

Abstract
Stress is usually correlated with an increased release of glucocorticoids from the adrenal glands. Within the hippocampus, a structure long known to be involved in spatial learning, two corticosterone-binding receptors are identified: the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR). Activation of these receptors impairs or facilitates hippocampal long-term potentiation (LTP), respectively. Stress elicited by behavioral manipulations may interfere with cognitive modulations of LTP during learning experiments. Here, we explore the influence of two stress-inducing procedures, handling and swimming, on the maintenance of dentate gyrus LTP in the rat induced by a weak tetanization of the perforant path. Manipulations started 15 min after tetanization. Handling alone resulted in a complete reversal of LTP. Handling followed by a 2 min swim in a water tank elicited prolonged protein synthesis but not β-adrenergic-dependent LTP compared with either control or handled animals. Blockade of the GRs but not of the MRs prevented the reversal of LTP by handling. Inactivation of the MRs but not of the GRs hindered LTP prolongation by swimming. Because the activated receptor complexes act as transcription factors, MR- and GR-related proteins may play a role in the maintenance of LTP. The data suggest a complex interplay of corticosterone-binding receptors on modulations of hippocampal LTP and thus, of stress on learning and functional plasticity in general.
Keywords: hippocampal long-term potentiation, swim stress, corticosterone, glucocorticoid receptor, mineralocorticoid receptor, dentate gyrus, β-adrenoceptors, protein synthesis, handling, reinforcement, early LTP, late LTP
Introduction
Stress is well known to modulate hippocampal long-term potentiation (LTP) as well as learning and memory (Kim and Diamond, 2002) and is usually evoked when animals are introduced into an apparatus used for testing spatial cognition (Morris, 1984; Kant et al., 1988; Sandi et al., 1997; Akirav et al., 2001). A bimodal effect of stress on learning and memory has been proposed (Diamond et al., 1992; Cahill and McGaugh, 1998; McGaugh, 2000; McGaugh and Roozendaal, 2002). At the cellular level, elevated stress impairs LTP in the CA1 and dentate gyrus (DG) regions of the hippocampus, both in vivo (Filipini et al., 1991; Pavlides et al., 1993; Xu et al., 1997; Pavlides et al., 2002) and in vitro (Foy et al., 1987; Shors et al., 1989; Kim et al., 1996; Zhou et al., 2000). Stress is correlated by increased release of corticosterone from the adrenal glands. Central actions of corticosterone are mediated by two corticosterone-binding receptors: the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR), the latter showing a 10-fold higher affinity for corticosterone than the former (De Kloet et al., 1993; van Steensel et al., 1996). Both receptor types are found in the hippocampus, which is also the brain region with the highest level of glucocorticoid receptor binding (McEwen et al., 1968; Reul and De Kloet, 1985; Reul et al., 1989). Whereas activation of GRs impairs hippocampal LTP, activation of MRs results in the facilitation of LTP (Pavlides et al., 1995; Kim and Yoon, 1998; Smigra et al., 1998; Pavlides and McEwen, 1999). Both receptor complexes regulate the expression of a variety of genes by directly binding to the DNA or via protein-protein interactions with other transcription factors (Diamond et al., 1990; Zhang et al., 1991; Cato et al., 1992a,b; Drouin et al., 1992, Herman, 1993; Malkoski et al., 1997).
Long-term potentiation can be divided into two major phases: protein synthesis-independent early LTP (3-4 hr) and protein synthesis-dependent late LTP with a duration of at least up to 8 hr (Krug et al., 1984; Frey et al., 1988, 2001; Matthies et al., 1990). The combination of a weakly stimulated input (eliciting early LTP) with a strong tetanization (inducing late LTP) of a second independent input within a time window of 30 min results in a prolongation of early LTP into late LTP in the first input, which is protein synthesis dependent (Frey and Morris, 1997, 1998). This late-associative interplay between two separate synaptic inputs has been explained by processes of “synaptic tagging” (Frey and Morris, 1997): the formation of a transient synaptic tag at a weakly stimulated input that has the potential to capture and process plasticity proteins whose synthesis is induced by a strong input, consequently reinforcing early LTP into long-lasting late LTP.
Similar behavioral reinforcements of an electrically induced early LTP in vivo by appetitive or aversive emotional stimuli were found under mild stress conditions (Izquierdo and Medina, 1995; Seidenbecher et al., 1997), depending on protein synthesis and on β-adrenergic activation. Akirav and Richter-Levin (2002) point out that a fast excitatory input from the basolateral amygdala (BLA) under high stress can serve as an “emotional tag,” resulting in an enhancement of DG-LTP.
This study aimed to examine stress effects on early DG-LTP within a vulnerable time window, with emphasis on the activation of corticosterone-binding and β-adrenergic receptors. Stress was elicited by behavioral manipulations (i.e., handling and swimming), which are related to spatial training paradigms.
Materials and Methods
Electrode and cannula implantation
Male Wistar rats (8 weeks of age) were anesthetized with Nembutal (40 mg/kg, i.p.). A monopolar recording electrode was implanted stereotaxically into the granule cell layer of the dentate gyrus [coordinates: antero-posterior (AP), -2.8; lateral (L), 1.8 from bregma, 3.2-3.5 ventral from dura] and a bipolar stimulation electrode was implanted into the perforant path (coordinates: AP, -6.9; L, 4.1, 2.2-2.5 ventral from dura) of the right hemisphere; coordinates are based on the atlas of Paxinos and Watson (1998). Each electrode consisted of an insulated stainless-steel wire 125 μm in diameter. During preparation, test pulses were delivered to optimize the population-spike amplitude (PSA). The animals were allowed at least 1 week to recover from surgery. For the pharmacological experiments, a cannula (coordinates: AP, -0.8; L, 1.6 from bregma) was additionally implanted into the right lateral ventricle.
