP toxicity; (2) activated MMP-9, which plays a pathophysiological role in BBB alteration, was observed as early as 4 hr after 3-NP treatment after increased levels of the MMP-9 pro-form in the injured striatum 2 hr after insult; (3) SOD1 overexpression protects against 3-NP-induced striatal swelling and damage along with MMP-9 expression, whereas SOD1 depletion exacerbates edema formation. These findings are supported by immunohistochemistry, Western blot analysis, and zymography. Furthermore, Evans blue extravasation, as an indicator of BBB disruption, showed a contemporary increase and spatial relationship with MMP-9 expression.
This is the first report to characterize the involvement of MMP-9 in early BBB disruption and striatal damage after 3-NP injection. Pro-MMP-9 as well as pro-MMP-2 was detected in the control striatum, and pro-MMP-9 was increased in the injured striatum as early as 2 hr after 3-NP treatment. Although there is no report of MMP expression in 3-NP-induced lesions, these results agree with our previous reports; both pro-MMP-2 and -9 were detected in the normal brain, and pro-MMP-9 was significantly increased as early as 2-3 hr after permanent and transient focal cerebral ischemia in mice (Fujimura et al., 1999; Gasche et al., 1999).
In the present study, oxidative stress occurred as early as 4 hr and nucleic acid oxidation was significantly increased 24 hr after 3-NP treatment using HEt in situ detection and 8OHG immunohistochemistry (Fig. 1). Expression of MMPs is regulated by multiple pathways such as inflammatory cytokines, oncogene products, and growth factors (Yong et al., 1998). It has been proposed that ROS regulate MMP activity in vitro (Shingu et al., 1994; Rajagopalan et al., 1996) as well as in vivo (Morita-Fujimura et al., 2000; Gasche et al., 2001; Gu et al., 2002). Oxidative stress plays a substantial role in 3-NP neurotoxicity. It has been reported that systemic 3-NP administration increases production of hydroxyl free radicals in the striatum (Schulz et al., 1996), that systemic 3-NP neurotoxicity is attenuated in SOD1-Tg mice (Beal et al., 1995), and that ROS-linked excitotoxicity is increased in the intact striatum of decorticated mice after 3-NP treatment (Kim et al., 2000a; Kim and Chan, 2001). Results from our current study using intrastriatal 3-NP administration in SOD1-Tg and SOD1-KO mice corroborate those findings. Overexpression of SOD1 protects striatal tissue from 3-NP-induced damage, whereas low levels of SOD1 activity exacerbate edema levels, suggesting that ROS production is an important component in this injury paradigm. There have also been reports that oxidative stress promotes BBB disruption in vivo (Chan et al., 1984, 1991; Kondo et al., 1997; Gasche et al., 2001; Aoki et al., 2002; Grzeschik et al., 2003) and in vitro (Imaizumi et al., 1996; Schroeter et al., 1999; Mujumdar et al., 2001; Zaragoza et al., 2002). The present data show that oxidized HEt signals, which indicate ROS production, are spatially correlated with MMP-9 cellular expression 4 hr after 3-NP treatment (Fig. 5). Pro-MMP-9 increased as early as 2 hr after 3-NP injection in the striatum along with increased levels of oxidized HEt signals, but the activated form of MMP-9 appeared at 4 hr, after maximal ROS production in the affected striatum (Figs. 1, 4, 6A). Moreover, MMP-9 immunoreactivity was decreased in SOD1-Tg mice (Fig. 6B), suggesting that oxidative stress may be involved, in part, in the mechanisms of MMP-9 induction and activation and subsequent BBB disruption in the injured striatum after 3-NP treatment.
Our data showing that the appearance of activated MMP-9 (Figs. 4, 6) a few hours after 3-NP treatment was accompanied by early BBB disruption (Fig. 2), and that MMP-9 expression showed a spatial relationship with Evans blue extravasation (Fig. 5), suggest that MMP-9 may play an active role in producing early vasogenic edema caused by 3-NP treatment in the present animal model. 3-NP was administered directly into the striatum to induce striatal degeneration with the greatest reproducibility, because lesions induced by systemic 3-NP treatment have been reported to be quite variable (Beal et al., 1993). Our recent reports have shown that 3-NP intrastriatal injection produces BBB damage or striatal swelling in the rat as early as 3-6 hr after treatment (Sato et al., 1997, 1998), suggesting that vasogenic edema may occur earlier than cellular death in the injured striatum in this model. The present results on BBB alteration are also in accordance with recent data showing that acute systemic 3-NP intoxication (3-NP, 20 mg/kg, s.c.) induces striatal vasogenic edema, as revealed by diffusion-weighted magnetic resonance imaging and IgG detection, within a few hours after a second systemic injection (Nishino et al., 1997). A study using chronic (4 weeks) 3-NP intoxication (total dose: 180 mg/kg, s.c.) has also shown prominent IgG extravasation in the injured striatum. Interestingly, IgG immunoreactivity is evident even in the absence of striatal lesions (Nishino et al., 1995), suggesting that mild BBB disruption precedes cell death in the striatum.
