

Abstract
At Aplysia sensory-to-motor neuron synapses, the inhibitory neuropeptide Phe-Met-Arg-Phe-NH2 (FMRFa) produces depression, and serotonin (5-HT) produces facilitation. Short-term depression has been found to result from the activation of a phospholipase A2. The released arachidonate is metabolized by 12-lipoxygenase to active second messengers. We find that FMRFa leads to the phosphorylation and activation of p38 mitogen-activated protein (MAP) kinase. Short-term depression and the release of arachidonate are blocked by the specific p38 kinase inhibitor SB 203580. Both the inhibitor and an affinity-purified antibody raised against recombinant Aplysia p38 kinase injected into sensory neurons prevented long-term depression, which depends on the phosphorylation of translation factors cAMP response element-binding protein 2 (CREB2) and activating transcription factor 2. Facilitation produced by 5-HT, on the other hand, inactivates p38 kinase. Chromatin immunoprecipitation assays indicate that p38 kinase activates CREB2. p38 kinase also is pivotal in the bidirectional regulation of synaptic plasticity: when the kinase is inhibited, brief treatment with 5-HT that normally produces only short-term facilitation now results in long-term facilitation. Conversely, in sensory neurons injected with the activated kinase, long-term facilitation is blocked, and brief exposure to FMRFa, which normally results in short-term depression, results in long-term depression. We conclude that p38 kinase, which itself is bidirectionally regulated by FMRFa and 5-HT, acts as a modulator of synaptic plasticity by positively regulating depression and serving as an inhibitory constraint for facilitation.
Keywords: Aplysia, histone deacetylase, long-term depression, long-term facilitation, phospholipase A2, p38 MAP kinase
Introduction
The capacity for bidirectional modulation is an important feature of synapses that undergo plastic changes (Lisman, 1989; Dudak and Bear, 1993). In Aplysia the facilitatory neurotransmitter serotonin (5-HT) strengthens the connections between sensory and motor neurons to produce facilitation, whereas the inhibitory neuropeptide Phe-Met-Arg-Phe-NH2 (FMRFa) produces depression by weakening them. In short-term facilitation, 5-HT activates the cAMP-dependent protein kinase (PKA) to phosphorylate and close S-type K+ channels, thereby enhancing transmitter output transiently (Siegelbaum et al., 1982; Shuster et al., 1985). In long-term facilitation, PKA catalytic subunits are imported into the nucleus to phosphorylate the transcription activator cAMP response element-binding protein 1 (CREB1), which then induces a cascade of gene expression consisting of immediate response genes that then activate downstream genes encoding proteins needed for new synaptic growth (Kandel and Schwartz, 1982; Kandel, 2001). Guan et al. (2002) found that the induction of the immediate response gene for the transcription factor CCAAT enhancer-binding protein (C/EBP) (Alberini et al., 1994) requires phosphorylated CREB1 and the recruitment of the CREB-binding protein (CBP) to the C/EBP promoter. The intrinsic histone acetylase activity of CBP then modifies chromatin structure, initiating gene expression. Martin et al. (1997) reported that the extracellular signal-regulated kinase (ERK) (p42) mitogen-activated protein (MAP) kinase is also activated and imported into the nucleus to activate C/EBP (Yamamoto et al., 1999). Long-term facilitation also requires the reversal of several inhibitory constraints: removal of both the repressor CREB2 and histone deacetylase HDAC5 from the promoter of C/EBP (Bartsch et al., 1995; Guan et al., 2002), degradation of PKA regulatory subunits mediated by the induction of ubiquitin C-terminal hydrolase (Hegde et al., 1997; Chain et al., 1999), and endocytosis of a cell-adhesion molecule enabling new synapses to form (Bailey et al., 1997).
What are the signaling pathways underlying depression? In short-term depression, FMRFa causes the release and metabolism of arachidonate leading to the opening of the K+S channel (Piomelli et al., 1987; Belardetti et al., 1989). The long-term form of depression, like long-term facilitation, depends on both transcription and translation (Montarolo et al., 1988), but the transcription factor responsible is CREB2 instead of CREB1 (Guan et al., 2002). In addition to inducing gene transcription, FMRFa also represses C/EBP, a gene crucial for long-term facilitation, by recruiting CREB2 to replace CREB1 and by deacetylating histones at the C/EBP promoter (Guan et al., 2002). Thus, CREB2 is therefore a transcription activator for one set of genes and a repressor for another.
p38 MAP kinase is a key regulator in stress, inflammation, development, and cell death (Nebreda and Porras, 2000; Harper and LoGrasso, 2001; Johnson and Lapadat, 2002; Cowan and Storey, 2003). Although the enzyme has been implicated in hippocampal synaptic transmission (Bolshakov et al., 2000; Armstrong et al., 2002; Rush et al., 2002), a process analogous to long-term depression in Aplysia, the underlying mechanisms are still unknown. We now find that FMRFa activates p38 kinase to phosphorylate and activate CREB2 and activating transcription factor 2 (ATF2). Inhibiting the kinase blocks long-term depression and enhances long-term facilitation; conversely, activating the enzyme enhances long-term depression and blocks long-term facilitation. Thus, in Aplysia p38 kinase is a bidirectional regulator of synaptic plasticity.
Materials and Methods
Animals and tissue extracts. Aplysia californica (70-100 gm), raised at the Mariculture Facility of the University of Miami (Miami, FL), after shipment were rested in seawater for 1 week before an experiment. The animals were exposed to 9 μm FMRFa or 50 μm 5-HT for 10 min and then anesthetized with MgCl2. Pleural ganglia were removed and immediately homogenized in lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerolphosphate, 1 mm Na3VO4, and 1 μg/ml leupeptin). The extracts were centrifuged at 3000 × g for 10 min. Proteins in the supernatants were separated by SDS-PAGE and transferred to nitrocellulose membranes for immunoblotting with antibody raised against phospho-p38 kinase (Cell Signaling Technology, Beverly, MA), stripped, and reblotted with anti-Aplysia p38 kinase antibody.
Electrophysiology and intracellular pressure injection. Aplysia sensory and motor neurons were cultured as described by Montarolo et al. (1988). Electrical recording was made using intracellular microelectrodes filled with 3.0 m KCl using an Axoclamp-2A amplifier in bridge mode. EPSPs were recorded from cocultured L7 motor neurons after extracellular stimulation of sensory neurons. For intracellular injection of antibodies or proteins, micropipettes were pulled to have an initial resistance of 50-70 MΩ when filled with 0.5 m NaCl and beveled to 30-40 MΩ. A picospritzer (General Valve, Fairchild, NJ) was used for pressure injection. SB 203580 was from Calbiochem (San Diego, CA). The anti-ATF2 antibody was from Cell Signaling Technology.
Statistical analysis. For multiple comparisons, we used a one-way ANOVA and the post hoc Newman-Keuls test. The Mann-Whitney U test was used when two groups were compared. Error values and error bars reflect SEM.