Electrophysiological recording
Rats were placed into a recording box (40 × 40 × 40 cm), and the electrodes were connected to a swivel by a flexible cable. This allowed the freely moving animals ad libitum access to food and water. The responses were amplified (differential amplifier, Inh; Science Products, Hochheim, Germany), transformed by an analog-to-digital interface (CED 1401+; Cambridge Electronic Design, Cambridge, UK), and stored on a personal computer. Biphasic constant current pulses (0.1 msec per half wave) were applied to the perforant path to evoke DG field potentials of ∼40% of the maximum PSA. Because the spike is required to induce LTP, the preparation was optimized to obtain a population spike. This influences the dipole of the field EPSP in the hilus, making the recording of the PSA more preferable than that of the EPSP. After registering a stable baseline for 1 hr, LTP was induced by weak tetanic bursts (three bursts of 15 pulses of 200 Hz with 0.2 msec duration of each stimulus and 10 sec interburst interval; same stimulus intensity as for PSA testing). Two minutes and then every 15 min after tetanization, five test stimuli (10 sec interpulse interval) were delivered and the mean values of field potentials were stored for 8 hr. A 24 hr value was obtained the next day. For analysis and presentation, the 15 min recordings were averaged in groups of four to yield 1 hr values. The 2 min value served as control to determine whether a sufficient initial potentiation (with no more than 25% difference between individual animals) had been obtained.
Tetanization and experimental manipulations were always performed between 10:00 and 11:00 A.M. to avoid interferences with the diurnal rhythm of corticosterone titers.
Stress procedures and experimental groups
The swim stress apparatus was a circular plastic water tank 1.82 m in diameter and 58 cm in height filled with water up to a level of 38 cm. The water temperature was 25 ± 2°C. Water was made opaque by a white latex fluid (Sakret, Giessen, Germany). For behavioral manipulations, all animals were used only once.
Control group. Animals in this group received a weak tetanus and were then left undisturbed in the recording chamber.
Swimming group. Rats in the swimming group were placed in the maze 15 min after tetanus for a 2 min swim. They were then dried with a towel and transferred back into the recording chamber. Before swimming, the electrodes were protected from water immersion by petroleum jelly.
Handling group. The handling group was identical to the swimming group in every respect, except that animals were not transferred into the water tank.
Hormone analysis
Blood samples of all groups were collected at the same time of day after decapitation of the animals. Blood samples were taken from parallel groups of animals that had not been implanted with electrodes. However, the experimental and handling procedures were exactly the same as for the implanted animals, with the exception of the absence of recording or LTP induction. Rats were killed 15 min after either swimming or handling. Trunk blood was sampled within 25-30 sec from opening the cache and handling the animal. Blood was allowed to coagulate on ice in an Eppendorf tube. Then the blood was centrifuged, and the serum was stored at -20°C. Samples were analyzed by radioimmunoassay (RIA) within 4-8 weeks. RIA was performed as described previously (Stefanski et al., 2001).
Pharmacology
Glucocorticoid receptors were blocked by mifepristone, and mineralocorticoid receptors were blocked by spironolactone (150 ng, i.c.v. each; Sigma, St. Louis, MO). Both substances were dissolved in ethanol and then brought up to 1 ml volume with 0.9% saline with a final concentration of 50 ng/μl (2% ethanol). The control solution consisted of 0.9% saline (2% ethanol). The solutions were injected at 1 μl/min to a total volume of 3 μl via a Hamilton syringe. Propranolol (Sigma), a β-adrenergic receptor antagonist, was liquefied in 0.9% saline at 2 μg in a volume of 5 μl, which was applied over a period of 5 min. Injectors were placed into the cannula 30 min before tetanus, and the solutions were injected 1 min after tetanus. The injectors were left in place until the behavioral manipulations were performed.
Anisomycin (Sigma), a reversible protein synthesis inhibitor, was first dissolved in 15 μl of 1N HCl solution and then treated with 1N NaOH to create a pH of 7.0. The solution was subsequently made up to a 50 μl volume with 0.9% sodium chloride. After recording of the baseline, the substance (240 μg, i.c.v.; 5 μl over a 5 min period) or the vehicle was applied. After 1 hr, the animals received the weak tetanus.
Statistics
For group comparisons of overall differences in LTP between groups, the general linear model for repeated measures was chosen (slight differences in the degrees of freedom result from a few missing values). Least significant difference multiple-comparison (LSD) post hoc tests were used for multiple group comparisons. Differences in hormone levels were evaluated by the Mann-Whitney U test after an overall comparison with the Kruskal-Wallis H test. The 24 hr values for the drug-treated and vehicle-treated groups were compared by one-way ANOVA and LSD post hoc tests. All tests were two-tailed, and the level of significance was set at p ≤ 0.05.
Results
Swimming and handling modulated DG-LTP in opposite directions
A 2 min swim subsequent to handling resulted in prolonged LTP, up to 24 hr when compared with control animals (F(1,13) = 7.95; p = 0.014). Handling alone, in contrast, reversed early LTP and led to a suppression of PSA amplitudes below baseline (F(1,13) = 9.07; p = 0.01) (Fig. 1A). This bidirectional modulation of LTP became most obvious when swimming animals were compared with handled animals (F(1,12) = 21.77; p = 0.001). An overall difference, justifying the separate analyses, could be found between all three groups (F(2,19) = 13.97; p < 0.0001). Handling had no effect on baseline values, as indicated by a baseline control group (F(1,12) = 0.20; p > 0.1), whereas swimming slightly depressed baseline values (F(1,10) = 7.71; p < 0.05) (Fig. 1B). No difference could be found between the baseline values for the swimming and the handling group (F(1,10) = 4.59; p > 0.05).
Figure 1.