Regulation of MMP activity is a very tight process involving natural tissue inhibitors of MMPs (TIMPs). MMP-9 activity is under the control of TIMP-1, the expression of which is not addressed in the current study. Thus, although we cannot exclude the possibility that TIMP-1 modulates MMP-9 activity in 3-NP-induced injury, our study shows that MMP inhibition prevents early BBB opening and attenuates striatal swelling and lesion volume 24 hr after insult (Fig. 3). These results, together with those showing the temporal-spatial relationship between MMP-9 expression and Evans blue extravasation, indicate that MMP-9 may be involved in early BBB opening and progressive striatal tissue disruption. The reduced edema levels in the SOD1-Tg animals concomitant with decreased MMP-9 expression further illustrate this relationship. It is important to note that, although BBB disruption was prevented almost totally by MMP inhibition, the lesion volume was reduced by ∼65%. This certainly illustrates the involvement of other pathophysiological pathways leading to progressive striatal injury after 3-NP treatment. Indeed, oxidative stress-related mitochondrial dysfunction is known to contribute directly to irreversible cellular damage in this setting. According to this concept, we can speculate that MMP-9, by promoting BBB disruption, contributes to acute striatal tissue damage, along with oxidative cellular damage. It is noteworthy that MMP-9 involvement in pathologies involving ROS, such as stroke, has been demonstrated in a model of focal ischemia in MMP-9-deficient mice (Asahi et al., 2000). There are few treatments for progressive neurodegenerative disorders. Therefore, an increased understanding of the mechanisms involved in neurodegenerative diseases may provide strategies to ameliorate them. In 3-NP-induced neurotoxicity, there is some evidence suggesting the involvement of astrocytes and endothelial cells, which are components of the BBB. Although 3-NP typically provokes neuronal cell death, some studies report astrocytic dysfunction (Deshpande et al., 1997; Fukuda et al., 1998) and electron lucency of endothelial cell cytoplasm, as well as striatal white matter changes that include axonal swelling, dendritic myelin changes, and tissue sponge changes (Gould and Gustine, 1982; Hamilton and Gould, 1987; Beal et al., 1993). Recently, it was reported that extracellular proteolytic activation of MMP-9 by nitric oxide can disrupt the extracellular matrix and may contribute to cell detachment and lead to a form of apoptosis-like death (Gu et al., 2002). Therefore, we can speculate that MMP-9 activation after 3-NP treatment produces BBB disruption, which causes acute striatal damage and may promote cell death signaling by detachment of neuronal cells from supporting cells such as endothelial cells or astrocytes. Further studies are needed to elucidate the respective role of these cells in MMP expression or activation in the setting of 3-NP-induced neurodegeneration.
Although MMP and the BBB have not yet been evaluated in HD, there have been reports that in AD, which is the most common neurodegenerative disease, calcium-dependent MMP is expressed at higher levels in the hippocampus compared with controls (Backstrom et al., 1992) and that MMPs are increased in enriched astrocyte and mixed hippocampal cultures after treatment with β-amyloid peptides, which is a pathognomonic finding in the brains of patients with AD (Deb and Gottschall, 1996). In the pathophysiology of AD, BBB disruption is a clear contributing factor (Huber et al., 2001). Recently, it was reported that some HD brains showed an Alzheimer-type pathological lesion (Jellinger, 1998). Thus, circumstantial evidence, such as experimental results and a resemblance between some cases of AD and HD, indicates that the role of MMP in neurodegenerative diseases, including HD, may require attention.
Results from our study lend strong support to the notion that ROS production followed by MMP-9 activation and subsequent BBB disruption play a key pathophysiological role in 3-NP-induced injury. Additional studies are needed to better understand the involvement of MMPs in neurodegenerative disorders and to eventually develop new therapeutic strategies targeting this family of enzymes.
Footnotes
This work was supported by National Institutes of Health Grants NS 14534, NS 25372, NS 36147, and NS 38653, an American Heart Association Bugher Foundation award, BK21 Project for Medical Science, Yonsei University, the Swiss National Science Foundation (Grant 3200B0-100738), and the R&D fund of the Geneva University Hospital. P.H.C. is a recipient of the Javits Neuroscience Investigator Award. We thank Jane O. Kim, Liza Reola, and Bernard Calagui for technical assistance, Paige Bracci for statistical analysis, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.