Molecular cloning of Aplysia p38 kinase. Poly(A +) RNA was purified from Aplysia nervous tissue with a MicroPoly(A) Pure Kit (Ambion, Austin, TX), and cDNA was synthesized with the First Strand cDNA Synthesis Kit for RT-PCR (Roche Molecular Biochemicals, Indianapolis, IN). Primers for degenerate RT-PCR were designed from two regions of the human p38 kinase gene: PVGSGAYG for the 5′ region and WMHYNQTVD for the 3′, which are nearly identical to Drosophila p38a but differ from Ap-ERK-MAP kinase. After the Ap-p38 coding region was cloned, the entire 5′ and 3′ cDNA ends were obtained with the SMART RACE cDNA Amplification Kit (Clontech, Palo Alto, CA). The full-length cDNA was amplified by Advantage Polymerase Mix (Clontech), and several clones were analyzed to confirm the sequence. Affinity-purified antibody against Ap-p38 kinase was raised by Zymed Laboratories (San Francisco, CA).
Preparation of Aplysia phospho-p38 kinase. The coding region of App38 kinase was engineered into pGEX-6P-1 (Amersham Biosciences, Piscataway, NJ), and the recombinant glutathione S-transferase (GST)-App38 kinase was induced and purified from Escherichia coli BL21 with a GST*Bind Purification Kit (Novagen, Madison, WI). The recombinant kinase was phosphorylated with active MAP kinase kinase 6 (MKK6) (Upstate Biotechnology, Lake Placid, NY). The activated Ap-p38 kinase was cut from GST by PreScission Protease (Amersham Biosciences), which together with free GST was removed by GST*Bind Resin.
Kinase assay. Human ATF2 (2 μg) (Cell Signaling Technology) was incubated with phosphorylated Ap-p38 kinase in kinase buffer (Cell Signaling Technology) supplemented with 0.2 mm ATP at 30°C for 30 min. Proteins from the reaction mixture were separated by SDS-PAGE, and the phosphorylated transcription factor was quantified by immunoblotting using anti-phospho-ATF2 (Cell Signaling Technology).
Assay for arachidonate. Pleural ganglia (seven) were dissected out and placed in ice-cold ethanol (20 μl) for 20 min. Octadeutero-arachidonate (1 ng; Biomol Research Labs, Plymouth Meeting, PA) was added, the ethanol extracts were adjusted to pH 3.0 with 3% formic acid, and the lipids were fractionated on a C18 solid-phase extraction column (Waters, Milford, MA) that had been washed with water, ethanol, and water again (5 ml each) before the samples were applied. The columns were then washed with 15% ethanol, water, and petroleum ether (5 ml), and the lipids were eluted with freshly redistilled ethyl acetate (4 × 1 ml). Eluates were dried under reduced pressure and then dissolved in 350 μl of acetonitrile/water/acetic acid (50:50:0.1; v/v) mobile phase for purification by RP-HPLC. The mobile phase was used to elute a NOVA-Pak C18 column (Waters) at 0.7 ml/min. The eluted lipids were esterified by reaction with acetonitrile/diisopropylethyl amine/pentafluorobenzyl-bromide (50:10:5) at room temperature for 10 min. Arachidonate was measured by gas chromatography-mass spectrometry of the pentafluorobenzyl ester and quantified by comparison with a contemporaneous standard curve. The samples were then dried, resuspended in decane, and analyzed on a Hewlett Packard 5988 GC/MS in the negative ion-chemical ionization mode.
Chromatin immunoprecipitory assay. Assays were performed as described by Guan et al. (2002). Nervous tissue was dissected out and fixed (4% paraformaldehyde, 30% sucrose, 0.1% Triton X-100, and PBS) at 4°C for 3 hr. Glycine was then added to a concentration of 0.125 m, and the nervous tissue was washed three times with PBS. Ganglia were desheathed in PBS and homogenized in lysis buffer (0.25% Triton X-100, 0.5% NP-40, 10 mm EDTA, 0.5 mm EGTA, 10 mm Tris-HCl, pH 8.0, 1 mm PMSF). Nuclei were collected and resuspended in 1 mm EDTA, 0.5 mm EGTA, 10 mm Tris-HCl, pH 7.5, 1 mm PMSF. Samples were sonicated, and chromatin was purified by cesium chloride gradient centrifugation and dialyzed against 10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 0.5 mm EGTA, and 5% glycerol. The average size of the DNA fragments was ∼600 base pairs long. The same amount of chromatin was used to perform immunoprecipitations with the specific antibodies [chromatin immunoprecipitation (ChIP) assay]. The presence of the C/EBP promoter was analyzed by quantitative PCR with the promoter-specific primer pair AACGCGTATGAATATGTGGAGTGG and TCTATCTTGGCGGTTTGCGTTAC. α-P 32 cytidine triphosphate was added for body labeling of PCR product. The Aplysia histone H4 promoter was analyzed with primer pairs GCTTCGCCTCGCTCTGTCTCC and GCGAATGGCGGGCTTGGTAATG. The anti-HDAC5 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA), and the anti-acetylated histone antibodies were from Upstate Biotechnology.
Results
Characterization of Ap-p38 kinase
Using RT-PCR and the rapid amplification of cDNA ends methods we cloned p38 kinase from Aplysia CNS cDNA (GenBank accession number AF508042). The inferred amino acid sequence is 80% identical to that of the human enzyme and contains a typical TGY MAP kinase dual phosphorylation site. Four isoforms of the kinase are known:α,β,γ, andδ. Onlyα andβ are expressed in vertebrate brain (Lee et al., 2000), and only these two isoforms are inhibited by the specific p38 kinase inhibitor SB 203580 (Nebreda and Porras, 2000). Phylogenetic analysis indicates that Ap-p38 belongs to the α-β p38 kinase subfamily (Fig. 1A).
Figure 1.

Characterization of Aplysia p38 MAP kinase. A, A phylogenetic comparison using clustal analysis with the DNA-Star program shows that the Aplysia enzyme belongs to the α-β subfamily of p38 kinases. B, The activated mammalian p38 kinase upstream kinase, MKK6, can phosphorylate recombinant Ap-p38 kinase in vitro. The phosphorylated enzyme was detected with a commercial anti-human phospho-p38 (Pp38) antibody, and the total amount of kinase was detected with an anti-Ap-p38 antibody (Fig. 2). C, When phosphorylated by MKK6, the recombinant Ap-p38 kinase can phosphorylate human ATF2 in vitro, which was detected with an anti-phospho ATF2 antibody, and the phosphorylation of ATF2 was blocked by the p38 kinase inhibitor SB 203580 (0.5 μm). Inactivated Ap-p38 kinase did not phosphorylate ATF2. D, Inhibition by SB 203580 was dose dependent. The same amount of ATF2 was incubated with activated Ap-p38 kinase and varying concentrations of the inhibitor. The phosphorylation of ATF2 was detected by anti-phospho-ATF2 antibody, and the activity of p38 kinase was assessed densitometrically by the phosphorylation of ATF2. With increasing concentrations of SB 203580, kinase activity was gradually inhibited with an IC50 of ∼50 nm.
We used the mammalian p38-upstream kinase MKK6 to phosphorylate recombinant Ap-p38 kinase, and the phosphorylated enzyme can be detected by a commercial antibody against the mammalian phospho-p38 kinase (Fig. 1B). Ap-p38 kinase phosphorylated by MKK6 can then phosphorylate the human transcription factor ATF2 (Fig. 1C), an established substrate of the vertebrate enzyme (Ono and Han, 2000), and this kinase activity was blocked by SB 203580 (Fig. 1C). To show that the in vivo kinase behaves like the cloned enzyme, we purified activated Ap-p38 kinase from Aplysia nervous tissue extracts by immunoprecipitation with the anti-phospho-p38 kinase antibody. ATF2 could also be phosphorylated by the in vivo kinase, and that kinase activity was blocked by SB 203580 (data not shown). The IC50 of the inhibitor for the Aplysia enzyme is in the 50 nm range (Fig. 1D), similar to that reported for mammalian homologs.