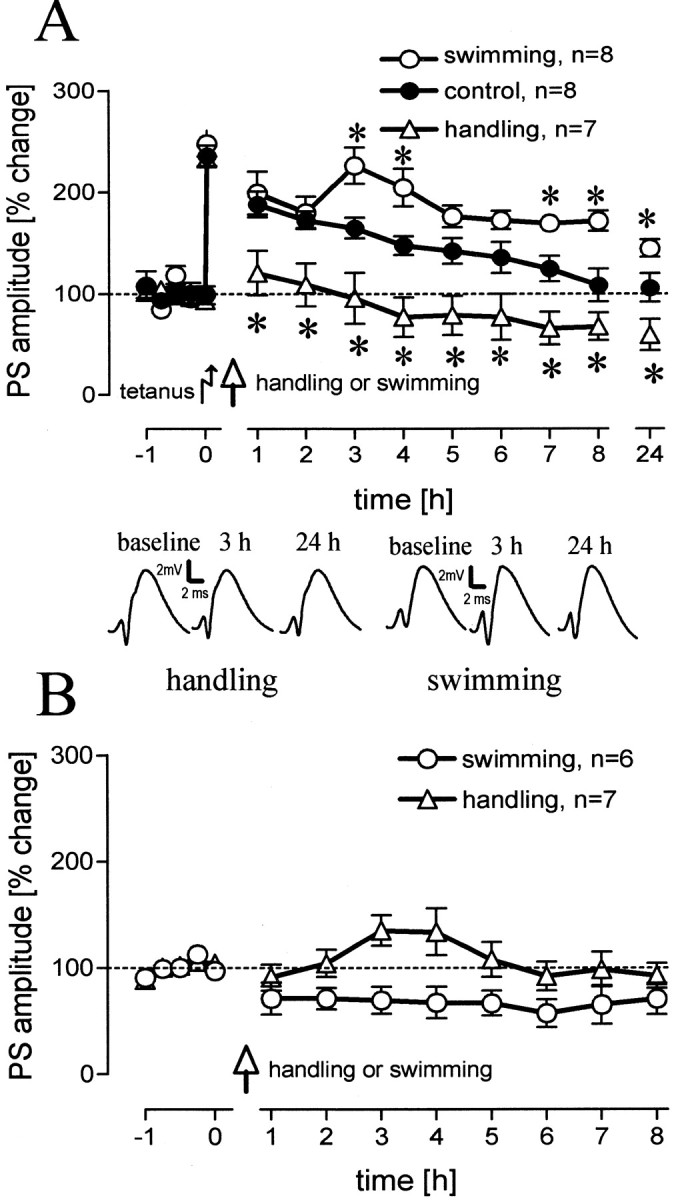
A, Population spike (PS) amplitudes (percentage change from baseline values) over 8 hr and a 24 hr value for rats that were handled 15 min after tetanization compared with unmanipulated animals (p = 0.024, 0.011, 0.016, 0.006, 0.007, 0.021, 0.003, 0.055, 0.021, levels for increasing time points) and for animals that experienced a 2 min swim compared with unmanipulated animals (p = 0.021, 0.012, 0.023, 0.005, 0.05, levels for increasing time points). Comparing the handled animals with the swimming animals reveals significant differences for all time points (p = 0.012, 0.007, 0.004, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001). B, Baseline control groups were used for both behavioral manipulations. Means ± SEM are given. Asterisks indicate significant time point differences. The insets show representative analog traces for a handled and a swimming animal at the indicated time points. Horizontal dashed lines indicate the 100% level.
Blockade of GRs but not of MRs prevented impairment of LTP by handling
We measured the titers of serum corticosterone in animals 15 min after the different behavioral manipulations and found significant overall differences (χ 2 = 18.59; df = 2; p < 0.001). Group comparisons (Fig. 2) revealed that handling caused an increase in corticosterone that was threefold that seen for the control group (U = 0, p < 0.01), whereas swimming augmented the titers nearly sixfold, also with respect to the control group (U = 0; p < 0.01). Handled animals showed significantly lower corticosterone titers than swimming animals (U = 0; p < 0.01). This suggests an impact of corticosterone on the maintenance of LTP, depending on the animals' manipulation.
Figure 2.

Levels of serum corticosterone for unmanipulated animals and for animals 15 min after handling or swimming. Asterisks indicate significant group differences.
The pharmacological analysis of the handling group alone revealed an overall difference between groups treated with a GR-antagonist (GR-ant), an MR-antagonist (MR-ant), or the vehicle (F(2,13) = 11.75; p = 0.001). Application of a GR-ant prevented impairment of LTP compared with vehicle-treated (Fig. 3A) animals (F(1,8) = 18.60; p < 0.01) and led to normal early LTP that was not distinguishable from that in the control group (F(1,12) = 2.71; p > 0.1). Injection of an MR-ant, however, had no effect on the impairment of LTP when compared with vehicle controls (Fig. 3B).
Figure 3.

Population spike (PS) amplitudes (percentage change from baseline values) over 8 hr and a 24 hr value for rats that were handled 15 min after tetanus and injected with a GR-antagonist (A) (n = 6; p = 0.005, 0.004, 0.013, 0.001, 0.012, 0.002, 0.003, 0.001, 0.013, levels for increasing time points) or an MR-antagonist (B) compared with vehicle-treated rats. Rats that experienced a 2 min swim after tetanus and injection of a GR-antagonist showed no differences compared with vehicle-treated rats (C), whereas injection of an MR-antagonist impaired LTP (D) (p = 0.007, 0.007, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001, levels for increasing time points). E, Injection of a mixture of GR-MR-antagonists also impaired LTP (p = 0.008, 0.0001, 0.0001, 0.0001, 0.0001, 0.0001, 0.001, 0.001, levels for increasing time points). F, Baseline control groups were used for both antagonists. Means ± SEM are given. Asterisks indicate significant time point differences. The diagram insets show representative analog traces for GR-ant-treated rats (A) and MR-ant treated rats (D) at the indicated time points. Dashed lines indicate the 100% level.
Inactivation of MRs but not GRs hindered LTP prolongation by swimming
An overall comparison between all drug- and vehicle-treated swim groups (including the anisomycin- and propranolol-treated groups) revealed a significant difference (F(5,36) = 15.12; p < 0.0001). Application of the GR-ant in animals that experienced a 2 min swim had no influence on LTP prolongation (Fig. 3C), whereas an acute blockade of MR receptors completely impaired LTP in swimming rats (F(1,9) = 43.76; p < 0.001), with a significant difference at all time points (Fig. 3D) from the second hour onward. It is known that acute blockade of MRs results in an increased release of corticosterone. This could lead to an overactivation of GRs that would then mask the effect of MR blockade on LTP. For this reason, a group treated with a mixture of the GR-ant and MR-ant was tested. We found no difference between the MR-ant and the GR-ant-MR-ant group (Fig. 3E), but a difference was found between vehicle-treated rats and the GR-ant-MR-ant group (F(1,9) = 20.18; p < 0.01) that was similar to that ascertained between the vehicle-treated rats and the MR-ant group. Mifepristone (F(1,10) = 1.36; p > 0.1) as well as spironolactone (F(1,10) = 0.25; p > 0.1) did not operate on baseline values, as reflected by the lack of effect on a baseline control group (Fig. 3F).