Correspondence should be addressed to Dr. Pak H. Chan, Neurosurgical Laboratories, Stanford University, 1201 Welch Road, MSLS P304, Stanford, CA 94305-5487. E-mail: phchan@leland.stanford.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238733-10$15.00/0
References
- Albin RL, Greenamyre JT ( 1992) Alternative excitotoxic hypotheses. Neurology 42: 733-738. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Dedeoglu A, Albers DW, Klivenyi P, Carlson EJ, Epstein CJ, Beal MF ( 2001) Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp Neurol 167: 189-195. [DOI] [PubMed] [Google Scholar]
- Aoki T, Sumii T, Mori T, Wang X, Lo EH ( 2002) Blood-brain barrier disruption and matrix metalloproteinase-9 expression during reperfusion injury: mechanical versus embolic focal ischemia in spontaneously hypertensive rats. Stroke 33: 2711-2717. [DOI] [PubMed] [Google Scholar]
- Asahi M, Asahi K, Jung J-C, del Zoppo GJ, Fini ME, Lo EH ( 2000) Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab 20: 1681-1689. [DOI] [PubMed] [Google Scholar]
- Backstrom JR, Miller CA, Tokes ZA ( 1992) Characterization of neutral proteinases from Alzheimer-affected and control brain specimens: identification of calcium-dependent metalloproteinases from the hippocampus. J Neurochem 58: 983-992. [DOI] [PubMed] [Google Scholar]
- Beal MF ( 1995) Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 38: 357-366. [DOI] [PubMed] [Google Scholar]
- Beal MF ( 2000) Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci 23: 298-304. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT ( 1993) Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 13: 4181-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Henshaw R, Matthews RT, Chan PH, Kowall NW, Epstein CJ, Schulz JB ( 1995) 3-Nitropropionic acid neurotoxicity is attenuated in copper/zinc superoxide dismutase transgenic mice. J Neurochem 65: 919-922. [DOI] [PubMed] [Google Scholar]
- Benov L, Sztejnberg L, Fridovich I ( 1998) Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 25: 826-831. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordán J, Lee CC, Miller RJ ( 1996) Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci 16: 1324-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH, Schmidley JW, Fishman RA, Longar SM ( 1984) Brain injury, edema, and vascular permeability changes induced by oxygen-derived free radicals. Neurology 34: 315-320. [DOI] [PubMed] [Google Scholar]
- Chan PH, Yang GY, Chen SF, Carlson E, Epstein CJ ( 1991) Cold-induced brain edema and infarction are reduced in transgenic mice overexpressing CuZn-superoxide dismutase. Ann Neurol 29: 482-486. [DOI] [PubMed] [Google Scholar]
- Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ ( 1998) Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci 18: 8292-8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CJ, Edmondson DE, Singer TP ( 1979) Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem 254: 5161-5167. [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P ( 1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262: 689-695. [DOI] [PubMed] [Google Scholar]
- Deb S, Gottschall PE ( 1996) Increased production of matrix metalloproteinases in enriched astrocyte and mixed hippocampal cultures treated with beta-amyloid peptides. J Neurochem 66: 1641-1647. [DOI] [PubMed] [Google Scholar]
- Deshpande SB, Fukuda A, Nishino H ( 1997) 3-Nitropropionic acid increases the intracellular Ca2+ in cultured astrocytes by reverse operation of the Na+-Ca2+ exchanger. Exp Neurol 145: 38-45. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Halley M, Busto R, Globus MY ( 1992) Intraventricular infusion of N-methyl-d-aspartate. 1. Acute blood-brain barrier consequences. Acta Neuropathol (Berl) 84: 621-629. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH ( 1999) Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res 842: 92-100. [DOI] [PubMed] [Google Scholar]
- Fukuda A, Deshpande SB, Shimano Y, Nishino H ( 1998) Astrocytes are more vulnerable than neurons to cellular Ca2+ overload induced by a mitochondrial toxin, 3-nitropropionic acid. Neuroscience 87: 497-507. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Fujimura M, Morita-Fujimura Y, Copin J-C, Kawase M, Massengale J, Chan PH ( 1999) Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab 19: 1020-1028. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Copin J-C, Sugawara T, Fujimura M, Chan PH ( 2001) Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab 21: 1393-1400. [DOI] [PubMed] [Google Scholar]