We next affinity purified an antibody raised against a peptide of Ap-p38 kinase that detected a single component of ∼Mr 38,000 both in extracts of Aplysia pleural ganglia (Fig. 2A) and in preparations of the recombinant protein (Fig. 1B). This antibody also blocked kinase activity (Fig. 2B).
Figure 2.

Antibody against Aplysia p38 kinase. A, An affinity-purified antibody raised against a peptide (PEAEKYDQSFEEMELG) from the Aplysia kinase specifically recognized a single component of approximately Mr 38 kDa, the size expected of p38 kinase, from homogenates of Aplysia pleural ganglia. Preincubation of the antibody with the peptide used as immunogen blocked the signal. B, Recombinant Ap-p38 kinase activated by mammalian MKK6 was incubated with human ATF2 and ATP, with or without Ap-p38 antibody. Kinase activity, monitored by the phosphorylation of ATF2 by Western blot with anti-phospho-ATF2 antibody, was completely blocked by the Ap-p38 antibody.
Ap-p38 kinase is regulated bidirectionally by FMRFa and 5-HT
To determine the role of p38 kinase in synaptic plasticity, Aplysia were exposed to either FMRFa (9 μm) or 5-HT (50 μm) for 10 min. Pleural ganglia were then homogenized, and the extracted proteins were separated by SDS-PAGE and immunoblotted with the anti-phospho-p38 antibody (to detect activated enzyme) and with the anti-Ap-p38 kinase antibody (to detect total enzyme). Exposure to FMRFa increased the phosphorylation of the kinase, whereas exposure to 5-HT decreased its phosphorylation (Fig. 3). Thus the activity of the enzyme is under bidirectional regulation by the two neurotransmitters that produce depression and facilitation.
Figure 3.

p38 kinase is bidirectionally regulated by 5-HT and FMRFa. Treatment with FMRFa (9 μm, 10 min) increased the phosphorylation of p38 kinase in pleural ganglion neurons, and treatment with 5-HT (50 μm, 10 min) decreased the phosphorylation. As control, the total amount of kinase was detected by immunoblotting with anti-Ap-p38 kinase. A, Representative immunoblots from four independent experiments are shown. B, Group data showing the regulation of phosphorylation by the two transmitters. Treatment with FMRFa stimulated the phosphorylation of p38 kinase, and 5-HT diminished the phosphorylation. The ratio of phospho-p38 kinase to the nonphosphorylated form was determined in each group of four experiments. The bars represent the mean values obtained with each transmitter divided by the contral ratio. Asterisks indicate significance p < 0.05.
Ap-p38 kinase mediates short-term depression by releasing arachidonate To study the physiological function of Ap-p38 kinase in synaptic plasticity, we first tested whether the enzyme is involved in short-term depression. EPSPs were recorded from Aplysia sensory-to-motor neuron cocultures, which had been incubated with or without SB 203580 for 30 min before and during treatment with one 5 min pulse of FMRFa. To monitor short-term changes. EPSPs were recorded 5 min later. Incubation of the cells with the inhibitor of p38 kinase blocked the short-term depression produced by FMRFa (Fig. 4A).
Figure 4.
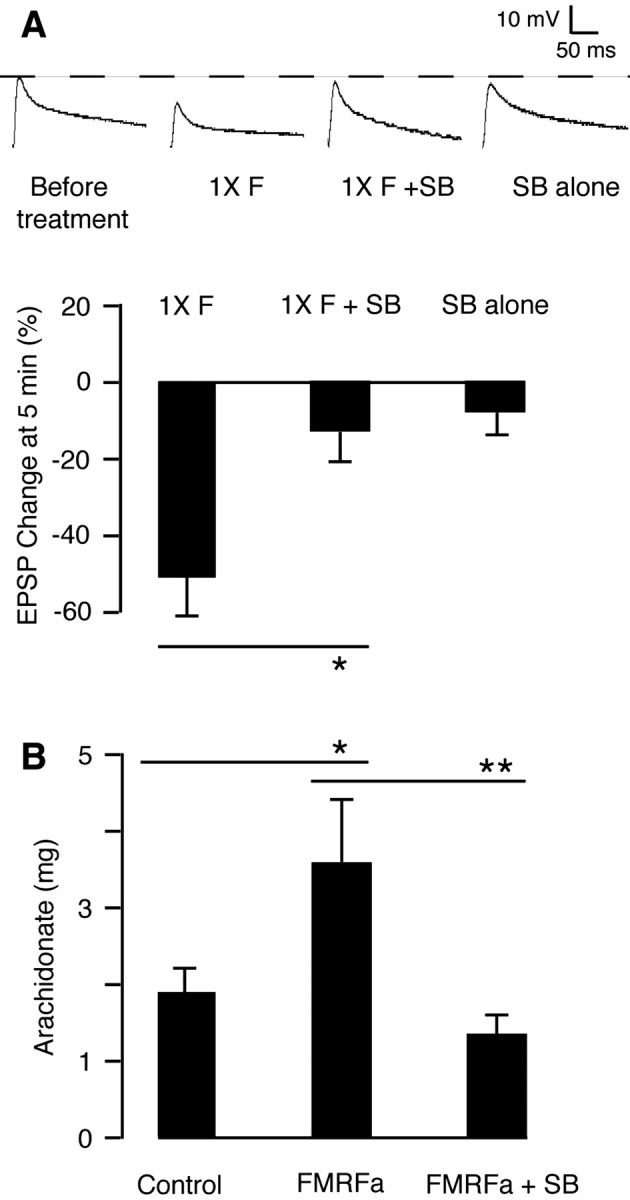
Ap-p38 kinase mediates short-term depression by releasing arachidonate. A, Ap-p38 kinase mediates short-term depression. The cells were treated with one 5 min pulse of FMRFa (1×F), which depressed the EPSP recorded at 5 min (-50.9 ± 10.6%; n = 8). Preincubation with the p38 kinase inhibitor SB 203580 (3 μm, 30 min) blocked the FMRFa-induced depression (1×F + SB; -12.7 ± 7.5%; n = 15; p < 0.05). The inhibitor alone (SB alone) had no short-term effect on the EPSP (-7.3 ± 5.6%; n = 7). Data were analyzed by ANOVA. Histograms show the percentage changes in the mean (±SE) of the EPSP amplitudes 5 min after the treatment. B, Ap-p38 kinase mediates the release of arachidonate. The FMRFa-induced release of arachidonate in pleural ganglia (Control, 1944.6 ± 305.7 pg, n = 7; FMRFa, 3658 ± 696.4 pg, n = 7) was blocked by 1 μm SB 203580 (FMRFa + SB, 1421 ± 249.3 pg, n = 6, p < 0.01). Data were analyzed by ANOVA.
Short-term depression resulting from the application of FMRFa is mediated by 12-lipoxygenase metabolites of arachidonate (Piomelli et al., 1987; Belardetti et al., 1989). We now find that the release of arachidonate in response to FMRFa was blocked by SB 203580 (Fig. 4B), indicating that the kinase activates a phospholipase A2 (PLA2).