The blockade of GRs had no significant effect compared with vehicle controls but displayed a slight enhancement of LTP up to 6 hr. In contrast, the blockade of MRs led to a suppression of early LTP similar to that in handled animals. The MR-ant-treated swimming group exhibited a significantly lower potentiation than control animals (F(1,12) = 20.07; p < 0.01) with significant differences from the second hour onward up to 7 hr (p < 0.01 each; p < 0.05 for the 6 and 7 hr time points).
Reinforcement of LTP was dependent on protein synthesis but not on β-adrenergic activation
The stress response to swimming leads not only to an increased level of blood corticosterone but also to a release of adrenaline and noradrenaline. These substances do not readily cross the blood-brain barrier. The main source of noradrenaline in the brain is the locus ceruleus, and there are several lines of evidence that the activation of β-adrenergic receptors plays a role in the prolongation of early LTP by behavioral reinforcement. Therefore, we tested the influence of β-adrenergic activation with a group of animals treated with propranolol, an unspecific β-adrenergic antagonist (Fig. 4A). We could not state significant differences between this group and the vehicle-treated group.
Figure 4.
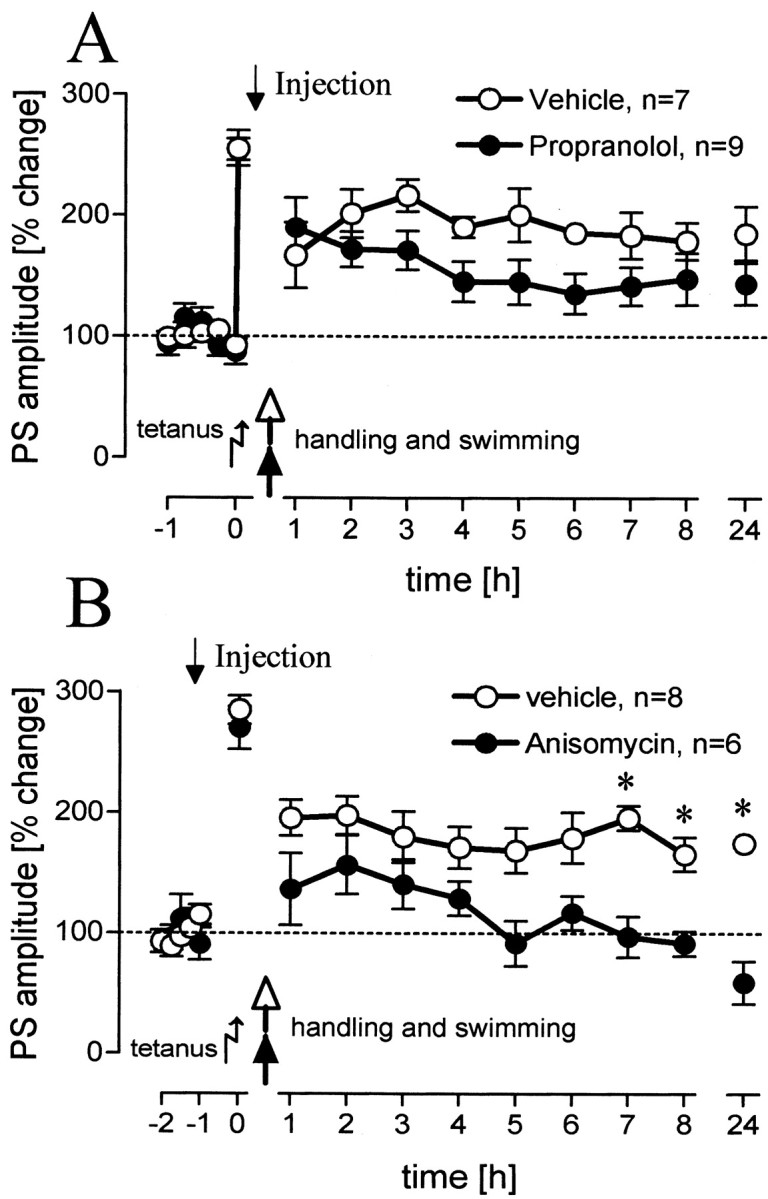
Population spike (PS) amplitudes (percentage change from baseline values) over 8 hr and a 24 hr value for rats that experienced a 2 min swim after tetanus and were injected with a β-adrenergic antagonist (A) or a protein-synthesis inhibitor (B) (p = 0.004, 0.03, 0.0001, levels for increasing time points). Means ± SEM are given. Asterisks indicate significant time point differences. Dashed lines indicate the 100% level.
To determine whether the late phase of the LTP reinforced by swimming is protein synthesis dependent, anisomycin, a protein synthesis inhibitor, was applied before behavioral manipulation. Anisomycin prevented the prolongation of early LTP by swimming (Fig. 4B), indicated by a significant difference with the vehicle-treated rats (F(1,10) = 10.09; p = 0.01). Figure 5 gives an overview of the differences at the 24 hr time point between treated rats in the swimming groups. In previous studies from our laboratory, it was shown that propranolol (Seidenbecher et al., 1997) and anisomycin (Frey et al., 2001) at the same doses and under the same protocol used in this study did not affect baseline values; therefore, no baseline control experiments have been conducted for these substances.
Figure 5.

Differences between the population spike amplitudes at the 24 hr time point for the drug-treated swimming groups. Vehicle post, Injection after tetanization; vehicle pre, injection before tetanization, control for the anisomycin-treated group. Means ± SEM are given. **p < 0.01; *p < 0.05.