- Gould DH, Gustine DL ( 1982) Basal ganglia degeneration, myelin alterations, and enzyme inhibition induced in mice by the plant toxin 3-nitropropionic acid. Neuropathol Appl Neurobiol 8: 377-393. [DOI] [PubMed] [Google Scholar]
- Greene JG, Sheu SS, Gross RA, Greenamyre JT ( 1998) 3-Nitropropionic acid exacerbates N-methyl-d-aspartate toxicity in striatal culture by multiple mechanisms. Neuroscience 84: 503-510. [DOI] [PubMed] [Google Scholar]
- Grzeschik SM, Maier CM, Chan PH ( 2003) Effects of cold injury-induced trauma in manganese superoxide dismutase-deficient mice. J Neurotrauma, in press. [DOI] [PubMed]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA ( 2002) S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297: 1186-1190. [DOI] [PubMed] [Google Scholar]
- Hamilton BF, Gould DH ( 1987) Correlation of morphologic brain lesions with physiologic alterations and blood-brain barrier impairment in 3-nitropropionic acid toxicity in rats. Acta Neuropathol (Berl) 74: 67-74. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Mann DM, Wester P, Winblad B ( 1986) An integrative hypothesis concerning the pathogenesis and progression of Alzheimer's disease. Neurobiol Aging 7: 489-502. [DOI] [PubMed] [Google Scholar]
- He F, Zhang S, Qian F, Zhang C ( 1995) Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid). Neurology 45: 2178-2183. [DOI] [PubMed] [Google Scholar]
- Huber JD, Egleton RD, Davis TP ( 2001) Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci 24: 719-725. [DOI] [PubMed] [Google Scholar]
- Imaizumi S, Kondo T, Deli MA, Gobbel G, Joó F, Epstein CJ, Yoshimoto T, Chan PH ( 1996) The influence of oxygen free radicals on the permeability of the monolayer of cultured brain endothelial cells. Neurochem Int 29: 205-211. [DOI] [PubMed] [Google Scholar]
- Jellinger KA ( 1998) Alzheimer-type lesions in Huntington's disease. J Neural Transm 105: 787-799. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Golde TE, Cohen ML, Younkin SG ( 1991) Serum amyloid P in Alzheimer's disease. Implications for dysfunction of the blood-brain barrier. Ann NY Acad Sci 640: 145-148. [DOI] [PubMed] [Google Scholar]
- Kim GW, Chan PH ( 2001) Oxidative stress and neuronal DNA fragmentation mediate age-dependent vulnerability to the mitochondrial toxin, 3-nitropropionic acid, in the mouse striatum. Neurobiol Dis 8: 114-126. [DOI] [PubMed] [Google Scholar]
- Kim GW, Chan PH ( 2002) Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab 22: 798-809. [DOI] [PubMed] [Google Scholar]
- Kim GW, Copin J-C, Kawase M, Chen SF, Sato S, Gobbel GT, Chan PH ( 2000a) Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab 20: 119-129. [DOI] [PubMed] [Google Scholar]
- Kim GW, Sugawara T, Chan PH ( 2000b) Involvement of oxidative stress and caspase-3 in cortical infarction after photothrombotic ischemia in mice. J Cereb Blood Flow Metab 20: 1690-1701. [DOI] [PubMed] [Google Scholar]
- Kim GW, Lewén A, Copin J-C, Watson BD, Chan PH ( 2001) The cytosolic antioxidant, copper/zinc superoxide dismutase, attenuates blood-brain barrier disruption and oxidative cellular injury after photothrombotic cortical ischemia in mice. Neuroscience 105: 1007-1018. [DOI] [PubMed] [Google Scholar]
- Kim GW, Kondo T, Noshita N, Chan PH ( 2002) Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice. Implications for the production and role of superoxide radicals. Stroke 33: 809-815. [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Stetler-Stevenson WG ( 1994) Quantitative zymography: detection of picogram quantities of gelatinases. Anal Biochem 218: 325-329. [DOI] [PubMed] [Google Scholar]
- Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH ( 1997) Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci 17: 4180-4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Backstrom JR, Cullen MJ, Miller CA, Atkinson RD, Tokes ZA ( 1996) Matrix metalloproteinases in the neocortex and spinal cord of amyotrophic lateral sclerosis patients. J Neurochem 67: 251-259. [DOI] [PubMed] [Google Scholar]
- Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M ( 1991) 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci 18: 492-498. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Didion SP ( 1996) Glutamate-induced disruption of the blood-brain barrier in rats. Role of nitric oxide. Stroke 27: 965-969. [DOI] [PubMed] [Google Scholar]