Inhibition of Ap-p38 kinase blocks long-term depression and enhances long-term facilitation
We next studied the role of Ap-p38 kinase in long-term depression. EPSPs were recorded from Aplysia sensory-to-motor neuron cocultures with cells incubated in the presence of SB 203580 for 30 min before and during treatment with FMRFa. EPSPs were recorded 24 hr later to monitor long-term changes. As reported previously, five 5 min pulses of FMRFa depressed the EPSP at 24 hr (Montarolo et al., 1988). When Ap-p38 kinase was inhibited by SB 203580, depression failed to develop (Fig. 5A), indicating that the kinase mediates long-term depression.
Figure 5.
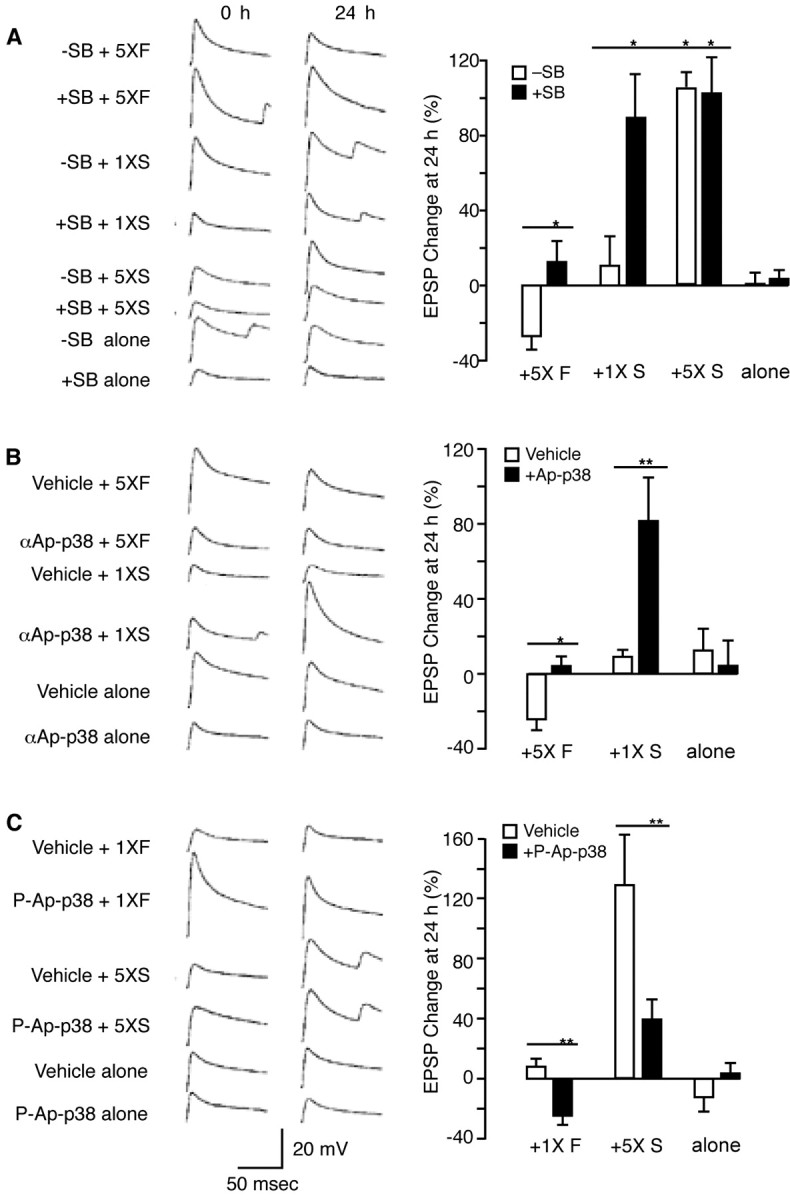
Bidirectional function of p38 kinase. A, SB 203580 blocked long-term depression and enhanced long-term facilitation: representative examples of EPSP traces before and 24 hr after treatment, and summary data. To induce long-term depression, the sensory-to-motor neuron cocultures were treated with five 5 min pulses of 1 μm FMRFa (5× F), which resulted in depressed EPSPs at 24 hr (-26.8 ± 6.6%; n = 12). This long-term depression was blocked by incubation with SB 203580 (3 μm) 30 min before and throughout the treatment with FMRFa (5×F+ SB; 12.6 ± 10.8%; n = 10; p < 0.05). For long-term facilitation, the cells were treated with five 5 min pulses or a single 5 min pulse of 5-HT (10 μm). A single pulse of 5-HT (1× S) had no long-term effect (10.6 ± 15.3%; n = 7), but long-term facilitation was established in cells preincubated with the inhibitor (3 μm, 30 min) and then treated with one pulse of 5-HT (1× S + SB; 88.9 ± 22.5; n = 10; p < 0.01). The long-term facilitation produced in this way was comparable with that resulting from five pulses of 5-HT (5× S; 103.5 ± 7.5%; n = 8). Five pulses of 5-HT in cells incubated in 3 μm SB 203580 (5× S + SB) did not enhance the long-term facilitation further (102.2 ± 18.7%; n = 7). Cells treated with the inhibitor alone (SB alone; 3.1 ± 4.7%; n = 11) had no long-term effect. B, Injection of Ap-p38 antibody blocked long-term depression and enhanced long-term facilitation: EPSP traces and summary data. The inactivating antibody against Ap-p38 kinase (Fig. 2) was injected into pleural sensory neurons before treatment with FMRFa or 5-HT. Five 5 min pulses of FMRFa coupled with injection of the vehicle (0.4 m potassium acetate and 10 mm Tris-HCl, pH 7.4) (5× F + vehicle) induced long-term depression at 24 hr (-24.2 ± 5.4%; n = 13); the long-term depression was blocked by injecting the antibody (5× F +αAp-p38; 3.3 ± 10.9%; n = 9; p < 0.05). A single pulse of 5-HT coupled with injection of the vehicle (1× S + vehicle) had no long-term effect (8.9 ± 3.7%; n = 14) but produced long-term facilitation after the Ap-p38 antibody was injected (1× S +αAp-p38; 81.2 ± 22.2%; n = 7; p < 0.01). No effect on the EPSP at 24 hr was observed with the injection of vehicle alone (12.5 ± 11.2%; n = 4) or antibody alone (4.1 ± 13.3%; n = 7). C, Injection of activated Ap-p38 kinase enhanced long-term depression and blocked long-term facilitation: EPSP traces and summary data. A single pulse of FMRFa coupled with the injection of vehicle (1× F + vehicle) had no long-term effect (7.6 ± 5.2%; n = 8). Injection of phospho-Ap-p38 kinase into sensory neurons coupled with a single pulse of FMRFa (1×F + p-Ap-p38) induced long-term depression (-24.6 ± 5.91%; n = 11; p < 0.01). Five pulses of 5-HT coupled with injection of vehicle (5× S + vehicle) induced long-term facilitation (127.5 ± 29.7%; n = 13); long-term facilitation was blocked by injecting phospho-Ap-p38 kinase (5× S + p-Ap-p38; 38.3 ± 12.5; n = 13%; p < 0.01). No effect at 24 hr was observed with injection of vehicle alone (-12.3 ± 9.2%; n = 6) or injection of phospho-Ap-p38 kinase alone (3.3 ± 6.8%; n = 12). Data were analyzed by ANOVA. Histograms show percentage changes in the mean (±SE) of EPSP amplitudes 24 hr after the treatment.