Discussion
We found bidirectional effects of behavioral manipulations on the maintenance of hippocampal LTP. Although handling 15 min after induction of early LTP resulted in an impairment of LTP, a 2 min swim, also 15 min after induction, resulted in prolongation of LTP to up to 24 hr. Because both of the behavioral manipulations increased the titers of circulating corticosterone, we studied the role of corticosterone receptors on LTP modulation by behavioral manipulation. The handling-dependent LTP impairment was reversed by blockade of glucocorticoid receptors and left unaffected by blockade of mineralocorticoid receptors. The LTP prolongation observed after swimming, however, was unaffected by blockade of GRs, whereas blockade of MRs resulted in an impairment of LTP comparable with that seen in untreated handled animals.
A possible explanation for the pattern of results obtained can be found in the finding by Gesing et al. (2001) that swim stress leads to a significant upregulation, within 24 hr, of MRs in the hippocampus (cf. Reul et al., 2000). Under basal conditions, as a result of their differential binding capacity, all MRs but not GRs will be occupied. After swimming, both the upregulation of MRs and the increased levels of corticosterone would result in a shift in the balance of MR-GR occupation, so that MR-induced modulatory effects on LTP would then overcome those of the GRs. The failure to observe an MR-antagonist effect in handled animals suggests no change in corticosterone-binding receptors. Accordingly, the activation of GRs may exceed that of MRs and lead to the impairment in LTP observed, which can be reversed by inhibition of the GRs but is left unaffected by inhibition of the MRs. This is supported by the finding that blockade of the MRs before swimming also impairs LTP, comparable with that of animals that were only handled. Therefore, the MR effect triggered by the swimming experience may overcompensate the GR effect and leads not only to a reversal but also to prolongation of LTP. Such a mechanism may at least function during acute high stress and acute MR inhibition. During chronic stress and subchronic systemic injection of spironolactone, Herman and Spencer (1998) identified increased levels of GR mRNA in CA1 and DG similar to that observed in adrenalectomized rats. They concluded that activation of MRs tonically inhibits GR biosynthesis in the hippocampus. A diminution or an increase in MR-over GR-mediated neuronal effects may regulate the neuronal responsiveness during stressful situations and may adjust behavioral adaptation (De Kloet et al., 1993; Joels and De Kloet, 1994). However, De Kloet et al. (1999) pointed out that the impairment of LTP and the facilitation of long-term depression (LTD) during stress does not merely depend on the occupation of corticosteroid receptors but on the history of activation of different inputs during previous events (e.g., activation of transcription factors), so that even during mild stress unrelated to a learning paradigm, the synaptic strength can be decreased. Such mechanisms may play a role in the reversal of LTP observed in our handled group, where the animals had experienced handling during the daily keeping routine.
The activated corticosterone receptors regulate gene expression in two ways: by transactivation, which requires homodimerization and binding of homodimers to the DNA, and through transrepression, by interaction of receptor monomers with other transcription factors, which does not require DNA binding (Heck et al., 1994). For CA1 pyramidal neurons, DNA binding is required to activate voltage-gated calcium channels by corticosterone (Karst et al., 2000). Our finding, that the LTP reinforcement is protein synthesis dependent, suggests that the protein products mediated by the MR activation play a role in the transformation processes of synapses related to late LTP in DG-granule neurons. In previous studies on the CA1 region of the hippocampus (Xu et al., 1997), it has been found that behavioral stress (exposure to an elevated platform) facilitates the induction of LTD and blocks the induction of LTP in anesthetized rats. This effect could be reversed by application of a GR-antagonist as well as a protein synthesis inhibitor before stress (Xu et al., 1998a). Overall, these results suggest a role of GRs in the control of long-lasting CA1 synaptic plasticity, because a strong tetanization protocol was used in this study to induce a protein synthesis-dependent LTP. The pattern observed in our handled group, where reversal of early LTP, which is protein synthesis independent, can be blocked by a GR-antagonist, suggests that the activation of GRs is also involved in cellular processes not related to gene expression in the DG. Rapid actions of GRs on cellular signaling pathways, such as activation of MAP kinase (mitogen-activated protein kinase), adenylyl cyclase, or protein kinase C (Cato et al., 2002), and other effects increasing the intracellular Ca2+ levels (Kerr et al., 1992; Kim and Yoon, 1998; Bhargava et al., 2000) have been described previously.
The prolongation of LTP by swimming does not depend on the activation of β-adrenergic receptors. Interestingly, the latter is reported for behavioral reinforcement experiments. Seidenbecher et al. (1997) pointed out that appetitive stimuli, such as water delivery to water-deprived rats, aversive stimuli, such as footshocks, or voluntary spatial exploration of a novel environment (Straube et al., 2003), when given around a weak tetanus, resulted in a β-adrenergic-dependent prolongation of DG-LTP. A β-adrenergic-dependent reinforcement of DG-early LTP can also be elicited by electrical stimulation of the BLA, a structure that is seen to be involved in the processing of emotionally arousing information (Akirav and Richter-Levin, 1999, 2002; Frey et al., 2001). In contrast, evidence for a CA1-LTP-impairing effect of the amygdala has been raised (Kim et al., 2001).
The main difference between these reports and our experiments is the highly stressful context in which our study was conducted. Increased extracellular levels of corticosterone within the hippocampus could already be observed a few minutes after the onset of stress (Linthorst et al., 2000) and may interfere with the β-adrenergic processes induced by arousing novelty. Some mechanisms of an interplay between β-adrenergic receptor activation and glucocorticoid receptors have been reported for different brain regions (Duman et al., 1989; Schmidt et al., 2001; Roozendaal et al., 2002) with little evidence of mutual impairment in the dentate gyrus. Smigra et al. (1998) found in dentate granule cells of adrenalectomized but not intact rats that the LTP-enhancing effect of MR activation depends on β-adrenergic activity.