- Miller RD, Monsul NT, Vender JR, Lehmann JC ( 1996) NMDA- and endothelin-1-induced increases in blood-brain barrier permeability quantitated with Lucifer yellow. J Neurol Sci 136: 37-40. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Kimura T, Hirata S, Fujise N, Ono T, Ishizuka K, Nakabayashi J ( 2000) Role of blood vessels in producing pathological changes in the brain with Alzheimer's disease. Ann NY Acad Sci 903: 46-54. [DOI] [PubMed] [Google Scholar]
- Morita-Fujimura Y, Fujimura M, Gasche Y, Copin J-C, Chan PH ( 2000) Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury-induced brain trauma. J Cereb Blood Flow Metab 20: 130-138. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Aru GM, Tyagi SC ( 2001) Induction of oxidative stress by homocyst(e)ine impairs endothelial function. J Cell Biochem 82: 491-500. [DOI] [PubMed] [Google Scholar]
- Nag S ( 1992) Vascular changes in the spinal cord in N-methyl-d-aspartate-induced excitotoxicity: morphological and permeability studies. Acta Neuropathol (Berl) 84: 471-477. [DOI] [PubMed] [Google Scholar]
- Nishino H, Shimano Y, Kumazaki M, Sakurai T ( 1995) Chronically administered 3-nitropropionic acid induces striatal lesions attributed to dysfunction of the blood-brain barrier. Neurosci Lett 186: 161-164. [DOI] [PubMed] [Google Scholar]
- Nishino H, Kumazaki M, Fukuda A, Fujimoto I, Shimano Y, Hida H, Sakurai T, Deshpande SB, Shimizu H, Morikawa S, Inubushi T ( 1997) Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-brain barrier: involvement of dopamine toxicity. Neurosci Res 27: 343-355. [DOI] [PubMed] [Google Scholar]
- Nishino H, Hida H, Kumazaki M, Shimano Y, Nakajima K, Shimizu H, Ooiwa T, Baba H ( 2000) The striatum is the most vulnerable region in the brain to mitochondrial energy compromise: a hypothesis to explain its specific vulnerability. J Neurotrauma 17: 251-260. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS ( 1996) Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest 98: 2572-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown Jr RH, Scott RW, Snider WD ( 1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13: 43-47. [DOI] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC ( 1998) Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29: 1020-1030. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE ( 1998) Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 29: 2189-2195. [DOI] [PubMed] [Google Scholar]
- Sato S, Gobbel GT, Honkaniemi J, Li Y, Kondo T, Murakami K, Sato M, Copin J-C, Chan PH ( 1997) Apoptosis in the striatum of rats following intraperitoneal injection of 3-nitropropionic acid. Brain Res 745: 343-347. [DOI] [PubMed] [Google Scholar]
- Sato S, Gobbel GT, Honkaniemi J, Li Y, Kondo T, Murakami K, Sato M, Copin J-C, Sharp FR, Chan PH ( 1998) Decreased expression of bcl-2 and bcl-x mRNA coincides with apoptosis following intracerebral administration of 3-nitropropionic acid. Brain Res 808: 56-64. [DOI] [PubMed] [Google Scholar]
- Schroeter ML, Mertsch K, Giese H, Muller S, Sporbert A, Hickel B, Blasig IE ( 1999) Astrocytes enhance radical defense in capillary endothelial cells constituting the blood-brain barrier. FEBS Lett 449: 241-244. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Henshaw DR, MacGarvey U, Beal MF ( 1996) Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem Int 29: 167-171. [DOI] [PubMed] [Google Scholar]
- Shingu M, Isayama T, Yasutake C, Naono T, Nobunaga M, Tomari K, Horie K, Goto Y ( 1994) Role of oxygen radicals and IL-6 in IL-1-dependent cartilage matrix degradation. Inflammation 18: 613-623. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Noshita N, Lewén A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH ( 2002) Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci 22: 209-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR ( 1998) Matrix metalloproteinases and diseases of the CNS. Trends Neurosci 21: 75-80. [DOI] [PubMed] [Google Scholar]
- Zaragoza C, Soria E, López E, Browning D, Balbín M, López-Otín C, Lamas S ( 2002) Activation of the mitogen activated protein kinase extracellular signal-regulated kinase 1 and 2 by the nitric oxide-cGMP-cGMP-dependent protein kinase axis regulates the expression of matrix metalloproteinase 13 in vascular endothelial cells. Mol Pharmacol 62: 927-935. [DOI] [PubMed] [Google Scholar]
- Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ ( 1999) Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154: 1423-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JW, Gottschall PE ( 1997) Zymographic measurement of gelatinase activity in brain tissue after detergent extraction and affinity-support purification. J Neurosci Methods 76: 15-20. [DOI] [PubMed] [Google Scholar]