Inhibition of p38 kinase also enhanced the development of long-term facilitation. When cells received only a single 5 min pulse of 5-HT, no effect was seen at 24 hr, but inhibition of Ap-p38 by SB 203580 together with just a single pulse of 5-HT resulted in a robust increase in the EPSP at 24 hr, comparable with the long-term facilitation produced by five pulses of 5-HT. Prolonged treatment together with the inhibitor did not augment long-term facilitation further, however, indicating that when p38 kinase is inhibited, a single pulse of 5-HT results in maximal facilitation. The inhibitor alone had no long-term effect (Fig. 5A).
To inhibit the kinase in a more specific way, our affinity-purified Ap-p38 antibody, which we showed inhibited Ap-p38 kinase activity (Fig. 2B), was pressure injected into sensory neurons. As with SB 203580, the antibody blocked the development of long-term depression normally induced by five pulses of FMRFa and enhanced long-term facilitation by producing the 24 hr response after only a single pulse of 5-HT. Injection of antibody alone in the absence of FMRFa had no long-term effect (Fig. 5B).
Activation of Ap-p38 kinase facilitates long-term depression and blocks long-term facilitation
Inhibiting Ap-p38 kinase blocked long-term depression and facilitated long-term facilitation. Does activating the kinase have a reciprocal effect? Although one pulse of FMRFa itself had no long-term effect, one pulse of FMRFa together with pressure-injected, preactivated Ap-p38 kinase did induce long-term depression. Activated Ap-p38 in the absence of FMRFa had no long-term effect, however. Conversely, injection of the phosphorylated kinase blocked the long-term facilitation that is induced by five 5 min pulses of 5-HT (Fig. 5C). Injection of denatured Ap-p38 kinase had no effect on long-term depression or long-term facilitation (data not shown). The system thus acts symmetrically: activating the kinase favors long-term depression and inhibits long-term facilitation; its inhibition blocks long-term depression and favors long-term facilitation.
Ap-p38 kinase regulates long-term synaptic plasticity by phosphorylating CREB2
We reported previously that FMRFa acts through CREB2 to block the formation of long-term facilitation and that inhibition of CREB2 enhances long-term facilitation and blocks long-term depression (Bartsch et al., 1995; Guan et al., 2002). We now show that Ap-p38 kinase phosphorylates CREB2 (Fig. 6A). To determine whether this phosphorylation activates the transcription factor, we tested whether the kinase mediates its binding to the promoter. ChIP assays showed that inhibiting p38 kinase with SB 203580 prevented the FMRFa-induced recruitment of CREB2 to the C/EBP promoter (Fig. 6B). Inhibition of the kinase also blocked the recruitment of HDAC5 as well as the FMRFa-induced deacetylation of histones (Fig. 6B,C). SB 203580 also blocked the inhibitory effect of FMRFa on the 5-HT-induced acetylation of histones (Fig. 6C). Thus activation of p38 kinase inhibits long-term facilitation by phosphorylating and activating CREB2 and by inducing histone deacetylation at the C/EBP promoter.
Figure 6.
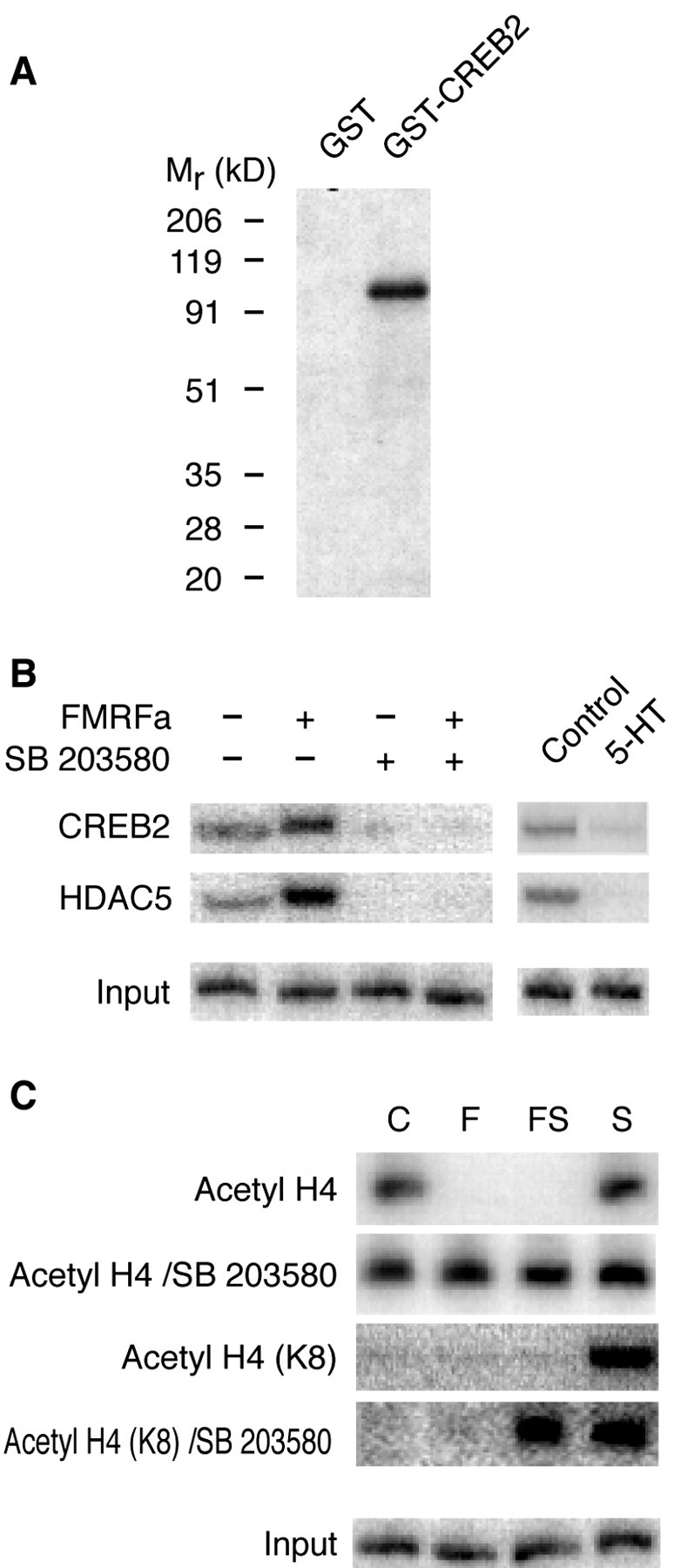
Ap-p38 kinase activates CREB2 and induces histone deacetylation. A, Ap-p38 kinase phosphorylates CREB2. GST or the GST-CREB2 fusion protein was incubated with activated Ap-p38 and γ-labeled 32P-ATP at 30°C for 30 min. The products were analyzed by running a gel and then exposing to an x-ray film. The kinase phosphorylated the GST-CREB2 fusion protein but not GST. B, Ap-p38 kinase activity is critical for recruiting CREB2 and HDAC5 to the C/EBP promoter. With ChIP assays, a basal level of CREB2 and HDAC5 was detected at the promoter in unstimulated neurons. Treatment with FMRFa (10 μm) for 90 min induced more CREB2 and HDAC5 to bind. SB 203580 (1 μm) lowered the basal amount of both CREB2 and HDAC5, an effect similar to that of a 90 min treatment with 5-HT. The inhibitor also blocked the FMRFa-induced recruitment of CREB2 and HDAC5. As control, chromatin samples were also analyzed before immunoprecipitation (Input) to show that equal amounts of starting material were applied. C, Ap-p38 kinase activity is necessary for FMRFa to induce histone deacetylation. Basal histone H4 acetylation (Acetyl H4) was detected at the C/EBP promoter. Although treatment with 5-HT (S) increased H4 acetylation, FMRFa, either alone (F) or together with 5-HT (FS), decreased the acetylation. SB 203580 blocked the FMRFa-induced histone deacetylation at the promoter (Acetyl H4/SB 203580). In addition, 5-HT (S) induced the specific acetylation of lysine 8 of H4 [Acetyl H4 (K8)], and FMRFa cotreatment (FS) blocked this acetylation. SB 203580 blocked this inhibitory effect of FMRFa [Acetyl H4 (K8)/SB 203580]. As control, chromatin samples were also analyzed before immunoprecipitation (Input). All of the samples were also analyzed with the primers specific to the promoter of Aplysia histone H4, a gene with a strong basal expression but no response to either 5-HT or FMRF (Guan et al., 2002). No changes were observed after any of the treatments (data not shown).