Because we found no evidence for the requirement of β-adrenergic activation for DG-LTP reinforcement in our study, an interplay between corticosterone-binding receptors and other types of receptors or transmission systems is very likely and remains to be investigated. This could involve the serotonergic (Shakesby et al., 2002) or the cholinergic system, which has been implicated in DG-LTP reinforcement (Frey et al., 2001). An involvement of the septohippocampal cholinergic system in an adaptive response to stress has been reported (Gilad, 1987). This includes a direct activation of hippocampal cholinergic terminals by corticosterone within minutes after a stressful event, with increased release of newly synthesized acetylcholine (Gilad, 1987; Gilad et al., 1987). In addition, a modulatory effect of the BLA on DG-LTP via cholinergic medial septum pathways seems likely (Spanis et al., 1999; Frey et al., 2001).
The novelty of the swim situation very likely contributes to our results. It has been found that perception of a novel environment under low-stress conditions prolongs early DG-LTP within a certain time window around a weak tetanus (Straube et al., 2003). A post-tetanus (2 min) exposure to 1 min of novelty prolongs early LTP, whereas a lasting novelty exposure impairs LTP. Similar results are found in the CA1: previously induced LTP by a strong tetanization protocol is reversed by lasting novelty exposure (Xu et al., 1998b), but the induction of CA1-LTP is facilitated in a dopamine-dependent manner by a brief (5 min) exposure (Li et al., 2003). Thus, the short-lasting swim experience in our study may also contribute to the facilitation of LTP. In addition, evidence has been raised that MRs are mediating behavioral reactivity during novel situations (Oitzl et al., 1994), whereas activation of GRs by stress during learning tasks is seen to facilitate the consolidation of information (Sandi et al., 1997; De Kloet et al., 1999; Shors, 2001). In comparing these studies, it is important to keep in mind that different mechanisms may underlie the effects of corticosteroid on the induction of LTP or on the modulation of an already induced LTP, as in our study.
It is well established that glucocorticoids modulate spatial learning (Oitzl and De Kloet, 1992; Sandi, 1998; Roozendaal et al., 1999), and that genomic processes are involved (Oitzl et al., 2001). There is much evidence that GRs are involved in the consolidation of acquired spatial information in the water maze and the Y-maze, whereas MR modulates the behavioral reactivity to novel stimuli (Oitzl et al., 1992, 1993; Conrad et al., 1999; De Kloet et al., 1999). However, in other learning paradigms, pharmacological effects were less distinct. Douma et al. (1998) reported impaired reference memory in a hole board task after MR but not GR blockade, whereas working memory was impaired only after a combined inhibition of MRs and GRs. However, the combined analysis of glucocorticoid effects on hippocampal LTP and spatial learning and memory remains to be conducted.
Footnotes
This study was supported by the Land Saxony-Anhalt (LSA 3475A/1102M) as well as by the European Union Framework V NAPPY to J.U.F. We are grateful to Drs. D. v. Holst and V. Stefanski for analyzing the blood samples in the Animal Department of Physiology (Bayreuth, Germany). We thank Dr. L. de Hoz for critically reading this manuscript and G. Behnisch and J. Maiwald for technical assistance.
Correspondence should be addressed to Dr. Volker Korz, Leibniz-Institute for Neurobiology, Brenneckestrasse 6, D-39118 Magdeburg, Germany. E-mail: korz@ifn-magdeburg.de.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237281-07$15.00/0
References
- Akirav I, Richter-Levin G ( 1999) Priming stimulation in the basolateral amygdala modulates synaptic plasticity in the rat dentate gyrus. Neurosci Lett 270: 83-86. [DOI] [PubMed] [Google Scholar]
- Akirav I, Richter-Levin G ( 2002) Mechanisms of amygdala modulation of hippocampal plasticity. J Neurosci 22: 9912-9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akirav I, Sandi C, Richter-Levin G ( 2001) Differential activation of hippocampus and amygdala following spatial learning under stress. Eur J Neurosci 14: 719-725. [DOI] [PubMed] [Google Scholar]
- Bhargava A, Meijer OC, Dallman MF, Pearce D ( 2000) Plasma membrane calcium pump isoform 1 gene expression is repressed by corticosterone and stress in rat hippocampus. J Neurosci 20: 3129-3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L, McGaugh JL ( 1998) Mechanisms of emotional arousal and lasting declarative memory. Trends Neurosci 21: 294-299. [DOI] [PubMed] [Google Scholar]
- Cato AC, Ponta H, Herrlich P ( 1992a) Regulation of gene expression by steroid hormones. Prog Nucleic Acid Res Mol Biol 43: 1-36. [DOI] [PubMed] [Google Scholar]
- Cato AC, Konig H, Ponta H, Herrlich P ( 1992b) Steroids and growth promoting factors in the regulation of expression of genes and gene networks. J Steroid Biochem Mol Biol 43: 63-68. [DOI] [PubMed] [Google Scholar]
- Cato AC, Nestl A, Mink S ( 2002) Rapid actions of steroid receptors in cellular signaling pathways. Sci STKE 2002: RE9. [DOI] [PubMed] [Google Scholar]
- Conrad CD, Lupien SJ, McEwen BS ( 1999) Support for a bimodal role for type II adrenal steroid receptors in spatial memory. Neurobiol Learn Mem 72: 39-46. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Oitzl MS, Joels M ( 1993) Functional implications of brain corticosteroid receptor diversity. Cell Mol Neurobiol 13: 433-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kloet ER, Oitzl MS, Joels M ( 1999) Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci 22: 422-426. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Bennett MC, Fleshner M, Rose GM ( 1992) Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 2: 421-430. [DOI] [PubMed] [Google Scholar]
- Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR ( 1990) Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 249: 1266-1272. [DOI] [PubMed] [Google Scholar]
- Douma BR, Korte SM, Buwalda B, la Fleur SE, Bohus B, Luiten PG ( 1998) Repeated blockade of mineralocorticoid receptors, but not of glucocorticoid receptors impairs food rewarded spatial learning. Psychoneuroendocrinology 23: 33-44. [DOI] [PubMed] [Google Scholar]
- Drouin J, Sun YL, Tremblay S, Lavender P, Schmidt TJ, de Lean A, Nemer M ( 1992) Homodimer formation is rate limiting for high affinity binding by the glucocorticoid receptor. Mol Endocrinol 6: 1299-1309. [DOI] [PubMed] [Google Scholar]
- Duman RS, Strada SJ, Enna SJ ( 1989) Glucocorticoid administration increases receptor-mediated and forskolin-stimulated cyclic AMP accumulation in rat brain cerebral cortical slices. Brain Res 477: 166-171. [DOI] [PubMed] [Google Scholar]
- Filipini D, Gijsbers K, Birmingham MK, Kraulis I, Dubrovsky B ( 1991) Modulation by adrenal steroids of limbic function. J Steroid Biochem Mol Biol 39: 245-252. [DOI] [PubMed] [Google Scholar]