A basal level of p38 kinase activity is present in unstimulated sensory neurons (Fig. 3). Corresponding to that activity, there is a basal amount of CREB2 and HDAC5 at the C/EBP promoter at rest (Guan et al., 2002) (Fig. 6B). Inhibition with SB 203580 decreased this basal amount of CREB2 and HDAC5 (Fig. 6B)toa level similar to that produced by 5-HT, suggesting that inhibiting p38 kinase results in the dephosphorylation of CREB2. This, in turn, is responsible for removing CREB2 and HDAC5 to produce long-term facilitation.
Inhibition of ATF2 blocked long-term depression and facilitated long-term facilitation
ATF2, a transcription factor shown to be downstream of p38 kinase (Ono and Han, 2000), appears to have a function similar to that of CREB2 in long-term plasticity. As with CREB2, inhibition of ATF2 (by injecting a commercial ATF2 antibody that binds to the phosphorylation site of the transcription factor) blocked long-term depression (Fig. 7). Interestingly, inhibition of ATF2 also enhances the formation of long-term facilitation: injection of the ATF2 antibody coupled with a single pulse of 5-HT was sufficient to induce long-term facilitation (Fig. 7). Unlike CREB2, however, as revealed by the ChIP assays, ATF2 does not bind to the C/EBP promoter after treatment with FMRFa (data not shown).
Figure 7.

ATF2 antibody blocked long-term depression and enhanced long-term facilitation. A, By Western blotting, the anti-ATF2 antibody reacted with a single component in extracts of Aplysia nervous tissue. B, Role of ATF2 on synaptic plasticity: representative examples of EPSP traces and summary data. Five 5 min pulses of FMRFa coupled with vehicle (0.4 m potassium acetate and 10 mm Tris-HCl, pH 7.4) injection (5× F + Vehicle) induced long-term depression (-24.24 ± 5.43%; n = 13), which was blocked by injecting the ATF2 antibody (5× F+α ATF2; 19.1 ± 11.13%; n = 8; p < 0.01). One pulse of 5-HT coupled with vehicle injection (1× S + Vehicle) had no long-term effect (8.93 ± 3.71%; n = 14). Antibody injection (1× S +αATF2) produced long-term facilitation after one pulse of 5-HT (53.46 ± 10.24%; n = 13; p < 0.05). Vehicle alone (12.5 ± 11.24%; n = 4) or antibody alone (3.75 ± 11.13%; n = 8) had no long-term effect. Data were analyzed by ANOVA. Histograms show percentage changes in the mean (±SE) of EPSP amplitudes 24 hr after the treatment.
Discussion
p38 kinase regulates long-term synaptic plasticity bidirectionally
The sensory-to-motor neuron synapses of Aplysia show two opposing types of synaptic plasticity: facilitation and depression, both having a short-term and a long-term form. The core signaling pathway for long-term facilitation has been well described: activation of PKA by 5-HT leading to the phosphorylation of CREB1, the recruitment of CBP, and the acetylation of histones around the promoter of the early response gene C/EBP. Synthesis of the transcription activator C/EBP then would result in the induction of effector genes encoding proteins required for long-term facilitation (Kandel, 2001). Less is known about the pathway for depression that is induced by FMRFa. It is clear, however, that the molecular mechanism underlying depression is not the simple reversal of the reactions that produce facilitation. We find that FMRFa activates p38 MAP kinase and that 5-HT inhibits the kinase. In contrast, ERK (p42) MAP kinase is activated by both 5-HT and FMRFa (Michael et al., 1998; Yamamoto et al., 1999) (and data not shown). In addition to being regulated bidirectionally, p38 kinase can itself mediate bidirectional activities: inhibition of the kinase blocks long-term depression and converts short-term facilitation into long-term facilitation. Injecting pre-activated p38 kinase into sensory neurons blocked long-term facilitation and converted short-term depression into long-term depression. These results suggest that p38 kinase acts both as a mediator of depression and an inhibitory constraint on facilitation and that the second-messenger pathways from FMRFa and 5-HT both converge at p38 kinase to regulate synaptic plasticity bidirectionally (Fig. 8).
Figure 8.

p38 MAP kinase regulates synaptic plasticity bidirectionally. A, In the cytoplasm, the inhibitory neuropeptide FMRFa, through an as yet unidentified mechanism, activates p38 kinase. The facilitatory neurotransmitter 5-HT, on the other hand, inhibits the kinase, presumably through activation of PKA. B, Activated p38 kinase moves into the nucleus, where it phosphorylates transcription factors CREB2 and ATF2. Phosphorylated CREB2 binds to the C/EBP promoter and induces histone deacetylation to repress the expression of C/EBP. Thus the long-term facilitation induced by 5-HT is blocked. Phosphorylated CREB2 and ATF2, which are also transcription activators, can bind to promoters of yet unidentified genes to induce the expression of proteins important to long-term depression, presumably through induction of histone acetylation. As a result, long-term depression develops.
What are the molecular mechanisms underlying the bidirectional regulation of synaptic plasticity? In the hippocampus, both long-term depression and long-term potentiation (LTP) depend on the influx of Ca2+ (for review, see Bear and Malenka, 1994), and protein phosphorylation by a persistently active Ca2+/calmodulin-dependent protein kinase II is thought to be the core molecular mechanism underlying the induction phase of potentiation (Schulman and Hyman, 1999). The apparent paradox raised by this bidirectional modulation was explained by Lisman (1989), who suggested that the depression is caused by activating protein phosphatase-1, an enzyme also regulated by Ca2+ and calmodulin. In Lisman's model, both increases and decreases in synaptic transmission are produced by an influx of Ca2+, the direction depending on the extent and duration of the changes in the intracellular Ca2+ concentration and the affinities of the appropriate phosphatases and kinases (Mulkey et al., 1993; Hanson et al., 1994; for review, see Lisman 1994).
The molecular mechanisms underlying synaptic plasticity in Aplysia sensory neurons differ from those in the hippocampus. In the cytoplasm of a resting hippocampal pyramidal cell the concentration of Ca2+ is low, and both LTP and long-term depression depend on Ca2+ influx: a moderate increase will produce long-term depression and a large increase will produce LTP. In contrast, in Aplysia sensory neurons, there is a substantial basal activity of p38 kinase, and the direction of long-term synaptic plasticity depends on the inhibition or the further activation of the kinase: inhibition by 5-HT permits the formation of long-term facilitation, and activation by FMRFa mediates long-term depression. A novel aspect of this mechanism is that the direction of synaptic action appears to be determined by the state of phosphorylation and activation of p38 kinase: because at rest there is a substantial amount of activated p38 kinase, this enzyme seems to be pivotal.
Ap-p38 kinase activates PLA2 to mediate short-term depression
12-Lipoxygenase metabolites of arachidonate are the second messengers for short-term depression (Piomelli et al., 1987; Belardetti et al., 1989). We now provide evidence that p38 kinase mediates the FMRFa-induced release of arachidonate, presumably by activating a PLA2. Inhibition of p38 kinase activity blocked both short-term depression and the release of arachidonate (Fig. 4), indicating that FMRFa produces depression by activating the kinase. Activation of cytoplasmic PLA2 by p38-kinase phosphorylation has been described for several cell types (Leslie, 1997; Börsch-Haubold et al., 1997; Gudmundsdóttir et al., 2001). Although it seems likely that p38 kinase mediates short-term depression by activating a PLA2, we cannot rule out the possibility that phosphorylation of the lipoxygenase (Werz et al., 2000, 2001; Eon et al., 2001; Reddy et al., 2002) or the simultaneous phosphorylation of both a cPLA2 and a lipoxygenase (Werz et al., 2002) are necessary for producing the active arachidonate metabolites.
Because the short-term action of FMRFa is blocked by pertussis toxin (Shapiro et al., 1988; Volterra and Siegelbaum 1988), the activation of p38 kinase by FMRFa is likely to be mediated by a G-protein-coupled receptor. Although this route of activating the kinase has been well documented, its mechanism is not yet understood (Marinissen and Gutkind, 2001). More commonly, the kinase is activated by a member of the small GTPase family, as reported recently in hippocampal neurons (Zhu et al., 2002).
CREB2 and ATF2 are elements downstream of p38 kinase
CREB2 has been shown to be critical for both long-term facilitation and long-term depression: its inhibition blocks long-term depression and enhances long-term facilitation (Bartsch et al., 1995; Guan et al., 2002). We now find that ATF2 is also critical for both long-term facilitation and long-term depression: inhibition of ATF2 blocks long-term depression and enhances long-term facilitation, indicating that the activated ATF2 is required for long-term depression and that it also acts as an inhibitory constraint for long-term facilitation. Furthermore, we find that p38 kinase phosphorylates and activates both CREB2 and ATF2 (Figs. 1, 6). Because CREB2 and ATF2 must both be activated to produce long-term depression and repress long-term facilitation, they both may be required simultaneously to induce the expression of as yet unidentified genes, the expression of which is crucial for long-term depression, and to repress genes important for long-term facilitation; alternatively, the two transcription factors may induce or repress different genes in parallel or in sequence (Fig. 8).
p38 kinase blocks long-term facilitation by activating CREB2 and inducing histone deacetylation
We showed previously that FMRFa blocks long-term facilitation by inducing the displacement of CREB1 by CREB2 from the C/EBP promoter and the recruitment of HDAC5 to induce histone deacetylation to repress C/EBP (Guan et al., 2002). As a result, long-term facilitation is blocked (Fig. 5C). We now find that the ability of CREB2 to bind to the promoter depends on p38 kinase activity: inhibition of the kinase prevented CREB2 from binding (Fig. 6). The kinase is also essential for the histone deacetylation induced by FMRFa: inhibition of the enzyme blocked the FMRFa-induced recruitment of HDAC5 and the histone deacetylation at the promoter (Fig. 6). ATF2 phosphorylated by p38 kinase also inhibited long-term facilitation. Presumably, activated ATF2 blocks by repressing genes important for long-term facilitation, but because it does not bind to the C/EBP promoter, it must repress other genes important for long-term facilitation.
The formation of long-term facilitation requires the removal of basal p38 kinase activity. Reflecting the basal amount of kinase activity in unstimulated neurons are basal amounts of CREB2 bound to the promoter. This pivotal basal activity would provide an inhibitory threshold regulating the gene induction needed to form long-term facilitation. Inhibition of basal p38 kinase activity removes the basal recruitment of both CREB2 and HDAC5. To induce long-term facilitation, an facilitatory input must be strong enough not only to activate the PKA-CREB1 core pathway to induce histone acetylation, but also to reduce deacetylation by blocking the p38 kinase pathway (Fig. 3). As a result, only strong facilitatory inputs (for example, five pulses of 5-HT) can induce long-term facilitation, but when the inhibitory threshold is already lowered by the inhibition of p38 kinase, a weak facilitatory input (one pulse of 5-HT that normally results only in a short-term effect) is able to produce the long-term effect (Fig. 5).
p38 kinase is not a mirror image of PKA
Both p38 kinase and PKA mediate short-term and long-term forms of synaptic plasticity. The two second-messenger pathways are not mirror images, however. Unlike the facilitatory pathway in which activated PKA alone is sufficient to produce long-term facilitation (Schacher et al., 1988; Chain et al., 1999), neither activation nor inhibition of p38 kinase by itself has any effect on long-term synaptic plasticity (Figs. 4A,B, 5C). Inhibition of p38 kinase alone is not sufficient to produce long-term facilitation, because although the repression is relieved, PKA is not activated, and consequently CREB1 cannot be phosphorylated and the gene expression required for long-term facilitation will not be induced. Because activation of p38 kinase alone is insufficient to produce long-term depression, some as yet unknown additional steps may be required for long-term depression.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants MH48850 and NS29832 to J.H.S., and by Howard Hughes Medical Institute to E.R.K. Aplysia were provided by the National Resource for Aplysia Facility at the University of Miami under NIH Grant RR10294. We thank Todd Sacktor, Steven Siegelbaum, and Wayne Sossin for reading this manuscript critically and for helpful discussions. We also are grateful for the scientific guidance of Steven J. Feinmark and Dimitris Thanos.
Z.G. and J.-H.K. contributed equally to this work.