- Foy MR, Stanton ME, Levine S, Thompson RF ( 1987) Behavioral stress impairs long-term potentiation in rodent hippocampus. Behav Neural Biol 48: 138-149. [DOI] [PubMed] [Google Scholar]
- Frey S, Bergado-Rosado J, Seidenbecher T, Pape HC, Frey JU ( 2001) Reinforcement of early long-term potentiation (early-LTP) in dentate gyrus by stimulation of the basolateral amygdala: heterosynaptic induction mechanisms of late-LTP. J Neurosci 21: 3697-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Morris RG ( 1997) Synaptic tagging and long-term potentiation. Nature 385: 533-536. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RGM ( 1998) Weak before strong: dissociating synaptic tagging and plasticity-factor accounts of late-LTP. Neuropharmacology 37: 545-552. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H ( 1988) Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro Brain Res 452: 57-65. [DOI] [PubMed] [Google Scholar]
- Gesing A, Bilang-Bleuel A, Droste SK, Linthorst AC, Holsboer F, Reul JM ( 2001) Psychological stress increases hippocampal mineralocorticoid receptor levels: involvement of corticotropin-releasing hormone. J Neurosci 21: 4822-4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad GM ( 1987) The stress-induced response of the septo-hippocampal cholinergic system. A vectorial outcome of psychoneuroendocrinological interactions. Psychoneuroendocrinology 12: 167-184. [DOI] [PubMed] [Google Scholar]
- Gilad GM, Rabey JM, Gilad VH ( 1987) Presynaptic effects of glucocorticoids on dopaminergic and cholinergic synaptosomes. Implications for rapid endocrine-neural interactions in stress. Life Sci 40: 2401-2408. [DOI] [PubMed] [Google Scholar]
- Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, Cato ACB ( 1994) A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J 13: 4087-4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP ( 1993) Regulation of adrenocorticosteroid receptor mRNA expression in the central nervous system. Cell Mol Neurobiol 13: 349-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Spencer R ( 1998) Regulation of hippocampal glucocorticoid receptor gene transcription and protein expression in vivo J Neurosci 18: 7462-7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo I, Medina JH ( 1995) Correlation between the pharmacology of long-term potentiation and the pharmacology of memory. Neurobiol Learn Mem 63: 19-32. [DOI] [PubMed] [Google Scholar]
- Joels M, De Kloet ER ( 1994) Mineralocorticoid and glucocorticoid receptors in the brain. Implications for ion permeability and transmitter systems. Prog Neurobiol 43: 1-36. [DOI] [PubMed] [Google Scholar]
- Kant GJ, Michael HY, D'Angelo PC, Brown AJ, Eggleston T ( 1988) Maze performances: a direct comparison of food vs. water mazes. Pharmacol Biochem Behav 31: 487-491. [DOI] [PubMed] [Google Scholar]
- Karst H, Karten YJG, Reichardt HM, De Kloet ER, Schütz G, Joels M ( 2000) Corticosteroid actions in hippocampus require DNA binding of glucocorticoid receptor homodimers. Nat Neurosci 3: 977-978. [DOI] [PubMed] [Google Scholar]
- Kerr DS, Campbell LW, Thibault O, Landfield PW ( 1992) Hippocampal glucocorticoid receptor activation enhances voltage-dependent Ca 2+ conductances: relevance to brain aging. Proc Natl Acad Sci USA 89: 8527-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM ( 2002) The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev 3: 453-462. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Yoon KS ( 1998) Stress: metaplastic effects in the hippocampus. Trends Neurosci 21: 505-509. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF ( 1996) Behavioral stress modifies hippocampal plasticity through N-methyl-d-aspartate receptor activation. Proc Natl Acad Sci USA 93: 4750-4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee HJ, Han JS, Packard MG ( 2001) Amygdala is critical for stress-induced modulation of hippocampal long-term potentiation and learning. J Neurosci 21: 5222-5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug M, Lossner B, Ott T ( 1984) Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res Bull 13: 39-42. [DOI] [PubMed] [Google Scholar]
- Li S, Cullen WK, Anwyl R, Rowan MJ ( 2003) Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nat Neurosci 6: 526-531. [DOI] [PubMed] [Google Scholar]
- Linthorst ACE, Flachskamm C, Barden N, Holsboer F, Reul JMHM ( 2000) Glucocorticoid receptor impairment alters CNS responses to a psychological stressor: an in vivo microdialysis study in transgenic mice. Eur J Neurosci 12: 283-291. [DOI] [PubMed] [Google Scholar]
- Malkoski SP, Handanos CM, Dorin RI ( 1997) Localization of a negative glucocorticoid response element of the human corticotropin releasing hormone gene. Mol Cell Endocrinol 127: 189-199. [DOI] [PubMed] [Google Scholar]
- Matthies H, Frey U, Reymann K, Krug M, Jork R, Schroeder H ( 1990) Different mechanisms and multiple stages of LTP. Adv Exp Med Biol 268: 359-368. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Weiss JM, Schwartz LS ( 1968) Selective retention of corticosterone by limbic structures in rat brain. Nature 220: 911-912. [DOI] [PubMed] [Google Scholar]
- McGaugh JL ( 2000) Memory: a century of consolidation. Science 287: 248-251. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B ( 2002) Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol 12: 205-210. [DOI] [PubMed] [Google Scholar]
- Morris R ( 1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11: 47-60. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, De Kloet ER ( 1992) Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci 106: 62-71. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Josephy M, Spruijt BM ( 1993) An ACTH/MSH(4 -9) analog counteracts the behavioral effects of a mineralocorticoid receptor antagonist. Pharmacol Biochem Behav 44: 447-450. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Fluttert M, De Kloet ER ( 1994) The effect of corticosterone on reactivity to spatial novelty is mediated by central mineralocorticosteroid receptors. Eur J Neurosci 6: 1072-1079. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Reichardt HM, Joels M, De Kloet ER ( 2001) Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc Natl Acad Sci USA 98: 12790-12795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlides C, McEwen BS ( 1999) Effects of mineralocorticoid and glucocorticoid receptors on long-term potentiation in the CA3 hippocampal field. Brain Res 851: 204-214. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Watanabe Y, McEwen BS ( 1993) Effects of glucocorticoids on hippocampal long-term potentiation. Hippocampus 3: 183-192. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Watanabe Y, Magarinos AM, McEwen BS ( 1995) Opposing roles of type I and type II adrenal steroid receptors in hippocampal long-term potentiation. Neuroscience 68: 387-394. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Nivon LG, McEwen BS ( 2002) Effects of chronic stress on hippocampal long-term potentiation. Hippocampus 12: 245-257. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C ( 1998) The rat brain in stereotaxic coordinates. San Diego: Academic.
- Reul JM, De Kloet ER ( 1985) Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117: 2505-2511. [DOI] [PubMed] [Google Scholar]
- Reul JM, Pearce PT, Funder JW, Krozowski ZS ( 1989) Type I and type II corticosteroid receptor gene expression in the rat. Mol Endocrinol 3: 1674-1680. [DOI] [PubMed] [Google Scholar]
- Reul JM, Gesing A, Droste S, Stec IS, Weber A, Bachmann C, Bilang-Bleuel A, Holsboer F, Linthorst AC ( 2000) The brain mineralocorticoid receptor: greedy for ligand, mysterious in function. Eur J Pharmacol 405: 235-249. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Nguyen BT, Power AE, McGaugh JL ( 1999) Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci USA 96: 11642-11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Quirarte GL, McGaugh JL ( 2002) Glucocorticoids interact with the basolateral amygdala β-adrenoceptor-cAMP/PKA system in influencing memory consolidation. Eur J Neurosci 15: 553-560. [DOI] [PubMed] [Google Scholar]
- Sandi C ( 1998) The role and mechanisms of action of glucocorticoid involvement in memory storage. Neural Plast 6: 41-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C, Loscertales M, Guaza C ( 1997) Experience-dependent facilitating effect of corticosterone on spatial memory formation in the water maze. Eur J Neurosci 9: 637-642. [DOI] [PubMed] [Google Scholar]
- Schmidt P, Holsboer F, Spengler D ( 2001) Beta(2)-adrenergic receptors potentiate glucocorticoid receptor transactivation via G-protein beta gamma-subunits and the phosphoinositide 3-kinase pathway. Mol Endocrinol 15: 553-564. [DOI] [PubMed] [Google Scholar]
- Seidenbecher T, Reymann KG, Balschun D ( 1997) A post-tetanic time window for the reinforcement of long-term potentiation by appetitive and aversive stimuli. Proc Natl Acad Sci USA 94: 1494-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakesby AC, Anwyl R, Rowan MJ ( 2002) Overcoming the effects of stress on synaptic plasticity in the intact hippocampus: rapid actions of serotonergic and antidepressant agents. J Neurosci 22: 3638-3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ ( 2001) Acute stress rapidly and persistently enhances memory formation in the male rat. Neurobiol Learn Mem 75: 10-29. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Seib TB, Levine S, Thompson RF ( 1989) Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science 244: 224-226. [DOI] [PubMed] [Google Scholar]
- Smigra M, Nishiyama N, Saito H ( 1998) Mineralocorticoid receptor-mediated enhancement of neuronal excitability and synaptic plasticity in the dentate gyrus in vivo is dependent on the beta-adrenergic activity. J Neurosci Res 51: 593-601. [DOI] [PubMed] [Google Scholar]
- Spanis CW, Bianchin MM, Izquierdo I, McGaugh JL ( 1999) Excitotoxic basolateral amygdala lesions potentiate the memory impairment effect of muscimol injected into the medial septal area. Brain Res 816: 329-336. [DOI] [PubMed] [Google Scholar]
- Stefanski V, Knopf G, Schulz S ( 2001) Long-term colony housing in Long Evans rats: immunological, hormonal, and behavioral consequences. J Neuroimmunol 114: 122-130. [DOI] [PubMed] [Google Scholar]
- Straube T, Korz V, Frey JU ( 2003) Bidirectional modulation of long-term potentiation by novelty-exploration in rat dentate gyrus. Neurosci Lett 344: 5-8. [DOI] [PubMed] [Google Scholar]
- van Steensel B, van Binnendijk EP, Hornsby CD, van der Voort HT, Krozowski ZS, De Kloet ER, van Driel R ( 1996) Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J Cell Sci 109: 787-792. [DOI] [PubMed] [Google Scholar]
- Xu L, Anwyl R, Rowan MJ ( 1997) Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387: 497-500. [DOI] [PubMed] [Google Scholar]
- Xu L, Holscher C, Anwyl R, Rowan MJ ( 1998a) Glucocorticoid receptor and protein/RNA synthesis-dependent mechanisms underlie the control of synaptic plasticity by stress. Proc Natl Acad Sci USA 95: 3204-3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Anwyl R, Rowan MJ ( 1998b) Spatial exploration induces a persistent reversal of long-term potentiation in rat hippocampus. Nature 394: 891-894. [DOI] [PubMed] [Google Scholar]
- Zhang XK, Dong JM, Chiu JF ( 1991) Regulation of alpha-fetoprotein gene expression by antagonism between AP-1 and the glucocorticoid receptor at their overlapping binding site. J Biol Chem 266: 8248-8254. [PubMed] [Google Scholar]
- Zhou J, Zhang F, Zhang Y ( 2000) Corticosterone inhibits generation of long-term potentiation in rat hippocampal slice: involvement of brain-derived neurotrophic factor. Brain Res 885: 182-191. [DOI] [PubMed] [Google Scholar]