Correspondence should be addressed to James H. Schwartz, Center for Neurobiology and Behavior, Columbia University, 1051 Riverside Drive, New York, NY 10032. E-mail: jhs6@columbia.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237317-09$15.00/0
References
- Alberini CM, Ghirardi M, Metz R, Kandel ER ( 1994) C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell 76: 1099-1114. [DOI] [PubMed] [Google Scholar]
- Armstrong JN, Brust TB, Lewis RG, MacVicar BA ( 2002) Activation of presynaptic p2X7-like receptors depresses mossy fiber-CA3 synaptic transmission through p38 mitogen-activated protein kinase. J Neurosci 22: 5938-5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Kaang BK, Chen M, Martin KC, Lim CS, Casadio A, Kandel ER ( 1997) Mutation in the phosphorylation sites of MAP kinase blocks learning-related internalization of apCAM in Aplysia sensory neurons. Neuron 6: 913-924. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER ( 1995) Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell 83: 979-992. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC ( 1994) Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol 4: 389-399. [DOI] [PubMed] [Google Scholar]
- Belardetti F, Campbell WB, Falck JR, Demontis G, Rosolowsky M ( 1989) Products of heme-catalized transformation of the arachidonate derivative 12-HPETE open S-Type K+ channels in Aplysia. Neuron 3: 497-505. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F ( 2000) Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci 3: 1107-1112. [DOI] [PubMed] [Google Scholar]
- Börsch-Haubold AG, Kramer RM, Watson SP ( 1997) Phosphorylation and activation of cytosolic phospholipase A2 by 38-kDa mitogen-activated protein kinase in collagen-stimulated human platelets. Eur J Biochem 245: 751-759. [DOI] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH ( 1999) Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron 22: 147-156. [DOI] [PubMed] [Google Scholar]
- Cowan KJ, Storey KB ( 2003) Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol 206: 1107-1115. [DOI] [PubMed] [Google Scholar]
- Dudak SM, Bear MF ( 1993) Bidirectional long-term modification of synaptic effectiveness in adult and immature hippocampus. Nature 381: 71-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eon Y-W, Cho S-H, Hwang J-S, Yoon S-B, Na DS, Kang I-J, Kang SS, Song WK, Kim J-H ( 2001) Rac and p38 kinase mediate 5-lipoxygenase translocation and cell death. Biochem Biophys Res Commun 284: 126-132. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim J-H, Miniaci MC, Schwartz JH, Thanos D, Kandel ER ( 2002) Integration of long-term memory related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111: 483-493. [DOI] [PubMed] [Google Scholar]
- Gudmundsdóttir I, Halldórsson H, Magnúsdóttir K, Thorgeirsson G ( 2001) Involvement of MAP kinases in the control of cPLA2 and arachidonic acid release in endothelial cells. Atherosclerosis 156: 81-90. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Meyer T, Stryer L, Schulman H ( 1994) Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12: 943-956. [DOI] [PubMed] [Google Scholar]
- Harper SJ, LoGrasso P ( 2001) Signaling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cell Signal 13: 299-310. [DOI] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH ( 1997) Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89: 115-126. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R ( 2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298: 1911-1912. [DOI] [PubMed] [Google Scholar]
- Kandel ER ( 2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030-1038. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH ( 1982) Molecular biology of learning: modulation of transmitter release. Science 218: 433-443. [DOI] [PubMed] [Google Scholar]
- Lee SH, Park J, Che Y, Han PL, Lee JK ( 2000) Constitutive activity and differential localization of p38 alpha and p38 beta MAPKs in adult mouse brain. J Neurosci Res 60: 623-631. [DOI] [PubMed] [Google Scholar]
- Leslie CC ( 1997) Properties and regulation of cytosolic phospholipase A2 J Biol Chem 272: 16709-16712. [DOI] [PubMed] [Google Scholar]
- Lisman JE ( 1989) A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA 86: 9574-9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE ( 1994) The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci 17: 406-412. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS ( 2001) G-protein coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22: 368-376. [DOI] [PubMed] [Google Scholar]
- Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER ( 1997) MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron 18: 899-912. [DOI] [PubMed] [Google Scholar]
- Michael D, Martin KC, Seger R, Ning MM, Baston R, Kandel ER ( 1998) Repeated pulses of serotonin required for long-term facilitation activate mitogen-activated protein kinase in sensory neurons of Aplysia. Proc Natl Acad Sci USA 95: 1864-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarolo PG, Kandel ER, Schacher S ( 1988) Long-term heterosynaptic inhibition in Aplysia. Nature 333: 171-174. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CH, Malenka RC ( 1993) An essential role for protein phosphatases in the induction of long-term depression in hippocampus. Science 261: 1051-1055. [DOI] [PubMed] [Google Scholar]
- Nebreda AR, Porras A ( 2000) p38 MAP kinases: beyond the stress response. Trends Biochem Sci 25: 257-260. [DOI] [PubMed] [Google Scholar]
- Ono K, Han J ( 2000) The p38 signal transduction pathway: activation and function. Cell Signal 12: 1-13. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Volterra A, Dale N, Sieglebaum SA, Kandel ER, Schwartz JH, Belardetti F ( 1987) Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cell. Nature 328: 38-43. [DOI] [PubMed] [Google Scholar]
- Reddy MA, Thimmalapura P-R, Lantig L, Nadler JL, Fatima S, Natarajan R ( 2002) The oxidized lipid and lipoxygenase product 12(S)hydroxyeicosatetraenoic acid induces hypertrophy and fibronectin transcription in vascular smooth muscle cells via p38 MAPK and cAMP response element-binding protein activation. J Biol Chem 277: 9920-9928. [DOI] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R ( 2002) Group I metabotropic glutamate receptor (mGluR)-dependent long-term mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci 22: 6121-6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacher S, Castellucci VF, Kandel ER ( 1988) cAMP evokes long-term facilitation in Aplysia sensory neurons that requires new protein synthesis. Science 240: 1667-1669. [DOI] [PubMed] [Google Scholar]
- Schulman H, Hyman SE ( 1999) Intracellular signaling. In: Fundamental neuroscience (Zigmond MJ, Bloom FE, Landis SC, Squire LR, eds), pp 269-316. New York: Academic.
- Shapiro E, Piomelli D, Feinmark S, Vogel SS, Chin GJ, Schwartz JH ( 1988) The role of arachidonic acid metabolites in signal transduction in an identified neural network mediating presynaptic inhibition in Aplysia. Cold Spring Harbor Symp Quant Biol 53: 425-433. [DOI] [PubMed] [Google Scholar]
- Shuster MJ, Camardo JS, Siegelbaum SA, Kandel ER ( 1985) Cyclic AMP-dependent protein kinase closes the serotonin-sensitive K+ channels of Aplysia sensory neurones in cell-free membrane patches. Nature 313: 392-395. [DOI] [PubMed] [Google Scholar]
- Siegelbaum SA, Camardo JS, Kandel ER ( 1982) Serotonin and cyclic AMP close single K+ channels in Aplysia sensory neurones. Nature 299: 413-417. [DOI] [PubMed] [Google Scholar]
- Volterra A, Siegelbaum SA ( 1988) Role of two different guanine mucleotide-binding proteins in the antagonistic modulation of the S-type K+ channel by cAMP and arachidonic acid metabolites in Aplysia sensory neurons. Proc Natl Acad Sci USA 85: 7810-7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werz O, Klemm J, Rådmark O ( 2000) 5-Lipoxygenase is phosphorylated by p38 kinase-dependent MAPKAP kinases. Proc Natl Acad Sci USA 97: 5261-5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werz O, Klemm J, Rådmark O, Samuelsson B ( 2001) p38 MAP kinase mediates stress-induced leukotriene synthesis in a human B-lymphocyte cell line. J Leukocyte Biol 70: 830-838. [PubMed] [Google Scholar]
- Werz O, Szellas D, Steinhiber D, Rådmark O ( 2002) Arachidonic acid promotes phosphorylation of 5-lipoxygenase at Ser-271 by MAPK-activated protein kinase 2 (MK2). J Biol Chem 277: 14793-14800. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Hegde AN, Chain DG, Schwartz JH ( 1999) Activation and degradation of the transcription factor C/EBP during long-term facilitation in Aplysia. J Neurochem 73: 2415-2423. [DOI] [PubMed] [Google Scholar]
- Zhu J, Qin Y, Zhao M, Van Aelst L, Malinow R ( 2002) Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110: 443-455. [DOI] [PubMed] [Google Scholar]